

Abstract
Glioblastoma is the most frequent and most aggressive brain tumor in adults. Solute carrier family 8 member 2 (SLC8A2) is only expressed in normal brain, but not present in other human normal tissues or in gliomas. Therefore, we hypothesized that SLC8A2 might be a glioma tumor suppressor gene and detected the role of SLC8A2 in glioblastoma and explored the underlying molecular mechanism. The glioblastoma U87MG cells stably transfected with the lentivirus plasmid containg SLC8A2 (U87MG-SLC8A2) and negative control (U87MG-NC) were constructed. In the present study, we found that the tumorigenicity of U87MG in nude mice was totally inhibited by SLC8A2. Overexpression of SLC8A2 had no effect on cell proliferation or cell cycle, but impaired the invasion and migration of U87MG cells, most likely through inactivating the extracellular signal-related kinases (ERK)1/2 signaling pathway, inhibiting the nuclear translocation and DNA binding activity of nuclear factor kappa B (NF-κB), reducing the level of matrix metalloproteinases (MMPs) and urokinase-type plasminogen activator (uPA)-its receptor (uPAR) system (ERK1/2-NF-κB-MMPs/uPA-uPAR), and altering the protein levels of epithelial to mesenchymal transitions (EMT)-associated proteins E-cardherin, vimentin and Snail. In addition, SLC8A2 inhibited the angiogenesis of U87MG cells, probably through combined inhibition of endothelium-dependent and endothelium-nondependent angiogenesis (vascular mimicry pattern). Totally, SLC8A2 serves as a tumor suppressor gene and inhibits invasion, angiogenesis and growth of glioblastoma.
Keywords: angiogenesis, glioblastoma, growth, invasion, SLC8A2, U87MG
INTRODUCTION
Gliomas are the most common and lethal type of adult primary brain tumors, accounting for more than 46% of all brain tumors (Louis et al., 2007). Glioblastoma is the highest grade glioma (WHO grade IV) and comprises 50–60% of all gliomas (Zhang et al., 2015). Despite comprehensive treatment, the prognosis of patients with glioblastoma remains poor, with the overall 5-year survival rate of less than 5% (Vigneswaran et al., 2015). The dismal prognosis of glioblastoma patients is associated with the resistance to chemotherapy and radiation as well as the high recurrence after surgical resection (Wang et al., 2015; Zhang et al., 2015). In general, the urgent need for exploring novel therapeutic targets of glioblastoma has been highlighted.
A characteristic molecular feature of most gliomas is the combination of various genetic aberrations, leading to both inactivation of tumor suppressor genes and amplification of oncogenes. Loss of heterozygosity (LOH) at the long arm of chromosome 19 (LOH 19q) is a common genetic alteration in human gliomas, whereas such a deletion is rare in most other common malignancies (Reifenberger and Louis, 2003; von Deimling et al., 2000). LOH 19q is associated with higher grade and poorer prognosis of gliomas, indicating glial-specific tumor suppressor genes in this chromosome region. Several deletion mapping studies of gliomas have narrowed the 19q commonly deleted region to an interval within the cytogenetic band 19q13.3 (Hartmann et al., 2002). Solute carrier family 8 member 2 (SLC8A2), also called sodium/calcium exchanger 2 (NCX2), is located on chromosome 19q13.32 (Qu et al., 2010). SLC8A2 encodes a Na+/Ca2+ exchanger mediates cellular Ca2+ efflux/influx and thus contributes to intracellular Ca2+ homeostasis (Quednau et al., 2004). Cytosolic Ca2+ plays a crucial role in intracellular signaling pathways, some of which are relevant to tumorigenesis and tumor progression (Berridge et al., 2003). In the previous study, from gene expression databases, we found that the expression of SLC8A2 was abundant in normal brain, but not present at any significant levels in gliomas (Qu, Jiao et al., 2010). We also found that DNA methylation played a key role in the transcriptional silencing of SLC8A2 in gliomas (Qu et al., 2010). Accordingly, the hypothesis was that SLC8A2 might be a tumor suppressor gene involved in gliomas development.
In the present study, to test this hypothesis, we constructed the human glioblastoma cell line U87MG stably transfected with SLC8A2 and assessed the role of SLC8A2 as a glioblastoma suppressor via in vivo and in vitro experiments.
MATERIALS AND METHODS
Cell line and culture
Human umbilical vein endothelial cells (HUVECs) and human glioblastoma cell line U87MG were obtained from the Cell Bank of the Chinese Academy of Science. The cells were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Invitrogen Life Technologies, Barcelona, Spain) supplemented with 10% fetal bovine serum (FBS, HyClone), penicillin (100 U/ml) and streptomycin (100 μg/ml) in a humidified 5% CO2 at 37°C.
Antibodies
Rabbit anti-ERK1/2 MAPK, rabbit anti-phospho-ERK1/2 MAPK (Thr202/Tyr204), rabbit anti-p38 MAPK, rabbit anti-phospho-p38 MAPK (Thr180/Tyr182), rabbit anti-SAPK/JNK, rabbit anti-phospho-SAPK/JNK (Thr183/Tyr185), rabbit anti-AKT, rabbit anti-phospho-AKT, rabbit anti-p65, rabbit anti-IkB, rabbit anti-MMP-2, rabbit anti-MMP-9, rabbit anti-lamin B1, rabbit anti- -β-catenin were from Cell Signaling Technology (USA). Rabbit anti-phospho-IkB was from Abcam (UK). Horsera-dish peroxidase-labeled goat anti-rabbit IgG, horsera-dish peroxidase-labeled goat anti-mouse IgG, mouse anti-GAPDH, and mouse anti-actin were from Multi-Sciences Biotech Co. Ltd (China). Rabbit anti-SLC8A2 was from MBL International Corporation (Japan).
Plasmid construction and stable transfection
To construct a recombinant lentivirus plasmid expressing the SLC8A2 gene, the open reading frame of the SLC8A2 was cloned into pENTR223.1 vector (Invitrogen, USA ) to generate Lenti-SLC8A2-IRES-EGFP. U87MG cells were seeded into 6-well plates and the next day Lenti-SLC8A2-IRES-EGFP was put into the plates to co-culture for 24 h. Then cells were split at a 1:5 dilution and exposed for 2–3 weeks in Blasticidin (Calbiochem, USA; Cat.No.400052) containing medium (2 μg/μl) and colonies were picked for the resistance to Blasticidin. Expression of SLC8A2 was confirmed by Western blot using a monoclonal antibody against SLC8A2. The U87MG stably transfected with Lenti-EGFP served as negative control. In the following statement, U87MG-SLC8A2 cells stand for U87MG cells stably transfected with SLC8A2. The control, U87MG-NC cells, for U87MG cells stably transfected with Lenti-EGFP. For further validation, the expression of SLC8A2 was confirmed by Western blot (Fig. 1A).
Fig. 1. SLC8A2 inhibits in vivo growth of xenograft tumors of U87MG cells.
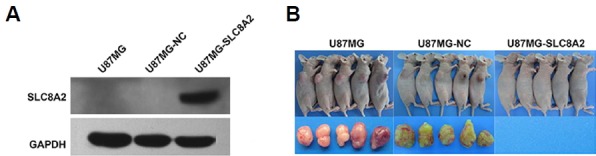
(A) The U87MG-SLC8A2 cells which were stably transfected with Lenti-SLC8A2-IRES-EGFP were confirmed by Western blot to overexpress SLC8A2 protein, while the U87MG and U87MG-NC (control cell) were not. GAPDH served as the loading control. (B) Effect of SLC8A2 on growth of xenograft tumors of U87MG cells. The 4 × 106 cells of the U87MG, U87MG-NC and U87MG-SLC8A2 in 0.1 ml PBS were injected subcutaneously into the right flank of 4-week-old nude mice. Each group contains five mice. After 4 weeks, no mouse developed tumor in U87MG-SLC8A2 cell group, while all five mice developed tumors in both U87MG-NC and U87MG cell group.
In vivo xenograft experiments in nude mice
Fifteen 4-week-old BALB/c nude mice were obtained from Animal Center of Soochow University (China). All animal operations were performed in strict accordance with the guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health. This study was approved by the Research Ethic Committee of Soochow University. The mice were randomly divided into three groups (n = 5 for each group): U87MG, U87MG-NC, and U87MG-SLC8A2. The cells were injected subcutaneously into the right flank of each mouse at a density of 4 × 106 cells. Tumor growth was monitored twice a week. At the end of 4 weeks, tumors from mice were dissociated and evaluated.
Cell proliferation assay
Cell growth was measured using the cell counting kit-8 (CCK-8, Dojindo Molecular Technologies, Gaithersburg, USA) according to the manufacturer’s protocol. Briefly, cells (2.5 × 103/well in 100 μl medium) of the three groups were seeded into the 96-well plates and 10 μl CCK-8 solution (Sigma, USA) was added into each well at the same time point every day for 6 d. After incubation for 90 min, the optical density (OD) at 450 nm was measured with averages from six replicates using a microplate reader.
Cell cycle assay by flow cytometry
Cells of the three groups were plated into a 6-well plate at a density of 1.5 × 105 cells/ml with a total volume of 2 ml. After cultured for 48 h at 37°C, cells were then collected and centrifuged at 300 × g for 5 min. Cell pellets were then resuspended in ice-cold ethanol (75%) and fixed for at least 24 h at −20°C. To determine cellular DNA content, fixed cells were centrifuged at 300 × g for 5 min and washed with PBS for one time. Re-suspend the cell pellets in 100 μl Rnase A (KeyGEN, China) at 37°C for 30 min, and then stain the cells with propidium iodide (PI) (KeyGEN) for 30 min in dark. Samples were analyzed by flow cytometry on Cytomics FC 500 (Beckman Coulter, USA) with the excitation of an argon laser at 488 nm. The percentage of cells in G1, S, and G2/M phases of cell cycle were analyzed with (Multiple) Software Version: CXP 2.2.
Cell migration and invasion assays
The cells (2 × 104/well) were suspended in 100 μl of serum-free medium and seeded into the upper chambers of the 24-well Transwell inserts with 8 μm pores (Corning, USA). DMEM with 10% FBS was added in the lower chambers. Following 48 h of incubation at 37°C, the cells on the upper surface of the filter were mechanically removed with a cotton swab, and the migrated cells on the lower surface were fixed with crystal violet cell colony staining kit (GenMed, USA). The stained cells were counted in five randomly selected fields per filter under a microscope (100 × magnification). The cell invasion potential was assessed as described above, except that the Transwell inserts were coated with Matrigel (60 μl/well, BD Bioscience, USA). Each experiment was performed in triplicate wells and repeated three times.
RNA isolation and reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells using TRIzol kit (Invitrogen, USA) and RT-PCR was then conducted following the manufacturer’s instructions. Gene primers were listed as follows: uPA: 5′-ACTCCAAAGGCAGCAATGAACT-3′ (forward), 5′-GTGCTGCCCTCCGAATTTCT-3′ (reverse); uPAR: 5′-CACAAAACTGCCTCCTTCCT-3′ (forward), 5′-AATCCCCGTTG GTCTTACAC-3′ (reverse); MMP-2 : 5′-CAGGCTCTTCTCCTTT CACAAC-3 (forward), 5′-AAGCCACGGCTTGGTTTTCCTC-3′ (reverse); MMP-9: 5′-TGGGCTTAGATCATTCCTCAGT-3′ (forward), 5′-TTCCCATCCTTGAACAAATACA-3′ (reverse); COX-2: 5′-ACAACATTCCCTTCCTTC-3′ (forward), 5′-CCTTATTTCCTTT CACACC-3′ (reverse); HIF-1α: 5′-CCTGAGCCTAATAGTCCC-3′ (forward), 5′-GGTGGCATTAGCAGTAGG-3′ (reverse); VEGF: 5′-CGAAACCATGAACTTTCTGCTGTC-3′ (forward) and 5′-TCACCGCCTCGGCTTGTCACAT-3′ (reverse); GAPDH: 5′-AAT GGGCAGCCGTTAGGAAA-3′ (forward), 5′-GCCCAATACGA CCAAATCAGAG-3′ (reverse). The relative expression of each gene was calculated by comparison with the housekeeping gene GAPDH.
Western blot
Cells were washed three times with pre-colded PBS solution and whole cell proteins were extracted by incubation with the radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China). The nuclear extracts were prepared using a NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, USA) according to the manufacturer’s instructions. Both protease and phosphatase inhibitor (Roche, Switzerland) were contained in the protein extracts. Protein concentration was determined using a BCA Protein Assay Kit (Beyotime). Equal amounts of protein (50 μg) were separated by sodium dodecyl sulfate-poly-acrylamide gel electrophoresis and electrophoretically transferred to poly-vinylidene difluoride membranes. The membranes were blocked with 5% non-fat dry milk for 1 h at room temperature and then incubated overnight at 4°C with the primary antibodies. After washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at 37°C. Immunoblots were visualized by Enhanced Chemiluminescence Detection Kit for HRP (Biological Industries, Israel) using Kodak X-OMAT LS film (Eastman Kodak, USA). The blots were subsequently stripped through incubation in TBST buffer and re-probed for GAPDH, lamin B1, or β-actin as a loading control. Quantitative data were obtained using Quantity One Software (Bio-Rad).
Gelatin zymography assay
Cells (about 70–80% cell density of confluent culture) were washed and cultured with serum-free DMEM for 18 h. Then the supernatant was collected and concentrated with the 30 kDa amicons (Millipore, USA). The enzymatic activity of MMP-2 was determined by SDS-PAGE as follows. Serum-free culture medium containing 25 μg protein per sample was prepared in non-denaturing loading buffer (0.5 M Tris-HCl, pH 6.8, 10% SDS, 0.1% bromophenol blue, and 10% glycerol) and were size-fractionated in 10% SDS-polyacrylamide gel impregnated with 0.1% gelatin. The gels were then washed with 2.5% Triton X-100 three times for 20 min at room temperature to remove SDS and then incubated in a developing buffer (50 mM Tris-HCl buffer (pH 7.4), 20 mM NaCl, 10 mM CaCl2, and 0.1 NaN3) for 40 h at 37°C. Subsequently, gels were fixed and stained with 10% 2-propanol and 10% acetic acid containing 0.5% Coomassie Blue R250. Gelatinase activity was visualized as clear bands within the stained gel. Gels were scanned, and a densitometric analysis was performed using the image analysis program Quantity One (Bio-Rad).
Human umbilical vein endothelial cells (HUVECs) tube formation assay
To evaluate the effect of SLC8A2 on the functional activity of angiogenesis in vitro, HUVECs tube formation assays were performed. Briefly, HUVECs cells were suspended in serum-free conditioned medium (CM) obtained from culture supernatant of U87MG, U87MG-NC or U87MG-SLC8A2 cells. 300 μl Matrigel (BD, Biosciences) were pipetted into 24-well plates and allowed to polymerize for 1 h at 37°C. HUVECs cells were then seeded onto 24-well plates coated with the polymerized Matrigel at a density of 5 × 103 cells/well in 500 μl of the indicated CM and incubated at 37°C in humidified 5% CO2 for 6 h. The capillary tube-like structures formed by HUVECs was observed and photographed under an inverted microscope.
Three-dimensional (3D) matrigel culture
Three-dimensional culture was used to evaluate the ability of cells to form vascular mimicry structure. U87MG, U87MG-NC, or U87MG-SLC8A2 cells (1.8 × 104) were re-suspended in 200 ml serum-free DMEM and added to a Matrigel-coated 48-well plate. The concentration of Matrigel used was 60%. After incubation at 37°C for 4 h, the capacity of vascular mimicry formation was observed under an inverted microscope.
Microarray assay and functional enrichment analysis
Total RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s protocol. Gene expression was analyzed using an Affymetrix GeneChip® Human Genome U133 Plus 2.0 Array. Samples for array hybridization were prepared as described in the Affymetrix geneChip® expression Technical Manual. The differentially expressed genes selected out from the microarray data set were subjected to Kyoto encyclopedia of genes and genomes (KEGG) pathway and Gene Ontology (GO) term enrichment analysis to identify altered biological functions.
Statistical analysis
Statistical analyses were performed using SPSS version 19.0 (SPSS, USA). Data are expressed as mean values ± SD. All data were analyzed by Student’s t-test or one-way ANOVA. Differences were considered statistically significant with an alpha valve of P < 0.05.
RESULTS
SLC8A2 inhibits in vivo growth of xenograft tumors of U87MG cells
We constructed the U87MG cells stably transfected with SLC8A2 to assessed the role of SLC8A2 in glioblastoma. Western blot analysis indicated that the protein level of SLC8A2 was significantly up-regulated in U87MG-SLC8A2 group as compared with untreated cells (U87MG) and negative control (U87MG-NC) group, verifying that U87MG cells stably transfected with SLC8A2 were well constructed (Fig. 1A). Then, we established xenograft mouse models to investigate the biological effect of overexpressing SLC8A2 on tumorigenicity of U87MG cells in vivo. Interestingly, our result showed that no mouse developed tumor in U87MG-SLC8A2 group, while all five mice developed tumors in both U87MG and U87MG-NC groups (Fig. 1B). These results indicated that SLC8A2 overexpression could inhibit the tumorigenicity of U87MG cell in vivo.
SLC8A2 does not affect the proliferation and cell cycle progression of U87MG cells in vitro
We then investigated the biological function of SLC8A2 on the proliferation of U87MG cells. CCK-8 assay was performed to determine the growth rate of the U87MG, U87MG-NC and U87MG-SLC8A2 cells in 6 d. As shown in Fig. 2A, no significant difference was noted among the three groups. In the following cell cycle analysis, we found that the percentages of the cells in the G0/G1 phase among U87MG, U87MG-NC and U87MG-SLC8A2 cells had no significant difference. And there was also no significant difference in the S and G2/M phase among the three groups (Fig. 2B). These data indicated that SLC8A2 could not affect the cell proliferation of U87MG cells in vitro.
Fig. 2. SLC8A2 does not affect the proliferation and cell cycle progression of U87MG cells in vitro.
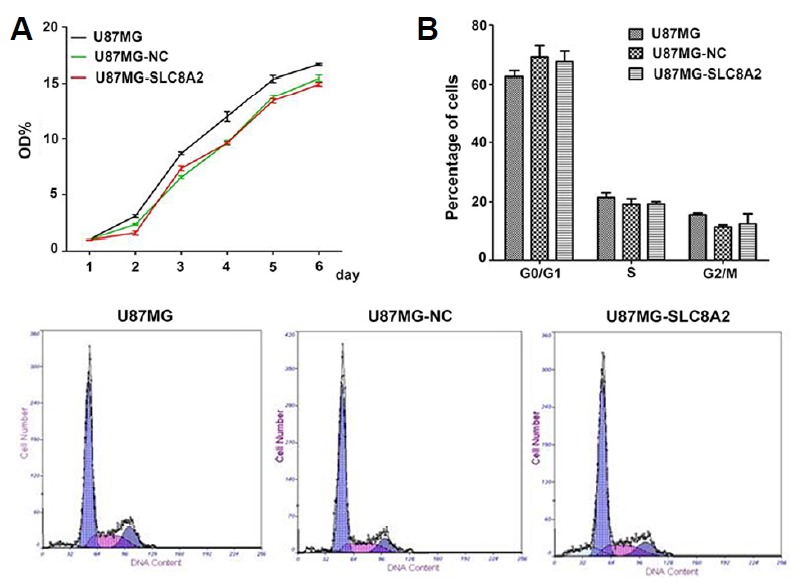
(A) Effect of SLC8A2 on proliferation of U87MG cells in vitro. The U87MG, U87MG-NC and U87MG-SLC8A2 cells were subjected to CCK-8 assay for 6 d. There was no significant difference among three groups. (B) Effect of SLC8A2 on cell cycle progression of U87MG cells in vitro. The U87MG, U87MG-NC and U87MG-SLC8A2 were subjected to cell cycle analysis by flow cytometry. Percentages of cells in G0/G1, S and G2/M of the three groups were scored and depicted. There was no significant difference among three groups.
SLC8A2 inhibits the invasion and migration of U87MG cells
By using Transwell assays, the effect of SLC8A2 on the invasion and migration of U87MG cells was evaluated. Cells from each group that moved through the Transwell chambers and the Matrigel-coated Transwell chambers were counted. As showed in the Fig. 3, the number of migrated and invaded cells in U87MG-SLC8A2 cells was significantly less than that in both U87MG and U87MG-NC, suggesting that SLC8A2 could inhibit the invasion and migration of U87MG cells.
Fig. 3. SLC8A2 inhibits the invasion and migration of U87MG cells.
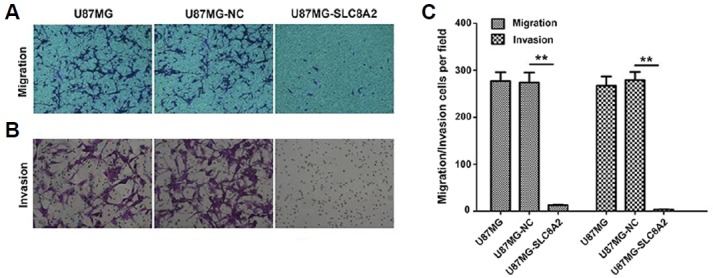
(A) Migration and (B) Matrigel invasion assays were performed using U87MG, U87MG-NC and U87MG-SLC8A2 cells. Representative images were shown. Data are presented as the mean number of the migration and invasion cells per filed counted from five randomly selected fields under a microscope (100 × magnification). **P < 0.01. There was no significant difference between the U87MG and the U87MG-NC groups.
SLC8A2 inhibits the expression of genes related to invasion and migration in U87MG cells
To further explore the molecular mechanisms of SLC8A2 on tumor invasion and migration, we analyzed the mRNA transcription of several important genes related to invasion and migration in glioblastoma. Matrix metalloproteinases (MMPs) and fibrinolytic system members were excessively expressed in glioblastoma and have direct association with the invasion and migration of glioblastoma (Gondi et al., 2004; Lakka et al., 2001; Nakano et al., 1995). Urokinase-type plasminogen activator PA (uPA) and its receptor uPAR are key components of the fibrinolytic system. As showed in Fig. 4A, the mRNA levels of MMP-2, MMP-9, uPA and uPAR were significantly reduced in U87MG-SLC8A2 cells as compared with that in U87MG-NC and U87MG cells.
Fig. 4. Effect of SLC8A2 on the mRNA transcription and protein expression related to invasion and migration as well as the enzyme activity of MMP-2 in U87MG cells.
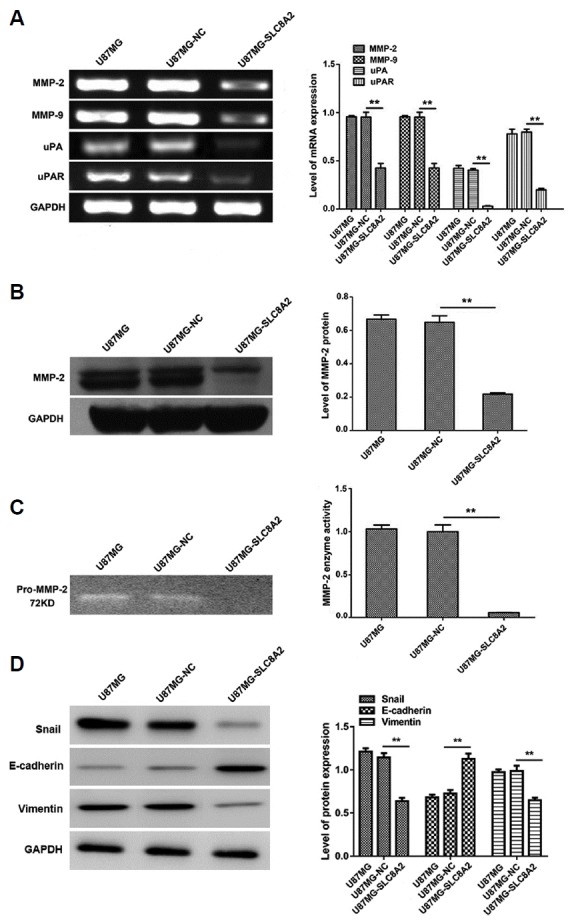
One representative result of three independent preparations is shown. (A) RT-PCR was conducted to detect the mRNA expression of MMP-2, MMP-9, uPA and its receptor uPAR in U87MG, U87MG-NC and U87MG-SLC8A2 cells. GAPDH served as the internal control. (B) The protein levels of MMP-2 in U87MG, U87MG-NC and U87MG-SLC8A2 cells were examined by Western blot. GAPDH served as the loading control. (C) The activity of MMP-2 secreted in the DMEM was determined by gelatin zymography. (D) The protein levels of Snail, E-cardherin and vimentin in U87MG, U87MG-NC and U87MG-SLC8A2 cells were examined by Western blot. GAPDH served as the loading control. **P < 0.01. There was no significant difference between the U87MG and the U87MG-NC groups.
In addition, Western blot analysis revealed that SLC8A2 significantly down-regulated the protein level of MMP-2 (Fig. 4B). Moreover, the gelatin zymogram analysis showed that the enzymatic activity of MMP-2 which was secreted in the serum-free DMEM was almost blocked by SLC8A2, which was consistent with the decrease in protein abundance of MMP-2 (Fig. 4C). These findings suggest that the anti-invasive effect of SLC8A2 is related to the inhibition of MMPs and uPA expression in U87MG cells.
Besides, the protein expression levels of epithelial to mesenchymal transitions (EMT)-associated proteins E-cardherin, vimentin and Snail were also detected. As shown in Fig, 4D, the protein levels of vimentin and Snail were significantly decreased, whereas the protein level of E-cardherin was significantly increased in U87MG-SLC8A2 cells as compared with that in U87MG-NC and U87MG cells.
SLC8A2 inhibits U87MG migration and invasion through ERK1/2-NF-κB-MMPs/uPA-uPAR signals
The kB binding site of nuclear factor kappa B (NF-κB) was identified in the promoter regions of MMP-2, MMP-9, uPA, and uPAR genes (Li et al., 2007; Yan and Boyd, 2007). The activation of NF-κB pathways could trigger their mRNA transcription (Li et al., 2007). To evaluate whether SLC8A2 regulated NF-κB pathway, the protein levels of total p65, inhibitor of kB (IkB), phospho-IkB (p-IkB), p65 in the nucleus were detected by Western blot. As shown in Fig. 5A, the protein level of total p65 was not altered in the three groups. However, the p-IkB level was reduced, the total IkB level was increased, as a matter of course, the p65 level in the nucleus was reduced in the U87MG-SLC8A2 cells as compared to the control, U87MG-NC cells (Fig. 5B). There was no difference between U87MG-NC and U87MG cells. These findings indicated that SLC8A2 could inhibit the transference of p65 from cytoplasm into nucleus, thus impaired the activation of NF-κB.
Fig. 5. Effect of SLC8A2 on NF-kB and MAPK (ERK1/2, p38, JNK, AKT) pathways in U87MG cells.

(A) The protein levels of total-IkB, p-IkB and total p65 in U87MG, U87MG-NC and U87MG-SLC8A2 cells were determined by Western blot. GAPDH served as the loading control. **P < 0.01. (B) The protein level of p65 in nuclear in U87MG, U87MG-NC and U87MG-SLC8A2 cells was determined by Western blot. Lamin B1 served as the nuclear control. **P < 0.01. (C) The protein levels of p-ERK1/2, ERK1/2, p-p38, p38, p-JNK, JNK, p-AKT and AKT are determined by Western blot. GAPDH served as the loading control. Blots shown in the experiment are typical of three independent experiments. **P < 0.01.
Mitogen-activated protein kinase (MAPK) and AKT pathways were frequently extensively deregulated in glioblastoma and their excessive activation led to abnormal activation of NF-κB and overexpression of MMP-2, MMP-9, uPA, and uPAR genes. MAPK has been classified into three major subfamilies: extracellular signal-related kinases (ERK) 1/2, c-Jun N-terminal kinase (JNK), and p38 (Keyse, 2000; Sui et al., 2014). The protein levels of AKT and MAPKs (ERK, JNK, p38) were investigated in U87MG-SLC8A2 cells. Among all kinases tested, ERK1/2 phosphorylation was selectively and dramatically decreased without altering its protein amount in U87MG-SLC8A2 cells, whereas the phosphorylation of JNK, p38 and AKT showed no significant change (Fig. 5C), indicating that SLC8A2 might reduce the migration and invasion of U87MG cells by inactivating the ERK1/2 MAPK pathway. Collectively, SLC8A2 inhibited U87MG invasion through ERK1/2-NF-κB-MMPs/uPA-uPAR signals.
Effect of SLC8A2 on angiogenesis of U87MG in vitro
Interaction between tumor cells and endothelial cells in glioblastoma was stimulated by Transwell-based co-culture of U87MG and HUVECs cells. To determine the effect of SLC8A2 overexpression on angiogenesis in glioblastoma microenvironment, endothelial cell migration, invasion and tube formation assays were performed. HUVECs cells were suspended in serum-free CM obtained from culture supernatant of U87MG, U87MG-NC or U87MG-SLC8A2 cells. Transwell assays revealed that the migration and invasion of endothelial cells were significantly inhibited by SLC8A2 overexpression as compared with U87MG and U87MG-NC group. And no significant difference was observed between U87MG-NC and U87MG cells (Fig. 6A). The above findings indicated that SLC8A2 overexpression significantly inhibited U87-induced migration and invasion of endothelial cells. Additionally, HUVECs tube formation assay showed that the tube formation of capillary-like structures by HUVECs cocultured with U87MG-SLC8A2 cells was significantly suppressed compared with U87MG and U87MG-NC group (Fig. 6B). Furthermore, the expression of vascular endothelial growth factor (VEGF), an angiogenic factor important for the induction of angiogenesis and blood vessel formation, was detected to investigate the molecular mechanism underlying the inhibition of angiogenesis by SLC8A2. We found that SLC8A2 significantly inhibited the expression of VEGF189, VEGF165, and VEGF121 (Fig. 6C), suggesting the inhibitory effect of SLC8A2 on VEGF-VEGF receptor system where tumor cells interacts with endothelial cells. Moreover, SLC8A2 overexpression significantly down-regulated the expression levels of some tumor angiogenesis-related genes, such as cyclooxygenase-2 (COX-2) (Fig. 6C) and hypoxia-inducible factor 1α (HIF-1α) (Fig. 6D).
Fig. 6. Effect of SLC8A2 on angiogenesis of U87MG in vitro.
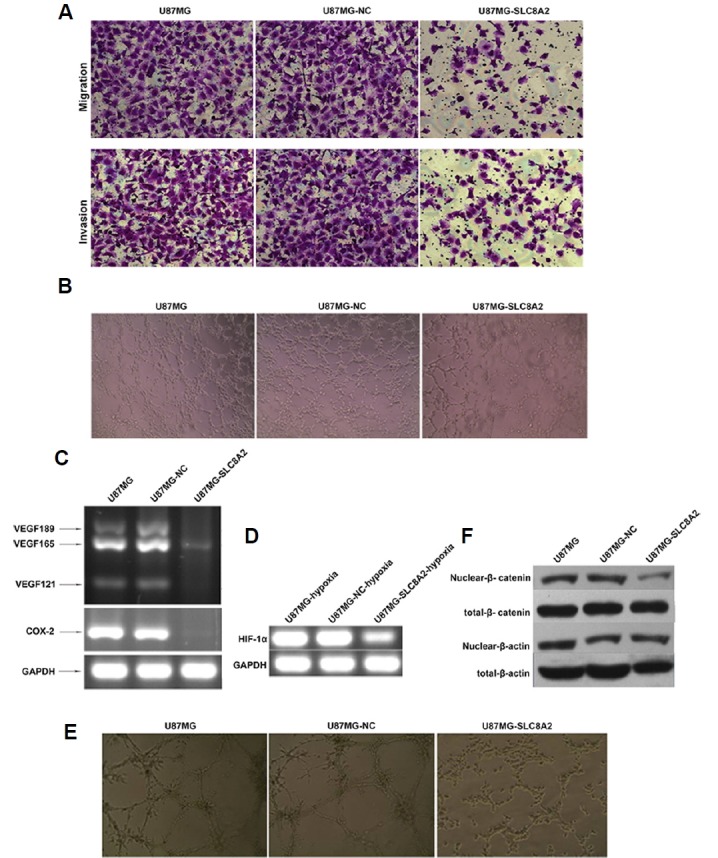
HUVECs cells were suspended in serum-free conditioned medium obtained from culture supernatant of U87MG, U87MG-NC or U87MG-SLC8A2 cells. (A) Cell migration and invasion of endothelial cells cocultured with U87MG, U87MG-NC and U87MG-SLC8A2 cells were evaluated by Transwell assays. (B) The tube formation of capillary-like structures by HUVECs co-cultured with U87MG, U87MG-NC, and U87MG-SLC8A2 cells were evaluated by HUVECs tube formation assay. (C) The expression of VEGF189, VEGF165, and VEGF121, COX-2, and (D) HIF-1α in U87MG, U87MG-NC and U87MG-SLC8A2 cells was detected by RT-PCR. GAPDH served as the internal control. (E) The three-dimensional Matrigel tube formation assay was used to investigate the capacity of vascular mimicry formation. (F) The protein levels of nuclear-β-catenin and total-β-catenin were examined by Western blot. The β-actin served as the loading control.
We then evaluated the effect of SLC8A2 on the vascular mimicry (VM) formation of U87MG cells using the 3D culture model. As shown in Fig. 6E, U87MG-SLC8A2 cells cultured in 3D vascular glue failed to form a complete vascular-like structure, and the cell morphology could not change plasticity, whereas U87MG and U87MG-NC cells displayed stronger ability of VM formation, with better vascular cavity structure. These results indicated that SLC8A2 may inhibit U87MG cells to form VM. COX-2 and Wnt/β-catenin were shown to play important biological roles in the formation of VMs (Paulis et al., 2010; Rong et al., 2016). β-catenin is a critical component of Wnt/β-catenin pathway. The attenuation of the Wnt/β-catenin pathway was due to the loss of nuclear β-catenin. Here, we found that, consistent with the decrease of COX-2, SLC8A2 overexpression inhibited protein levels of nuclear-β-catenin, indicating that SLC8A2 may inhibit Wnt/β-catenin pathway (Fig. 6F).
Effect of SLC8A2 overexpression on gene expression profile and signaling
To investigate the molecular mechanism of SLC8A2 gene expression in suppressing glioblastoma, total RNA was isolated from U87MG-NC and U87MG-SLC8A2 cells and analysis of gene expression profiling was performed using the Affymetrix GeneChip® Human Genome U133 Plus 2.0 Array. The array contains 47,000 transcripts, representing 38,500 well-characterized human genes. Analysis identified significant expression differences of 2853 genes after SLC8A2 overexpression, with 1555 genes being upregulated and 1298 genes being down-regulated (fold change > 2.0, P < 0.01), as compared with the U87MG-NC group. The heatmap for cluster analysis of differentially expressed genes was shown in Fig. 7.
Fig. 7. The heatmap for cluster analysis of differentially expressed genes.
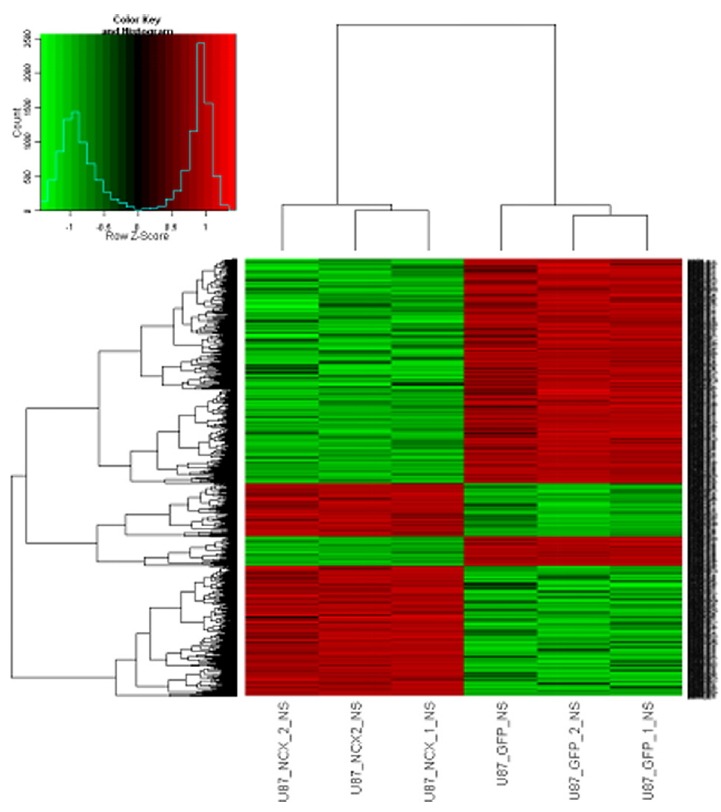
The fold change in gene expression levels between U87-NC and U87-SLC8A2 cells. Fold change>2.0, P < 0.01. Red, up-regulation of gene expression; Green, down-regulation of gene expression.
KEGG pathway enrichment analysis showed that these differentially expressed genes were involved in a total of 98 significantly altered pathways including MAPK, Wnt, and TGF-β, as well as biological processes including cell adhesion, tightness junction, apoptosis, cell cycle, and nucleotide excision repair (Enrichment test P value <0.01, false discovery rate (FDR) P value <0.01). The 24 pathways with most significant changes were listed in Supplementary Table 1.
The results of Gene Ontology (GO) term enrichment analysis revealed that these differentially expressed genes were associated with cell proliferation, cell communication and cell migration (Enrichment test P value <0.01, FDR P value <0.01). The first 7 GO terms with most significant changes were listed in Supplementary Table 2.
DISCUSSION
The glioblastoma cell line U87MG, originally derived from a human glioblastoma, is highly proliferative and tumorigenic both in vivo and in vitro. Our previous study showed that SLC8A2 was totally silenced in U87MG cells (Qu et al., 2010). Thereby, the stably SLC8A2-transfected U87MG cell line, U87MG-SLC8A2, was constructed. The xenograft experiments in nude mice showed that SLC8A2 could inhibit the tumorigenicity of U87MG cell in vivo, verifying that SLC8A2 possesses tumor-suppressive functions. However, we found that SLC8A2 did not significantly affect the cell proliferation and cell cycle of U87MG in vitro. The inconsistent biological function of SLC8A2 in vitro and in vivo might be the interplay between SLC8A2 and the complex tumor ecosystem, which was also called the tumor microenvironment. Minor alterations in single component of the tumor ecosystem may cause a dramatic reorganization of the whole system (Sounni and Noel, 2013). We assume that SLC8A2 may interfere with certain element of the tumor ecosystem and thus counteracted growth of U87MG xenograft in vivo.
Malignant gliomas including glioblastoma are characterized by local invasion (Jan et al., 2010) and there is rarely a clear border between the tumor and the surrounding normal brain parenchyma, both of which make it an important contribution to the ineffectiveness of current treatment modalities. Understanding the mechanisms of glioblastoma invasion and migration is therefore vital for designing effective therapeutic interventions to prevent the spread of this disease (Claes et al., 2007; Griscelli et al., 2000; Lefranc et al., 2009). The invasion process of glioblastoma is complicated, involving a series of sequential steps. The degradation of the extracellular matrix (ECM) by the proteolytic enzymes is the first step of this process. MMPs and uPA-uPAR system are the key proteases produced by glioma cells involving the degradation of ECM, and uPA may cleaves and activates the MMPs in a directly or indirectly way (Chintala et al., 1999; Rao, 2003; Kwiatkowska and Symons, 2013). High levels of MMPs and uPA/uPAR are closely related to the malignant gliomas progression (Lakka et al., 2001; Nakano et al., 1995). Silence of uPA, uPAR and MMP-9 inhibited glioma cell invasion (Gondi et al., 2004). The EMT is a key step toward cancer invasion and metastasis(Guo et al., 2014). In the present study, we found that the expression of MMP-2, MMP-9, uPA and uPAR was dramatically inhibited by SLC8A2 in U87MG cells. In addition, the activity of MMP-2 was blocked, which was consistent with its decreased protein level. As the basic expression level of MMP-9 in U87MG cells in vitro was low, it was quite hard to determine the change of its activity. Besides, the protein levels of EMT-associated proteins E-cardherin, vimentin and Snail were altered after SLC8A2 knockdown in U87MG cells in vitro. These results suggested that SLC8A2 inhibited the invasive behavior of U87MG cells, at least partially, probably by regulating the aberrant expression level and activity of MMPs and uPA-uPAR, as well as altering the protein levels of EMT-associated proteins.
NF-κB pathway is constitutively activated and plays a key role in the tumorigenesis, tumor invasion, metastasis, and angiogenesis of human gliomas (Gray et al., 2014; Li et al., 2007). It is an effective strategy to suppress invasion of glioma cells by blocking NF-κB activity (Li et al., 2007). NF-κB is a sequence-specific transcription factor that belongs to the reticuloendotheliosis (Rel) family proteins (Karin et al., 2002; Ravi and Bedi, 2004). The NF-κB family contains five closely related DNA-binding proteins: RelA (p65), RelB, c-Rel, NF-κB1 (p50) and NF-κB2 (p52) (Verma et al., 1995). Stimulation including the cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-1 leads to the rapid phosphorylation, ubiquitination, and ultimately proteolytic degradation of NF-κB intrinsic inhibitor IκB, which frees NF-κB-p65 to translocate from cytoplasm to the nucleus and activate the transcription of its target genes (Rothwarf and Karin, 1999; Yamamoto and Gaynor, 2004). Reports showed that constitutive NF-κB activity in cancer cells can be inhibited through down-regulation of the NF-κB intrinsic inhibitor IκB (Li et al., 2007). The functional NF-κB-binding sites were identified in the promoter regions of MMP-9, uPA and uPAR (Li et al., 2007; Yan and Boyd, 2007). The activated NF-κB factor in nucleus may functionally bind to their promoters and directly regulates their mRNA transcription (Li et al., 2007). Here, we found that the phosphorylation of IkB (p-IkB) and the p65 level in the nucleus were significantly reduced in the U87MG-SLC8A2 cells as compared with the control, indicating that SLC8A2 could inhibit the transference of p65 from cytoplasm into nucleus, thus impaired the activation of NF-κB in U87MG cells. This might be the molecular mechanism by which SLC8A2 reduced the expression levels of the proteolytic enzymes, MMP-2, MMP-9, uPA and uPAR (NF-κB-MMPs/uPA-uPAR).
MAPK family constitutes important mediators of signal transduction pathways that serve to coordinate the cellular response to a variety of extracellular stimuli. Based on structural differences, it has been classified into three major subfamilies: ERK1/2, JNK, and p38 (Keyse, 2000; Sui et al., 2014). In general, these subfamilies are present in their non-activated forms in the cytoplasm. Activation of them has been demonstrated to be involved in the invasion and migration in gliomas (Guo et al., 2013). In addition, MAPKs are the up-stream modulators of the NF-κB pathway, and also play an important role in regulating the expression of MMPs and uPA-uPAR system (Chen et al., 2005; Jung et al., 2012; Lee et al., 2013; Shi et al., 2013). Several studies have revealed that the cytosolic Ca2+ frequently interacted with the MAPK pathways and thus contributed to the progression of glioma (Lee et al., 2008). Thus the inhibition of MAPK pathways may serve as an effective treatment strategy for glioma. Among all kinases tested, we found that ERK1/2 phosphorylation was selectively and dramatically inhibited without altering its protein by SLC8A2 in U87MG cells, whereas the phosphorylation of JNK, and p38 showed no significant change, indicating that SLC8A2 specifically blocked the activation of the ERK1/2 pathway through a certain mechanism, which has no effect on the other pathways.
Evidences have accumulated that AKT pathway could also regulate the NF-κB pathway (Chen et al., 2005) and play a key role in regulation of the invasive phenotype in the glioma cells (Molina et al., 2010; Pu et al., 2004). Therefore, we investigated whether SLC8A2 affected the AKT in glioblastoma cell U87MG. Of note, no alteration in the protein level of phospho-AKT was detected in the U87MG-SLC8A2 cells.
Glioblastoma is one of the most angiogenic malignancies and anti-angiogenic targeted therapy for glioblastoma has been under increase investigation (Futamura et al., 2007; Szarvas et al., 2009; Verhoeff et al., 2009). Our study found that SLC8A2 inhibited migration, invasion, and the tube formation of endothelial cells, as well as down-regulated the expression of VEGF189, VEGF165, and VEGF121. In addition, COX-2 and HIF-1α, tumor angiogenesis-related genes which can regulate VEGF expression (Kimba et al., 2005), were also suppressed by SLC8A2 overexpression. These results suggested that SLC8A2 inhibited endothelium-dependent angiogenesis of glioblastoma. A new microvascular circulation not involving endothelial cells, termed VM, by which highly aggressive tumor cells can form vessel-like structures themselves (Arbab et al., 2015), is reported as one part of the vascularization of glioblastoma. COX-2, MMPs, HIF-1α, VEGFR2, Wnt/β-catenin, ERK1/2 and other molecular or signaling pathways play an important biological role in VM formation (Paulis et al., 2010; Rong et al., 2016). For example, knockdown of COX-2 by siRNA or abrogating activity of COX-2 by celecoxib (COX-2 inhibitor) led to impairment of VM formation (Rong et al., 2016). Here, we found that SLC8A2 down-regulated expression of COX-2, HIF-1α, MMPs (MMP-2, MMP-9) and VEGFs (VEGF121, VEGF165, VEGF189), as well as inhibited Wnt/β-catenin and ERK1/2 signaling, indicating that SLC8A2 may inhibit angiogenesis by suppressing vascular mimicry. Accordingly, we hypothesized that SLC8A2 may be a common regulator of endothelium-dependent and endothelium-nondependent angiogenesis (vascular mimicry pattern), which then affected the formation of glioma blood vessels. The “combined inhibition” advantage of SLC8A2 rendered it a new target for anti-angiogenic therapy for glioblastoma.
In this study, gene expression microarray identified 2853 differentially expressed genes following SLC8A2 overexpression, with 1555 genes being up-regulated and 1298 genes being down-regulated. Bioinformatic analysis showed that these differentially expressed genes were associated with 98 pathways including MAPK, Wnt, and TGF-β, as well as biological functions including migration, adhesion, cell cycle, proliferation, communication, apoptosis, tightness junction, and nucleotide excision repair. Consistent with the bioinformatic analysis, results of in vitro experiments we performed above indicated that the molecular regulatory mechanism of SLC8A2 in inhibiting cell migration, invasion, and angiogenesis of U87MG was related to inactivation of the ERK1/2 MAPK and Wnt/β-catenin pathways. These findings provide new clues to the further study of the function and molecular mechanism of SLC8A2.
In summary, we found that SLC8A2 could inhibit the tumorigenicity of U87MG cell in vivo, confirming its tumor-suppressor role in glioblastoma. We also observed that SLC8A2 decreased the migrative and invasive capacity of U87MG cells, most likely through inactivating the ERK1/2 MAPK signaling pathway, inhibiting the nuclear translocation and DNA binding activity of NF-κB, and reducing the level of MMPs and uPA-uPAR system (ERK1/2-NF-κB-MMPS/uPA-uPAR). In addition, SLC8A2 may be a common negative regulator of endothelium-dependent and endothelium-nondependent angiogenesis (vascular mimicry pattern) of U87MG cells. Collectively, our findings suggest that SLC8A2 serves as a tumor suppressor gene and inhibits invasion, angiogenesis and growth of glioblastoma.
Supplementary data
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (81072058), the Natural Science Foundation of Jiangsu Province (BK2010230).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Arbab A.S., Jain M., Achyut B.R. Vascular mimicry: the next big glioblastoma target. Biochem Physiol. 2015;4 doi: 10.4172/2168-9652.1000e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M.J., Bootman M.D., Roderick H.L. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Chen P.N., Hsieh Y.S., Chiou H.L., Chu S.C. Silibinin inhibits cell invasion through inactivation of both PI3K-Akt and MAPK signaling pathways. Chemico-Biol Int. 2005;156:141–150. doi: 10.1016/j.cbi.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Chintala S.K., Tonn J.C., Rao J.S. Matrix metalloproteinases and their biological function in human gliomas. Int J Dev Neurosci. 1999;17:495–502. doi: 10.1016/s0736-5748(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Claes A., Idema A.J., Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol. 2007;114:443–458. doi: 10.1007/s00401-007-0293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futamura M., Kamino H., Miyamoto Y., Kitamura N., Nakamura Y., Ohnishi S., Masuda Y., Arakawa H. Possible role of semaphorin 3F, a candidate tumor suppressor gene at 3p21.3, in p53-regulated tumor angiogenesis suppression. Cancer Res. 2007;67:1451–1460. doi: 10.1158/0008-5472.CAN-06-2485. [DOI] [PubMed] [Google Scholar]
- Gondi C.S., Lakka S.S., Dinh D.H., Olivero W.C., Gujrati M., Rao J.S. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA). inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol. 2004;1:165–176. doi: 10.1017/s1740925x04000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray G.K., McFarland B.C., Nozell S.E., Benveniste E.N. NF-kappaB and STAT3 in glioblastoma: therapeutic targets coming of age. Exp Rev Neurotherapeutics. 2014;14:1293–1306. doi: 10.1586/14737175.2014.964211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griscelli F., Li H., Cheong C., Opolon P., Bennaceur-Griscelli A., Vassal G., Soria J., Soria C., Lu H., Perricaudet M., et al. Combined effects of radiotherapy and angiostatin gene therapy in glioma tumor model. Proc Natl Acad Sci USA. 2000;97:6698–6703. doi: 10.1073/pnas.110134297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G., Yao W., Zhang Q., Bo Y. Oleanolic acid suppresses migration and invasion of malignant glioma cells by inactivating MAPK/ERK signaling pathway. PLoS one. 2013;8:e72079. doi: 10.1371/journal.pone.0072079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q., Zhao Y., Chen J., Hu J., Wang S., Zhang D., Sun Y. BRAF-activated long non-coding RNA contributes to colorectal cancer migration by inducing epithelial-mesenchymal transition. Oncol Lett. 2014;8:869–875. doi: 10.3892/ol.2014.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C., Johnk L., Kitange G., Wu Y., Ashworth L.K., Jenkins R.B., Louis D.N. Transcript map of the 3.7-Mb D19S112-D19S246 candidate tumor suppressor region on the long arm of chromosome 19. Cancer Res. 2002;62:4100–4108. [PubMed] [Google Scholar]
- Jan H.J., Lee C.C., Shih Y.L., Hueng D.Y., Ma H.I., Lai J.H., Wei H.W., Lee H.M. Osteopontin regulates human glioma cell invasiveness and tumor growth in mice. Neuro-oncology. 2010;12:58–70. doi: 10.1093/neuonc/nop013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.S., Jung K., Kim D.H., Kim H.S. Selective inhibition of MMP-9 gene expression by mangiferin in PMA-stimulated human astroglioma cells: involvement of PI3K/Akt and MAPK signaling pathways. Pharmacol Res. 2012;66:95–103. doi: 10.1016/j.phrs.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Karin M., Cao Y., Greten F.R., Li Z.W. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Keyse S.M. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- Kimba Y., Abe T., Wu J.L., Inoue R., Fukiki M., Kohno K., Kobayashi H. Mutant IkappaBalpha suppresses hypoxia-induced VEGF expression through downregulation of HIF-1alpha and COX-2 in human glioma cells. Oncol Res. 2005;15:139–149. doi: 10.3727/096504005776367898. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska A., Symons M. Signaling determinants of glioma cell invasion. Adv Exp Med Biol. 2013;986:121–141. doi: 10.1007/978-94-007-4719-7_7. [DOI] [PubMed] [Google Scholar]
- Lakka S.S., Bhattacharya A., Mohanam S., Boyd D., Rao J.S. Regulation of the uPA gene in various grades of human glioma cells. Int J Oncol. 2001;18:71–79. [PubMed] [Google Scholar]
- Lee S.Y., Lee H.Y., Kim S.D., Jo S.H., Shim J.W., Lee H.J., Yun J., Bae Y.S. Lysophosphatidylserine stimulates chemotactic migration in U87 human glioma cells. Biochem Biophys Res Commun. 2008;374:147–151. doi: 10.1016/j.bbrc.2008.06.117. [DOI] [PubMed] [Google Scholar]
- Lee E.J., Lee S.J., Kim S., Cho S.C., Choi Y.H., Kim W.J., Moon S.K. Interleukin-5 enhances the migration and invasion of bladder cancer cells via ERK1/2-mediated MMP-9/NF-kappaB/AP-1 pathway: involvement of the p21WAF1 expression. Cell Signal. 2013;25:2025–2038. doi: 10.1016/j.cellsig.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Lefranc F., Rynkowski M., DeWitte O., Kiss R. Present and potential future adjuvant issues in high-grade astrocytic glioma treatment. Adv Tech Stand Neurosurg. 2009;34:3–35. doi: 10.1007/978-3-211-78741-0_1. [DOI] [PubMed] [Google Scholar]
- Li L., Gondi C.S., Dinh D.H., Olivero W.C., Gujrati M., Rao J.S. Transfection with anti-p65 intrabody suppresses invasion and angiogenesis in glioma cells by blocking nuclear factor-kappaB transcriptional activity. Clin Cancer Res. 2007;13:2178–2190. doi: 10.1158/1078-0432.CCR-06-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., Burger P.C., Jouvet A., Scheithauer B.W., Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina J.R., Hayashi Y., Stephens C., Georgescu M.M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia. 2010;12:453–463. doi: 10.1593/neo.10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A., Tani E., Miyazaki K., Yamamoto Y., Furuyama J. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in human gliomas. J Neurosurg. 1995;83:298–307. doi: 10.3171/jns.1995.83.2.0298. [DOI] [PubMed] [Google Scholar]
- Paulis Y.W., Soetekouw P.M., Verheul H.M., Tjan-Heijnen V.C., Griffioen A.W. Signalling pathways in vasculogenic mimicry. Biochim Biophys Acta. 2010;1806:18–28. doi: 10.1016/j.bbcan.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Pu P., Kang C., Li J., Jiang H. Antisense and dominant-negative AKT2 cDNA inhibits glioma cell invasion. Tumour Biol. 2004;25:172–178. doi: 10.1159/000081099. [DOI] [PubMed] [Google Scholar]
- Qu M., Jiao H., Zhao J., Ren Z.P., Smits A., Kere J., Nister M. Molecular genetic and epigenetic analysis of NCX2/SLC8A2 at 19q13.3 in human gliomas. Neuropathol Appl Neurobiol. 2010;36:198–210. doi: 10.1111/j.1365-2990.2010.01070.x. [DOI] [PubMed] [Google Scholar]
- Quednau B.D., Nicoll D.A., Philipson K.D. The sodium/calcium exchanger family-SLC8. Pflugers Arch. 2004;447:543–548. doi: 10.1007/s00424-003-1065-4. [DOI] [PubMed] [Google Scholar]
- Rao J.S. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3:489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- Ravi R., Bedi A. NF-kappaB in cancer--a friend turned foe. Drug Resist Updat. 2004;7:53–67. doi: 10.1016/j.drup.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Reifenberger G., Louis D.N. Oligodendroglioma: toward molecular definitions in diagnostic neuro-oncology. J Neuropathol Exp Neurol. 2003;62:111–126. doi: 10.1093/jnen/62.2.111. [DOI] [PubMed] [Google Scholar]
- Rong X., Huang B., Qiu S., Li X., He L., Peng Y. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget. 2016;7:83976–83986. doi: 10.18632/oncotarget.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwarf D.M., Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Science’s STKE. 1999;1999:Re1. doi: 10.1126/stke.1999.5.re1. [DOI] [PubMed] [Google Scholar]
- Shi M.D., Shih Y.W., Lee Y.S., Cheng Y.F., Tsai L.Y. Suppression of 12-O-tetradecanoylphorbol-13-acetate-induced MCF-7 breast adenocarcinoma cells invasion/migration by alpha-tomatine through activating PKCalpha/ERK/NF-kappaB-dependent MMP-2/MMP-9 expressions. Cell Biochem Biophys. 2013;66:161–174. doi: 10.1007/s12013-012-9465-8. [DOI] [PubMed] [Google Scholar]
- Sounni N.E., Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem. 2013;59:85–93. doi: 10.1373/clinchem.2012.185363. [DOI] [PubMed] [Google Scholar]
- Sui X., Kong N., Ye L., Han W., Zhou J., Zhang Q., He C., Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344:174–179. doi: 10.1016/j.canlet.2013.11.019. [DOI] [PubMed] [Google Scholar]
- Szarvas T., Jager T., Droste F., Becker M., Kovalszky I., Romics I., Ergun S., Rubben H. Serum levels of angiogenic factors and their prognostic relevance in bladder cancer. Pathol Oncol Res. 2009;15:193–201. doi: 10.1007/s12253-008-9107-z. [DOI] [PubMed] [Google Scholar]
- Verhoeff J.J., van Tellingen O., Claes A., Stalpers L.J., van Linde M.E., Richel D.J., Leenders W.P., van Furth W.R. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer. 2009;9:444. doi: 10.1186/1471-2407-9-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma I.M., Stevenson J.K., Schwarz E.M., Van Antwerp D., Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Vigneswaran K., Neill S., Hadjipanayis C.G. Beyond the World Health Organization grading of infiltrating gliomas: advances in the molecular genetics of glioma classification. Ann Transl Med. 2015;3:95. doi: 10.3978/j.issn.2305-5839.2015.03.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Deimling A., Fimmers R., Schmidt M.C., Bender B., Fassbender F., Nagel J., Jahnke R., Kaskel P., Duerr E.M., Koopmann J., et al. Comprehensive allelotype and genetic anaysis of 466 human nervous system tumors. J Neuropathol Exp Neurol. 2000;59:544–558. doi: 10.1093/jnen/59.6.544. [DOI] [PubMed] [Google Scholar]
- Wang H., Xu T., Jiang Y., Xu H., Yan Y., Fu D., Chen J. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia. 2015;17:239–255. doi: 10.1016/j.neo.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Gaynor R.B. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Yan C., Boyd D.D. Regulation of matrix metalloproteinase gene expression. J Cell Physiol. 2007;211:19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- Zhang M., Ye G., Li J., Wang Y. Recent advance in molecular angiogenesis in glioblastoma: the challenge and hope for anti-angiogenic therapy. Brain Tumor Pathol. 2015;32:229–236. doi: 10.1007/s10014-015-0233-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.