

Abstract
Pre-mRNA splicing further increases protein diversity acquired through evolution. The underlying driving forces for this phenomenon are unknown, especially in terms of gene expression. A rice alternatively spliced transcript detection microarray (ASDM) and RNA sequencing (RNA-Seq) were applied to differentiate the transcriptome of 4 representative organs of Oryza sativa L. cv. Ilmi: leaves, roots, 1-cm-stage panicles and young seeds at 21 days after pollination. Comparison of data obtained by microarray and RNA-Seq showed a bell-shaped distribution and a co-lineation for highly expressed genes. Transcripts were classified according to the degree of organ enrichment using a coefficient value (CV, the ratio of the standard deviation to the mean values): highly variable (CVI), variable (CVII), and constitutive (CVIII) groups. A higher index of the portion of loci with alternatively spliced transcripts in a group (IAST) value was observed for the constitutive group. Genes of the highly variable group showed the characteristics of the examined organs, and alternatively spliced transcripts tended to exhibit the same organ specificity or less organ preferences, with avoidance of ‘organ distinctness’. In addition, within a locus, a tendency of higher expression was found for transcripts with a longer coding sequence (CDS), and a spliced intron was the most commonly found type of alternative splicing for an extended CDS. Thus, pre-mRNA splicing might have evolved to retain maximum functionality in terms of organ preference and multiplicity.
Keywords: alternatively spliced transcript detection microarray, coefficient value, rice, RNA sequencing, transcriptome
INTRODUCTION
Eukaryotic organisms have evolved ingenious mechanisms to increase protein diversity. At the genome level, diversity is amplified through various mechanisms of gene duplication, such as whole-genome (WG) duplication and segment and tandem duplications, during evolution (Freeling, 2009). At the protein level, alternative RNA splicing provides another layer of diversity, whereby transcripts of many multi-exonic genes are processed with the inclusion or exclusion of particular exons in the final mRNA molecule (Berget et al., 1977; Chow et al., 1977). It has been reported that ~95% of multi-exonic genes are alternatively spliced in humans; in contrast, the number is ~33% in rice (Pan et al., 2008; Zhang et al., 2010). Pre-mRNA splicing is regulated by a system of trans-acting RNA-binding proteins that bind to cis-acting sites on the primary transcript, followed by recruitment of spliceosomal proteins (Ser/Arg-rich proteins, SRs) to the target site. By recruiting small nuclear ribonucleoproteins (snRNPs) that form the spliceosome, SRs are well known for their role in alternative splicing (Glisovic et al., 2008; Lunde et al., 2007). Although many RNA isoforms can be generated from a single locus, the majority of loci generate only two or three isoforms in abundance.
Splicing isoforms consist of several groups corresponding to patterns of exon truncation or extension, intron retention or inclusion/exclusion of entire exons in a mature transcript (Cambell et al., 2006; Kriventseva et al., 2003). Such splice modes have been classified by several research groups. For example, Lewis et al. (2003) proposed classifying splice modes based on splice site usage and effects on the coding sequence. These authors reported that splice sites can be introduced or lost according to the mode of splice type, such as an intra-exon splice, an imperfect exon skip, alternate sites, exon inclusion, and a perfect exon skip; changes in coding region were also considered. Campbell et al. (2006) classified alternative splice forms into 9 types, including alternate acceptor/donor sites and retained or skipped exons in rice. Several web tools are also available for detecting alternative splicing (Huang et al., 2003; Koscielny et al., 2009; Zhu et al., 2014). Expression Atlas (http://www.ebi.ac.uk/gxa) is a value-added database that provides information about gene, protein and splice variant expression in different cell types, organs, and developmental stages (Petryszak et al., 2014). Indeed, splicing events are often differentially regulated across tissues and during development, suggesting that individual isoforms may have specific spatial or temporal roles. Such analyses of alternative splice forms might provide clues about which genes are under alternative splicing pressure during development or caused by various biotic or abiotic stresses.
Rice (Oryza sativa) is an important crop that supplies food more than 3 billion people. Based on the entire genome sequence and map-based sequence, the genome size of rice is approximately 389 Mb. In addition, the recent International Rice Genome Sequencing Project (IRGSP) 1.0 version annotated approximately 38,000 protein-coding sequences (Price et al., 2002; Project, International Rice Genome Sequencing 2005; Salunkhe et al., 2011), among which 2,478 non-redundant sets of rice transcription factors (TFs) (Gao et al., 2006; Perez-Rodriguez et al., 2010; Priya and Jain, 2013) involving major families such as AP2-EREBP (165), bHLH (160), NAC (144), and MADS (90) were found. As the genome size of rice is relatively small, it has been used as a model monocot plant to provide information at the genome scale for numerous biology fields, including organogenesis, development, and stress responses. The attributes associated with the small genome of rice have allowed researchers in these fields to perform proteomic analysis (Choudhary et al., 2009; Degenkolbe et al., 2013; Rabello et al., 2008), transcriptomic analysis using microarrays (Degenkolbe et al., 2009; 2013; Rabbani et al., 2003; Rensink and Buell, 2005), and genetic analysis using next-generation sequencing (NGS) (Huang et al., 2012). Moreover, to identify genes related to agronomic characters, various cultivars have recently been sequenced using NGS followed by mapping to reference sequences (Arai-Kichise et al., 2014; Huang et al., 2010; Yamamoto et al., 2010).
Over the last two decades, DNA microarray technology has been extensively applied in gene expression studies in various areas of biology, such as transcriptome changes during shoot, root, and callus development (Che et al., 2006; Jiao et al., 2009; Ma et al., 2005; Su et al., 2007; Wang et al., 2010). Microarrays have also been used to determine gene expression profiles of whole plants or various organs during stress or physiological changes (Kreps et al., 2002; Lenka et al., 2011; Minh-Thu et al., 2013; Rabbani et al., 2003; Sarkar et al., 2014; Seki et al., 2002; Shankar et al., 2014; Shinozaki and Yamaguchi-Shinozaki, 2000). In these analyses, several versions of rice chips have been used, such as the GeneChip (25 bp) from Affymetrix (www.affymetrix.com) and customized chips (60 bp) from Agilent (www.agilent.com) or NimbleGen (sequencing.roche.com). A spotting technology-based NSF45K array (50–70 bp) has also been employed to distinguish alternatively spliced transcripts (Jung et al., 2008). Recently, NGS without prior annotation information was utilized to analyze organs under various physiological conditions (Kyndt et al., 2012; Wakasa et al., 2014; Yang et al., 2015a; 2015b; Zhai et al., 2013). However, due to the lack of issues associated with probe redundancy and annotation, RNA sequencing (RNA-Seq) simplifies data interpretation and is rapidly replacing microarray technology.
In this study, we designed feature probes for an alternatively spliced transcript detection microarray (ASDM) for rice. The 40,139 transcripts/36,176 loci in ASDM can be detected in the current Rice Annotation Project (RAP) database containing 44,552 transcripts/37,868 loci. ASDM was applied to differentiate transcriptomes among the leaf, root, 1-cm panicle and young seed. In a parallel analysis, the results were compared with those obtained by RNA-Seq. Both technologies revealed the significance of alternative splicing in terms of organ enrichment. The several benefits of alternatively splicing with organ bias are discussed.
MATERIALS AND METHODS
Plant materials and RNA isolation
All four organs in this study were prepared from Oryza sativa L. cv. Ilmi. Leaves and roots were harvested separately at 14 days after sowing in soil. Flowers before pollination were collected in the range of 0.5–1.5-cm panicles (P1cm). Seeds at 20–30 days after pollination (S21DAP) were collected at the internal ripening stage of the grains. Total RNA was isolated from tissues using Hybrid-R™ (GeneAll, Korea) extraction following the manufacturer’s manual. For experimental reproducibility, all samples were prepared twice from distinct plants and immediately frozen in liquid nitrogen.
Microarray design
An ASDM for rice was designed based on the genome information of IRGSP_1_0_representative_2014_06_25 (http://rapdb.lab.nig.ac.jp). Four 60-nt-long feature probes as representative features (normally ending with ‘-01’) were designed for each gene, starting 60 bp upstream of the end of the stop codon and shifting 30 bp, such that 4 probes covered 60 bp of the coding sequence (CDS) and 90 bp of the 3′ untranslated region (UTR). For loci with alternatively spliced transcripts, additional probes were designed to distinguish alternatively spliced transcripts. Unique exons (UniqExon) or regions (UniqExonU or UniqExonD) were identified by comparing genome coordinates between alternatively splice transcripts. Regions created by joining adjacent exons were designated as ‘UniqExonJ’. Genes from chloroplasts (123) and mitochondria (74) and selection markers such as gfp, gus, hyg, bar, and kan were included. In total, 175,192 probes were designed, with an average probe size of 60 nt and a Tm between 75 and 85°C. The genome version contains 37,868 loci and 44,552 transcripts, and ASDM covers 36,176 loci and 40,139 transcripts. The microarray was manufactured at Agilent (Agilent Technologies, United States, http://www.agilent.com).
Microarray analysis
Probes were labeled using Agilent Low RNA Input Linear Amplification Kit PLUS following the manufacturer’s protocol (http://www.agilent.com). Total RNA (500 ng) was used to generate labeled cRNA by incorporating cyanine CTP. The microarray was scanned using an Agilent DNA microarray scanner (G2505C), and raw intensity data were extracted with the feature extraction software. The data were processed with the limma package in R (http://www.bioconductor.org/), and background adjustment and normalization were performed with the background Correct and normalize Between Arrays functions, respectively, in the program. plot Densities functions were used to analyze the distribution of probe intensities for 8 microarray data. Frequency densities before and after normalization were plotted against log2-based intensities (Supplementary Figs. S1a and S1b). The microarray intensities ranged from a minimum 23 to a maximum 545,883. Consistency between the microarrays was tested using leaves (Supplementary Fig. S1c). Log2-based intensities of 2 leaf samples were compared. The linear model was Y = 0.998x + 0.018, and the Pearson correlation coefficient was 0.996. Multiple analysis was performed after filtering out control probes and low-expression probes (Ritchie et al., 2015). The package adopted the linear modeling approach implemented by lmFit and the empirical Bayesian statistics implemented by eBayes. Genes with adjusted-P-values or false discovery rates (FDRs) below 0.05 were collected, and those with log2 scale values higher than 1 or less than −1 in at least one organ compared to that of the leaf were further selected. The data were processed with the limma package based on R statistical language.
Gene Ontology (GO) analysis
Biological term enrichment among CVI, CVII, CVIII-i, and CVIII-ii genes was assessed using GoMiner (Ashburner et al., 2000; Zeeberg et al., 2003), as described previously (Minh-Thu et al., 2013). To identify a tentative ortholog of a rice gene in the Arabidopsis genome, BLASTP analysis was performed for the two species; genes with a score of 70 or higher were considered tentative counterparts. The microarray contains 27,448 genes, and 21,556 of these match Arabidopsis genes with scores equal to or greater than 70; these genes were used as the total gene set in GoMiner. Among the 21,556 rice genes in this study, significant changes in biological processes, molecular functions and cellular components were found for 1,342, 406 and 308, respectively. As the relationships of GO terms are hierarchical, the top terms (66) in each clade for the 1,342 biological process terms were collected and subjected to hierarchical clustering using limma language in the Bioconductor project (http://master.bioconductor.org/). FDR values were obtained from 100 randomizations. GO terms for which the FDR was less than 0.05 in at least at one group were collected. For presenting GO terms in hierarchical clusters, unique GO terms were first selected. The FDR values ranging from less than 0.05 were scaled 0.5 to 5 such that the more enriched terms have values close to 5 and darker color codes in the heatmap function (http://master.bioconductor.org/). Other non-significant FDR values were transformed to 0 using Perl scripting language. To reduce redundancy, the top terms in each clad were selected, and hierarchical clustering was performed with the Bioconductor R package (http://master.bioconductor.org/).
RNA sequencing
mRNA was purified using the TruSeq RNA sample preparation kit (http://www.illumina.com/) following the company’s protocol. Following purification, the mRNA was fragmented, and first-strand cDNA was generated using reverse transcriptase and random primers. Second-strand cDNA synthesis was then performed using DNA polymerase I and RNase H. These cDNA fragments were subjected to an end-repair process, the addition of a single A base, and adaptor ligation. The products were purified and enriched by polymerase chain reaction (PCR) to create a cDNA library in a bridged amplification reaction that occurred on the surface of the flow cell. A flow cell containing millions of unique clusters was loaded into a HiSeq 2000 for automated cycles of extension and imaging. Each sequencing cycle was carried out in the presence of all four nucleotides, generating a series of images with each representing a single base extension at a specific cluster. Libraries were constructed for the leaf, root, P1cm, and S21DAP. Repetitions were performed for all samples, except roots. In the initial sequencing, 2.7–0 Gb was sequenced at each end. These results generated 16–43 × 106 paired-end reads.
Data processing
Programs such as TopHat, Cufflinks and HTSeq were used to process sequence reads. Differential exon usage was examined with the DEXseq package in Bioconductor. Briefly, the rice genome sequence (IRGSP-1.0_2014-06-25) in the RAP database (http://rapdb.lab.nig.ac.jp) was indexed using Bowtie 2 with an FM Index based on the Burrows-Wheeler transform (Langmead et al., 2009). Reads in FASTQ files were aligned using TopHat (v2.0.4) with default parameters (Kim et al., 2013). TopHat aligned the RNA-Seq reads to genomes using the short read aligner Bowtie 2 and then analyzed the mapping results to identify splice junctions between exons. In the program, samtools (version 1.2) was used for indexing and sorting of bam and sam files. For transcriptome assembly and isoform quantitation from the RNA-Seq reads, Cufflinks was used and tested for comparison (Trapnell et al., 2012). Prior to statistical analysis of exons using the DEXseq package, per-gene read counts were performed with htseq-count in HTSeq-0.6.1 using the annotated gtf file (Anders et al., 2015). To extract exons among transcripts, non-overlapping exonic regions were defined with dexseq_prepare_annotation.py, and then the per-exon read counts were extracted with the script dexseq_count.py in the DEXSeq package (Anders et al., 2012). For each exon of each gene, the data contain the number of reads in each sample that overlap with the exon. If an exon’s boundary is not the same in all transcripts, the exon is separated into two or more parts, referred to as “counting bin”. A read that overlaps with several counting bins of the same gene is counted for each of these. The program returned one file for each biological replicate with the exon counts. Finally, differential exon usage was assessed with the DEXseq package in Bioconductor (http://bioconductor.org). In the package, generalized linear models (GLMs) for model read counts and parameters are calculated from a negative binomial (NB) distribution of these count data (McCullagh and Nelder, 1989).
Reverse transcription PCR (RT-PCR) and real-time PCR
For RT-PCR, 5 μg of total RNA was reverse transcribed using oligo (dT) and a RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific™, USA). PCR was performed with one-hundredth of the first-strand cDNA mixture and gene-specific primers. The PCR conditions used were as follows: 10 min at 94°C and 30 cycles of 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C. The 20 μl contained 10 μl Solg™ 2X Real-Time PCR Smart mix [Solgent, Korea], 2 μl cDNA, 1 μl primers, and 7 μl water, and real-time PCR was performed using the CFX96 touch real-time PCR detection system (Bio-Rad, USA) with three technical replicates. Expression was assessed by evaluating threshold cycle (CT) values. The relative expression level of tested genes was normalized to the tubulin gene and calculated using the 2−ΔΔCT method (Livak et al., 2001). The sequences of all primers used in this study can be found in Supplementary Table S11.
RESULTS
Target probes were designed based on unique sequences among alternatively spliced transcripts
It has been reported that the 3′ regions of genes in plants are more specific than the 5′ regions (Eveland et al., 2008). To reflect these features, four 60-nt-long feature probes were designed for each gene, starting 60 bp upstream of the end of the stop codon and shifting 30 bp, such that the four probes covered 60 bp of the CDS and 90 bp of the 3′ UTR (arrows collectively indicate R for locus Os08g0482700 in Fig. 1A). To distinguish alternatively spliced transcripts, additional probes were designed based on unique sequence variations among alternatively spliced transcripts. For example, two transcripts, Os08t0482700-01 (encoding cupredoxin domain-containing protein) and Os08t0482700-02 (encoding conserved hypothetical protein), are alternatively spliced from mRNA produced by the Os08g0482700 locus. In addition, probes were designed to represent the unique features of each transcript. The Os08t0482700-01 transcript is composed of two exons and a 546-bp CDS contained within these exons. In contrast, the Os08t0482700-02 transcript is composed of one exon and has a 228-bp CDS located within this exon. The Os08t0482700-01 transcript has unique junction sites that join the spliced transcripts, and a 60-bp feature probe (UniqExonJ) was designed based on two 30-bp-long regions in the CDS for each exon. Thus, the probe represents a part of the unique exon (Os08t0482700-01_UE) of Os08t0482700-01 and is used to represent Os08t 0482700-01 alone. When representative probes cannot distinguish between alternatively spliced transcripts, ‘-01_UE’ can be used for expression of the ‘-01’ transcript. The Os08 t0482700-02 transcript was compared to Os08t0482700-01; a portion of the 5′ UTR, the CDS and a portion of the 3′ UTR are unique, and these regions were designated as UniqExonU, UniqExon, and UniqExonD, respectively. Three 60-bp target probes were designed according to these regions to represent Os08t0482700-02.
Fig. 1. Design of the feature probes of the rice alternatively spliced transcript detection microarray (ASDM).
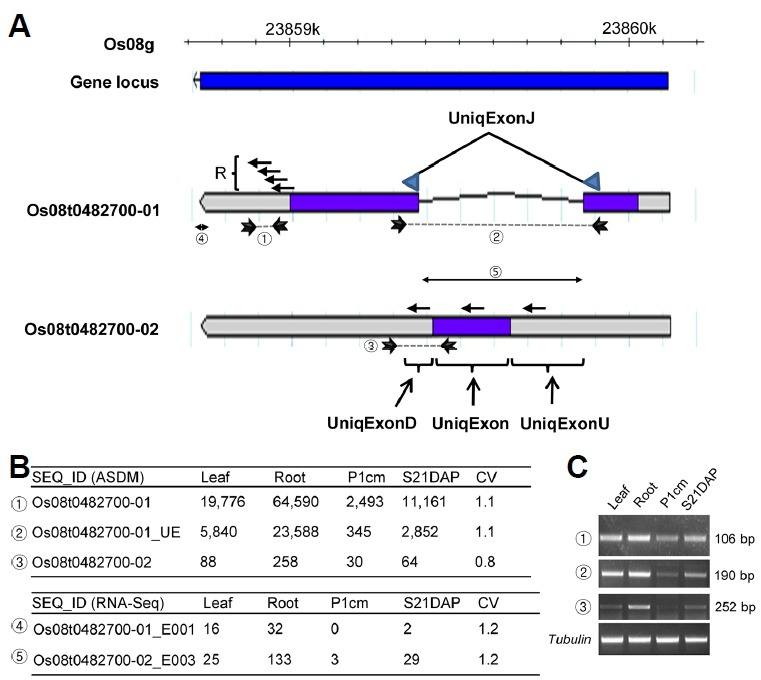
(A) Probes are positioned and over-scaled based on the genome features in RAP-DB (rapdb.dna.affrc.go.jp). Four probes are collectively indicated as R for the locus Os08g0482700 covering 60 bp of the CDS and 90 bp of the 3′ UTR. Additionally, a 60-bp feature probe (UniqExonJ) was designed by joining 30-bp-long spliced transcripts from two regions (arrow heads) in the CDS for each exon for Os08t0482700-01. Thus, the probe represents a unique junction of two exons of Os08t0482700-01 and is indicated as Os08t0482700-01_UE in the intensity file. The Os08t0482700-02 transcript has three unique regions compared to Os08t0482700-01: part of the 5′ UTR, the CDS and part of a 3′ UTR, designated as UniqExonU, UniqExon, and UniqExonD, respectively. Three 60-bp-long target probes were designed according to these regions. (B) Signal intensities and transcript numbers for rice ASDM (top) and RNA-Seq (bottom), respectively. Exons in RNA-Seq are designated according to the exon partition by dexseq_prepare_annotation.py (Anders et al., 2012). Exon bins are indicated by suffixing E and numeral generated by exonic_part_number attribute (Supplementary Table S4). (C) Confirmation of expression by semi-quantitative RT-PCR.
A total of 37,868 genes are representatively annotated in the IRGSP and RAP databases (http://rapdb.lab.nig.ac.jp), and 5,254 loci encode 11,938 alternatively spliced transcripts (Table 1). Among these alternatively spliced transcripts, 7,502 transcripts were designed with the unique exon features indicated above. Ultimately, 175,192 feature probes representing 40,139 of the 44,552 transcripts were designed and designated ASDM.
Table 1.
Summary of loci in IRGSP v1.0 and design features of the rice ASDM
| Chr | Loci | Alternative locia) | Alternative transcriptb) | Total transcriptsc) | Designed locid) | Designed alternative locie) | Designed alternative transcriptsf) | Designed total_transcriptsg) |
|---|---|---|---|---|---|---|---|---|
| Os01 | 5,273 | 778 | 1,765 | 6,260 | 5,070 | 522 | 1,093 | 5,641 |
| Os02 | 4,216 | 642 | 1,464 | 5,038 | 4,056 | 423 | 908 | 4,541 |
| Os03 | 4,550 | 761 | 1,735 | 5,524 | 4,407 | 478 | 1,010 | 4,939 |
| Os04 | 3,359 | 473 | 1,083 | 3,969 | 3,200 | 347 | 729 | 3,582 |
| Os05 | 3,027 | 437 | 985 | 3,575 | 2,914 | 284 | 610 | 3,240 |
| Os06 | 3,159 | 406 | 919 | 3,672 | 3,042 | 278 | 592 | 3,356 |
| Os07 | 2,870 | 362 | 829 | 3,337 | 2,753 | 260 | 547 | 3,040 |
| Os08 | 2,588 | 350 | 799 | 3,037 | 2,456 | 241 | 514 | 2,729 |
| Os09 | 2,111 | 285 | 641 | 2,467 | 2,012 | 205 | 434 | 2,241 |
| Os10 | 2,115 | 260 | 582 | 2,437 | 1,981 | 174 | 369 | 2,176 |
| Os11 | 2,424 | 247 | 543 | 2,720 | 2,240 | 161 | 340 | 2,419 |
| Os12 | 2,176 | 253 | 593 | 2,516 | 2,045 | 166 | 356 | 2,235 |
|
| ||||||||
| Summary | 37,868 | 5,254 | 11,938 | 44,552 | 36,176 | 3,539 | 7,502 | 40,139 |
RAP-DB annotation based on the genome IRGSP_1_0_representative_2014_06_25 was used
The number of loci with alternatively spliced transcripts
The number of transcripts due to alternative splicing at these loci
The total number of transcripts, both single and alternatively spliced transcript(s)
The number of loci included in this ASDM
The number of loci with alternative splicing included in this ASDM
The number of transcripts with alternative splicing included in this ASDM
The number of transcripts with single or alternative splicing included in this ASDM
Confirmation of organ preferential genes with alternative splicing transcripts
We applied the rice ASDM to detect alternatively spliced genes among the leaf, root, panicle at 0.5–1.5 cm (P1cm) and seed at 20–30 days after pollination (S21DAP). The 175,193 feature probes for 41,953 transcripts were normalized as described in the Methods section (Supplementary Figs. S1a and S1b). Consistency for intensities between microarrays using total RNA from leaves was higher than 0.99, suggesting that the experiment was very reproducible (Supplementary Fig. S1c). The data have been deposited at National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO); GSE87536. Normalized intensities are presented in Supplementary Table S1.
To confirm the detection of alternatively spliced transcripts by ASDM, several genes were tested. As shown in Fig. 1B (ASDM), among the four organs, the strongest intensity for Os08t0482700-01 were found in the root. The Os08t04827 00-01_UE transcript represented by the unique junction Os08t0482700-01 confirmed this result. The intensities of Os08t0482700-01 and Os08t0482700-01_UE showed a similar pattern among the organs, being highly expressed in the root, followed by the leaf, S21DAP and P1cm. Although Os08t0482700-02 exhibited lower expression compared to Os08t0482700-01, similar to Os08t0482700-01 and Os08t0 482700-01_UE, it also was highly expressed in the root. Semi-quantitative PCR confirmed these patterns of expression (Fig. 1C). Primers were designed to represent the unique regions of each transcript, and as expected, these primers amplified segments presenting a tendency similar to the microarray data shown in Fig. 1B.
We also examined Os04g0612500, Os10g0471100 and Os07g0673400 in the leaf, P1cm and S21DAP samples, respectively (Supplementary Fig. S2). The probe intensities of the microarray showed the highest expression of Os04g06 12500 in the leaf, which was confirmed by the result for Os04t0612500-01_UE (encoding a 36.4-kDa proline-rich protein) (Supplementary Fig. S2A). The probe designed using the 5′ and 3′ ends of Os04t0612500-02 also revealed expression in the leaf. RT-PCR using primers for Os04t 0612500-01, Os04t0612500-01_UE and Os04t0612500-02 amplified products of the expected sizes. Through evaluation of mRNA levels by real-time PCR, approximately 300 times higher expression in the leaf compared to the other tissues was found. Real-time PCR results also indicated strong expression in the leaf.
The microarray data showed that transcript Os10t04711 00-01 (encoding a protein with high similarity to proteins involved in cuticular wax biosynthesis) is specifically expressed in the P1cm stage (Supplementary Fig. S2B). The intensity of Os10t0471100-01_UE also confirmed that this transcript is highly expressed at this stage. The degree of intensity of Os10t0471100-02 (CER1) was lower than that of Os10t0471100-01, and this transcript was also found to be exclusively expressed in the P1cm sample. In addition, the mRNA levels detected by real-time PCR showed enhanced expression of this transcript in P1cm.
The microarray probe intensities showed the highest expression of Os07g0673400 in S21DAP, and the result for Os07t0673400-01 (encoding Rossmann-like alpha/beta/alpha sandwich fold domain) confirmed this result (Supplementary Fig. S2C). The probe designed using the 5′ end of Os07t0673400-02 also revealed expression in the S21DAP sample. RT-PCR with primers representing Os07t0673400-01, Os07t0673400-01_UE and Os07t0673400-02 generated products of the expected sizes. The mRNA levels detected by real-time PCR showed heightened expression of this transcript at the S21DAP stage. These data suggest that ASDM can detect expression of loci in the tested organs as well as alternatively spliced transcripts.
Genome-wide comparison of rice ASDM and RNA-Seq suggest co-linearity between the two technologies
In a parallel analysis, RNA-Seq was performed using the Illumina HiSeq 2000 platform, as indicated in the Methods section. Approximately 17–43 million reads were obtained from 30–80 Gb raw reads (Supplementary Table S2). These data were aligned to the reference genome 15–22 million times with TopHat, a splice-aware aligner (Kim et al., 2013). Unmapped hits varied widely; the lowest number of reads (1.1 M reads) was found in the seed and the highest number (17.6 M) in one of the leaf samples. The distribution of raw RNA-Seq data is shown in Supplementary Fig. S3. The dataset was loaded and normalized with the estimate Size Factors function in the DEXSeq package (Anders et al., 2012). In Supplementary Fig. S3, the exon dispersion estimates, fitted mean-dispersion values function and shrunken values are indicated by black points, red lines and blue points, respectively. Finally, median values for transcripts were obtained. The leaf sample provided the number of transcripts aligning more than once in the genome and showed the highest number of transcripts (27,852), with the panicle at 1 cm sample displaying the lowest (25,507) (Supplementary Table S3). The transcripts with maximum counts for the leaf, root, P1cm, and S21DAP were Os11t0707000-01 (119,487.5, encoding ribulose-bisphosphate carboxylase activase), Os12t0571100-01 (54,236, encoding metallothionein-like protein type 1), Os02t0121300-01 (34,784, encoding cyclophilin 2) and Os01t0762500-00 (224,247, encoding glutelin subunit), respectively. The intensities of these transcripts by ASDM were also very high, at 313,918 (Os11t0707000-01), 416,636.6 (Os12t0571100-01), 244,628.4 (Os02t0121300-01), and 251,958.8 (Os01t0762500-00), respectively, suggesting co-linearity between the two technologies. In terms of transcripts with low numbers, among the 40,139 transcripts in the microarray, 12,287–14,632 transcripts were counted less than once by RNA-Seq. The median value of these transcripts by ASDM was 100.5.
We tested genome-wide co-linearity between the microarray intensities and RNA-Seq counts. The log2-based average intensities of the leaf microarray data and the log2-based counts for the organs were compared (Fig. 2). Pearson’s correlation from two methods ranged 0.30–0.36 for the leaf, root, P1cm and S21DAP, suggesting correlation between the data. We also downloaded publicly available RNA-Seq data from TENOR (http://tenor.dna.affrc.go.jp), and data for the leaf and root at 10 days after germination were compared to those in ASDM. Pearson correlation coefficients ranged 0.25–0.27. The p-values of the F-statistics were also less than 2.2e-16 and significant (Supplementary Fig. S4). The distributions were similar to those for the leaf and root, as described above.
Fig. 2. Data comparison between ASDM intensities and RNA-Seq counts.
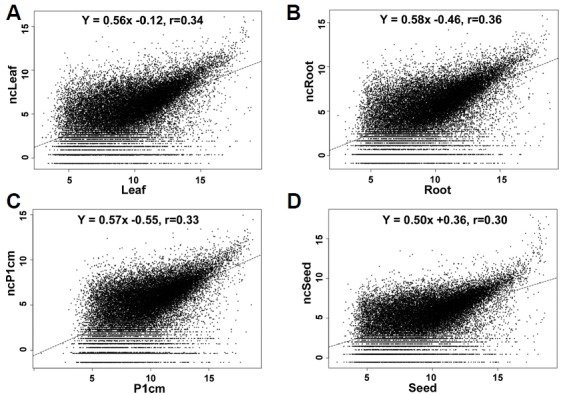
The values are represented on the x- and y-axes, respectively. Pearson’s correlation from two methods ranged from 0.30 to 0.36 for (A), leaf (B), root (C), P1cm and (D), S21DAP samples.
A list of exons of the transcripts was generated from a GTF file in IRGSP-1.0 subversion 2016-03-09 with a python script, as described in the Methods section (Anders et al., 2012). The overlapping exon regions are marked as “counting bins” by the program, and the read counts are distinguished by the statistical model in the program. The bins of transcripts such as Os08t0482700-01 and Os08t0482700-02 for the root (Fig. 1B, RNA-Seq), Os04t0612500-1 and Os04t0612500-2 for the leaf Os10t0471100-1 and Os10t0471100-2 for P1cm and Os07t0673400-1 and Os07t0673400-2 for S21DAP (Supplementary Fig. S2, RNA-Seq in b panel) are listed in Supplementary Table S4. The pattern of the exon counts for these transcripts was compared to the ASDM data, with a very similar result for each organ, even though expression values can vary depending on the technology employed.
Application of rice ASDM and RNA-Seq technologies to distinguish between genes that are enriched in organs and those that are constitutively expressed across organs
Among 45,128 transcripts, the number with normalized counts below 1 ranged from 12,000 to 15,000, and the median intensity of ASDM for these transcripts was 100.5. Based on the microarray data, the transcripts (32,718) with intensity greater than 300 (3* the median intensity of the transcripts, where RNA-Seq counts were greater than 1) in at least one organ were chosen for further analysis. The average values are presented in Supplementary Table S5. Among these transcripts, 21,497 were for a single exon, and 11,129 were for loci with alternatively spliced transcripts. To identify genes differentially expressed in each organ, we compared normalized intensities between distinct organs by adapting a statistical analysis approach using values of coefficient of variation (CV), which is the standard deviation of the mean and is used as a standardized measure of dispersion for probability distribution or frequency distribution (Everitt, 1998). Thus, a higher CV value suggests high variation among samples and is likely to be organ preferential in our data. This technique was also adopted by a rice gene expression database, CREP (Collections of Rice Expression Profiling, http://crep.ncpgr.cn, Wang et al., 2010), and presented in our previous paper (Chae et al., 2016). CREP contains 190 Affymetrix GeneChip Rice Genome Arrays using RNA from 39 tissues collected throughout the life cycle of two indica varieties: Zhenshan 97 and Minghui 63. To delineate CV values in our dataset, we attempted to compare our results with those in CREP. We also estimated the distribution of the 100 genes most constitutively expressed in CREP in our ASDM and RNA-Seq datasets, as described previously (Chae et al., 2016). The mean values of these genes were 0.18 (sd 0.02), 0.11 (sd 0.05) and 0.34 (sd 0.19) for CREP, ASDM and RNA-Seq, respectively (Supplementary Fig. S5).
Based on these CV values in ASDM, genes were tentatively categorized into 3 groups, as shown in Fig. 3A: CVI (higher than 1.1), CVII (higher than 0.7), and CVIII (below 0.7). We classified these groups as an organ-preferential group (CVI, 5,410), an organ-enriched group (CVII, 6,547), and a nonspecific group (CVIII, 20,761). CVIII was sub-divided as -i (highest), -ii (medium) and -iii (lowest) according to the degree of expression according to ASDM. As the CV values of RNA-Seq were comparable to those of ASDM, the CV values from RNA-Seq are also categorized as those from ASDM in Fig. 3B and Supplementary Table S3: CVI (higher than 1.4), CVII (higher than 0.8), and CVIII (below 0.8). The number of transcripts for the CVI, CVII and CVIII categories were 5,721, 8,574 and 18,371, respectively. CVIII was also sub-divided as -i (highest), -ii (medium) and -iii (lowest), at 3,467, 5,858, and 9,046, respectively.
Fig. 3. Coefficient of variation, CV, is the ratio of the standard deviation to the mean of a transcript, which was used to compare the distribution of transcript expression according to organ in the ASDM and RNA-Seq datasets.
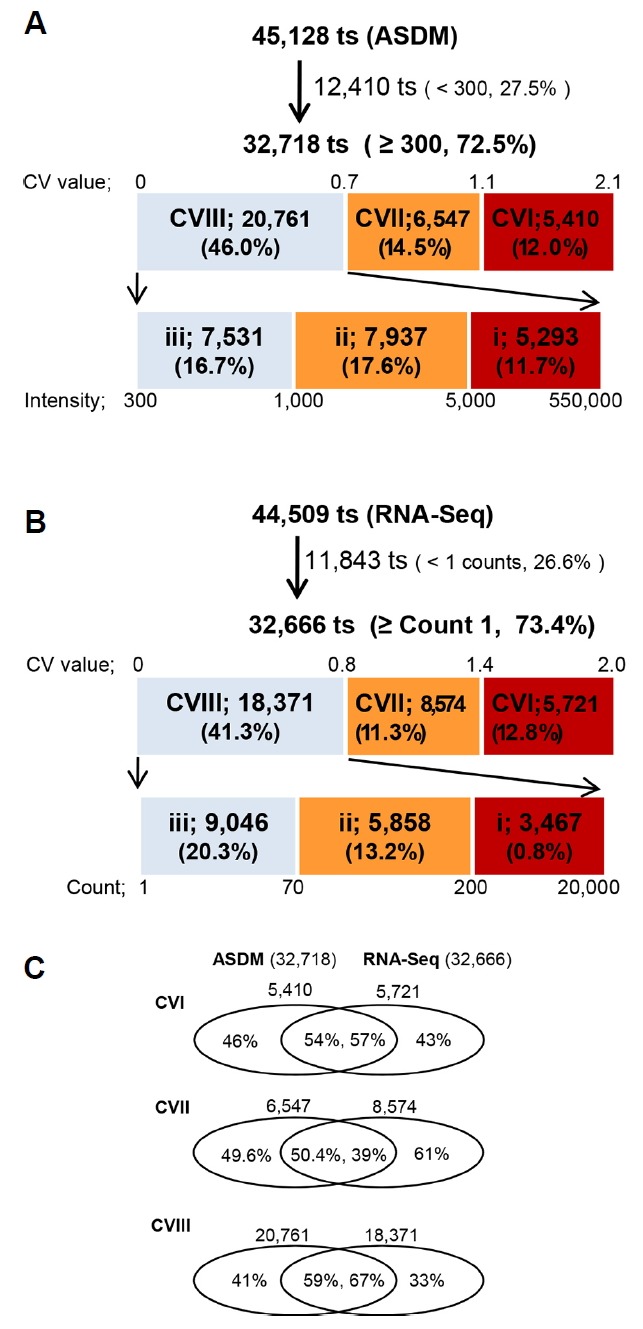
(A) Based on these CV values, genes were categorized into three groups for the ASDM dataset: CVI (higher than 1.1), CVII (higher than 0.7 to lower than 1.1), and CVIII (below 0.7). We classified these groups as an organ-preferential group (CVI, 5,410), an organ-enriched group (CVII, 6,547), and a non-specific group (CVIII, 20,761). (B) Similarly, the RNA-Seq dataset was also classified: CVI (higher than 1.4), CVII (higher than 0.8 to lower than 1.4), and CVIII (below 0.8). (C) Commonly detected numbers of transcripts are denoted as percentages of the effective number of transcripts for ASDM and RNA-Seq datasets, respectively.
When the intensities and counts were set ≥ 300 and ≥ 1 for ASDM and RNA-Seq, respectively, the effective number of expressed genes was approximately 37,000 for both technologies. Genes commonly detected using these technologies were searched according to CV group (Fig. 3C): approximately 40–50% of transcripts were commonly detected for each of the CVI and CVII groups, whereas approximately 60% of transcripts were detected for CVIII. For each technology, the number of transcripts commonly detected for most CV groups was greater than the number of excluded transcripts.
Hierarchical clustering of genes with a single transcript and alternatively spliced transcripts
As the genes of the CVI group for ASDM showed organ enrichment, those with a single transcript (4,172) and alternatively spliced transcripts (1,169) in this group were delineated by hierarchical clustering (Fig. 4). Highly expressed genes (618) in the leaf with a single transcript were enriched for a metabolic process represented by photosynthesis (Supplementary Table S6). These genes were Os07t0556200-01 (encoding Rieske [2Fe-2S] region domain-containing protein), Os02t0578400-01 (encoding photosystem II oxygen-evolving complex protein, PsbQ family protein), and Os07t0105600-01 (encoding photosystem II oxygen-evolving complex protein, PsbQ family protein). At least 6 C2C2-CO-like and putative zinc finger TFs that promote flowering induction in Arabidopsis under long photoperiods, such as Os06t0264200-00, Os09t0240200-01 and Os03t0711100-01, were highly expressed in the leaf. In the root, 665 genes were highly expressed and clustered into 3-4 groups. The root system development-related genes Os07t0128800-00 (encoding PR1a protein), Os05t0135200-01 (encoding plant peroxidase domain-containing protein), and Os02t0662000-01 (encoding Rcc3 protein) were highly expressed. Five AP2-EREBP family members, including Os03t0341000-01, Os01t 0752500-00 and Os03t0191900-01, were also strongly expressed. It is worth noting that approximately 30 non-protein coding transcripts, such as Os02t0791650-00, Os05t0400301-00, and Os02t0178901-00, were similarly highly expressed. For P1cm, the reproductive process was strongly activated, with Os10t0489900-01 (encoding CDT1a protein), Os03t0800200-00 (encoding protein argonaute MEL1), Os03t0137800-01 (encoding cyclin-dependent kinase inhibitor family protein) being highly expressed, as were the MADS box TFs Os05t0423400-01 (encoding PISTILLATA-like MADS box protein), Os02t0682200-01 (OsMADS6), and Os07t0605200-01 (OsMADS18). Highly expressed genes in S21DAP clustered into 3 groups. Processes related to seed storage metabolites such as amylopectin, amino sugar, and lipid localization-related processes were enriched, including Os08t0520900-00 (encoding iso amylase), Os02t0456100-01 (encoding glutelin), and Os03t 0808500-00 (encoding plant lipid transfer protein/Par allergen family protein), and starch biosynthesis-related genes such as Os06t0229800-01 (encoding starch synthase IIA.) and Os06t0160700-01 (encoding starch synthase I chloroplast precursor, EC 2.4.1.21). CCAAT-containing TFs Os10t 0191900-00, Os10t0397900-00, and Os01t0102400-01 and NAC domain TFs such as Os03t0327800-01, Os01t03931 00-01 and Os01t0104500-01 were also highlighted.
Fig. 4. Hierarchical clustering of CVI-group genes based on ASDM with 4,172 single transcript (ts) loci and with 1,169 alternatively spliced transcripts according to organ.
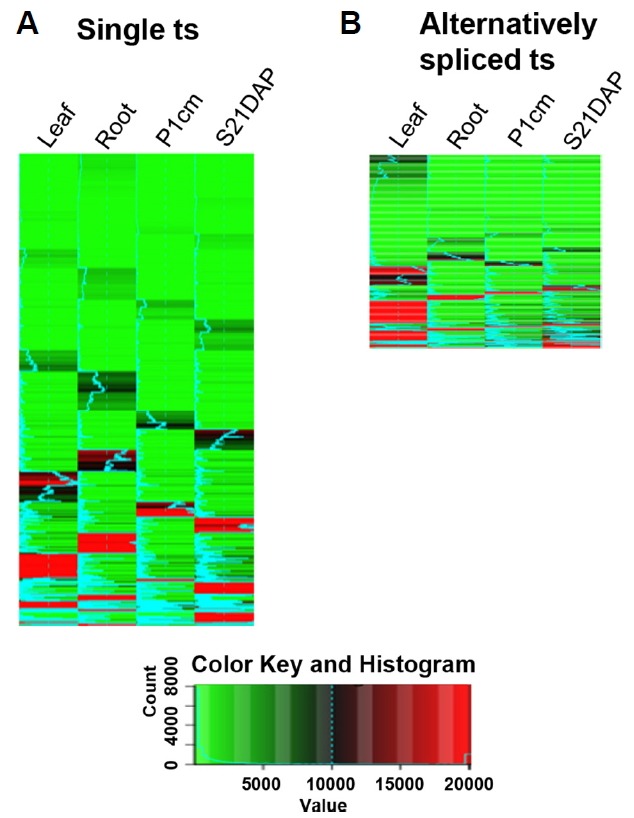
The matrix was calculated with the hclust and dist functions in R, and the image was generated with heatmap.2 in Bioconductor. Gene lists are provided in Supplementary Tables S6 and S7.
Highly expressed genes with alternatively spliced transcripts in the leaf clustered into 4-5 groups (Supplementary Table S7). Metabolic process, cellular component organization and biogenesis were enriched by the genes Os12t 0277500-01 (encoding RuBisCO subunit binding-protein, alpha subunit chloroplast precursor) and Os12t0420200-01 (encoding NAD(P)-binding domain-containing protein). In the leaf, alternatively spliced transcripts for TFs such as Os04t0497700-01 (encoding C2C2-CO-like, CONSTANS-like protein) and Os06t0348800-01 (encoding G2-like) were found. Two to three groups were strongly expressed in the root, coding for proteins that are induced under decreased oxygen levels, such as Os11t0210500-01 (encoding alcohol dehydrogenase) and Os03t0183000-01 (encoding AP2 domain-containing protein RAP2.6). Transporters such as Os10t0444700-02 (encoding phosphate transporter 6), Os02t0650300-01 (encoding iron-phytosiderophore transporter for iron homeostasis) were also highly expressed. With regard to P1cm, the reproductive process and developmental process were enriched, with Os08t0538700-01 (encoding retinoblastoma-related protein) and Os08t0442700-01 (encoding similar to SERK1) highly expressed. Os07t 0108900-02 (encoding MADS domain-containing protein for sexual reproduction) was highly expressed compared to Os07t0108900-01; Os02t0258300-01 (encoding zinc finger FYVE/PHD-type domain-containing protein) was also highly expressed. Four C3H family members, such as Os03t0329200-01 and Os03t0328900-01, were also highly expressed. For S21DAP, genes involved in the generation of precursor metabolites and energy, such as Os01t0663400-01 (encoding aspartic proteinase oryzasin 1 precursor) and Os08t0345800-01 (encoding glucose-1-phosphate adenylyltransferase), were enriched. Two prolamins, Os05t0329100-01 (encoding prolamin) and Os05t0329300-01, were also highly expressed.
To examine biological term enrichment, GO analyses were performed for genes of the CVI, CVII, CVIII-i, and CVIII-ii groups from the ASDM dataset (Ashburner et al., 2000; Zeeberg et al., 2003). This comparison was performed for loci of whole genes and those with single (16,999) or alternatively spliced (5,844) transcripts (Supplementary Figs. S6a and S6b). GO analysis of loci of whole genes by RNA-Seq was also performed (Supplementary Fig. S6c). These data suggested that GO analysis of the genes with single transcripts and alternatively transcripts reveal the representative characteristics of organs, even though subtle differences between the ASDM and RNA-Seq datasets were observed.
The portion of genes with alternatively spliced transcripts is increased among genes that are constitutively expressed across organs
In the rice genome, there are 5,254 loci with more than two alternative transcripts (http://rapdb.lab.nig.ac.jp); among these, 3,539 loci were included in the current microarray. We determined the influence of organ specificity on the number of alternatively spliced transcripts or the influence of the degree of expression on the genome. As shown in Table 2, the ratio of loci with alternatively spliced transcripts was 0.14 (5,254/37,869) in the current genome annotation and 0.15 (5,244/36,141) in ASDM; these ratios were designated as the index of loci with alternatively spliced transcripts (IAST) in the group. IAST of expressed genes was 0.19, slightly higher than those of the genome and ASDM. These values were 0.14, 0.22 and 0.24 for CVI, CVII, and CVIII, respectively. The CVI group was subdivided into the leaf, root, P1cm, and S21DAP, with values of 0.26, 0.08, 0.13, and 0.09, respectively. For the CVII group, these values were 0.18–0.28 and generally higher than those in the CVI group. When CVIII was divided into -i (highest), -ii (medium) and -iii (lowest) according to the degree of expression, IAST values of these groups ranged from 0.31–0.34. The IAST values appeared to increase from CVI to CVIII, suggesting that the ratio of genes with alternative splicing increased as the genes were expressed throughout each organ. We tested this hypothesis by performing a literature review using the web-based database CREP (Wang et al., 2010) and projected the IAST values of the 100 genes that were most constitutively expressed in CREP. Interestingly, IAST was 0.43 (Supplementary Table S8 and Consti100 in Table 2) for these constitutively expressed genes, higher than the values reported above.
Table 2.
The portion of loci with alternatively spliced transcripts among organ-preferential transcripts based on ASDM analysis
| ts_totala) | loci_totalb) | loci_single_ts c) | loci_as_ts d) | loci_single_ts/loci_total | loci_as_ts/loci_totale) | ts_variant_from_as_loci | |
|---|---|---|---|---|---|---|---|
| Genome | 44,553 | 37,869 | 32,615 | 5,254 | 0.86 | 0.14 | 11,938 |
| ASDM | 38,399 | 36,141 | 30,897 | 5,244 | 0.85 | 0.15 | 7,502 |
| CV total | 32,718 | 26,573 | 21,486 | 5,087 | 0.81 | 0.19 | |
| CVI | 5,410 | 4,862 | 4,172 | 690 | 0.86 | 0.14 | |
| CVII | 6,547 | 5,883 | 4,607 | 1,276 | 0.78 | 0.22 | |
| CVIII | 20,761 | 16,735 | 12,707 | 4,028 | 0.76 | 0.24 | |
| CVI_Leaf | 1,755 | 1,397 | 1,030 | 367 | 0.74 | 0.26 | |
| CVI_Root | 1,716 | 1,638 | 1,501 | 137 | 0.92 | 0.08 | |
| CVI_P1cm | 771 | 720 | 629 | 91 | 0.87 | 0.13 | |
| CVI_S21DAP | 1,168 | 1,108 | 1,012 | 96 | 0.91 | 0.09 | |
| CVII_Leaf | 1,636 | 1,433 | 1,029 | 404 | 0.72 | 0.28 | |
| CVII_Root | 1,713 | 1,534 | 1,203 | 331 | 0.78 | 0.22 | |
| CVII_P1cm | 1,980 | 1,821 | 1,488 | 333 | 0.82 | 0.18 | |
| CVII_S21DAP | 1,218 | 1,127 | 887 | 240 | 0.79 | 0.21 | |
| CVIII_i | 5,293 | 4,786 | 3,154 | 1,632 | 0.66 | 0.34 | |
| CVIII_ii | 7,937 | 7,302 | 5,056 | 2,246 | 0.69 | 0.31 | |
| CVIII_iii | 7531 | 6848 | 4497 | 2351 | 0.66 | 0.34 | |
| Consti100f) | 100 | 100 | 57 | 43 | 0.57 | 0.43 |
These ratios are designated as the index of loci with alternatively spliced transcripts in the group (IAST). These values are 0.14, 0.22 and 0.24 in CVI, CVII, and CVIII, respectively
Total number of transcript variants at RAP-DB
Total number of genes
Number of genes that produce a transcript
Number of genes that have alternatively spliced transcripts
IAST, a fraction of the number of loci with alternatively spliced transcript among the total gene loci
List of 100 constitutively expressed genes in Supplementary Table S8, reported by Wang et al.(2010), was used.
We also assessed IAST for CV groups based on RNA-Seq analysis (Table 3). The IAST values for CVI, CVII, and CVIII were 0.09, 0.12 and 0.21, respectively, and comparable to those based by ASDM. The values among the CVI and CVII organs were relatively higher in the leaf and lower in the root, P1cm, and S21DAP samples, but the values were somewhat lower than those based on ASDM. The IAST values among the CVIII subgroups were higher than those among other groups, as shown for ASDM, and were also less than those for 100 constitutive genes (Consti100 in Table 2). These data confirm the observation in the ASDM analysis, whereby IAST of genes constitutively expressed throughout organs was higher than that of organ-enriched genes.
Table 3.
The portion of loci with alternatively spliced transcripts among organ-preferential transcripts based on RNA-Seq analysis
| ts_totala) | loci_totalb) | loci_single_tsc) | loci_as_tsd) | loci_single_ts/loci_total | loci_as_ts/loci_totale) | |
|---|---|---|---|---|---|---|
| CV total | 32,666 | 26,759 | 22,071 | 4,688 | 0.82 | 0.18 |
| CVI | 5,721 | 5,161 | 4,700 | 461 | 0.91 | 0.09 |
| CVII | 8,574 | 7,495 | 6,603 | 892 | 0.88 | 0.12 |
| CVIII | 18,371 | 14,477 | 11,378 | 3,099 | 0.79 | 0.21 |
| CVI_Leaf | 1,938 | 1,570 | 1,280 | 290 | 0.82 | 0.18 |
| CVI_Root | 1,648 | 1,565 | 1,493 | 72 | 0.95 | 0.05 |
| CVI_P1cm | 706 | 668 | 631 | 37 | 0.94 | 0.06 |
| CVI_S21DAP | 1,429 | 1,362 | 1,301 | 61 | 0.96 | 0.04 |
| CVII_Leaf | 2,542 | 2,143 | 1,815 | 328 | 0.85 | 0.15 |
| CVII_Root | 2,127 | 1,850 | 1,616 | 234 | 0.87 | 0.13 |
| CVII_P1cm | 2,197 | 2,014 | 1,857 | 157 | 0.92 | 0.08 |
| CVII_S21DAP | 1,708 | 1,531 | 1,389 | 142 | 0.91 | 0.09 |
| CVIII_i | 3,467 | 2,678 | 2,048 | 630 | 0.76 | 0.24 |
| CVIII_ii | 5,858 | 4,746 | 3,805 | 941 | 0.80 | 0.20 |
| CVIII_iii | 9,046 | 7,760 | 6,675 | 1,085 | 0.86 | 0.14 |
These ratios are designated as the index of loci with alternatively spliced transcripts in the group (IAST) as indicated in the ASDM dataset. These values are 0.09, 0.12 and 0.21 in CVI, CVII, and CVIII, respectively
Total number of transcript variants at RAP-DB
Total number of genes
Number of genes that produce a transcript
Number of genes that have multiple (alternatively spliced) transcripts
IAST, a fraction of the number of loci with alternatively spliced transcript among the total gene loci.
A transcript confers organ preference in a locus, and the alternatively spliced transcript(s) confers the same preference or reduced organ specificity
We tested organ preference among alternatively spliced transcripts of the CVI group enriched in each organ. The number of these transcript variants were 527, 179, 120 and 124 from 367, 137, 91 and 96 loci of the leaf, root, P1cm, S21DAP, respectively (Table 4). We tested CV groups and organ preference between a major transcript and its alternatively spliced transcripts. For example, two transcripts, Os04t0612500-01 and Os04t0612500-02, are alternatively spliced from pre-mRNA of Os04g0612500 (Supplementary Fig. S2A). Os04t0612500-01 is classified as a CVI_Leaf and Os04t0612500-02 is also preferential to the leaf thus these transcripts showed the same bias for the leaf; the gene can be grouped as loci_CVI_same_organ. Os10t0471100-01 and Os10t0471100-02 from Os10g0471100 is an example of a grouping for P1cm (Supplementary Fig. S2B). Similarly, the lists of transcript variants that are classified in the same organ were 305, 81, 57 and 54 from 147, 39, 28 and 26 loci for the leaf, root, P1cm, S21DAP, respectively (Supplementary Table S9). In contrast, a number of transcript variants were found for which the major transcript belongs to CVI but the alternatively spliced transcript(s) belongs to as_ts_Not_CVI; CVII, CVIII or a group expressed in low amount. For example, Os07t0673400-01 from Os07g0673400 (Supplementary Fig. S2C) belongs to the seed-preferential group, and its alternatively spliced transcript, Os07t0673400-02 belongs to CVII and is classified as_ts_Not_CVI. Os08t0482700-02 from Os08g0482700 is expressed in a low amount and may be an additional example of as_ts_Not_CVI (Fig. 1 and Supplementary Table S4). The numbers of as_ts_Not_CVI were 222, 98, 63 and 70 for the leaf, root, P1cm and S21DAP. Interestingly, none of the alternatively spliced transcript variants classified as CVI showed other organ specificity or bias, and these might be grouped as as_ts_CVI_diff_organ (Table 4). These finding suggest that any two alternatively spliced transcripts in the CVI group tended to show the same organ preference or the other alternatively spliced transcript showed less organ preference.
Table 4.
Summary of CVI transcripts from loci that have alternatively spliced transcripts
| CVI_organ | as_tsa) | loci_as_tsb) | as_ts_CVI_organc) | loci_CVI_same_organd) | as_ts_CVI_diff_organe) | as_ts_Not_CVIf) |
|---|---|---|---|---|---|---|
| CVI_Leaf | 527 | 367 | 305 | 147 | 0 | 222 |
| CVI_Root | 179 | 137 | 81 | 39 | 0 | 98 |
| CVI_P1cm | 120 | 91 | 57 | 28 | 0 | 63 |
| CVI_S21DAP | 124 | 96 | 54 | 26 | 0 | 70 |
The number of transcript variants in each CVI organ
Number of loci of these transcripts
Number of CVI transcript variants that are grouped in the same organ
Number of loci of these organs
Number of CVI alternatively spliced transcript variants that are grouped in different CVI organ(s)
Number of transcripts that have the alternatively spliced transcript in CVII, CVIII or a group expressed in low amounts.
A longer CDS transcript among alternatively spliced transcripts at a locus tends to be more expressed
We tested the extent of expression among spliced transcripts within a locus by comparing transcripts with the highest and lowest expression values. To simplify expression variability, we compared the log2-based ratio between transcripts with the highest and lowest expression according to the ASDM dataset. The log ratio of the highest and lowest values is shown for all tested organs in Supplementary Fig. S7. For example, Os12t0291100-01 exhibited 14.6-fold higher expression than that of the lowest, Os12t0291100-02 (468,018.3/32,040.6), in the leaf at locus Os12t0291100 (encoding ribulose bisphosphate carboxylase small chain). The log2-based values are presented according to organ for the CVI group (Supplementary Fig. S7). For instance, CVI_Leaf showed 323 loci with alternatively spliced sites. The mean log2-based value for transcripts with the highest and lowest expression was 3.3 (sd = 1.9). The other organs exhibited similar mean ranges. We also expanded the evaluation to the CVIII group (Supplementary Fig. S8) using 3,382 of 4,028 loci to determine values. The mean value was 2.7, similar to that of the CVI organ-preferential group, suggesting that reciprocal expression was complementary among transcripts within a locus, regardless of organ specificity. We confirmed similar variation in tests based on RNA-Seq analysis (data not shown).
Campbell et al. (2006) classified types of alternative splicing into 9 groups based on the results of Program to Assemble Spliced Alignments (PASA): alternate acceptor, alternate donor, alternate terminal exon, retained exon and skipped exon, initiation within an intron, termination within an intron, and spliced intron and retained intron. We categorized the type of alternative splicing to assess splice types and variation in expression between transcripts with the highest and lowest expression. PASA was applied first to whole transcripts, detecting 5,028 alternative splices for 2,438 of 4,143 loci in the genome and 3,598 alternative splices for 2,415 loci in the microarray (Supplementary Fig. S9 and Supplementary Table S10).
It has been reported that expression varies among alternatively spliced transcripts. Nonsense-mediated mRNA decay (NMD) has been postulated as the target of RNA degradation (Lewis et al., 2003). When an alternative splice is introduced, the stop codon is classified, and the corresponding mRNA isoform is labeled an NMD candidate, resulting in lower expression compared to the isoform with a longer CDS. If the spliced transcript escapes NMD, the result is a transcript with a shorter CDS. To examine how CDS length, due to alternative splicing, affects the degree of expression, transcripts with alternative splicing are sorted by the degree of expression and the CDS lengths compared according to organ and CV group. As shown in Table 5, among alternatively spliced transcripts, longer CDS transcripts appeared to be more expressed than shorter ones for all organs and CV groups. A paired t-test showed a p-value of 0.029, suggesting that these differences are significant.
Table 5.
Number of genes with a longer CDS among alternatively spliced transcripts with different degrees of expression
| Highest intensitya) | Lowest intensityb) | |
|---|---|---|
| CVI_Leaf | 63 | 54 |
| CVI_Root | 23 | 10 |
| CVI_P1cm | 16 | 13 |
| CVI_S21DAP | 21 | 13 |
| CVII_Leaf | 72 | 41 |
| CVII_Root | 76 | 35 |
| CVII_P1cm | 63 | 40 |
| CVII_S21DAP | 48 | 30 |
| CVIII_i | 227 | 172 |
| CVIII_ii | 408 | 250 |
| CVIII_iii | 492 | 287 |
|
| ||
| Total | 1,509 | 945 |
Genes with alternatively spliced transcripts were collected, and the degrees of intensity were sorted and then assessed to determine which transcript has a longer CDS. A paired t-test between the highest and lowest expressed transcripts within locus with a longer CDS showed a p-value of 0.029, suggesting that the differences are significant
The number of transcripts with highest intensity and a longer CDS
The number of transcripts with lowest intensity and a longer CDS
DISCUSSION
Genome-wide gene expression profiling using microarray and RNA-Seq technologies has provided abundant information regarding the basic biological mechanisms underlying development, stress responses, and other processes (Gordon et al., 2014; Ma et al., 2005; Severin et al., 2010; Shinozaki and Yamaguchi-Shinozaki, 2000; Wakasa et al., 2014). In the current version of rice ASDM, 60-bp-long oligomers were used, and 40,953 of the 44,552 transcripts deposited in IRGSP 1.0 were included. We improved the feature probe design to assess alternative splicing. Because long oligomers can be designed for spliced regions composed of two exons, the approach may have advantages over short oligomer-based microarrays. Several loci, such as Os08g0482700, Os04g0612500, Os10g0471100, and Os07g0673400, with alternatively spliced transcripts were discerned in four organs: the leaf, root, early stage of the panicle at 1 cm and seed at 20–30 days after pollination (Fig. 1 and Supplementary Fig. S2). Genome-wide comparison with RNA-Seq data suggested co-linearity of transcript expression between rice ASDM and RNA-Seq, even though the counts for RNA-Seq were somewhat compressed (Fig. 2 and Supplementary Fig. S3), which might be due to saturation problems associated with hybridization-based microarrays. The results for distribution showed a wide range of variation for genes with low expression values according to both technologies. Lower reproducibility has been reported for gene classes that are less strongly expressed, even for RNA-Seq data, despite the excellent overall correlation (Mortazavi et al., 2008). Several factors, such as probe design for ASDM and PCR amplification for RNA-Seq, can affect the number of genes determined to be expressed in low amounts, which appears to be the case with our ASDM and RNA-Seq datasets. Nonetheless, these technologies do exhibit colinearity for highly expressed genes. In a study comparing RNA-Seq with a model organism, Saccharomyces cerevisiae, high consistency between the two platforms for gene expression-level analysis (correlation ≥ 0.91) was reported (Nookaew et al., 2012), and in a study of human T cell activation, Zhao et al. (2014) reported that RNA-Seq performed better in detecting low-abundance transcripts and demonstrated a broader dynamic range than microarray. Although analysis of RNA-Seq data requires extensive bioinformatic skills and computer resources (CPU and RAM), with improvement in cost and analytical complexity, RNA-Seq is being utilized more frequently.
ASDM and RNA-Seq provide similar GO profiles for the transcripts, as based on CV values
CV values for constitutive expression vary according to the number of organ samples. In this report, CV values of 0.7 and 0.8 were tentatively used for ASDM and RNA-Seq, respectively (Fig. 3). Wang et al. (2010) and our previous study (Chae et al., 2016) reported values of 0.1 and 0.3 using 39 tissues and 7 representative organs/tissues, respectively. For both technologies, the variation in and criteria for constitutive expression increased as the number of samples decreased. Because values vary depending on the approach, the medium range of CVII groups was adopted as an intermediate for each technology. These demarcations showed that analysis of genes with alternatively spliced transcripts can be achieved for both in a similar manner. The effective number of expressed transcripts was approximately 33,000 for ASDM and RNA-Seq (Fig. 3C), and the genes commonly detected ranged from 40 to 70%, more than the number transcripts exclusive for each technology in each CV group. Among the organs examined, higher numbers for constitutively expressed genes in both datasets were found for commonly expressed group. In particular, the results of GO analysis of the CV group between ASDM and RNA-Seq were quite similar (Supplementary Fig. S6), suggesting that both technologies are applicable for transcriptome analysis. Hierarchical clustering of the CVI group among the organs showed representative transcripts and TFs. For example, representative processes were observed, such as photosynthesis-related terms for the leaf, metallothionein for the root, cdc2 kinase for P1cm and glutelin and starch-debranching enzyme for S21DAP (Yuan et al., 2008; Yun et al., 2011). GO analysis was conducted on the loci of entire genes, followed by GO analyses of single-transcript and alternatively spliced transcript loci. In general, GO enrichment according to loci with alternatively spliced transcripts was in agreement with those with a single transcript.
Expressed TFs with a single transcript in P1cm included MADS box TFs Os05t0423400-01 (encoding PISTILLATA-like MADS box protein), Os02t0682200-01 (OsMADS6), and Os07t0605200-01 (OsMADS18). In a previous report, prolonged expression of Os02t0682200-01 and Os07t0605200-01 to the stage after pollination was observed (Chae et al., 2016), suggesting that these genes are involved in panicle development. MADS box and AP TFs are crucial for floral morphology (Coen and Meyerowitz 1991; Pelaz et al., 2000; Yun et al., 2013). Highly expressed genes in the S21DAP sample included those coding for CCAAT-containing TFs, such as Os10t0191900-00, Os10t0397900-00, and Os01t 0102400-01, and NAC domain TFs, such as Os03t0327800-01, Os01t0393100-01 and Os01t0104500-01. NAC genes, which constitute one of the largest families of plant-specific TFs and are present in a wide range of species, have been implicated in the regulation of many developmental processes, including seed development, embryo development, and shoot apical meristem formation (Duval et al., 2002; Kim et al., 2007a; Sperotto et al., 2009). These NAC TFs are also enhanced during the seed stage according to the Hana database (http://evolver.psc.riken.jp).
Analysis of the transcriptome using ASDM might be useful for alternatively spliced transcripts that include TFs. Indeed, a TF with an alternatively spliced transcript in P1cm, Os07t 0108900-02 (encoding MADS-domain-containing protein for sexual reproduction), was highly expressed compared to Os07t0108900-01. In rice, florigen and Hd3a, associated with the 14-3-3 protein in the cytoplasm, translocate to the nucleus and regulate expression of the MADS box-containing TF gene Os07g0108900 (OsMADS15, Tsuji et al., 2013), which is a homolog of an Arabidopsis floral identity gene APETALA1. Overexpression of the gene results in precocious phenotypes with regard to early internode elongation, shoot-borne crown root development, reduced plant height and early flowering at the seedling stage (Lu et al., 2012). As both transcripts contain a MADS box, an expression vector may need to be designed to discern between them. Os02t0258300-01 (encoding zinc finger FYVE/PHD-type domain-containing protein) was also highly expressed, as were four C3H family members, such as Os03t0329200-01 and Os03t0328900-01. In the S21DAP sample, Os12t 0621100-01, Os12t0621100-02 (encoding C2C2-YABBY TF) and Os03t0799600-01 (encoding PHD-containing TF) were significantly expressed from loci with alternatively spliced transcripts. Some YABBY genes are expressed in the apical-ventral region during vascular tissue development and early embryogenesis (Itoh et al., 2016; Liu et al., 2007), suggesting that these TFs are involved in early seed development.
Constitutively expressed genes are under greater alternative splicing pressure
Pre-mRNA or alternative splicing is found in eukaryotic organisms, some of which become highly developed through various processes. Many trans- and cis-acting factors have been identified, and one basic question with regard to alternative splicing pressure for common and organ-preferential genes remains. To address this question, we examined the influence of organ preference on the number of alternatively spliced transcripts and the influence of the degree of expression on the genome. Ratios were designated as IAST. The IAST value of expressed genes was 0.19, slightly higher than those of the genome and ASDM. These values were 0.14, 0.22 and 0.24 for CVI, CVII, and CVIII, respectively (Table 2). When CVIII was divided according to the extent of expression, IAST ranged from 0.31–0.34. IAST values also were higher in groups with greater constitutive expression, culminating in 0.43 for the most constitutive 100 genes in the literature (Wang et al., 2010). These observations were also confirmed by RNA-Seq data (Table 3). Although IAST values for CVI, CVII, and CVIII were 0.09, 0.12 and 0.21, respectively, slightly lower than those based on ASDM, the tendency toward increase with constitutively expressed groups and variations among organs were similar in these independent analyses.
To date, it remains unclear why constitutively expressed genes would have a higher IAST. All exon boundaries require canonical splice sites (e.g., GT-AG, GC-AG, AT-AC), and all bases in the genome can be exposed to mutation (Burset et al., 2000; Lorkovic et al., 2005; Mount and Steize 1981). DNA damage events can occur at a high frequency in higher eukaryotes, such that a single mammalian cell can accumulate ~10,000 abasic (apurinic) lesions per day (Lindahl and Nyberg, 1972). For comparison, mutation rates in microbes with DNA-based chromosomes are close to 1/300 per genome per replication (Drake et al., 1998). A high level of transcription has been associated with elevated spontaneous mutations and recombination rates in eukaryotic organisms (Datta and Jinks-Robertson, 1995). For example, the effect of transcription on the rate of spontaneous mutation in the yeast S. cerevisiae was examined using a lys2 frameshift allele under the control of a highly inducible promoter. As the rate of spontaneous reversion was shown to increase when the mutant gene was highly transcribed, transcriptionally active DNA and enhanced spontaneous mutation rates are correlated in yeast. Several hypotheses have been proposed to explain transcription-associated mutagenesis, e.g., transcription impairs replication fork progression in a directional manner, and DNA lesions accumulate under high-transcription conditions (Kim et al., 2007). Genes constitutively expressed across organs might have higher rates of mutation than organ-preferential genes, which may undergo reduced transcription. However, a high rate transcription might not be the sole factor explaining this phenomenon. Subdivision of CVIII by intensity, i.e., CVIII-i, -ii, and -iii, did not appear to result in variability (Tables 2 and 3). Alternatively, the neutral theory of molecular evolution may offer an explanation, such that organ-preferential genes within cells of differentiated organs might be under pressure toward alternative splicing (Kimura, 1968). Specifically, the products of alternative splicing may be detrimental to organogenesis and development. Indeed, positions within introns corresponding to known functional elements involved in pre-mRNA splicing, including the branch site, splice sites, and polypyrimidine tract, show reduced levels of genetic variation in human polymorphism data (Lomelin et al., 2010).
Alternatively spliced transcripts do not show a distinct preference of organ
According to our analysis, a transcript confers organ preference and alternatively spliced transcript(s) confer(s) the same preference or show less organ specificity. However, alternatively spliced transcripts do not exhibit additional organ distinctness or preference (Table 4). In other words, among CVI genes that show strong organ preference, none of alternatively spliced forms belong to the CVI group of the other organ. These finding suggest that multiple specificity may not be allowed for any two alternatively spliced transcripts in the CVI group. However, alternative splicing may be allowed for genes that are less detrimental to organogenesis and development, as mentioned above with regard to the observation of lower IAST in CVI compared to CVIII. Nonetheless, alternatively spliced transcripts that escape an organism’s surveillance evolve additional functionality and may be more abundant in other organs, resulting in CVII or CVIII. These aspects may place more constraint on pre-mRNA splicing of CVI genes, with lower IAST, as shown in Tables 2 and 3.
Numerous alternatively spliced transcripts have been implicated in flowering (Marquardt et al., 2014; Wang et al., 2014), circadian clock (Filichkin et al., 2010; Seo et al., 2011), and abiotic stress (Shang et al., 2017). In these analyses, alternatively spliced transcripts have a similar biological function. In the example of OsDREB2B expressed under drought conditions, two splice variants are differentially expressed in response to heat and drought stresses in rice (Matsukura et al., 2010). The transcript of OsDREB2B1 is more abundant under normal growth conditions; it contains a 53-base pair exon 2 insertion in the mature mRNA that introduces a shift in open reading frame. In plants exposed to high temperature stress, OsDREB2B2, in which exon 2 is spliced out in the mature mRNA, dominates. Genome-wide research in salt stress has revealed several features of pre-mRNA splicing in plants under abiotic stress; the expression level of these stress-induced variants is quite low relative to that of the major splicing variant(s), and most stress-induced splicing variants are associated with intron retention (Cui and Xiong, 2015; Ding et al., 2014) with a premature CDS. These observations might be in line with the allowance of alternative splicing for maximum function by avoiding expression of other alternatively spliced transcript(s) under unwanted circumstances.
Pre-mRNA splicing might be involved in balancing maximization of function and additional functionality to avoid distinct functions. For alternatively spliced variants of mRNAs, there is no way to distinguish functional differences that may be at risk of a waste of resources. Differential regulation through the components of the spliceosome might be possible, which would provide additional complexity in gene regulation. In this regard, pre-mRNA splicing might be involved in unique strategies contrasting those shown in gene families that provide direct functional distinctness by using unique promoters to respond to different signals (Freeling, 2009). As an example of a gene family, two transcripts, Os10t0505700-01 and Os11t0620300-01 (Nonspecific lipid-transfer protein 2) belong to the same ortholog group, OG5_137065 (orthomcl.org). However, Os10t0505700-01 is leaf preferential, while Os11t0620300-01 is seed preferential.
Among alternatively spliced transcripts, those with a longer CDS show higher expression
Our data also suggest that reciprocal expression is complementary among transcripts within a locus, regardless of organ specificity (Liu et al., 2011). The log2-based values of the highest and lowest expressed transcripts were 2.5–2.7 (sd = 1.9) (Supplementary Figs. S7 and S8). For CV classes, we found that longer CDS-containing transcripts largely showed higher expression (Table 5). More frequently, those with a longer CDS had the highest expression, as opposed to those with the lowest expression (1,509 compared with 945, respectively). Among the 9 alternatively spliced groups, spliced_intron was the form most frequently found among transcripts with high expression and a longer CDS (data not shown). In a survey of human transcripts, a stop codon >50 nt upstream of the final exon–exon splice junction of an inferred mRNA isoform is called a premature termination codon (Lewis et al., 2003), rendering them apparent targets for NMD. Coupling of alternative splicing and NMD may indicate that the cell possesses a large number of irrelevant mRNA isoforms that must be eliminated. When an alternative splice is introduced, the stop codon is classified, and the corresponding mRNA isoform is labeled an NMD candidate, resulting in lower expression compared to the isoform with a longer CDS.
There may be additional mechanisms for maintaining alternative splicing. Reciprocal or complementary expression patterns have been reported, whereby a copy among gene duplicates is expressed in a certain organ or tissue type and another copy is expressed to a different extent in other organs or tissue types (Liu et al., 2011). Approximately 30% of whole-genome duplicate pairs and 38% of tandem duplicate pairs show reciprocal expression patterns in Arabidopsis. This result suggests that a balancing pressure exists among these duplicated genes and that the balance is broken when the genes undergo sub-functionalization due to partitioning of the ancestral expression pattern or ‘new functionalization’ due to acquisition of a new expression pattern (Wegmann et al., 2008). Such expression balancing might be subject to alternative splicing pressure. Indeed, commonly expressed genes are under evolutionary pressure toward alternative splicing, whereas organ-preferential genes might be less subjected to alternative splicing. Favorable conditions for the occurrence of alternative splicing might be the result of constant expression and balancing of alternatively spliced isoforms under a developmental process. Based on our data, a large fraction of isoforms for transcripts with low expression and a longer CDS demonstrate reciprocal expression with other alternatively spliced transcripts. In a sense, reciprocal expression might be a common phenomenon for gene families and also for alternatively spliced transcripts of a gene.
In this study, rice ASDM and RNA-Seq were applied to differentiate alternatively spliced transcripts between representative rice organs, including the leaf, root, panicle at 1 cm, and young seed. Transcripts were classified according to organ enrichment, and the results are collectively explained in terms of evolutionary pressure. The portion of loci with multiple transcripts due to alternative splicing was higher among constitutively expressed genes than organ-preferential loci. The allowance of alternative splicing is maximized by avoiding expression of the other alternative splicing transcript under unwanted circumstances. Among loci, transcripts with a longer CDS tended to be more highly expressed than those with a shorter CDS. These genome-wide technologies might be useful for studying transcriptomes and their biological significance with regard to pre-mRNA splicing.
Supplementary data
ACKNOWLEDGMENTS
We thank Dr. Brian J Haas at Broad Institute (Cambridge, MA) for the PASA program. This work was supported by grants from the Next-Generation BioGreen 21 Program (YKK, grant no. PJ01107401; BHN, grant no. PJ01105703) and RDA the Republic of Korea.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Anders S., Reyes A., Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai-Kichise Y., Shiwa Y., Ebana K., Shibata-Hatta M., Yoshikawa H., Yano M., Wakasa K. Genome-wide DNA polymorphisms in seven rice cultivars of temperate and tropical japonica groups. PLoS One. 2014;9:e86312. doi: 10.1371/journal.pone.0086312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. Gene Ontology, tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget S.M., Moore C., Sharp P.A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci USA. 1977;74:3171–3175. doi: 10.1073/pnas.74.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burset M., Seledtsov I.A., Solovyev V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M.A., Haas B.J., Hamilton J.P., Mount S.M., Buell C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics . 2006;7:327. doi: 10.1186/1471-2164-7-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae S.H., Kim J.S., Jun K.M., Pahk Y.-M., Kim M.-J., Lee S.-B., Park H.-M., Lee T.-H., Nahm B.H., Kim Y.-K. Analysis of representative organ-specific genes and promoters of rice using a 3′ORF-oriented long oligomer microarray. J Plant Biol. 2016;59:579–593. [Google Scholar]
- Che P., Lall S., Nettleton D., Howell S.H. Gene expression programs during shoot, root, and callus development in Arabidopsis tissue culture. Plant Physiol. 2006;141:620–637. doi: 10.1104/pp.106.081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Chow L.T., Gelinas R.E., Broker T.R., Roberts R.J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell. 1977;12:1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- Coen E.S., Meyerowitz E.M. The war of the whorls, genetic interactions controlling flower development. Nature. 1991;353:31–37. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- Cui P., Xiong L. Environmental Stress and Pre-mRNA Splicing. Molecular Plant. 2015;8:1302–1303. doi: 10.1016/j.molp.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Datta A., Jinks-Robertson S. Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science. 1995;268:1616–1619. doi: 10.1126/science.7777859. [DOI] [PubMed] [Google Scholar]
- Degenkolbe T., Do P.T., Kopka J., Zuther E., Hincha D.K., Köhl K.I. Identification of drought tolerance markers in a diverse population of rice cultivars by expression and metabolite profiling. PLoS One. 2013;22:e63637. doi: 10.1371/journal.pone.0063637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenkolbe T., Do P.T., Zuther E., Repsilber D., Walther D., Hincha D.K., Köhl K.I. Expression profiling of rice cultivars differing in their tolerance to long-term drought stress. Plant Mol Biol. 2009;69:133–153. doi: 10.1007/s11103-008-9412-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F., Cui P., Wang Z.Y., Zhang S.D., Ali S., Xiong L.M. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genomics. 2014;15:431. doi: 10.1186/1471-2164-15-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J.W., Charlesworth B., Charlesworth D., Crow J.F. Rates of Spontaneous Mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval M., Hsieh T.F., Kim S.Y., Thomas T.L. Molecular characterization of AtNAM, a member of the Arabidopsis NAC domain super family. Plant Mol Biol. 2002;50:237–248. doi: 10.1023/a:1016028530943. [DOI] [PubMed] [Google Scholar]
- Eveland A.L., McCarty D.R., Koch K.E. Transcript Profiling by 3′-Untranslated Region Sequencing Resolves Expression of Gene Families1. Plant Physiol. 2008;146:32–44. doi: 10.1104/pp.107.108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt B. The Cambridge Dictionary of Statistics. Cambridge University Press; Cambridge, UK: 1998. [Google Scholar]
- Filichkin S.A., Priest H.D., Givan S.A., Shen R., Bryant D.W., Fox S.E., Wong W.K., Mockler T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010;20:45–58. doi: 10.1101/gr.093302.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M. Bias in plant gene content following different sorts of duplication, tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol. 2009;60:433–453. doi: 10.1146/annurev.arplant.043008.092122. [DOI] [PubMed] [Google Scholar]
- Gao G., Zhong Y., Guo A., Zhu Q., Tang W., Zheng W., Gu X., Wei L., Luo J. DRTF, a database of rice transcription factors. Bioinformatics. 2006;22:1286–1287. doi: 10.1093/bioinformatics/btl107. [DOI] [PubMed] [Google Scholar]
- Glisovic T., Bachorik J.L., Yong J., Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Letters. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S.P., Priest H., Des Marais DLSchackwitz W., Figueroa M., Martin J., Bragg J.N., Tyler L., Lee C.R., Bryant D., et al. Genome diversity in Brachypodium distachyon, deep sequencing of highly diverse inbred lines. Plant J. 2014;79:361–374. doi: 10.1111/tpj.12569. [DOI] [PubMed] [Google Scholar]
- Huang H.D., Horng J.T., Lee C.C., Liu B.J. ProSplicer, a database of putative alternative splicing information derived from protein, mRNA and expressed sequence tag sequence data. Genome Biol. 2003;4:R29. doi: 10.1186/gb-2003-4-4-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Niu B., Gao Y., Fu L., Li W. CD-HIT Suite, a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Kurata N., Wei X., Wang Z., Wang A., Zhao Q., Zhao Y., Liu K., Lu H., Li W. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501. doi: 10.1038/nature11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh J., Sato Y., Sato Y., Hibara K., Shimizu-Sato S., Kobayashi H., Takehisa H., Sanguinet K.A., Namiki N., Nagamura Y. Genome-wide analysis of spatiotemporal gene expression patterns during early embryogenesis in rice. Development. 2016;143:1217–1227. doi: 10.1242/dev.123661. [DOI] [PubMed] [Google Scholar]
- Jiao Y., Tausta S.L., Gandotra N., Sun N., Liu T., Clay N.K., Ceserani T., Chen M., Ma L., Holford M. A transcriptome atlas of rice cell types uncovers cellular, functional and developmental hierarchies. Nat Genet. 2009;41:258–263. doi: 10.1038/ng.282. [DOI] [PubMed] [Google Scholar]
- Jung K.-H., Bartley L.E., Cao P., Canlas P.E., Ronald P.C. Analysis of alternatively spliced rice transcripts using microarray data. Rice . 2008;2:9020. [Google Scholar]
- Kim N., Abdulovic A.L., Gealy R., Lippert M.J., Jinks-Robertson S. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair (Amst) 2007;6:1285–1296. doi: 10.1016/j.dnarep.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.G., Kim S.Y., Park C.M. A membrane-associated NAC transcription factor regulates salt-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Planta. 2007a;226:647–654. doi: 10.1007/s00425-007-0513-3. [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2, accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- Koscielny G., Le Texier V., Gopalakrishnan C., Kumanduri V., Riethoven J., Nardone F., Stanley E., Fallsehr C., Hofmann O., Kull M. ASTD: the alternative splicing and transcript diversity database. Genomics. 2009;93:213–220. doi: 10.1016/j.ygeno.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Kreps J.A., Wu Y., Chang H.S., Zhu T., Wang X., Harper J.F. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002;130:2129–2141. doi: 10.1104/pp.008532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva E.V., Koch I., Apweiler R., Vingron M., Bork P., Gelfand M.S., Sunyaev S. Increase of functional diversity by alternative splicing. Trends in Genet. 2003;19:124–128. doi: 10.1016/S0168-9525(03)00023-4. [DOI] [PubMed] [Google Scholar]
- Kyndt T., Denil S., Haegeman A., Trooskens G., De Meyer T., Van Criekinge W., Gheysen G. Transcriptome analysis of rice mature root tissue and root tips in early development by massive parallel sequencing. J Exp Bot. 2012;63:2141–2157. doi: 10.1093/jxb/err435. [DOI] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenka S.K., Katiyar A., Chinnusamy V., Bansal K.C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotech J. 2011;9:315–327. doi: 10.1111/j.1467-7652.2010.00560.x. [DOI] [PubMed] [Google Scholar]
- Lewis B.P., Green R.E., Brenner S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T., Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- Liu H.L., Xu Y.Y., Xu Z.H., Chong K. A rice YABBY gene, OsYABBY4, preferentially expresses in developing vascular tissue. Dev Genes Evol. 2007;217:629–637. doi: 10.1007/s00427-007-0173-0. [DOI] [PubMed] [Google Scholar]
- Liu S.-L., Baute G.J., Adams K.L. Organ and cell type–specific complementary expression patterns and regulatory neofunctionalization between duplicated genes in Arabidopsis thaliana. Genome Biol Evol. 2011;3:1419–1436. doi: 10.1093/gbe/evr114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lomelin D., Jorgenson E., Risch N. Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 2010;20:311–319. doi: 10.1101/gr.094151.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorkovic Z.J., Lehner R., Forstner C., Barta A. Evolutionary conservation of minor U12-type spliceosome between plants and animals. RNA. 2005;11:1095–1107. doi: 10.1261/rna.2440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.-J., Wei H., Wang Ya., Wang H.-M., Yang R.-F., Zhang X.-B., Tu J.-M. Overexpression of a transcription factor OsMADS15 modifies plant architecture and flowering time in rice (Oryza sativa L.) Plant Mol Biol Rep. 2012;30:1461–1469. [Google Scholar]
- Lunde B.M., Moore C., Varani G. RNA-binding proteins, modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Chen C., Liu X., Jiao Y., Su N., Li L., Wang X., Cao M., Sun N., Zhang X., et al. A microarray analysis of the rice transcriptome and its comparison to Arabidopsis. Genome Res. 2005;15:1274–1283. doi: 10.1101/gr.3657405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S., Raitskin O., Wu Z., Liu F., Sun Q., Dean C. Functional consequences of splicing of the antisense transcript COOLAIR on FLC transcription. Mol Cell. 2014;54:156–165. doi: 10.1016/j.molcel.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukura S., Mizoi J., Yoshida T., Todaka D., Ito Y., Maruyama K., Shinozaki K., Yamaguchi-Shinozaki K. Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol Genet Genom. 2010;283:185–196. doi: 10.1007/s00438-009-0506-y. [DOI] [PubMed] [Google Scholar]
- McCullagh P., Nelder J.A. Generalized linear models. 2nd ed. Chapman & Hall/CRC; Boca Raton, FL: 1989. [Google Scholar]
- Minh-Thu P.T., Hwang D.J., Jeon J.S., Nahm B.H., Kim Y.K. Transcriptome analysis of leaf and root of rice seedling to acute dehydration. Rice . 2013;6:38. doi: 10.1186/1939-8433-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Mount S.M., Steize J.A. Sequence of U1 RNA from Drosophila melanogaster, implications for U1 secondary structure and possible involvement in splicing. Nucleic Acids Res. 1981;9:6351–6383. doi: 10.1093/nar/9.23.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nookaew I., Papini M., Pornputtapong N., Scalcinati G., Fagerberg L., Uhlén M., Nielsen J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays, a case study in Saccharomyces cerevisiae. Nucleic Acids Res. 2012;40:10084–10097. doi: 10.1093/nar/gks804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Pelaz S., Ditta G.S., Baumann E., Wisman E., Yanofsky M.F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature. 2000;405:200–203. doi: 10.1038/35012103. [DOI] [PubMed] [Google Scholar]
- Perez-Rodriguez P., Riano-Pachon D.M., Correa L.G., Rensing S.A., Kersten B., Mueller-Roeber B. PlnTFDB, updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010;38:D822–7. doi: 10.1093/nar/gkp805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryszak R., Burdett T., Fiorelli B., Fonseca N.A., Gonzalez-Porta M., Hastings E., Huber W., Jupp S., Keays M., Kryvych N., et al. Expression Atlas update--a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42:D926–32. doi: 10.1093/nar/gkt1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A.H., Cairns J.E., Horton P., Jones H.G., Griffiths H. Linking drought-resistance mechanisms to drought avoidance in upland rice using a QTL approach, progress and new opportunities to integrate stomatal and mesophyll responses. J Exp Bot. 2002;53:989–1004. doi: 10.1093/jexbot/53.371.989. [DOI] [PubMed] [Google Scholar]
- Priya P., Jain M. RiceSRTFDB: a database of rice transcription factors containing comprehensive expression, cis-regulatory element and mutant information to facilitate gene function analysis. Database (Oxford) 20132013 doi: 10.1093/database/bat027. bat027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Project, International Rice Genome Sequencing. The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Rabbani M.A., Maruyama K., Abe H., Khan M.A., Katsura K., Ito Y., Yoshiwara K., Seki M., Shinozaki K., Yamaguchi-Shinozaki K. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol. 2003;133:1755–1767. doi: 10.1104/pp.103.025742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabello A.R., Guimaraes C.M., Rangel P.H., da Silva F.R., Seixas D., de Souza E., Brasileiro A.C., Spehar C.R., Ferreira M.E., Mehta A. Identification of drought-responsive genes in roots of upland rice (Oryza sativa L) BMC Genomics . 2008;9:485. doi: 10.1186/1471-2164-9-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensink W.A., Buell C.R. Microarray expression profiling resources for plant genomics. Trends Plant Sci. 2005;10:603–609. doi: 10.1016/j.tplants.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salunkhe A.S., Poornima R., Prince K.S.J., Kanagaraj P., Sheeba J.A., Amudha K., Suji K., Senthil A., Babu R.C. Fine mapping QTL for drought resistance traits in rice (Oryza sativa L.) using bulk segregant analysis. Mol Biotechnol. 2011;49:90–95. doi: 10.1007/s12033-011-9382-x. [DOI] [PubMed] [Google Scholar]
- Sarkar N.K., Kim Y., Grover A. Coexpression network analysis associated with call of rice seedlings for encountering heat stress. Plant Mol Biol . 2014;84:125–143. doi: 10.1007/s11103-013-0123-3. [DOI] [PubMed] [Google Scholar]
- Seki M., Narusaka M., Ishida J., Nanjo T., Fujita M., Oono Y., Kamiya A., Nakajima M., Enju A., Sakurai T. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002;31:279–292. doi: 10.1046/j.1365-313x.2002.01359.x. [DOI] [PubMed] [Google Scholar]
- Seo PJPark MJLim MHKim SGLee MBaldwin I.T., Park C.M. A self-regulatory circuit of CIRCADIANCLOCK-ASSOCIATED1 underlies the circadian clock regulation of temperature responses in Arabidopsis. Plant Cell 2011. 2011;24:2427–2442. doi: 10.1105/tpc.112.098723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin A.J., Woody J.L., Bolon Y.T., JosephB Diers B.W., Farmer A.D., Muehlbauer G.J., Nelson R.T., Grant D., Specht J.E., et al. RNA-Seq Atlas of Glycine max, a guide to the soybean transcriptome. BMC Plant Biol. 2010;10:160. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang X., Cao Y., Ma L. Alternative splicing in plant genes: a means of regulating the environmental fitness of plants. Int J Mol Sci. 2017;18:432. doi: 10.3390/ijms18020432. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar A., Srivastava A.K., Yadav A.K., Sharma M., Pandey A., Raut V.V., Das M.K., Suprasanna P., Pandey G.K. Whole genome transcriptome analysis of rice seedling reveals alterations in Ca 2 ion signaling and homeostasis in response to Ca 2 deficiency. Cell Calcium. 2014;55:155–165. doi: 10.1016/j.ceca.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Shinozaki K., Yamaguchi-Shinozaki K. Molecular responses to dehydration and low temperature, differences and cross-talk between two stress signaling pathways. Curr Opin Plant Biol. 2000;3:217–223. [PubMed] [Google Scholar]
- Sperotto R.A., Ricachenevsky F.K., Duarte G.L., Boff T., Lopes K.L., Sperb E.R., Grusak M.A., Fett J.P. Identification of upregulated genes in flag leaves during rice grain filling and characterization of OsNAC5, a new ABA-dependent transcription factor. Planta. 2009;230:985–1002. doi: 10.1007/s00425-009-1000-9. [DOI] [PubMed] [Google Scholar]
- Su N., He K., Jiao Y., Chen C., Zhou J., Li L., Bai S., Li X., Deng X.W. Distinct reorganization of the genome transcription associates with organogenesis of somatic embryo, shoots, and roots in rice. Plant Mol Biol. 2007;63:337–349. doi: 10.1007/s11103-006-9092-0. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji H., Taoka K., Shimamoto K. Florigen in rice, complex gene network for florigen transcription, florigen activation complex, and multiple functions. Curr Opin Plant Biol. 2013;16:228–235. doi: 10.1016/j.pbi.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Wakasa Y., Oono Y., Yazawa T., Hayashi S., Ozawa K., Handa H., Matsumoto T., Takaiwa F. RNA sequencing-mediated transcriptome analysis of rice plants in endoplasmic reticulum stress conditions. BMC Plant Biol . 2014;14:101. doi: 10.1186/1471-2229-14-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Xie W., Chen Y., Tang W., Yang J., Ye R., Liu L., Lin Y., Xu C., Xiao J., Zhang Q. A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 2010;61:752–766. doi: 10.1111/j.1365-313X.2009.04100.x. [DOI] [PubMed] [Google Scholar]
- Wang Z., Wu Z., Raitskin O., Sun Q., Dean C. Antisense-mediated FLC transcriptional repression requires the p-TEFb transcription elongation factor. Proc Natl Acad Sci USA. 2014;111:7468–7473. doi: 10.1073/pnas.1406635111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann D., Dupanloup I., Excoffier L. Width of gene expression profile drives alternative splicing. PLoS One. 2008;3:e3587. doi: 10.1371/journal.pone.0003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T., Nagasaki H., Yonemaru J., Ebana K., Nakajima M., Shibaya T., Yano M. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genomics . 2010;11:267. doi: 10.1186/1471-2164-11-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Chen X., Zhu C., Peng X., He X., Fu J., Ouyang L., Bian J., Hu L., Sun X., et al. RNA-seq reveals differentially expressed genes of rice (Oryza sativa) spikelet in response to temperature interacting with nitrogen at meiosis stage. BMC Genomics . 2015;16:959. doi: 10.1186/s12864-015-2141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.Y., Hao D.L., Song Z.Z., Yang G.Z., Wang L., Su Y.H. RNA-Seq analysis of differentially expressed genes in rice under varied nitrogen supplies. Gene. 2015;555:305–317. doi: 10.1016/j.gene.2014.11.021. [DOI] [PubMed] [Google Scholar]
- Yuan J., Chen D., Ren Y., Zhang X., Zhao J. Characteristic and expression analysis of a metallothionein gene, OsMT2b, down-regulated by cytokinin suggests functions in root development and seed embryo germination of rice. Plant Physiol. 2008;146:1637–1650. doi: 10.1104/pp.107.110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M.-S., Umemoto T., Kawagoe Y. Rice debranching enzyme isoamylase3 facilitates starch metabolism and affects plastid morphogenesis. Plant Cell Physiol. 2011;52:1068–1082. doi: 10.1093/pcp/pcr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun D., Liang W., Dreni L., Yin C., Zhou Z., Kater M.M., Zhang D. OsMADS16 genetically interacts with OsMADS3 and OsMADS58 in specifying floral patterning in rice. Mol Plant. 2013;6:743–756. doi: 10.1093/mp/sst003. [DOI] [PubMed] [Google Scholar]
- Zeeberg B.R., Feng W., Wang G., Wang M.D., Fojo A.T., Sunshine M., Narasimhan S., Kane D.W., Reinhold W.C., Lababidi S. GoMiner, a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Guo G., Hu X., Zhang Y., Li Q., Li R., Zhuang R., Lu Z., He Z., Fang X., et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010;20:646–654. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Hultin-Rosenberg L., Forshed J., Branca R.M., Orre L.M., Lehtiö J. SpliceVista, a tool for splice variant identification and visualization in shotgun proteomics data. Mol Cell Proteomics. 2014;13:1552–1562. doi: 10.1074/mcp.M113.031203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai R., Feng Y., Wang H., Zhan X., Shen X., Wu W., Zhang Y., Chen D., Dai G., Yang Z., et al. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genomics . 2013;14:19. doi: 10.1186/1471-2164-14-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Fung-Leung W.-P., Bittner A., Ngo K., Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One. 2014;9:e78644. doi: 10.1371/journal.pone.0078644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.