

Abstract

Idiopathic pulmonary fibrosis (IPF) is a serious and deadly disease for which treatment options are limited. The recent approval of antifibrosis agent nintedanib represents one of the first therapeutic approaches for the treatment of IPF. Here, we report novel indolinone-based multikinase inhibitors that target angiogenesis and fibrosis pathways and may serve as potential therapeutics for IPF. KBP-7018 is a novel, tyrosine kinase-selective inhibitor with potent effects on three fibrotic kinases (c-KIT, PDGFR, and RET). The pharmacokinetics (PK) properties of KBP-7018 were favorable in mice, rats, and dogs. In a bleomycin (BLM)-induced mouse pulmonary fibrosis model, 10, 30, and 100 mg/kg daily doses (q.d.) of KBP-7018 improved the 28-day survival rate in a dose-dependent manner. The improved efficacy of KBP-7018 compared to nintedanib provided a certain level of chemical validation for the involvement of PDGFR, c-KIT, and RET in IPF. Thus, KBP-7018 represents a novel multikinase inhibitor with differentiated activity, highly enhanced selectivity, and acceptable PK profiles that will enter phase I clinical trials.
Keywords: idiopathic pulmonary fibrosis, multikinase inhibitors, SAR, PK properties, in vivo study
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease that has a median survival time of 2–3 years. Its therapeutic options were limited until the approval of pirfenidone and nintedanib (compounds 1 and 2, Figure 1) in 2014.1 Although both drugs received the breakthrough drug designation from the FDA to facilitate rapid approval, their efficacy in IPF is only moderate. Pirfenidone and nintedanib significantly slow the rate of disease progression. However, they do not halt or reverse the deterioration of IPF and therefore have shown no benefit in the overall survival of patients. Accordingly, a significant unmet medical need is in the management and reversal of IPF.2
Figure 1.

Chemical structures of pirfenidone 1, the lead compound nintedanib 2, and compound 3 (KBP-7018).
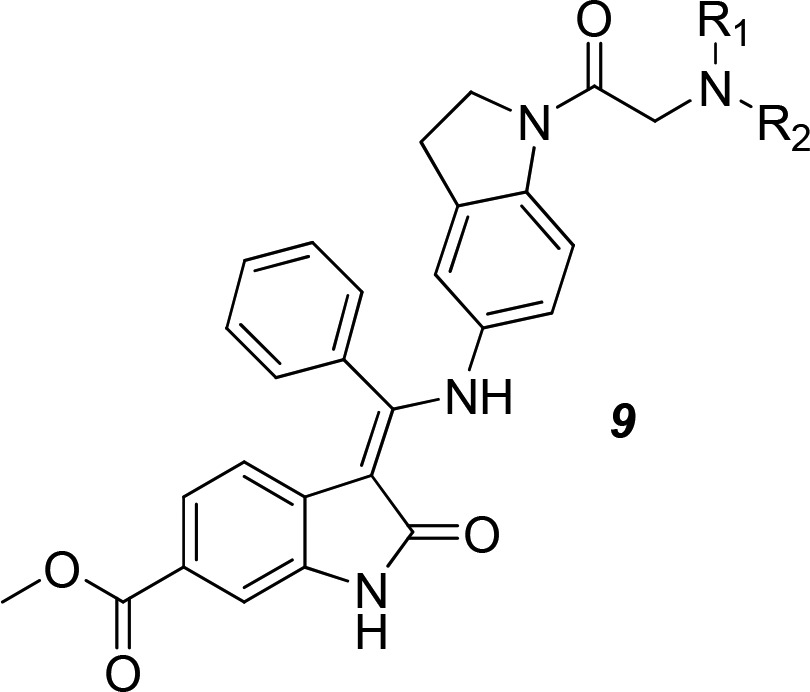
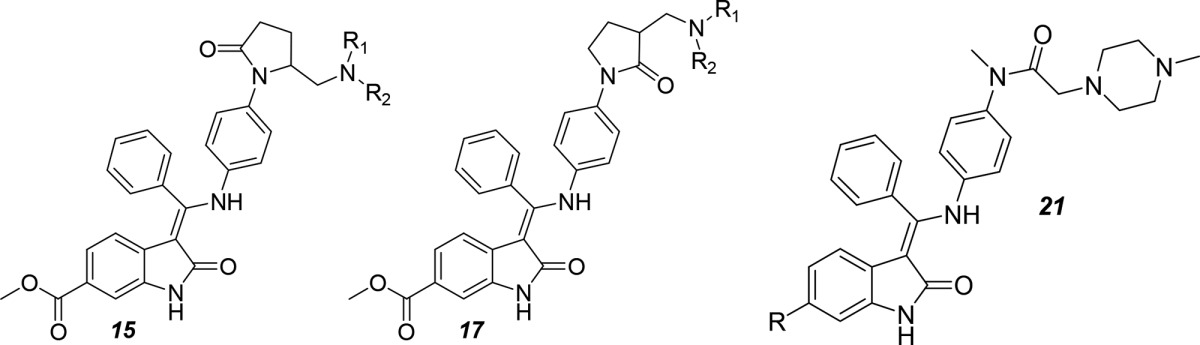
Although the etiology and pathology of IPF have not yet been elucidated, IPF is considered to result from epithelial cell injury and the subsequent unconstrained fibrotic process. Tyrosine kinases are believed to be involved in a range of signaling pathways required cellular homeostasis, including the pathogenesis of pulmonary fibrosis.3 Based on accumulating evidence from both in vitro studies and in vivo animal models, tyrosine kinases mediated lung myofibroblast proliferation via platelet-derived growth factor (PDGFRs), fibroblast growth factor receptors (FGFRs) and vascular endothelial growth factor receptors (VEGFRs). Nintedanib, a well-known multityrosine kinase inhibitor that blocks VEGFR1, VEGFR2, VEGFR3, FGFR1, FGFR2, FGFR3, PDGFα, and PDGFβ, was originally used as a treatment of nonsmall cell lung cancer.4−6 Remarkably, nintedanib was later recognized as a potent inhibitor of lung fibrosis in an animal model, which encouraged its study in clinical trials and the subsequent approval as a treatment for IPF.7,8 However, potential side effects, including the inhibition of the human ether-a-go-go related gene potassium channel (hERG K, IC50 2.6 μM) and the rather low oral bioavailability of nintedanib (4.7%) limited its application. More importantly, nintedanib has a very narrow therapeutic window, which prompted us to structurally modify this compound. Based on the SAR studies of nintedanib, 6-methoxycarbonyl-substituted indolinones were key structures that possessed a potent inhibitory effect on VEGFR, PDGFR, and FGFR. Therefore, the related indolinone in the parent compound was chosen as a starting point to optimize the inhibitory effect on kinases. We adopted a shape-based scaffold hopping approach to convert region 1 of nintedanib to a dihydroindole ring to yield series 9 and oxypyrrolidine to yield series 15 and 17, thus enabling rapid optimization. In addition, we introduced alternative isosteres in region 2 of nintedanib. Finally, several additional isosteres were prepared with a modification at the C-6 position of indole ring in the parent compound to produce series 21 with diverse chemotype-based selections. A detailed description of these compounds has been recently reported.9 Here, we report the design, synthesis, SAR, PK properties, and in vivo efficacy of indolinone-based kinase inhibitors using nintedanib as the lead compound. The optimization of the lead compound 2 to compound 3 (belonging to series 9, also named KBP-7018) produced a compound that targeted PDGFR, c-KIT, and RET with high selectivity. Changes in region 1 (the aryl ring) exerted a profound effect on the overall selective inhibitory effects on tyrosine kinase. A novel tyrosine kinase inhibitor 3 significantly reduced the inhibitory effects on hERG K compared with the parent compound (nintedanib). This compound will soon enter phase I clinical trials.
The synthesis of compounds 9 is shown in Scheme 1. The acylation of indoline 4 yielded the chloroacetamide 5 followed by a displacement reaction with various amines to yield compound 6. Next, the reduction of the nitro group in compound 6 afforded an amino group (compound 7), which was coupled to a known enol ether 8 to produce compounds 9.4 Products were obtained as hydrochlorides to improve their solubility for subsequent in vitro and in vivo tests.
Scheme 1. Preparation of Compound 9.

Reagents and conditions: (i) chloroacetyl chloride, dichloromethane (DCM), trimethylamine (TEA), −20 °C, 90%; (ii) HNR1R2, methyl cyanide (MeCN), K2CO3, reflux, 55%; (iii) methanol (MeOH), Pd/C, H2, room temperature (rt), 92%; (iv) MeOH, reflux, 67%.
A different synthetic route was developed to prepare the pyrrolidone derivatives of nintedanib by adapting known procedures, which was described in Scheme 2. The tosylation of starting alcohol 10 was followed by a displacement reaction to generate the amine group, resulting in compound 11. Coupling compound 11 with 4-nitrobromobenzene 12 under Buchwald conditions afforded an aryl lactam to produce compound 13. Afterward, the nitro group in compound 13 was hydrogenated to yield an aniline group in compound 14. The coupling of compound 14 and enol ether 8 provided the final compounds 15. Compound 17 was synthesized using a similar method as shown in Scheme 2, with the exception that compound 16 were substituted for its isomer 10.
Scheme 2. Preparation of Compounds 15 and 17.

Reagents and conditions: (i) tosyl chloride (TsCl), DCM, 4-dimethylaminopyridine (DMAP), TEA, 0 °C; (ii) HNR1R2, MeCN, K2CO3, reflux; (iii) Cs2CO3, tris(dibenzylideneacetone)dipalladium (Pd2(dba)3), Xantphos, dioxane, reflux; (iv) MeOH, Pd/C, H2, rt; (v) MeOH, reflux.
Several additional analogues of nintedanib were prepared by introducing modifications at the C-6 position4 (Scheme 3). The substituted lactam in compound 18 was condensed with trimethyl orthobenzoate in acetic anhydride to yield enol ether 19, which was coupled with the aniline in compound 20 under basic conditions to provide compounds 21a–g. Under well-established conditions, the bromo intermediate 21a was converted to the boronate 22, which was coupled to different aryl halides under Suzuki conditions to produce compounds 21g and 21h.10
Scheme 3. Preparation of Compound 21.

Reagents and conditions: (i) trimethyl orthobenzoate, acetic anhydride, reflux; (ii) MeOH, KOH, 50 °C; (iii) bis(pinacolato)diboron, potassium acetate (KOAc), PdCl2(bis(diphenylphosphino)ferrocene) (PdCl2(dppf)), dioxane, 100 °C; (iv) 21g: 2-bromofuran, Cs2CO3, PdCl2(dppf), dioxane, H2O, 100 °C; 21h: 3-chloro-4-(4-methoxybenzyl)-4H-1,2,4-triazole, Cs2CO3, PdCl2(dppf), dioxane, H2O, 100 °C, 30% followed by trifluoroacetic acid (TFA), 110 °C, microwave.
When evaluating compounds for their kinome selectivities, PDGFR was chosen as the key target due to its critical role in the initiation and progression of IPF. Therefore, the initial strategy was to improve the druglike properties and potency of nintedanib (2) against PDGFR. Derivatives of compound 9 containing dihydroindole groups were designed and synthesized by introducing a variety of amines (Table 1). In all cases, the inhibitory activities of each derivative toward FGFR and VEGFR were weaker than nintedanib, indicating that the dihydroindole core may not be optimal for improving the efficacy against FGFR and VEGFR. Fortunately, the compounds showed a reserved affinity for PDGFR, with IC50 values ranging from 10 to 100 nM. A compound with a small Me2N group (compound 9a) exhibited a reasonable inhibitory potency (IC50, 15 nM), whereas a compound with a larger amino group (9b) exhibited a loss of activity. Surprisingly, neither the removal of the basicity of the nitrogen (9c) nor the aromatization of the amino group (9d and 9e) had an impact on potency. The morpholinyl-bearing compound 3 (KBP-7018) also inhibited PDGFR activity, despite the lack of affinity for FGFR and a decreased inhibition of VEGFR. Taken together, basicity was not a prerequisite for high inhibitory potency against PDGFR, and as various basic groups with different pKa values were all tolerated, the dihydroindol core may play a key role in affinity to the hinge region of the PDGFR. Because an X-ray structure of PDGFR is not available, a detailed explanation of its activity is beyond the scope of the present study.
Table 1. SAR of Indoline Derivative 9a.

NA: not available. The data are presented with two significant figures due to standard experimental error.
The designed compounds were further evaluated for their abilities to inhibit the h-PDGF-bb-stimulated proliferation of 3T3 cells. Compounds 9b–f and 3 were potent in the ∼100 nM range, indicating sufficient cell permeability. Compounds with high potencies were further evaluated for their effects on the hERG K. To our surprise, a morpholino substituent at region 2 (compound 3, also known as KBP-7018) significantly reduced the inhibitory effects against hERG K compared to nintedanib. Furthermore, the substituents other than morpholine (e.g., 9b–e) also reduced the hERG K inhibitory effect.
Because of its high potency and minimal effects on hERG K, compound 3 (KBP-7018) is a promising kinase inhibitor. Based on the additional physicochemical properties of compound 3, such as its acceptable solubility and clean properties, we evaluated this compound in subsequent tests.
The lactams 15 and 17 showed similar SAR trends in PDGFR activity. However, the affinities for FGFR1 and VEGFR were increased compared with compounds 9b–f (Table 2). Both the S-enantiomer (15a) and R-enantiomer (15b) of piperizinyl compounds showed a comparable potency to nintedanib against both VEGFR and PDGFR, whereas the activity in FGFR1 was decreased. On the other hand, a Me2N analogue (15c) also showed decreased activities against PDGFR, FGFR1, and VEGFR compared with nintedanib. Furthermore, the isomeric piperazinyl analogue (17a), a racemate, did not show activity against PDGFR, whereas individual enantiomers (17b and 17c) were also active in cells with potencies similar to compounds 9b–e and 3. Disappointingly, the presence of the piperazine nucleus elicited a significant inhibitory effect of hERG K, such as in compound 15a (IC50, 0.95 μM against hERG K). Therefore, these compounds were not suitable for further optimization.
Table 2. SAR of the Indoline Derivatives 15, 17, and 21a.

NA: not available. The data are presented with two significant figures due to standard experimental error.
Because the ester group is regarded as a metabolic liability (Table 2), we next performed SAR studies by modifying the substituent in position 6 of the indolinone group with various isosteric groups. All isosteric heterocycles showed reasonable potencies against FGFR, PDGFR and VEGFR compared with nintedanib. The triazole derivative 21h provided the desired potency, whereas 5-oxazole-bearing compound 21f was the least potent. Most of the other compounds, such as 21c and 21h, had nanomolar potencies against PDGFR and reasonable activities against FGFR1 and VEGFR.
However, all tested compounds in this subseries exhibited significant toxicity, as evidenced by the lethality of an intravenous injection of 5 mg/kg of either compound 21c or 21d. Analogues modified at the C-6 position might be metabolized into methanoic acidic derivations in vivo, resulting in the observed toxicities.
Based on the findings described above, we concluded that KBP-7018 failed to inhibit FGFR1 and VEGFR2 but exhibited sufficient inhibitory activities against PDGFRβ, revealing a central role for KBP-7018 in fibrotic tissue. Consequently, we wondered that KBP-7018 may have a potent on other target kinases that are involved in regulating fibrosis. Accordingly, we used a 64-member kinome panel to test our hypothesis. Kinases were investigated based on the increased frequency in a kinase panel assay related to IPF. Among those kinases, KBP-7018 exhibited >80% inhibition of only 5 enzymes at 1 μM, whereas nintedanib (2) inhibited 19 enzymes, indicating the high selectivity of KBP-7018 compared with nintedanib (Table 3).
Table 3. Results of the Assessment of the Activity (%) of 1 μM KBP-7018 toward 64 Enzymes in a Kinase Panel Compared with Nintedanib (Compounds with <80% Inhibition Are Not Shown).
| % enzyme activitya |
|||||
|---|---|---|---|---|---|
| nintedanib | KBP-7018 | ||||
| ABL | 96% | FER | 90% | RET | 99% |
| ALK | 92% | FGR | 96% | FLT3 | 91% |
| BLK | 91% | FLT3 | 97% | FYN | 87% |
| BTK | 96% | FYN | 97% | c-KIT | 91% |
| INSR | 83% | HCK | 84% | PDGFRα | 88% |
| ITK | 85% | RET | 100% | ||
| c-KIT | 99% | c-SRC | 95% | ||
| YES | 99% | LCK | 100% | ||
| JAK3 | 89% | LYNa | 92% | ||
| PDGFRα | 100% | ||||
Data for the following 45 kinases are not shown: AKT1, AMPKa1, AURA/B, BRAF, BRK, CDK2, CHK1, CK 1d, CSK, DYRK1a, EGFR, EGFR (T790M), ERK2, GSK3, p70S6K, HER2/4, IGF1R, IKKβ, IRAK1/4, JAK1/2, JNK2/3, MAPKAPK2/5, c-MET, MSK1, MST2, NEK2, P38α/β, p70S6K, PDK1, PI3Kα/δ, PKCa, c-Raf, ROCK2, RSK1, SGK, SYK, ZAP70 (the kinase abbreviations shown in the Supporting Information).
KBP-7018 was chosen for further in vivo testing because of its cellular potency, reduced effect on hERG K, and attractive selectivity profiles. The PK properties of KBP-7018 were studied in mice, rats, and dogs. The oral bioavailability and the half-life of KBP-7018 were acceptable in all three species (Table 4), indicating its druglike profile.
Table 4. PK Properties of KBP-7018.
| species | dose (mg/kg) | t1/2 (h) | AUC (h·ng/mL) | F (p.o.) (%) |
|---|---|---|---|---|
| mouse | 50 | 4.3 | 17004 | 51 |
| rat | 10 | 4.8 | 6406 | 68 |
| dog | 20 | 3.3 | 2964 | 29 |
Finally, KBP-7018 was evaluated in a C57 mouse model of bleomycin (BLM)-induced chronic pulmonary fibrosis using nintedanib (2) as a positive control (Figure 2).11,12 All three doses (10, 30, 100 mg/kg, q.d.) significantly improved the 28-day survival rate in a dose-dependent manner. At 30 mg/kg/day, KBP-7018 showed a higher in vivo efficacy compared to nintedanib. Notably, after the oral administration of 100 mg/kg/day nintedanib, the mice exhibited significantly reduced survival compared with the mice that received the oral administration of the lower dose of 30 mg/kg/day, indicating a narrow therapeutic window for nintedanib. In contrast, mice displayed sufficient tolerance to KBP-7018 at doses ranging from 10 to 100 mg/kg.
Figure 2.
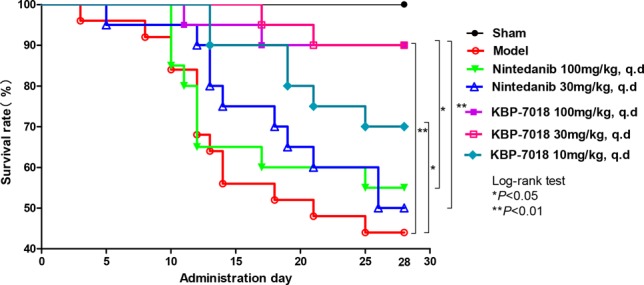
Efficacy of KBP-7018 in a mouse model of BLM-induced pulmonary fibrosis. Sham: saline. Model: intratracheally injection of a BLM solution (5.0 mg/kg in 50 μL).
The improved efficacy of KBP-7018 compared to nintedanib might be attributed to several aspects. First, KBP-7018 exhibited improved PK profiles compared with nintedanib, as supported by its acceptable oral bioavailability and half-life in all tested species. In a previous study, KBP-7018 showed a significantly lower efflux ratio against P-gp than nintedanib in a Caco-2 cell monolayer transport system,13 thereby increasing intestinal permeability and absorption in vivo. Second, KBP-7018 demonstrated a highly selectively inhibition of tyrosine kinases compared with nintedanib. Specifically, it exhibited a lower affinity for VEGFR2, a known anticancer target, resulting in a lower death rate in the mouse model of BLM-induced pulmonary fibrosis. Accordingly, KBP-7018 is superior to nintedanib and displays a wider therapeutic window (Figure 2). Finally, KBP-7018 had a less profound effect on hERG K, thus reducing the relating cardiac side effect.
Notably, KBP-7018 inhibited c-KIT and RET (which are implicated in angiogenesis and fibrosis14,15) with a potency in the nanomolar range (IC50 = 10 nM and 7.6 nM, respectively). Based on the results from the present study, KBP-7018 shows promising potency against IPF in a mouse model of BLM-induced pulmonary fibrosis. The superior in vivo activities of KBP-7018 indicate that the combination of PDGFR, c-KIT, and RET as potential therapeutic targets for IPF.
In conclusion, a novel series of indolinone-based multikinase inhibitors targeting the angiogenesis and fibrosis pathways were established. KBP-7018 showed sufficient potencies against three angiogenic kinases (c-KIT, PDGFR, and RET). The PK properties of KBP-7018 in mice, rats and dogs were favorable. In a mouse model of BLM-induced pulmonary fibrosis, KBP-7018 dose-dependently improved the 28-day survival rate when administered q.d. at doses of 10, 30, 100 mg/kg. Therefore, KBP-7018 represents a novel multikinase inhibitor with improved activity, high selectivity, and acceptable PK profiles. KBP-7018 will proceed to phase I clinical trials in the near future.
Glossary
Abbreviations
- BLM
bleomycin
- c-KIT
c-Kit proto-oncogene encoded protein tyrosine kinase
- hERG
human ether-a-go-go related gene
- IPF
idiopathic pulmonary fibrosis
- PDGFRα
platelet-derived growth factor receptor α
- P-gp
P-glycoprotein
- PK
pharmacokinetic
- RET
REarranged during Transfection (RET) proto-oncogene encoded protein tyrosine kinase
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00164.
Detailed descriptions of the experiments, in vitro kinase and cellular activity assays, PK and in vivo animal model, and experimental treatments (PDF)
Author Contributions
The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- du Bois R. M. Strategies for treating idiopathic pulmonary fibrosis. Nat. Rev. Drug Discovery 2010, 9, 129–40. 10.1038/nrd2958. [DOI] [PubMed] [Google Scholar]
- Hunninghake G. M. A new hope for idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2142–3. 10.1056/NEJMe1403448. [DOI] [PubMed] [Google Scholar]
- Grimminger F.; Gunther A.; Vancheri C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–33. 10.1183/09031936.00149614. [DOI] [PubMed] [Google Scholar]
- Roth G. J.; Heckel A.; Colbatzky F.; Handschuh S.; Kley J.; Lehmann-Lintz T.; Lotz R.; Tontsch-Grunt U.; Walter R.; Hilberg F. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J. Med. Chem. 2009, 52, 4466–80. 10.1021/jm900431g. [DOI] [PubMed] [Google Scholar]
- Hilberg F.; Roth G. J.; Krssak M.; Kautschitsch S.; Sommergruber W.; Tontsch-Grunt U.; Garin-Chesa P.; Bader G.; Zoephel A.; Quant J.; Heckel A.; Rettig W. J. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–82. 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- Roth G. J.; Binder R.; Colbatzky F.; Dallinger C.; Schlenker-Herceg R.; Hilberg F.; Wollin S. L.; Kaiser R. Nintedanib: from discovery to the clinic. J. Med. Chem. 2015, 58, 1053–63. 10.1021/jm501562a. [DOI] [PubMed] [Google Scholar]
- Macagno F.; Varone F.; Leone P. M.; Mari P. V.; Panico L.; Berardini L.; Richeldi L. New treatment directions for IPF: current status of ongoing and upcoming clinical trials. Expert Rev. Respir. Med. 2017, 11, 533–548. 10.1080/17476348.2017.1335601. [DOI] [PubMed] [Google Scholar]
- Hughes G.; Toellner H.; Morris H.; Leonard C.; Chaudhuri N. Real World Experiences: Pirfenidone and Nintedanib are Effective and Well Tolerated Treatments for Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2016, 5, 78. 10.3390/jcm5090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo H.; Wang A.; Zhang Q.. Indole full ketone derivative used as tyrosine kinase inhibitor. WO2014/086102A1, 2014.
- Treu M. K.; Reaiser Ulrich T.. WO2010/012747A1, 2010.
- Sato N.; Takasaka N.; Yoshida M.; Tsubouchi K.; Minagawa S.; Araya J.; Saito N.; Fujita Y.; Kurita Y.; Kobayashi K.; Ito S.; Hara H.; Kadota T.; Yanagisawa H.; Hashimoto M.; Utsumi H.; Wakui H.; Kojima J.; Numata T.; Kaneko Y.; Odaka M.; Morikawa T.; Nakayama K.; Kohrogi H.; Kuwano K. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir. Res. 2016, 17, 107. 10.1186/s12931-016-0420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Wang T.; Wang X.; Sun B. B.; Li J. Q.; Liu D. S.; Zhang S. F.; Liu L.; Xu D.; Chen Y. J.; Wen F. Q. Blockade of advanced glycation end product formation attenuates bleomycin-induced pulmonary fibrosis in rats. Respir. Res. 2009, 10, 55. 10.1186/1465-9921-10-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Huang Z.; Li H.; Zhang Q.; Tan X.; Lu F.; Li S. Characterization of preclinical in vitro and in vivo pharmacokinetics properties for KBP-7018, a new tyrosine kinase inhibitor candidate for treatment of idiopathic pulmonary fibrosis. Drug Des., Dev. Ther. 2015, 9, 4319–28. 10.2147/DDDT.S83055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montani D.; Perros F.; Gambaryan N.; Girerd B.; Dorfmuller P.; Price L. C.; Huertas A.; Hammad H.; Lambrecht B.; Simonneau G.; Launay J. M.; Cohen-Kaminsky S.; Humbert M. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2011, 184, 116–23. 10.1164/rccm.201006-0905OC. [DOI] [PubMed] [Google Scholar]
- Abdollahi A.; Li M.; Ping G.; Plathow C.; Domhan S.; Kiessling F.; Lee L. B.; McMahon G.; Grone H. J.; Lipson K. E.; Huber P. E. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 2005, 201, 925–35. 10.1084/jem.20041393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.