

Summary
Deterioration of metabolic health is a hallmark of aging and generally assumed to be detrimental to longevity. Exposure to a high-calorie diet impairs metabolism and accelerates aging; conversely, calorie restriction (CR) prevents age-related metabolic diseases and extends lifespan. However, it is unclear whether preservation of metabolic health is sufficient to extend lifespan. We utilized a genetic mouse model lacking Fabp4/5 that confers protection against metabolic diseases and shares molecular and lipidomic features with CR to address this question. Fabp4-deficient mice exhibit extended metabolic healthspan with protection against insulin resistance and glucose intolerance, inflammation, deterioration of adipose tissue integrity, and fatty liver disease. Surprisingly, however, Fabp-deficient mice did not exhibit any extension of lifespan. These data indicate that extension of metabolic healthspan in the absence of CR can be uncoupled from lifespan, indicating the potential for independent drivers of these pathways, at least in laboratory mice.
eTOC Blurbs
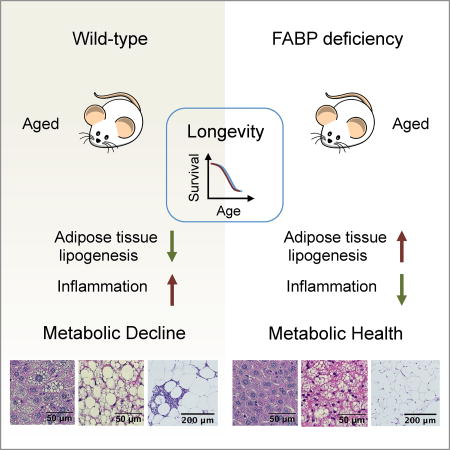
Deterioration of metabolic health is a hallmark of aging and thought to be detrimental to longevity. Charles et al. utilize FABP-deficient mice as a model to demonstrate that the preservation of metabolic health in this model persists throughout life even under metabolic stress, but does not lead to longevity.
Introduction
Deterioration of metabolic health including emergence of increased adiposity, insulin resistance, and dyslipidemia, is a key pathological manifestation of aging (Lopez-Otin et al., 2016), and contributes to age-related diseases, such as diabetes, cancer, cardiovascular disease, and neurodegenerative diseases. In the past two decades, much has been learned about the etiology and underlying mechanisms of the decline in metabolic health (Hotamisligil, 2017; Shulman, 2014). For example, metabolically driven chronic inflammation or metaflammation, ectopic fat deposition and metabolism, and organelle dysfunction all contribute to insulin resistance and abnormal glucose homeostasis. Metabolic stress associated with hyperlipidemia engages key inflammatory and stress signaling modules, including c-Jun N-terminal kinase (JNK), inhibitor of nuclear factor kappa-B kinase (IKK) and protein kinase C (PKC), interferes with metabolic physiology orchestrated by nutrient-sensing pathways, e.g. insulin signaling, adenine-monophosphate-activated protein kinase (AMPK), and mammalian target of rapamycin (mTOR), and ultimately leads to metabolic dysfunction and disease (Arruda and Hotamisligil, 2015; Hotamisligil, 2006). Dietary restriction regimens, prominently calorie restriction, on the other hand, delay the onset of aging and age-associated diseases and impact the aforementioned mechanisms in a corrective manner (Anderson and Weindruch, 2010; Fontana et al., 2010; Yang et al., 2016). In fact, mounting studies targeting these inflammatory modules and nutrient-sensing pathways lend support to the concept that increasing metabolic fitness may extend healthspan and lifespan across the phyla.
As demonstrated in the early genetic studies in round worms and flies, the mechanisms linking metabolic health and aging are finely controlled and context-dependent (Kenyon, 2010). For example, while blunting insulin/insulin-like growth factor (IGF) signaling extends lifespan, total ablation of this pathway is detrimental to metabolic health and even longevity (Accili et al., 1996; Joshi et al., 1996). Holzenberger et al. report that heterozygous knockout of IGF receptor increases the lifespan in female mice by 33%, but does not alter the lifespan of males, possibly due to severely impaired glucose tolerance (Holzenberger et al., 2003). Our group has shown that in mice, obesity-induced JNK activation suppresses insulin signaling and impairs metabolic health (Hirosumi et al., 2002), however, studies in fruit flies suggest that hyperactivation of JNK can increase lifespan by tuning down insulin signaling (Wang et al., 2005). These observations call into the question the direction and nature of the relationship between metabolic health and lifespan, as lipids and their metabolism regulate the action of these pathways (Hansen et al., 2013).
Fatty acid binding proteins (FABPs) are known as intracellular buffer proteins for fatty acids that are involved in lipid trafficking, metabolism, and signaling (Hotamisligil and Bernlohr, 2015). Of the nine FABP isoforms in mammals, Fabp4 and Fabp5 (also known as aP2 and mal1, respectively) carry out important immunometabolic functions and are the best-characterized mediators of metabolic diseases (Furuhashi and Hotamisligil, 2008; Hotamisligil and Bernlohr, 2015). The roles of Fabp4/5 in the pathogenesis of metabolic diseases appear to be mainly due to their functions in adipocytes and macrophages, although they are expressed more broadly (Furuhashi et al., 2008) and can also act as hormones through their secreted forms (Burak et al., 2015; Cao et al., 2013). Considering the redundancy of their lipid trafficking function and molecular compensation, these two genes have been studied in a double knockout mouse model (Maeda et al., 2005). Combined deficiency of Fabp4 and Fabp5 (hereafter referred to as Fabp-deficient) protects mice from high fat diet (HFD)-induced obesity, hepatic steatosis, insulin resistance and diabetes (Maeda et al., 2005). In the leptin-deficient mouse model, Fabp deficiency restores euglycemia and improves glucose tolerance and insulin sensitivity, despite severe obesity (Cao et al., 2006). Fabp deficiency also protects against atherosclerosis and extends survival in apolipoprotein E-deficient mice (Boord et al., 2004). Thus the Fabp-deficient mice provide a useful model to address the question whether preservation of metabolic health contributes to extension of lifespan.
Here, we examined metabolic function in multiple cohorts of Fabp-deficient mice in independent facilities. Collectively, our results show that Fabp deficiency remarkably attenuated age-related body weight gain and deterioration of glucose tolerance, insulin sensitivity, hepatosteatosis and abnormalities of tissue structure in female mice, with smaller effects observed in male mice. Lipidomic and gene expression profiling of adipose tissue revealed a remarkable similarity between the effects of Fabp deficiency and calorie restriction (CR), and CR also decreased the level of circulating Fabp4. However, examination of the lifespan and healthspan of Fabp-deficient mice of both sexes did not reveal any alteration of lifespan or preservation of muscular, cognitive, or cardiac functions with age. In summary, these results indicate that even a marked extension of metabolic healthspan does not necessarily lead to extension of the lifespan or the preservation of cardiac, muscular, and cognitive systems, at least in laboratory mice.
Results
Fabp deficiency protects against age-related weight gain and glucose intolerance
Weight gain during aging can contribute to the decline of insulin sensitivity and glucose tolerance and can contribute to the development of metabolic diseases (Longo et al., 2015). Given that Fabp4/5 deficiency protects against diet-induced obesity, insulin resistance, and diabetes in young mice, we asked whether it might also confer these benefits in aging mice in the absence of a dietary challenge. To this end, we evaluated the metabolic health of middle-aged (11–12 months) Fabp-deficient and control mice of both sexes, maintained throughout their life on regular chow diet. In both males and females, Fabp deficiency reduced body weight gain (female WT 37.2 ± 1.74 g, Fabp4/5−/− 29.4 ± 0.965 g, P < 0.01; male WT 46.2 ± 2.54 g, Fabp4/5−/− 38.9 ± 1.98 g, P < 0.05, Student’s t-test) (Figure 1A). In line with decreased adiposity (Figure 1A), female Fabp-deficient mice also displayed decreased circulating levels of leptin (Figure 1B). Male Fabp-deficient mice exhibited decreased circulating leptin, despite no statistically significant change in fat mass. Furthermore, Fabp deficiency improved glucose tolerance in middle-aged mice in both sexes (Figure 1C), associated with improved insulin sensitivity (Supplementary Figure 1A). In a second cohort, similar improvements in body weight were observed in Fabp-deficient mice analyzed at 15 months of age of both sexes (Supplementary Figure 1B). Improvements in systemic glucose metabolism were stronger in female Fabp-deficient mice (Supplementary Figure 1C). These results indicate that the metabolic protection is a robust and persistent phenotype with more marked effects in female mice.
Figure 1. Fabp deficiency protects against age-related weight gain and decline in glucose homeostasis.

Middle-aged (11~12-month old) mice of both sexes were investigated for metabolic phenotypes (N = 7–8 female, N = 6 male). A. Body weight and body composition measured by DEXA. B. Circulating levels of leptin (N = 5–6). C. Intraperitoneal glucose tolerance test (IPGTT). Open circle, WT; closed circle, Fabp4/5−/−. Areas under the curve (AUC) were plotted on the right. D. Homeostatic model assessment of insulin resistance (HOMA-IR) in aged (23-month-old and 8-month-HFD-fed) mice (N = 3 per group). Data were presented as mean ± SEM, and analyzed by unpaired two-way Student’s t-Test or two-way ANOVA post hoc Sidak test, *P < 0.05, **P < 0.01, ***P < 0.001. See also Figure S1.
In light of the pathogenic roles of Fabp4 and Fabp5 in diet-induced obesity, we next challenged a cohort of late-middle-aged mice (15 month-old, N = 10 per group of each sex) with a high fat diet for additional 8 months. This regimen is known to provoke additional metabolic stress and further exacerbate insulin resistance and impaired glucose metabolism. Consistent with the metabolic protection observed in chow-fed mice, aged high-fat diet-fed Fabp4/5−/− mice also exhibited improved systemic glucose intolerance (Figure 1D). Although the insulin sensitizing effect of Fabp deficiency in this setting was very marked in females, it did not reach statistical significance in males. However, euglycemia was maintained in Fabp4/5−/− mice in both sexes at the end of this regimen (Supplementary Figure 1D). These data provide strong evidence of the critical role for Fabp4 and Fabp5 in the deterioration of metabolic health in the setting of both aging and diet-induced obesity.
Fabp4/5 deficiency protects against age-related pathologies in metabolic tissues
We next performed histological analysis of liver, brown adipose tissue (BAT), and white adipose tissue (WAT). Ectopic fat accumulation and adipose tissue inflammation are known measures of age-associated pathology (Gregg et al., 2012; Sellayah and Sikder, 2014; Woods et al., 2012). Consistent with the observed improvement in metabolic health, Fabp deficiency abrogated hepatosteatosis in both sexes (Figure 2A). The ultra-structure of livers from aged mice showed that Fabp4/5 deficiency preserved the integrity of endoplasmic reticulum (ER) and mitochondria, with sparse formation of glycogen granules and lipid droplets (Figure 2B). In addition, Fabp deficiency markedly alleviated the age-related lipid-laden status of brown adipose tissue (Figure 2C), and mitigated inflammation indicated by the dramatically reduced formation of crown-like structures in WAT (Figure 2D).
Figure 2. Fabp deficiency protects against age-related pathologies in metabolic tissues.

Histological analysis of middle-aged (12-month old) mice (N = 6 per gender per genotype, except N = 4 for Fabp4/5−/− female mice). A. Histology and pathology grades of liver. Scale bar, 50 µm. Grade 0, normal; Grade 1, microvesicular steatosis; Grade 2, macrovesicular steatosis. B. Representative transmission electron microscopy (TEM) of liver sections from 2-year-old WT and Fabp4/5−/− mice. GG (glycogen granules), ER (endoplasmic reticulum), M (mitochondria), LD (lipid droplet). C. Histology and pathology grades of brown adipose tissue (BAT). Scale bar, 50 µm. Grade 0, normal; Grade 1, multilocular and sparse unilocular lipid droplets; Grade 2, multilocular and dense unilocular lipid droplets; Grade 3, unilocular lipid droplets. D. Histology and pathology grades of white adipose tissue (WAT). Scale bar, 100 µm. Grade 0, normal; Grade 1, sparse crown-like structures; Grade 2, large or multiple crown-like structures. See also Figure S2.
We also found that female Fabp-deficient mice exhibited significantly decreased circulating levels of proinflammatory cytokines interleukin-6 (IL-6) (Supplementary Figure 2A) and tumor necrosis factor alpha (TNF) (Supplementary Figure 2B). However, the level of these pro-inflammatory molecules in serum was not mitigated in the same manner in male Fabp-deficient mice. Furthermore, the improved brown fat structure was associated with increased energy expenditure in middle-aged mice (Supplementary Figure 2C), despite no significant difference in food intake (Supplementary Figure 2D). In addition to regulation of the resting energy expenditure, Fabp deficiency increased respiratory exchange ratio in male mice, reflecting a shifted fuel preference from lipids to carbohydrates (Supplementary Figure 2E). These data demonstrate that Fabp deficiency confers protection against age-associated pathologies of metabolic organs, although there are sex-related differences in the extent of these effects.
Caloric restriction mimics adipose tissue lipidome changes observed in Fabp deficiency
We reasoned that if Fabp4 and Fabp5 actions are involved in the age-related deterioration of metabolic systems, Fabp signaling pathways may also be antagonized by calorie restriction, which is a well-established modulator of health and life span. To test this hypothesis, we first examined the impact of short-term CR on circulating levels of Fabp4 and Fabp5 and adipose tissue de novo lipogenesis, an outcome closely linked to Fabp deficiency (Cao et al., 2008). C57BL/6 male mice were fed 40% calorie restricted diet (CR) or regular chow (ad libitum, AL) for 4 weeks. We observed that this regimen was associated with decreased serum Fabp4 (Figure 3A) (AL 102.9 ± 13.95 ng/mL, CR 36.25 ± 9.789 ng/mL, n = 5), without altering Fabp5 (Figure 3A). This was not due to a reduction in secretion from adipose tissue as we observed increased secretion of Fabp4 (2.0 ± 0.2 fold) and Fabp5 (2.0 ± 0.2 fold) from adipose tissue explants isolated from CR male mice (Supplementary Figure 3A Upper Panel). Therefore, the reduction in circulating Fabp4 observed in CR mice may be a reflection of decreased total adiposity or perhaps decreased secretion from a non-adipose source of Fabp4. The CR-induced Fabp4 secretion may also be sex dependent, as we did not observe any significant change in CR female mice (Supplementary Figure 3A Lower Panel). Conversely, CR increased the expression of Fabp5 in adipose tissue without altering Fabp4 (Figure 3B and Supplementary Figure 3B–C).
Figure 3. Caloric restriction mimics adipose tissue lipidome changes observed in Fabp deficiency.

A. Circulating levels of Fabp4 and Fabp5 in 4-week CR mice and ad libitum control mice (N = 5). B. Western blotting analysis of subcutaneous adipose tissue from 4-week CR mice and ad libitum control mice (N = 3). (C–G) Epididymal white adipose tissue (WAT) and liver samples from 20-month-old calorie restricted (CR Aged), ad libitum (AL Aged) control mice, 4-month-old wildtype (WT Young) and Fabp4/5 deficient (Fabp4/5−/− Young) mice were subjected to lipidomics analysis (N = 6). Levels of non-esterified fatty acids (C), C16:1n7 palmitoleate (D), diacylglycerols (E). F. Expression of genes involved in adipose tissue DNL. G. Illustration of the de novo lipogenesis pathway. Enzymes (blue) and fold change (CR/AL, red) of mRNAs are marked. H. Transcript levels of DNL enzymes in rat adipose tissue (N = 5–7). Data were retrieved from a previous microarray profiling study (Linford et al. 2007). Data were presented as mean ± SEM, and analyzed by unpaired two-way Student’s t-Test, *P < 0.05, **P < 0.01, ***P < 0.001. See also Figure S3.
De novo lipogenesis (DNL) is a key mechanism linking adipose tissue to systemic metabolism, and adipose tissue DNL is elevated in Fabp-deficient mice (Cao et al., 2008; Yore et al., 2014). Western blotting analysis of adipose tissue revealed that CR increased levels of key enzymes in the DNL pathway, including acetyl-CoA carboxylase (Acc), ATP-citrate lyase (Acly), and acetyl-CoA synthetase 2 (Acas2) (Figure 3B and Supplementary Figure 3B–C). Together with results of the Fabp4 secretion assay, these findings suggest that secreted Fabp4 may not be critical for regulation of DNL in adipose tissue. Next, we explored the transcriptional mechanism of elevated DNL in CR. We found that short-term CR increased the level of the cleaved form of sterol regulatory element-binding protein 1c (Srebp1c) in adipose tissue, indicative of increased Srebp1c activity (Supplementary Figure 3D). Though carbohydrate responsive element-binding protein (Chrebp) was readily detectable in liver, we did not detect Chrebp protein in adipose tissue in either dietary regimen. In contrast, we did not observe increased Srebp1c cleavage in the adipose tissue from young Fabp mice (Supplementary Figure 3E), suggesting an alternative activation of lipogenic pathway in this model. Together, these results suggest that short-term calorie restriction antagonizes important Fabp-regulated pathways.
Next, we examined whether long-term calorie restriction regulates the lipidome of adipose tissue in a pattern reminiscent of Fabp deficiency by profiling lipids in WAT and liver in a systematic manner. Tissue samples were collected from 19-month-old C57BL/6 male mice that had been fed a 40% calorie restricted diet (60% of calories consumed by ad libitum fed mice) since the age of 16 weeks, or from 20-month-old ad libitum control mice. We then compared these lipid profiles to those of 20-week-old C57BL/6 WT and Fabp4/5-deficient mice from a previous study (Cao et al., 2008) in a side-by-side manner. Similar to Fabp4/5 deficiency, CR increased the level of non-esterified fatty acids (NEFA) in WAT (Figure 3C). Palmitoleate (C16:1n7) levels are low in diet and thus its serum level serves as a marker for endogenous de novo synthesis of fatty acids, in this case derived from adipose tissue (Cao et al., 2008). We have previously shown that adipose tissue production of palmitoleate is enhanced in Fabp4/5 deficiency, and furthermore, that exogenous palmitoleate infusion is sufficient to improve insulin sensitivity in mice (Cao et al., 2008). Here, we found that CR resulted in a 3-fold increase in palmitoleate in WAT, similar to the degree of change observed in Fabp4/5 deficiency (Figure 3D). To our knowledge, this is the only example of a physiological stimulus that so robustly induces adipose tissue DNL and palmitoleate production (Bruss et al., 2010). In mammalian cells, newly synthesized fatty acids are mainly shunted for biosynthesis of diacylglycerol (DAG) and phospholipids (Berg, 2002). Consistent with our observation of increased accumulation of NEFAs, both CR and Fabp deficiency increased levels of DAG and phospholipids in WAT (Figure 3E and Supplementary Figure 3F). Interestingly, however, CR also induced decreases in the levels of NEFAs, palmitoleate, and DAGs in the liver, changes that were not observed in Fabp deficiency (Figure 3C–E).
Just as short-term CR increased the capacity of adipose tissue de novo lipogenesis (DNL) (Figure 3B), transcript profiling of adipose tissue showed that long-term CR induced more than 100-fold the expression levels of key DNL enzymes, e.g. Acly, Acas2, and Acc (Figure 3F), and in fact increased the transcript levels of every enzyme in the DNL pathway (Figure 3G). It has previously been reported that aging decreases the transcript levels of adipogenic genes (e.g. Fasn and Acc) in rat adipose tissue, which can be restored by CR (Linford et al., 2007). To understand these data in the context of aging, we performed computational analysis of this published dataset (NCBI GEO accession ID: GSE6718), and found that the transcriptional program for adipose tissue DNL was suppressed in aging, but was restored by CR (Figure 3H), which strongly supports the concept that adipose tissue DNL is transcriptionally regulated during aging and anti-aging interventions. Overall, our systematic approach verified that long-term CR induces a transcriptional program of DNL in adipose tissue, contributing to a lipid profile that resembles Fabp4/5 deficiency.
Fabp4/5 deficiency does not extend lifespan
Given the connections between metabolic health and longevity, we next asked whether Fabp4/5 deficiency increases lifespan. Interestingly, we observed that there was no significant difference between the survival curves of Fabp deficient and control mice of either sex (Figure 4A). This study was carried out in the Harvard TH Chan School of Public Health (HSPH) animal facility. The absence of an effect on lifespan was also independently reproduced at the San Antonio Nathan Shock Center (UT Health San Antonio) with mice bred at our institution (data not shown). Despite unaltered lifespan, middle-aged Fabp-deficient mice displayed a qualitatively younger-looking general physical appearance, as shown in the photographs (Figure 4B). In agreement with no change in lifespan, serum insulin-like growth factor (IGF-1) was not different between middle-aged Fabp4/5−/− and WT mice (Supplementary Figure 4A). A potential impact of Fabp deficiency on life span was also evaluated on mice that were subjected to HFD. Similar to animals of standard diet and, despite the profound resistance to diet-induced metabolic decline in aged mice (Figure 1E), Fabp deficiency did not alter survival of mice on the HFD feeding regimen (Supplementary Figure 4B). Hence, multiple studies in independent facilities and cohorts of mice all demonstrated identical life span between wild type and Fabp-deficient animals.
Figure 4. Fabp deficiency does not extend lifespan.

A. Kaplan-Meier survival curves of chow-fed Fabp4/5−/− (red) and WT (blue) mice (N = 40) for both sexes. Each data point represents one animal. B. Physical appearance of WT and Fabp4/5−/− mice. (C–E) Echocardiography studies of aged (30-month-old) WT (N = 6 female/8 male) and Fabp4/5−/− (N = 3 female/8 male) mice. Left ventricle mass adjusted by body weight (C), ejection fraction (D), and end diastolic dimension (E). F. Grip strength test (WT, N = 25 female/26 male); Fabp4/5−/−, N = 15 female/20 male). Data were presented as mean ± SEM, and analyzed by Log-rank test, or unpaired Student’s t-Test, *P < 0.05, **P < 0.01, ***P < 0.001. See also Figure S4, Table S1 and Table S2.
To investigate the effect of Fabp deficiency on the aging of other physiological systems, we performed further healthspan studies in aged (30-month-old) mice, including assessments of cardiac, muscular, and cognitive functions. Surprisingly, Fabp4/5-deficient mice of either sex did not exhibit a significant improvement on measures of cardiac function (Figure 4C–E). Echocardiography revealed potentially decreased heart function in aged female Fabp4/5−/− mice but not in male mice. Specifically, Fabp deficiency decreased body weight-adjusted left ventricle mass and end diastolic dimension in females, indicative of left ventricle atrophy, although ejection fraction was not significantly altered. Fabp deficiency did not improve measures of muscular function, such as grip strength (Figure 4F), and gait characteristics (Supplementary Tables 1–2) in mice of either sex. Instead, we observed decreased grip strength in Fabp-deficient female mice, although this parameter was unaltered in males, indicating the potential of a sex-specific decrease in neuromuscular function associated with Fabp4/5 deficiency. Fabp deficiency also did not result in significant improvements in measures of cognitive function, such as Y-maze test assessing learning and working memory (Supplementary Figure 4C), or the tail suspension test assessing anti-depressant activity (Supplementary Figure 4D). Together, these studies suggest that despite broad metabolic improvement, Fabp deficiency does not extend lifespan or promote the maintenance of cardiac, muscular, or cognitive systems.
Discussion
In this study, we have shown that the lipid chaperones Fabp4/5 are critical intermediate factors in the deterioration of metabolic systems during aging. Consistent with their roles in chronic inflammation and insulin resistance in young prediabetic mice, we found that Fabps promote the deterioration of glucose homeostasis, metabolic tissue pathologies, particularly in white and brown adipose tissue and liver, and local and systemic inflammation associated with aging. A systematic approach, including lipidomics and pathway-focused transcript analysis, revealed that caloric restriction and Fabp4/5 deficiency result in similar changes to the adipose tissue metabolic state, specifically enhanced expression of genes driving de novo lipogenesis and NEFA accumulation. Furthermore, caloric restriction was associated with reduced Fabp4 in circulation, providing a potential molecular mechanism underlying its metabolic benefit.
The extension of metabolic health by Fabp deficiency is long-lasting even in aged female mice and even after a high fat diet challenge (Figure 1E). We noticed sexual dimorphism in adiposity (Figure 1A), Fabp4 secretion in response to CR (Supplementary Figure 3A), glycemic control at late ages (Figure 1D, and Supplementary Figure 1C), systemic inflammation (Supplementary Figure 2A–B), and cardiomuscular functions (Figure 4C–F), suggesting that sex-related factors may impact these phenotypes in the context of aging. Interestingly, we did not observe a reduction in the secretory capacity for aP2 in adipose tissue during CR, suggesting that the reduction seen in serum levels may reflect overall adiposity or point to the possibility of an alternate source of secreted aP2. However, despite the remarkable protection in glycemic control, insulin sensitivity, inflammation, and tissue steatosis in Fabp-deficient mice, we did not observe any change in the life span curves. We also did not detect preservation of cardiac, muscular, and cognitive functions. In females, there was even a mild decline in cardiomuscular function associated with Fabp deficiency during aging. These observations support the concept that in higher organisms significant improvements in metabolic tissue inflammation, metabolic tissue integrity, and systemic metabolic homeostasis may not necessarily lead to increased longevity.
Interestingly, previous studies have suggested a link between the maintenance of glucose homeostasis and overall health in aging. For example, epidemiology and clinical trials in type 1 diabetic patients gave rise to the concept that tight glycemic control extends both health span and lifespan in patients (Rodriguez-Gutierrez and Montori, 2016). Metformin, a commonly prescribed drug for type 2 diabetes, increases lifespan and healthspan of male mice (Martin-Montalvo et al., 2013). However, emerging evidence from the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial has challenged this conclusion and found that intensive glycemic control had neutral effects on death and nonfatal cardiovascular events among persons with advanced type 2 diabetes, and may paradoxically increase cardiovascular-related death (Accord_Study_Group, 2016; Accord_Study_Group et al., 2011). Recent studies suggest that calorie restriction also extends lifespan in a context-dependent manner, though it uniformly improves metabolic health (Mattison et al., 2012; Mitchell et al., 2016; Pearson et al., 2008). Further supporting the concept that these endpoints can be uncoupled, exercise improves metabolic health, but does not increase lifespan in mice (Samorajski et al., 1985), and genetic studies targeting insulin/IGF or mTORC1 pathways suggest that extension of lifespan does not necessarily require the maintenance of glucose homeostasis (Lamming et al., 2012; Selman et al., 2008). Although these studies do not establish causality, it may also be the case in humans that metabolic health does not lead to longevity but to a sustained health span.
Our studies with Fabp-deficient mice now provide genetic evidence in animal models that prolonged metabolic health, particularly glucose and lipid homeostasis, may be uncoupled from lifespan and maintenance of cardiac, muscular, and cognitive systems, which partially recapitulates the human pathophysiology observed during intensive glycemic control. Furthermore, it is intriguing that there is a considerable overlap between the unique lipidomic profile, especially in adipose tissue, of Fabp-deficient animals with those that have been subject to caloric restriction. Future studies exploring the similarities and distinctions between these models in multiple sites may provide additional insights into specific pathways and their regulation of healthspan and lifespan. Further exploration of the disconnect between metabolic health and longevity may also shed light on better therapeutic approaches against diabetes and possibly other metabolic diseases that are associated with aging as a risk factor.
Experimental Procedures
Use and Treatment of Animals
Mice were produced and housed in a specific pathogen-free barrier facility certified by Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All procedures were approved by the Harvard Medical Area (HMA) Institutional Animal Care and Use Committees (IACUC) and the University of Texas Health Science Center at San Antonio (UTHSCSA) IACUC. Long-term calorie restriction: C57BL/6 male mice (n = 6) were housed individually, and fed on a calorie restricted (CR) chow (NIH31/NIA fortified 3.0 g pellets), or ad libitum (AL) chow (NIH31random weight chunk formula) before euthanization at the age of 19 months or 20 months. CR was initiated at 14 weeks of age at 10%, then 15 weeks of age at 25%, and finally at 16 weeks of age at 40%. Mice were euthanized after a 5-hour food withdrawal at 03:30 PM. Short-term calorie restriction: C57BL/6 male/female mice (n = 3 or 6) were housed individually, and fed on a 40% CR chow for 3.5–4 weeks as indicated, or ad libitum (AL) chow. Lifespan studies: Virgin C57BL/6 mice, i.e. Fabp4/5−/− and wild-type (WT) littermates, were generated from breeder pairs carrying heterozygous alleles for Fabps. Upon weaning, 80 mice of the same sex but different genotypes were housed together at a density of 3–4 mice/cage, and allowed to live out their normal lifespan. An independent lifespan study was carried out in UTHSCSA Barshop Institute for Longevity and Aging Studies. Mice with homozygous null mutations in Fabp4 and Fabp5, and WT littermates (N = 30 per genotype for females; N = 29 male WT, and N = 31 male Fabp4/5−/−) were generated in the Harvard TH Chan School of Public Health animal facility and shipped to UTHSCSA. Healthspan studies: The healthspan of mice was assessed in Healthspan and Functional Assessment Core of the San Antonio Nathan Shock Center (http://nathanshock.barshop.uthscsa.edu/healthspan-and-functional-assessment/), with mice bred at Harvard as described above (Details can be found in the section Lifespan studies). Metabolic studies: Cohorts of Fabp4/5−/− and littermate WT mice at the age of 11–12 months or 15–16 months were used for metabolic studies, including metabolic phenotyping studies, gross pathology and microscopic analysis of tissues. These experiments were performed at the animal facility of Harvard T.H. Chan School of Public Health. Metabolic cage (Oxymax, Columbus Instruments) and total body fat measurement by DEXA (PIXImus) were performed as previously described (Furuhashi et al., 2007). Glucose tolerance tests were performed by intra peritoneal injection of glucose at a dose of 1.0 g/kg after a 16-hour overnight fast. Insulin tolerance tests were performed by intraperitoneal injection of insulin at a dose of 1 U/kg after a 6-hour daytime food withdrawal. Glucose levels were read using Breeze2 glucose test strips (Bayer).
Statistical and Demographic Analysis
Data are presented as mean ± SEM. N indicates the number of animals per test group. Data passed the Shapiro-Wilk normality test. Student’s t-test (unpaired, two-tailed, equal variance) and post-hoc Sidak test were used for comparisons. P-values are shown in the figures. N.S., not significant, * P < 0.05, ** P < 0.01, *** P < 0.01, **** P < 0.0001. Data from global lipid profiling were processed in Prism 6 (GraphPad Software), and analyzed by Holm-Sidak-adjusted t tests. Demographic data were processed in Prism 6, and analyzed by Kaplan-Meier method and log-rank test.
Biomarker assays and ELISA
Serum samples were subjected to Meso Scale Discovery Assay System (MSD #K152BYC and K15012C) for quantification of leptin, TNF, and IL-6. Serum IGF-1 levels were measured by Mouse/Rat IGF-1 Quantikine ELISA Kit (R&D Systems, #MG100). Serum Fabp5 levels were measured by CircuLex Mouse FABP5/E-FABP/mal1 ELISA Kit (MBL CircuLex cat# CY-8056). Serum Fabp4 levels were measured by an in-house developed aP2 sandwich ELISA system. Fabp4 antibodies (Clone G9 and HA3) recognizing different epitopes were produced at Dana-Farber Cancer Institute Antibody Core Facility, and used as the capture antibody and detection antibody, respectively (Burak et al., 2015). After an overnight coating by 10 µg/mL Clone G9, a 96-well high-bind microplate (Corning 2592) was washed three times by 0.1% Tween-20 PBS, blocked by 5% BSA-PBS for 1 h, and incubated with 10 times diluted serum samples for 1 h. After 3 times wash, the plate was incubated with 200 ng/mL HRP-conjugated Clone HA3 antibody for 1 h. After 6 times wash, TMB buffer was added to the plate for color development and 0.18 M H2SO4 was added to terminate the reaction. Mouse A-FABP standard protein was obtained from Biovendor. The absorbance was read at 450 nm.
Histology and Electron Microscopy
Paraffin-embedded specimens were stained with H&E. Olympus CX41 microscope linked to a CCD camera was used to image H&E stained slides. Pathological scores were graded in a blinded fashion. For transmission electron microscopy: 2-year-old WT and Fabp4/5−/− female mice were anesthetized with 2 mg/ml xylazine and 2 mg/ml ketamine and perfused with saline followed by fixative buffer containing: 2.5% glutaraldehyde, 2.5% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4). After perfusion, small pieces (1–2 mm cubes) of liver were immersed in the same fixative buffer described above for at least 2 hours at 4 °C. The tissues were washed in 0.1M cacodylate buffer and post-fixed with 1% osmiumtetroxide (OsO4)/1.5% potassiumferrocyanide (K4Fe(CN)6) for 1 hr, washed in water three times and incubated in 1% uranyl acetate in maleate buffer for 1 hr followed by a three-time wash in maleate buffer and subsequent dehydration in grades of alcohol (10 min each; 50%, 70%, 90%, 2×10 min 100%). The following day, samples were embedded in TAAB 812 Resin mixture and polymerized at 60 °C for 48 hrs. After embedding, ultra-thin sections (about 90 nm) were generated using a Reichert Ultracut-S microtome, collected on to copper grids, stained with lead citrate and examined in a JEOL 1200EX – 80kV microscope. Images were recorded with an AMT 2k CCD camera at 4000× magnification.
Western Blot Analysis
Proteins were extracted from adipose tissue or liver samples with NP-40 lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 10 mM NaF, 2 mM Na3VO4, PMSF) with complete protease inhibitors (Sigma-Aldrich) and subjected to 4–12% gradient SDS-polyacrylamide gel electrophoresis (NuPAGE™ Novex™ 4–12% Bis-Tris Protein Gels, ThermoFisher). Proteins from PAGE gels were transferred to PVDF membranes. Membranes were blotted for FABP5 (Cell Signaling #39926, 1:1000), FABP4 (inhouse, clone HA3 (Burak et al., 2015) HRP conjugated, 1:10,000; or Proteintech #12802-1-AP, 1:5,000 in FABP secretion assay), ACC (Cell Signaling #3662, 1:1000; or #3676, 1:5,000 in adipose explant analysis), ACLY (Cell Signaling #4332, 1:1000, or 1:5,000 in adipose explant analysis), ACAS2 (Cell Signaling #3658, 1:5,000), SREBP1 (Santa Cruz, clone K-10, sc-367, 1:500), ChREBP (Santa Cruz, clone M-300, sc-33764, 1:500), GAPDH (Bioeasytech #BE0023, 1:5,000), HSP70 (Proteintech, #10995-1-AP, 1:5,000), and beta-Tubulin (Santa Cruz, sc-9104, 1:1000).
FABP Secretion Assay
Subcutaneous adipose depots were dissected for explant culture (subcutaneous depots were used because the size is comparable between CR and AL mice). Adipose tissue samples (one lobe per mouse) were washed in high-glucose pyruvate-supplemented DMEM (hereafter referred to as DMEM) twice and DMEM containing 10% FBS consecutively and minced into roughly 2 mm-size pieces with scissors. Explants were washed with DMEM with 10% FBS and incubated for 1 h in 500 µL DMEM with 10% FBS. Conditioned medium sample was prepared by mixing 100 µL CM with 20 µL 6× Laemmli’s buffer. Cell lysate sample was prepared by homogenizing explant in 1 mL lysis buffer, centrifuged at 12,000 rpm at 4 °C for 10 minutes, and mixing 100 µL CL with 20 µL 6× Laemmli’s buffer. Protein samples were denatured at 70 °C for 5 minutes and analyzed by Western blotting.
Quantitative Real-time RT-PCR
Total RNA was isolated using Trizol Reagent (Invitrogen) and cDNA was synthesized with iScript Reverse Transcription Supermix (Bio-Rad). Quantitative real-time PCR analysis was performed using Power SYBR Green Master Mix (Thermo-Fisher) in ViiA 7 Real-Time PCR systems (Applied Biosystems). Data were normalized to acidic ribosomal phosphoprotein P0 (Rplp0, 36B4) expression. The primer sequences are listed in Table S3.
Systematic Analysis of Lipid Profiles
Lipids from tissues were determined by TrueMass Profiling—TrueMass Lipomic Panel (Lipomics Technologies, West Sacramento, CA). Specifically, lipids were extracted from tissues in the presence of authentic internal standards by the Folch method with chloroform:methanol (2:1 v/v). Individual lipid classes were separated by liquid chromatography. (Agilent Technologies model 1100 Series). Each lipid class was transesterified in 1% sulfuric acid in methanol under a nitrogen atmosphere at 100 °C for 45 min. The resulting fatty acid methyl esters were extracted from the mixture with hexane containing 0.05% butylated hydroxytoluene and prepared for gas chromatography under nitrogen. Fatty acid methyl esters were separated and quantified by capillary gas chromatography (Agilent Technologies model 6890) equipped with a 30 m DB-88MS capillary column (Agilent Technologies) and a flame-ionization detector. Global lipid profiles from 4-month-old wildtype and Fabp4/5−/− deficient mice were previously reported (Cao et al., 2008), and were plotted in this study for the purpose of side-by-side comparison.
Supplementary Material
Highlights.
Fabp deficiency protects against age-related inflammation and metabolic diseases
Calorie restriction and Fabp deficiency share many molecular features
Unlike caloric restriction, Fabp deficiency does not extend lifespan
Life-long metabolic health may not be uniformly associated with extended longevity
Acknowledgments
We are grateful to the members of the Sabri Ülker Center and the Hotamisligil laboratory for their support and invaluable discussions. We thank Dr. Kathryn C. Claiborn and Dr. Martin P. McGrath for editorial advice. We thank Prof. James F. Nelson and Prof. Merry L. Lindsey for coordinating longevity and healthspan studies. We thank Prof. Suneng Fu, Prof. James Mitchell, and Alban Longchamp for supporting short-term calorie restriction studies. We thank Prof. William Mair for inspiring discussions. The work on FABPs in Hotamisligil lab work is supported in part by the grant from the National Institutes of Health (AI116901). KNC was supported by training grants 5T32ES016645 and 5T32CA009078.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
K.N.C. and G.S.H. conceived and designed the study. G.S.H., K.N.C., M.D.L., F.E., A.P.A, and K.I. designed and/or performed experiments. K.N.C., M.D.L., and G.S.H. interpreted the data. M.D.L. and G.S.H. wrote the manuscript.
References
- Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, Asico LD, Jose PA, Taylor SI, Westphal H. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. doi: 10.1038/ng0196-106. [DOI] [PubMed] [Google Scholar]
- Accord_Study_Group. Nine-Year Effects of 3.7 Years of Intensive Glycemic Control on Cardiovascular Outcomes. Diabetes Care. 2016;39:701–708. doi: 10.2337/dc15-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accord_Study_Group. Gerstein HC, Miller ME, Genuth S, Ismail-Beigi F, Buse JB, Goff DC, Jr, Probstfield JL, Cushman WC, Ginsberg HN, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med. 2011;364:818–828. doi: 10.1056/NEJMoa1006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, Weindruch R. Metabolic reprogramming, caloric restriction and aging. Trends Endocrinol Metab. 2010;21:134–141. doi: 10.1016/j.tem.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda AP, Hotamisligil GS. Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes. Cell Metab. 2015 doi: 10.1016/j.cmet.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JMT, J L, Stryer L. Biochemistry. New York: United States of America: W.H. Freeman and Company; 2002. The Biosynthesis of Membrane Lipids and Steroids; pp. 732–735. [Google Scholar]
- Boord JB, Maeda K, Makowski L, Babaev VR, Fazio S, Linton MF, Hotamisligil GS. Combined adipocyte-macrophage fatty acid-binding protein deficiency improves metabolism, atherosclerosis, and survival in apolipoprotein E-deficient mice. Circulation. 2004;110:1492–1498. doi: 10.1161/01.CIR.0000141735.13202.B6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruss MD, Khambatta CF, Ruby MA, Aggarwal I, Hellerstein MK. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am J Physiol Endocrinol Metab. 2010;298:E108–116. doi: 10.1152/ajpendo.00524.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burak MF, Inouye KE, White A, Lee A, Tuncman G, Calay ES, Sekiya M, Tirosh A, Eguchi K, Birrane G, et al. Development of a therapeutic monoclonal antibody that targets secreted fatty acid-binding protein aP2 to treat type 2 diabetes. Sci Transl Med. 2015;7:319ra205. doi: 10.1126/scitranslmed.aac6336. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Maeda K, Gorgun CZ, Kim HJ, Park SY, Shulman GI, Kim JK, Hotamisligil GS. Regulation of metabolic responses by adipocyte/macrophage Fatty Acid-binding proteins in leptin-deficient mice. Diabetes. 2006;55:1915–1922. doi: 10.2337/db05-1496. [DOI] [PubMed] [Google Scholar]
- Cao H, Sekiya M, Ertunc ME, Burak MF, Mayers JR, White A, Inouye K, Rickey LM, Ercal BC, Furuhashi M, et al. Adipocyte lipid chaperone AP2 is a secreted adipokine regulating hepatic glucose production. Cell Metab. 2013;17:768–778. doi: 10.1016/j.cmet.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Fucho R, Gorgun CZ, Tuncman G, Cao H, Hotamisligil GS. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118:2640–2650. doi: 10.1172/JCI34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, Kono K, Babaev VR, Fazio S, Linton MF, et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg SQ, Gutierrez V, Robinson AR, Woodell T, Nakao A, Ross MA, Michalopoulos GK, Rigatti L, Rothermel CE, Kamileri I, et al. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology. 2012;55:609–621. doi: 10.1002/hep.24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Flatt T, Aguilaniu H. Reproduction, fat metabolism, and life span: what is the connection? Cell Metab. 2013;17:10–19. doi: 10.1016/j.cmet.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542:177–185. doi: 10.1038/nature21363. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Bernlohr DA. Metabolic functions of FABPs-mechanisms and therapeutic implications. Nat Rev Endocrinol. 2015 doi: 10.1038/nrendo.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi RL, Lamothe B, Cordonnier N, Mesbah K, Monthioux E, Jami J, Bucchini D. Targeted disruption of the insulin receptor gene in the mouse results in neonatal lethality. EMBO J. 1996;15:1542–1547. [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linford NJ, Beyer RP, Gollahon K, Krajcik RA, Malloy VL, Demas V, Burmer GC, Rabinovitch PS. Transcriptional response to aging and caloric restriction in heart and adipose tissue. Aging Cell. 2007;6:673–688. doi: 10.1111/j.1474-9726.2007.00319.x. [DOI] [PubMed] [Google Scholar]
- Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, et al. Interventions to Slow Aging in Humans: Are We Ready? Aging Cell. 2015;14:497–510. doi: 10.1111/acel.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic Control of Longevity. Cell. 2016;166:802–821. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Maeda K, Cao H, Kono K, Gorgun CZ, Furuhashi M, Uysal KT, Cao Q, Atsumi G, Malone H, Krishnan B, et al. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005;1:107–119. doi: 10.1016/j.cmet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Madrigal-Matute J, Scheibye-Knudsen M, Fang E, Aon M, Gonzalez-Reyes JA, Cortassa S, Kaushik S, Gonzalez-Freire M, Patel B, et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016;23:1093–1112. doi: 10.1016/j.cmet.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gutierrez R, Montori VM. Glycemic Control for Patients With Type 2 Diabetes Mellitus: Our Evolving Faith in the Face of Evidence. Circ Cardiovasc Qual Outcomes. 2016;9:504–512. doi: 10.1161/CIRCOUTCOMES.116.002901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samorajski T, Delaney C, Durham L, Ordy JM, Johnson JA, Dunlap WP. Effect of exercise on longevity, body weight, locomotor performance, and passive-avoidance memory of C57BL/6J mice. Neurobiol Aging. 1985;6:17–24. doi: 10.1016/0197-4580(85)90066-1. [DOI] [PubMed] [Google Scholar]
- Sellayah D, Sikder D. Orexin restores aging-related brown adipose tissue dysfunction in male mice. Endocrinology. 2014;155:485–501. doi: 10.1210/en.2013-1629. [DOI] [PubMed] [Google Scholar]
- Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, Ramadani F, Okkenhaug K, Schuster E, Blanc E, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008;22:807–818. doi: 10.1096/fj.07-9261com. [DOI] [PubMed] [Google Scholar]
- Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–1141. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Woods JA, Wilund KR, Martin SA, Kistler BM. Exercise, inflammation and aging. Aging Dis. 2012;3:130–140. [PMC free article] [PubMed] [Google Scholar]
- Yang L, Licastro D, Cava E, Veronese N, Spelta F, Rizza W, Bertozzi B, Villareal DT, Hotamisligil GS, Holloszy JO, et al. Long-Term Calorie Restriction Enhances Cellular Quality-Control Processes in Human Skeletal Muscle. Cell Rep. 2016;14:422–428. doi: 10.1016/j.celrep.2015.12.042. [DOI] [PubMed] [Google Scholar]
- Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014;159:318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.