

Abstract
Background
Amplicon deep sequencing permits sensitive detection of minority clones and improves discriminatory power for genotyping multi-clone Plasmodium falciparum infections. New amplicon sequencing and data analysis protocols are needed for genotyping in epidemiological studies and drug efficacy trials of P. falciparum.
Methods
Targeted sequencing of molecular marker csp and novel marker cpmp was conducted in duplicate on mixtures of parasite culture strains and 37 field samples. A protocol allowing to multiplex up to 384 samples in a single sequencing run was applied. Software “HaplotypR” was developed for data analysis.
Results
Cpmp was highly diverse (He = 0.96) in contrast to csp (He = 0.57). Minority clones were robustly detected if their frequency was >1%. False haplotype calls owing to sequencing errors were observed below that threshold.
Conclusions
To reliably detect haplotypes at very low frequencies, experiments are best performed in duplicate and should aim for coverage of >10′000 reads/amplicon. When compared to length polymorphic marker msp2, highly multiplexed amplicon sequencing displayed greater sensitivity in detecting minority clones.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4260-y) contains supplementary material, which is available to authorized users.
Keywords: Plasmodium falciparum, Malaria, Amplicon sequencing, SNP, Haplotype clustering, Multi-clone infections, msp2, csp, cpmp, HaplotypR software
Background
In infection biology of malaria as well as of many other pathogens, detection of minority clones is a crucial task. In areas of high malaria transmission, most infected hosts harbour multiple clones of the same Plasmodium species. To better understand the epidemiology and infection dynamics of malaria, individual parasite clones are tracked over time to measure the acquisition, elimination and persistence of individual clones in a human host. The incidence of new clones per host serves as surrogate measure for the exposure of an individual and for the transmission intensity in a population [1]. Identification of new infections is also crucial in clinical trials of antimalarial drugs, where persisting clones need to be distinguished from new clones in post-treatment samples from patients with recurrent parasitaemia [2, 3]. For such diverse applications, genotyping methods based on length polymorphic markers had been applied for decades, particularly by targeting microsatellite markers or genes encoding parasite surface antigens such as merozoite surface proteins 1 and 2 (msp1, msp2) [4, 5].
Despite their wide use in many malaria research laboratories, length polymorphic markers have important limitations. For example, microsatellite typing suffers from frequent occurrence of stutter peaks, possibly resulting from polymerase slippage on stretches of simple tandem repeats. A cut-off requirement for a minimal peak height (e.g. 33% of the predominant peak [6]) is required to prevent scoring of artefact fragments. However, this cut-off makes it impossible to detect minority clones falling below the selected threshold. Another limitation of length polymorphic marker, particularly the highly polymorphic parasite surface antigens, consists in the usually large size differences between alleles. Major size differences lead to bias in amplification, preferring the shorter PCR fragments in samples that concurrently contain multiple P. falciparum infections [7].
Deep sequencing of short amplicons has the potential to overcome some of the shortfalls of length polymorphic genotyping markers, in particular the influence of fragment length of a marker on the detectability of minority clones. Earlier studies used two different approaches for genotyping of P. falciparum and P. vivax by amplicon deep sequencing: (i) Sequencing of the classical length polymorphic genotyping markers, such as P. falciparum msp1 and msp2 [8]. Alternatively, sequencing targeted non-repetitive regions that harbour extensive single nucleotide polymorphism (SNP), such as the P. falciparum circumsporozoite protein (csp) or P. vivax msp1 [9–11]. The strength of these approaches is that all SNPs within an amplicon are linked by a single sequence read, leading directly to haplotype identification. (ii) Sequencing of multiple loci of genome-wide distribution, whereby each locus comprises one SNP [12]. This latter approach is particularly suited for population genetic investigations, as these loci are not linked. The downside is that the haplotype of each infecting clone has to be reconstructed, which is difficult or even impossible for samples with a high number of co-infecting clones per host [13]. Thus, genotyping of samples containing multi-clone infections remains an unresolved challenge when multiple genome-wide loci are targeted.
In previous studies, amplicon deep sequencing was performed on two platforms, 454/Roche or Ion Torrent. In the past these technologies have produced longer sequences than the 37 bp reads obtained by the Illumina sequencing platform. Now Illumina MiSeq generates reads of up to 600 bp length (Illumina, MiSeq Reagent Kit v3). Sequencing error rates of 454/Roche and Ion Torrent technologies were high, owing to insertion and deletion (indel) errors occurring predominantly in homopolymeric regions [14–16]. Illumina sequencing is less susceptible to indel errors and has an overall smaller error rate [16].
The present report outlines a strategy and protocols for identifying highly diverse markers for SNP-based genotyping of P. falciparum by amplicon sequencing. The primary aim was to thoroughly assess the analytical sensitivity and specificity of amplicon sequencing in detecting minority clones. In epidemiological studies involving hundreds of samples sequencing costs per sample are crucial. Therefore we designed a highly multiplexed protocol, allowing sequencing of up to 384 barcoded P. falciparum amplicons in a single Illumina MiSeq run. Because multiple concurrent P. falciparum clones may differ greatly in density, sequencing analysis strategies need to identify alleles of very low abundance. To distinguish true minority clones from sequencing errors, quality checks were designed based on replicates of samples and integrated into the sequence analysis pipeline. The newly created data analysis software package was validated using experimental mixtures of P. falciparum in vitro culture strains, and tested on field samples.
Results
Marker selection
A protocol for deep sequencing and data analysis was developed for two molecular markers, namely the P. falciparum csp gene (PF3D7_0304600) and gene PF3D7_0104100, annotated in the malaria sequence database PlasmoDB as “conserved Plasmodium membrane protein” (cpmp). Results from these two markers were compared with classical length polymorphic genotyping using the highly diverse marker msp2. Sizes of msp2 fragments amplified for genotyping range from 180 to 515 bp in PNG using published primers (Additional file 1: Table S1). Marker csp has been used for deep sequencing in the past [9] and the previously published primers (Additional file 1: Table S1) were used. The csp amplicon spans the T-cell epitope of the circumsporozoite protein from nucleotide position 858 to 1186 of the 3D7 reference sequence.
The newly validated marker cpmp was identified by calculating heterozygosity in 200 bp windows of 3′411 genomic P. falciparum sequences from 23 countries available from the MalariaGEN dataset [17]. Genes from multi-gene families or regions of poor sequence alignments, often caused by length polymorphism of intragenic tandem repeats, were excluded from the list of potential markers. A 430 bp fragment of cpmp spanning nucleotide positions 1895 to 2324 scored highest in expected heterozygosity (He) and was prioritized as candidate for a highly diverse amplicon sequencing marker. He in the worldwide dataset was 0.93 for cpmp compared to 0.86 for csp (Table 1, Additional file 1: Figure S1 and S2). Genomes originating from Papua New Guinea (PNG) revealed 9 haplotypes in 22 genomes for cpmp and 3 haplotypes in 30 genomes for csp.
Table 1.
Diversity of markers cpmp and csp based on 3′411 genomes of the MalariaGen dataset
| Marker | He a | No. of SNPs | Fragment sizeb | No. of Haplotypes |
|---|---|---|---|---|
| cpmp | 0.930c | 20c | 383c | 82 of 980c,d |
| csp | 0.857 | 40 | 287 | 77 of 1323d |
aExpected heterozygosity
bFragment size without primer sequence
cTrimming of reads in the here presented experiments led to a reduction of variation (Characteristics for a shorter cpmp fragment size of 310 bp: He = 0.913, SNPs = 14 and number of haplotypes = 47)
dFrom 3411 genomes only genomes with non-ambiguous SNP calls in selected region were used
Assessment of sequence quality
Csp and cpmp amplicons from 37 field samples and 13 mixtures of P. falciparum culture strains HB3 and 3D7 were sequenced on Illumina MiSeq in paired-end mode. A total of 5′810′566 paired raw sequences were retrieved. Of all reads, 326′302 mapped to the phiX reference sequence. 4′989′271 paired sequence reads were successfully de-multiplexed to yield a set of amplicon sequences per individual sample. 4′411′214 reads could be assigned to individual amplicons. Median sequence coverage over all sequenced samples was 1′490 for cpmp (1st and 3rd quartiles: [537, 2183]) and 731 for csp (1st and 3rd quartiles: [524, 1092]). The discrepancy in median sequence coverage was deliberate and resulted from our pooling strategy to underrepresent csp amplicons to prevent their predominance in the sequencing library due to this amplicon’s shorter length (Additional file 1).
The quality of the sequence run was assessed by investigating the sequencing error rate in sequence reads of the spiked-in phiX control. The mean mismatch rate per nucleotide of phiX control reads with respect to the phiX174 genome was 5.2% (median 0.34%). The mismatch rate increased towards the end of sequence reads, up to 11% for forward reads and 54% for reverse reads (Additional file 1: Figure S3). To censor regions of high mismatch rates, forward and reverse sequence reads were trimmed before any further analyses to a length of 240 and 170 nucleotides, respectively. After trimming, the mean mismatch rate per nucleotide of phiX control reads was 0.50%.
As further quality check, the sequencing error rate was assessed in sequences of Linkers F and R (Additional file 1: Figure S4). These linker sequences never get amplified but are joined to the product in PCR, therefore any mismatch detected in these stretches will derive from either sequencing or initial primer synthesis. The average number of sequence mismatches in this part was 0.12% per sample per nucleotide (Additional file 1: Table S2 and Figure S5). The sequencing error rate also was assessed in regions corresponding to the primers of each marker (Additional file 1: Figure S4). Mismatches with respect to the known sequences of the PCR primers may derive from amplification errors or from errors in sequencing or primer synthesis during preparation of the sequencing library. The average number of mismatches in the primer regions was 0.28% for cpmp and 0.71% for csp per nucleotide per sample (Additional file 1: Figure S6 and Table S2).
Finally, the sequencing error rate was assessed in amplicons obtained from various mixtures of P. falciparum culture strains HB3 and 3D7. Potential sources of mismatches with respect to the reference sequence of strains 3D7 and HB3 include amplification error, sequencing error and errors due to de-multiplexing of samples [18]. The average number of sequence mismatches after trimming to lengths of 240 and 170 nucleotides respectively for forward and reverse reads was 0.38% for cpmp and 0.46% for csp (Fig. 1, Additional file 1: Table S2). This equates to 1–2 mismatches per read of 310 nucleotides. On average 87.5% of reads for cpmp and 85.5% for csp from mixtures of strains HB3 and 3D7 contained ≤2 mismatches per read with respect to the strains’ reference sequences. Together the analyses of phiX and HB3/3D7 sequences indicated an intrinsic sequence error rate of 0.4–0.5%. The error rate of the linker sequence suggested that one third of these errors were sequencing errors, while two thirds were amplification errors.
Fig. 1.
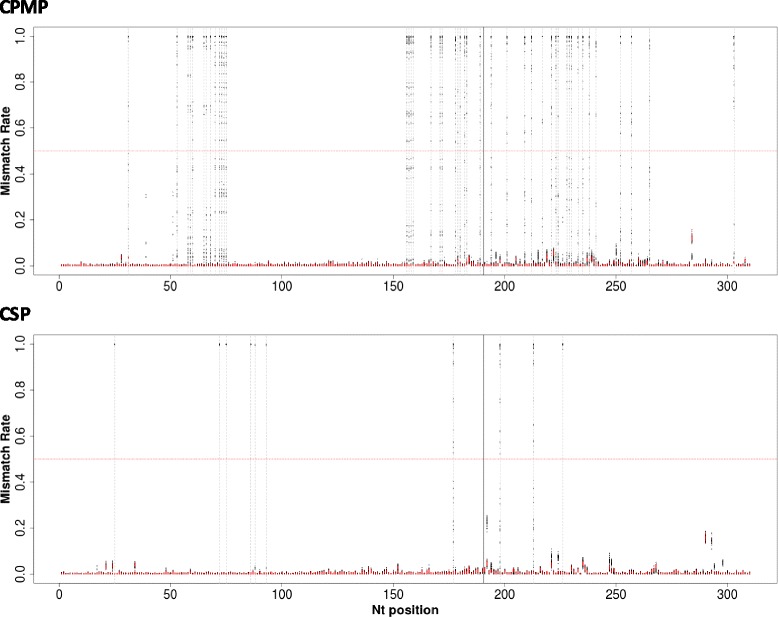
Mismatch rate per nucleotide position derived from all samples sequenced for markers cpmp and csp. Each data point represents the mean observed mismatch rate observed in all reads of one sample at the respective nucleotide position. Red data points: control samples (P. falciparum culture strains); black data points: field samples; X-axis: nucleotide position in sequenced fragment; Y-axis: mismatch rate with respect to the reference sequence (for control samples: sequences of strains 3D7 and HB3, for field samples, 3D7 sequence); dashed grey lines represent SNPs with a mismatch rate of >0.5 in >1 sample; red dotted horizontal line indicates a mismatch rate of 0.5; solid black vertical line: position of concatenation of forward and reverse reads
Limit of detection assessed in serial dilutions of parasite culture
To test the feasibility to also genotype blood samples of low parasite density, serial dilutions of P. falciparum strain 3D7 over 5 orders of magnitude (5–50′000 parasites/μl) were sequenced (Additional file 1: Table S3). The 3D7 haplotype was detected in all dilutions. However, sequence coverage for dilutions harbouring 5 and 50 parasites/μl was below 550 reads. This indicated that the desired equimolar representation of amplicons was not achieved by our pooling strategy (Additional file 1 Pooling of samples - Pool for PCR without visible product on agarose gel). Our approach did not fully counterbalance lower amounts of amplicon.
Assessment of minority clone detectability
Defined mixtures of P. falciparum strains HB3 and 3D7 were sequenced to assess the detectability of minority clones under controlled conditions. The minority clone was detected in all tested dilution ratios up to 1:3000 (Table 2, Additional file 1: Table S4 and S5). Reads comprising obvious PCR artefacts (indels and chimeras) were detected in these mixtures up to a frequency of 0.48% for marker cpmp and 6.2% for csp. Up to 8.4% of reads for cpmp and 10.8% for csp were singletons or failed to cluster with 3D7 or HB3 haplotypes. This proportion of reads is therefore most likely an estimate of the cumulative background noise of the methodology. These reads fell below the default cut-off criteria (details below) and were thus excluded.
Table 2.
Detectability of the minority clone in defined ratios of P. falciparum strains HB3 and 3D7
| Ratios in mixtures | cpmp | csp | Minimum Coverage HaplotypRc | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HB3:3D7 | 3D7a
% |
HB3a
% |
PCR artefacts % |
Back-groundb
% |
Coverage | 3D7a
% |
HB3a
% |
PCR artefacts % |
Back-groundb
% |
Coverage | |
| 1:1 | 34.6 | 57.4 | 0.48 | 7.53 | 40,768 | 34.7 | 50.5 | 5.79 | 9.01 | 9009 | 6 |
| 1:10 | 75.6 | 16.4 | 0.40 | 7.59 | 13,037 | 76.1 | 10.1 | 5.63 | 8.08 | 3341 | 30 |
| 1:50 | 88.8 | 3.15 | 0.06 | 7.95 | 4953 | 82.7 | 2.88 | 6.23 | 8.16 | 14,711 | 150 |
| 1:100 | 90.9 | 1.53 | 0.36 | 7.26 | 13,311 | 83.5 | 2.25 | 5.41 | 8.88 | 11,975 | 300 |
| 1:500 | 90.8 | 0.48 | 0.27 | 8.44 | 5649 | 84.0 | 0.46 | 4.76 | 10.8 | 3508 | 1500 |
| 1:1000 | 91.5 | 0.23 | 0.03 | 8.26 | 3039 | 85.7 | 0.22 | 5.09 | 9.02 | 1807 | 3000 |
| 1:1500 | 92.5 | 0.11 | 0.48 | 6.94 | 55,887 | 86.3 | 0.08d | 5.71 | 7.91 | 23,619 | 4500 |
| 1:3000 | 92.5 | 0.094 | 0.38 | 7.00 | 7417 | 85.0 | 0.04d | 5.87 | 9.10 | 2318 | 9000 |
a Percent of reads that cluster with 3D7 and HB3 reference sequences
b Singleton reads and reads that failed to cluster with 3D7 or HB3 haplotypes
c Theoretical minimum required coverage needed to detect minority clone by software HaplotypR with default cut-off values
d Haplotypes considered as noise by software HaplotypR (default cut-off: ≥3 reads per haplotype and a minority clone detection limit of 1:1000)
Simulations by bootstrap resampling were applied to estimate the probability to detect a minority clone at increasing sequencing coverage and decreasing ratios of the minority clone in a mixture of two strains. Resampling was repeated 1000 times and included only sequence data from mixtures of strains that were sequenced at a coverage of >3000 reads. At a coverage of 10′000 sampled reads the minority clone was robustly detected at ratios 1:1 to 1:1000 for cpmp and up to 1:500 for csp (Fig. 2, Additional file 1: Figure S7 and S8). The cut-off set for haplotype positivity required that a haplotype was detected ≥3 times and represented ≥0.1% of all reads from the respective blood sample. More stringent criteria to call a haplotype (i.e. a higher minimum number of reads) would require a higher coverage for the detection of minority clones. Thus, more stringency in haplotype definition on the one hand reduces sensitivity, but increases specificity by eliminating false haplotypes attributable to background noise (Additional file 1: Figure S7 and S8).
Fig. 2.
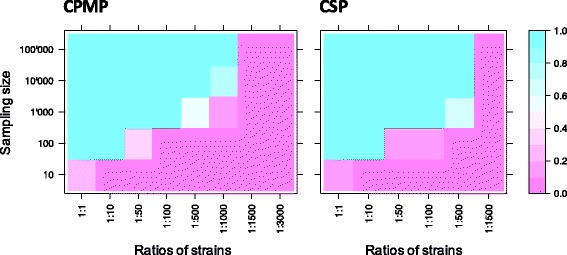
Simulation of minority clone detectability by bootstrapping for marker cpmp and csp. Cut-off for acceptance of a haplotype was a minimum coverage per haplotype of 3 reads and a minority clone detection limit of 1:1000. Samples were drawn from reads of defined mixtures of P. falciparum strain 3D7 and HB3. X-axis shows dilution ratios of strains 3D7 and HB3; Y-axis indicates the sampling size (number of draws from sequence reads) for each mixture of strains. Sampling was repeated 1000 times to estimate mean minority clone detectability
Specification of default cut-off settings in software HaplotypR
Cut-off values for the analysis of sequencing data were defined to support removal of background noise caused by sequencing and amplification errors. The following values represent minimal stringency and can be adjusted to higher stringency to increase specificity in the HaplotypR pipeline:
-
(i)
Cut-off settings for SNP calling were defined by a population-based approach. A SNP was required to be dominant (>50% of all reads) in ≥2 samples. A single dominant occurrence of a SNP is likely caused by amplification or sequencing error.
-
(ii)
Cut-off settings for haplotype calling required a haplotype to be supported by ≥3 reads in ≥2 samples (including independent replicates of the same sample). Per haplotype a minimum of 3 reads are needed to distinguish SNPs from sequencing errors, because a consensus sequence cannot be determined from 2 disparate reads alone. Random sequencing and amplification errors would unlikely lead repeatedly to a particular haplotype.
-
(iii)
Cut-off settings for calling minority clones required that a minority clone would represent at least 0.1% of all reads of a sample, which corresponds to a detection limit for minority clones of 1:1000. For the current project, the cut-off was justified by the results obtained from artificial mixtures of culture strains, which defined the technical limit of detection for a minority clone. This parameter may be set to more stringent values.
Application of these three default cut-off values to mixtures of culture strains had the effect that HaplotypR missed the minority clone for both markers in the greatest dilution ratio of the two strains tested (1:3000). For marker csp the minority clone fell below the cut-off even in the 1:1500 ratio (Table 2). No false-positive haplotypes were called after applying default cut-off criteria, even in samples with a very high coverage in the controlled mixtures (up to 55′000 reads) and in simulations by bootstrapping (up to 100′000 sampled reads) (Additional file 1: Table S4 and S5).
Validation of SNP calling
The above criteria were validated on reads from culture strains and primer sequences. The background sequencing error rate at each individual nucleotide position was measured to distinguish sequencing and amplification errors from true SNPs. Mismatch rates of up to 22% was measured in primer sequences (Additional file 1: Figure S6), and 18% in amplicons from culture strains (Fig. 1, Additional file 1: Table S2). None of these mismatches led to calling of a SNP after the above cut-off was applied (i.e. >50% of reads in ≥2 samples).
Validation of amplicon sequencing in field samples
37 P. falciparum samples from PNG were genotyped by amplicon sequencing. Dendrograms were produced for each marker from raw sequencing reads (Fig. 3, Additional file 1: Figure S9). Branch lengths in these dendrograms represent the number of SNPs that differ between any sequences compared. Branches with sequences belonging to the same haplotype (defined as “clusters”) are labelled in the same colour. Haplotype frequencies within each individual sample were determined from the reads of the sample before applying cut-offs (Fig. 3, panel “Quantification”). When analysing the genetic diversity in field sample, haplotypes were only counted as true haplotypes if both replicates pass the haplotype calling cut-off. This more stringent criterion was introduced to prevent erroneous over-estimation of multiplicity due to false haplotypes.
Fig. 3.
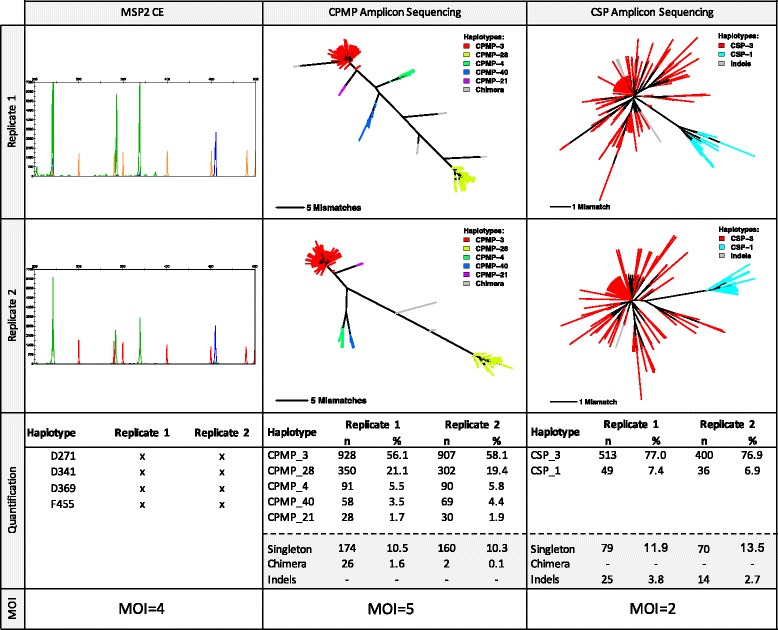
Comparison of genotyping by length-polymorphic marker msp2 and amplicon sequencing of cpmp and csp. Raw data from length-polymorphism- and SNP-based genotyping for one P. falciparum-positive field sample. Left panel: Capillary electropherograms (CE) for msp2 nested PCR products (duplicate experiments); X-axis: fragment length, Y-axis: peak heights (arbitrary intensity units); size standards: red/orange peaks; 3D7-type msp2 genotypes: green peaks; FC27-type msp2 genotypes: blue peaks. Middle and right panel: Dendrograms derived from sequence reads of marker cpmp (middle) and csp (right); coloured lines represent membership to a specific, colour-coded haplotype; Grey lines: sequence reads of PCR artefacts (later excluded by cut-off settings); line length: number of mismatches according to bar insert. Bottom panels: Read counts (n) and percentage of reads (%) per haplotype and final multiplicity call
All samples were genotyped for length polymorphic marker msp2 using capillary electrophoresis (CE) for fragment sizing. Msp2 genotyping was reproducible and consistent between different laboratories (Fig. 3, Additional file 1: Figure S9: left column). A mean multiplicity of infection (MOI) of 2.2 was observed in 37 field samples analysed by msp2 genotyping and 25/37 (67.5%) of samples harboured multiple clones (Fig. 4, Additional file 1: Table S6). Mean MOI and He were compared between the genotyping methods (Table 3, Fig. 4, Additional file 1: Table S6). The resolution of marker cpmp was slightly higher than that of msp2 with 27 cpmp haplotypes versus 25 msp2 alleles, He of 0.96 versus 0.95 and a higher mean MOI of 2.41 versus 2.19, respectively. Overall the two methods agreed well, with good concordance of MOI (Cohen’s Kappa 0.71, equal weights, z = 6.64, p-value = 3.04e-11). Compared to msp2 the discriminatory power of csp was substantially lower with only 4 csp haplotypes found in 37 samples, He of 0.57 and mean MOI of 1.54. Concordance between csp and msp2 MOI was poor (Cohen’s Kappa 0.38, equal weights, z = 4.48, p-value = 7.61e-6).
Fig. 4.
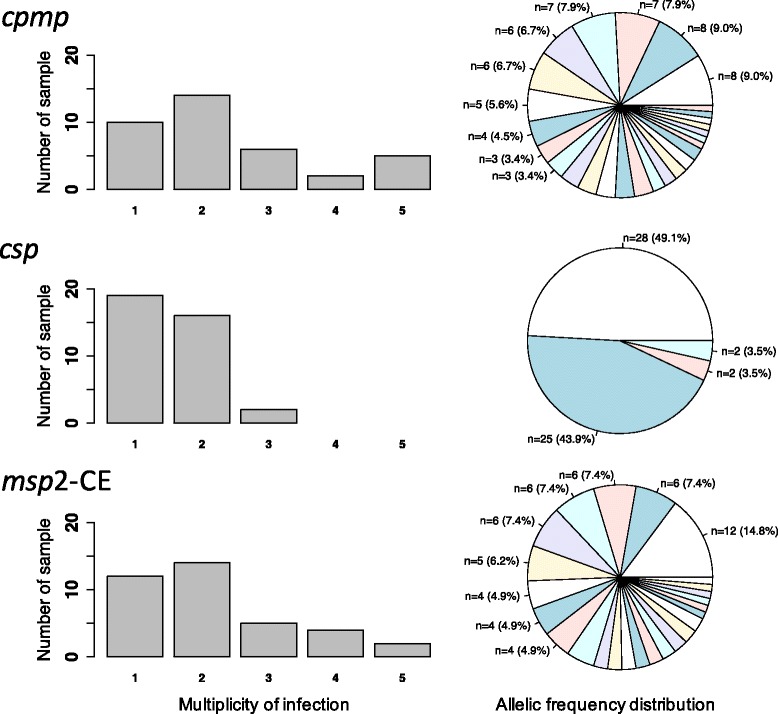
Frequency distribution of multiplicity of infection and allelic frequencies of cpmp, csp and msp2-CE. 37 P. falciparum positive samples from PNG were analysed for the 3 markers cpmp (27 haplotypes), csp (4 haplotypes) and msp2-CE (25 genotypes). Pie charts represent allelic frequency distribution for each marker in 37 samples
Table 3.
Summary of genotyping results from three molecular markers analysed in 37 field samples
| Marker | He | Mean MOI | Number SNPsa | Number Haplotypes | Concordance of MOI Κ |
|---|---|---|---|---|---|
| msp2 CE | 0.948 | 2.19 | NA | 25 | Reference |
| cpmp | 0.957 | 2.41 | 45 | 27 | 0.71b (good) |
| csp | 0.574 | 1.54 | 10c | 4 | 0.38d (poor) |
a With respect to the reference sequence of P. falciparum strain 3D7
b Cohen’s Kappa (2 raters, weights = equal): z = 6.64, p-value = 3.04e-11
c 4/10 SNPs are fixed within these 37 field samples
d Cohen’s Kappa (2 raters, weights = equal): z = 4.48, p-value = 7.61e-6
Reproducibility of amplicon sequencing in field samples
Csp and cpmp haplotypes obtained from 37 field samples were compared between replicates to investigate reproducibility of the molecular and bioinformatic analyses. For both replicates of the field samples the default cut-off criteria for haplotype calling (≥3 reads and minority clone detection limit of 1:1000) were applied. Concordance between replicates was very good with Cohen’s Kappa 0.84 (equal weights, z = 7.769, p-value = 7.99e-15) for cpmp and 0.91 (equal weights, z = 6.466, p-value = 1.01e-10) for csp. Comparison of replicates permitted to investigate the amount of false haplotype calls. True haplotypes should be detected in both replicates, unless the sequence depth is not sufficient for detecting a minority clone in one of the replicates. Cpmp minority clones that had a frequency > 1.0% of all reads were consistently detected with ≥3 reads in both replicates (Table 4, Additional file 1: Figure S10). For csp this was achieved for minority clones with a frequency of >0.70%. 18 cpmp haplotypes were detected with ≥3 reads in only one of the replicates. In three instances one of the replicate did not pass the cut-off criteria due to low coverage. For marker csp, 2 haplotypes with ≥3 reads were detected in one replicate only. In summary, a comparison of replicates indicated 15 potentially false haplotype calls for cpmp and 2 for csp. These calls stem from reads with a frequency < 1%, Therefore, performing replicates are essential to prevent erroneous over-estimation of multiplicity due to false haplotypes.
Table 4.
Concordance of haplotype calls in replicates of 37 field samples
| cpmp | csp | Passed cut-offa | |
|---|---|---|---|
| Haplotype frequency within sample ≥ 1% | |||
| present in both replicates | 87 | 57 | yes |
| present in single replicate only | 0 | 0 | no |
| Haplotype frequency within sample < 1% | |||
| present in both replicates at ≥ 3 reads b | 2 | 0 | yes |
| present in both replicates one ≥ 3 readsb and one < 3 readsb | 1c | 0 | yes/nod |
| present in single replicate at ≥ 3 readsb | 17e | 2 | yes/nod |
| present in both replicates at < 3 readsb | 1 | 0 | no |
| present in single replicate at < 3 readsb | 10 | 5 | no |
Bold rows indicate haplotypes that did pass cut-off criteria in both replicates
a Default cut-off criteria to accepted haplotype ≥3 reads and a minority clone detection limit of 1:1000
b Owing to default cut-off for haplotype call
c Second replicate had too low coverage to detect ≥3 reads
d Potential false haplotype calls as only one replicate passed cut-off criteria
e In 2 instances second replicate had too low coverage to detect minority clone
An attempt was made to investigate the influence of the number of PCR cycles performed during amplicon library preparation on the generation of artefacts. This was possible by using 25 and 15 cycles in the nested PCR for replicate 1 and 2, respectively. Cycle number had no influence on the proportion of singleton and indel reads. However, the proportion of chimeric cpmp reads was higher in replicate 1 using 25 cycles than in replicate 2 using 15 cycles (0.63% versus 0.13%, Student’s t-Test P-value = 0.0221). No chimeric csp reads were detected in the field samples (Table 5).
Table 5.
Mean proportion of singleton or chimeric reads and indels detected in both field sample replicates
| Marker | Replicate 1 | Replicate 2 | ||||
|---|---|---|---|---|---|---|
| Singletons % |
Indels % |
Chimera % |
Singletons % |
Indels % |
Chimera % |
|
| csp | 11.55 | 3.78a | 0.00 | 11.47 | 4.05a | 0.00 |
| cpmp | 9.76 | 0.073b | 0.631c | 9.74 | 0.034b | 0.130c |
a Marker csp: Indels Replicate 1 versus 2; Student’s t-Test: t = −1.336, df = 71.052, p-value = 0.1858
b Marker cpmp: Indels Replicate 1 versus 2; Student’s t-Test: t = 1.3304, df = 71.94, p-value = 0.1876
c Marker cpmp: Chimera Replicate 1 versus 2; Student’s t-Test: t = 2.3552, df = 55.4, p-value = 0.02208
Discussion
This report presents the development of a new genotyping methodology for P. falciparum based on amplicon deep sequencing. The search for new markers was prompted by severe limitations of length polymorphic markers, which represent the currently used standard for genotyping malaria parasites. A strong bias towards preferential amplification of shorter fragments in multi-clone infections was observed, so that larger fragments were lost even if only 5-fold underrepresented compared to shorter fragments from the same sample [7]. This called for an alternative approach that relies on haplotypes created from several SNPs rather than length polymorphism. With respect to minority clone detectability, amplicon sequencing overcame this pitfall of length polymorphism methods and also performed very well in field samples.
Amplicon sequencing showed an excellent resolution when using the novel genotyping marker cpmp (PF3D7_0104100). The strategy applied for down-selecting highly diverse regions in the genome suggested cpmp as the top candidate. Cpmp is most abundantly expressed in sporozoite stages [19], but the function of the encoded protein is unknown. The gene is under balancing selection with a Tajima’s D of 1.16 in Guinea and 1.05 in Gambia [20]. In this study, cpmp revealed a genetic diversity similar to the length polymorphic region of the widely-used marker msp2. 45 SNPs were observed in the 37 field samples of this study, leading to the designation of 27 haplotypes for marker cpmp. With increasing number of field samples processed, additional rare SNPs and even more haplotypes are likely to be found. The diversity of cpmp was high also in the global MalariaGEN dataset (He = 0.93); its resolution as genotyping marker in other geographic regions remains to be shown. In contrast, marker csp, analysed in parallel to cpmp and also used in earlier studies, showed a limited diversity with only 4 haplotypes detected in 37 field samples. Earlier studies reported similar low diversity for csp in regions of Asia Pacific [21]. Thus, csp is not suited to serve as a single genotyping marker in PNG. However, the global diversity of csp according to the MalariaGEN dataset seems to be high (He = 0.86), and high diversity has also been observed in African isolates [21].
Implementing amplicon sequencing required parallel development of a bioinformatics pipeline. A known problem in sequence analysis is the robust detection of minority clones from a background of experimentally induced artefacts. We addressed this problem with the design of HaplotypR, a software package dedicated to stepwise analyse of sequence reads for samples containing multiple clones. The HaplotypR pipeline can be divided into three steps: In the first step, this pipeline de-multiplexes and clusters raw sequence reads to clusters of related sequences, so called “representative haplotypes”. This step employs Swarm2 software, which expands pools of amplicons (identical sequence reads) by iteratively joining other pools of amplicons that are separated by a defined number of mismatches (e.g. one substitution, insertion or deletion) [22, 23]. This strategy permits unbiased clustering of sequence reads without the need to define a list of SNPs. This enables capturing of previously unknown SNPs without any adjustments to the pipeline. In the next step HaplotypR checks all representative haplotypes for presence of PCR artefacts (indels and chimeras), and labels and censors these. In the final step HaplotypR removes background noise by applying defined cut-offs and reports a list of final haplotypes calls.
Validation of HaplotypR was made possible by reads from serial dilutions of P. falciparum culture strain 3D7 and from controlled mixtures of strains HB3 and 3D7. On those control samples the impact of amplification and sequencing errors could be assessed. An increased frequency of sequence mismatches relative to the 3D7 reference sequence of up to 22% was observed at a few specific genomic locations including the sequences of amplified primers. To differentiate these sequencing errors from true genotypes of rare minority clones, we defined a SNP calling cut-off where a genotype was required to be dominant (>50% of all reads) in at least 2 samples. This cut-off is critical to distinguish true positive genotypes that are rare in the population from sequencing errors.
To prevent reporting of false haplotypes, HaplotypR pipeline applies two types of cut-offs: firstly, a cut-off for singleton exclusion, whereby a SNP or haplotype needed to be supported by more than one sample. It is unlikely that these cut-offs would remove true haplotypes, except if the sample size was very small. In this case, it is recommended to amplify and sequence samples in duplicate, as in this study. A true haplotype is expected to be present in both replicates and thus will not get excluded. Secondly, a cut-off for haplotype coverage was defined requiring that a haplotype is supported by a user-defined number of sequence reads. This flexible cut-off can be selected for each marker. The coverage cut-off removes false or weakly supported haplotypes and thus improves specificity. On the other hand, the ability to detect minority clones (i.e. sensitivity) will be limited by a cut-off based on coverage. Sequence reads from a minority clone were detected in all ratios up to 1:3000 in the mixtures of strains HB3 and 3D7. However, due to high background noise, false haplotypes with a frequency of up to 0.01% were also detected, making the definition of a cut-off to remove background noise obligatory. Applying these default cut-offs in HaplotypR decreased minority clone detectability from 1:3000 to 1:1000.
In an other publication a parasite density specific cut-off was applied in addition to a default cut-off [24].
The potential of amplicon sequencing for genotyping samples of very low parasitaemia was assessed in serial dilutions of strain 3D7. Sequence reads were retrieved from samples of a parasitaemia as low as 5 parasites/μl, however coverage was below 100 reads for the lowest level of parasitaemia. To reliably genotype samples spanning a wide range of parasitaemias, similar sequence coverage (and thus unbiased normalization of input material) for all samples is needed. The inexpensive strategy used to adjust amplicon concentrations of individual samples to equal levels prior to pooling for highly multiplexed sequencing still resulted in fluctuation in the sequence coverage, but a commercial DNA normalisation kit may improve equimolar pooling of samples [25, 26].
All samples in this study were sequenced in 2 replicates. This was done to assess the reproducibility of amplicon sequencing method of genotyping very low abundant minority clones, and to investigate the effect of nested PCR cycle number on artefacts. Analysing replicates of field samples revealed that haplotypes with a frequency of >1% were consistently detected in both replicates. In contrast, haplotypes with a frequency of <1% were frequently detected only in a single replicate. If minority clones of <1% frequency are to be reliably detected, amplifying and sequencing two or more replicates for each sample would be essential to call true haplotypes.
To detect minority clones with high sensitivity and specificity, samples need to be sequenced at high coverage and in replicates. As sensitivity may be adjusted by sequence coverage, choices have to be made in a trade-off between sequencing costs and sensitivity. The specific genotyping application can guide this choice. For example, in large scale field studies with many samples, a high degree of multiplexing of samples at moderate sequence coverage may be chosen to keep sequencing cost low. Furthermore, a less sensitive approach without performing replicates may be sufficient when detection of very rare minority clones is less of an issue. Another important application of genotyping of malaria parasites is the example of “recrudescence typing” during in vivo drug efficacy trials. To distinguish a new infection from one present as a minority clone prior to drug administration requires highest sensitivity and every clone must be reliably detected. In such cases a sequencing approach with less multiplexing is desired to achieve high coverage and maximal detection of minority clones.
The power of high sequencing coverage was shown for example in a study assessing the subclonal diversity in carcinomas [27]. Minority variants with a frequency of 1:10′000 were detected with a sequence depth of 100′000 reads per sample. Our results reported from malaria field samples does not have sufficient sequence depth to achieve such sensitivity, as median sequencing depth per sample was 1′490 reads for cpmp and 731 reads for csp owing to a high number of samples and of markers sequenced in parallel. A total of 352 samples were multiplexed in a single sequence run. Samples simultaneously processed but not included in the present analysis served for an unrelated research question. According to our protocol for PCR-based sequencing library preparation, costs per sample for Amp-Seq were twice that of msp2-CE genotyping [5]. Thus, the approach applied by us is cost effective as it permits parallel processing of several hundred samples, a range typically encountered in population-wide studies.
Targeted amplicon sequencing is not only used for investigating genetic diversity of Plasmodium parasites, but also widely applied in other fields, e.g. to study diversity of other pathogens, diversity in eco-systems or sequence alteration caused by CRISPER/Cas9 [8–11, 24, 28–31]. For pooling of multiple samples in one sequencing run, individual samples are generally either labelled by ligating a sequencing adapter that carries an index sequence [28] or by amplification with the sequencing adapter carrying an index and linker sequence [29, 30]. Data analysis follows two main strategies to retrieve haplotypes either by clustering of the full sequence read [10, 22, 24] or by SNP calling and optional haplotype inference [13, 28].
Conclusions
Short amplicon sequencing has the advantage that no multi-locus haplotype reconstruction is needed, as all SNPs are linked by a single paired-end read. This allows the reliable analysis of samples of very high MOI, a prerequisite for genotyping in areas of high malaria endemicity. An additional strength of this method is that previously undescribed or newly evolving haplotypes can be captured without any adjustment of the typing methodology or the HaplotypeR pipeline. The main limiting factor for the detection of minority clones was the sequence depth per sample. The sequence coverage in the present study was in the order of 1000 reads (median number of reads for cpmp was 1490 and for csp 731). This permitted detection of minority clones at a frequency of >0.3% of the total parasite load. To robustly detect minority clones at 0.1% frequency, a coverage of 10′000 reads is recommended. In addition, experiments should be performed in duplicate. The need to detect such low-abundance clones depends on the specific research question, which should guide experimental decision on number of samples and multiplexed amplicons as well as on the desired sequence depth.
The specification of amplification and sequencing errors presented here as well as the developed bioinformatic tools to handle such complex analytical tasks are relevant to all amplification-based genotyping methods of multiple clones or quasi-species within a sample. The newly developed pipeline can be used to analyse any amplicon sequencing based genotyping data irrespective of marker or organism.
Methods
Parasite genomic DNA
P. falciparum in vitro culture strains HB3 and 3D7 were mixed in 8 different proportions to generate well defined control samples with known MOI and well defined ratios of genomes. The ratios in these HB3-3D7 mixtures ranged from 1:1 to 1:3′000. Five additional control samples represented a dilution series of strain 3D7 with parasite densities ranging from 50′000 to 5 parasite/μl. Dilutions were prepared in human gDNA to reconstitute the nucleic acid concentration of a human blood sample. Details of parasite quantification were published previously [7]. Thirty-seven archived field samples from a cohort study conducted in East Sepik Province, Papua New Guinea (PNG) in 2008 were used to validate the performance of protocols for genotyping and data analysis in natural P. falciparum infections [32].
Genotyping using length polymorphic marker msp2
For determination of mean MOI field samples were genotyped using the classical P. falciparum genotyping marker msp2. Fluorescently labelled nested PCR products were sized by CE on an automated sequencer and analysed using GeneMapper software according to previously published protocols [5]. Each DNA sample was genotyped twice in independent laboratories to assess reproducibility of clone multiplicity (Fig. 3 and Additional file 1: Figure S9).
Amplicon deep sequencing marker selection and assay development
3′411 genomes from 23 countries, published by the Plasmodium falciparum Community Project (MalariaGEN), were screened to identify highly diverse markers for SNP-based genotyping [17]. The P. falciparum genomes were divided in 200 bp windows and He was calculated for each window as follows: where n is the number of clones and p i the frequency of allele i. Annotated genes (PlasmoDB v11.0) that overlapped with windows of high heterozygosity were selected for further evaluation. Genes belonging to gene families, such as var, rifin, stevor and surf families, were excluded from the list, as well as genes with high heterozygosity that is usually caused by length polymorphism (Additional file 2).
Primers for marker cpmp were designed manually. Location of primers was selected to flank a region of maximum diversity (Additional file 1: Figure S1 and S2). Amplicon sizes were limited to a maximum of 500 bp to conform to possible read lengths of the Illumina MiSeq platform. Quality control of primers was assessed with online tools for secondary structure and primer dimer interaction (https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/multiple-primer-analyzer.html) [33]. Primer sequences are listed in Additional file 1: Table S1 and He values for amplicons are shown in Additional file 1: Figure S1 and S2.
Sequencing library preparation
The sequencing library was generated by 3 rounds of PCR with KAPA HiFi HotStart ReadyMix PCR Kit as described earlier [30]. A first round of 25 cycles amplified the gene of interest. A second marker-specific nested PCR amplified the primary product with primers that carried a 5′ linker sequence. We compared different cycle numbers for this second round: 25 cycles for replicate 1 and 15 cycles for replicate 2. This comparison was done to test for effects of cycle number on sequence diversity caused by imperfect polymerase fidelity [34]. To allow pooling and later de-multiplexing of amplicons, a third and final amplification was performed using primers binding to the F and R Linker sequence at the 3′ end, that introduced a sample-specific molecular barcode sequence plus the Illumina sequence adapter at the 5′ end. The relative positions of all these elements are depicted in the schematic in Additional file 1: Figure S4. A detailed PCR protocol containing primer sequences, cycle conditions und pooling steps are described in Additional file 1.
PCR products were purified with NucleoMag beads. The expected fragment size of the sequencing library was confirmed by Agilent 2200 Tapestation System. DNA concentration of the sequencing library was quantified by Qubit Fluorometer (Thermo Fisher Scientific). Sequencing was performed on an Illumina MiSeq platform in paired-end mode using Illumina MiSeq reagent kit v2 (500-cycles) together with a Enterobacteria phage PhiX control (Illumina, PhiXControl v3).
Bioinformatic analysis pipeline “HaplotypR”
Sequence reads were mapped with bowtie2 (parameter: end-to-end and sensitive) [35] to the phiX174 genome (Accession: J02482.1) for assessing the quality of the sequencing run and calculating sequencing error per nucleotide position. Reads were then de-multiplexed to separate individual samples and different genotyping markers (Fig. 5). Primer sequences were truncated, the sequence was trimmed according to the quality of the phiX control sequence reads and paired reads were fused together.
Fig. 5.
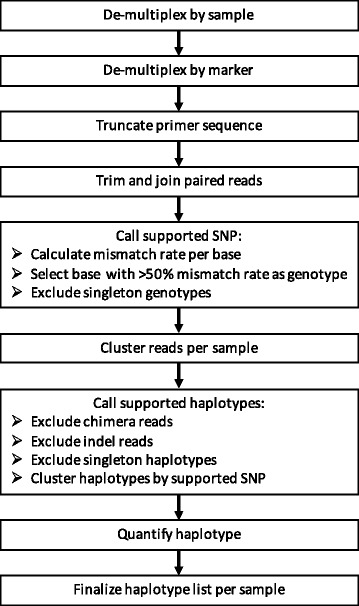
Bioinformatic analysis pipeline applied on highly multiplexed deep sequencing data
For analysis of control samples, fused reads were mapped to the corresponding primers and P. falciparum reference sequences of strains 3D7 and HB3 (Accession: AL844502.2, AL844501.2, AB121018.1, AANS01000117.1). Rates of mismatches to primer and reference sequences were calculated for each individual sample at each nucleotide position. A SNP was defined as a nucleotide position with a > 50% mismatch rate in the sequence reads from at least two independent samples.
For prediction of haplotypes, fused reads were clustered individually per sample with Swarm2 software (parameters: boundary = 3 and fastidious mode) [22, 23]. The centre of each cluster represents the most abundant sequence of the cluster and thus constitutes a predicted haplotype. The cluster size represents the within-sample clone frequency in the tested sample. Haplotypes with a cluster size of 1 were classified as singletons and considered background noise. Haplotypes were checked for PCR artefacts such as indels and chimeric reads. Indels are caused by polymerase slippage which occurred primarily at stretches of homo-polymers. Chimeric reads, caused by incomplete primer extension and inhomologous re-annealing, were identified with vsearch software (parameters: uchime_denovo mode, mindiffs = 3, minh = 0.2) [36]. To distinguish chimera haplotypes resulting from PCR artefacts from true recombined haplotypes, a population-wide approach (combining all samples of the entire study) is implemented in HaplotypR. A chimera was classified as such if a haplotype was identified as chimera by vsearch at all instances it occurred. On the other hand, if a chimera was detected in only some of the samples, it was not classified as chimera, but as a true haplotype. However, in such instances this haplotype was always flagged and the outcome “true chimera” or “true haplotype” was resolved by using replicates. This approach is justified, as it is expected that a true recombinant haplotype would be transmitted without its parent haplotypes.
The full analysis pipeline, named HaplotypR, was implemented as R package and is illustrated in Figure 5 (https://github.com/lerch-a/HaplotypR.git).
Estimated detectability of minority clones by sampling
Detectability of minority clones was estimated by bootstrapping from the reads of the control samples with defined HB3-3D7 strain ratios. Reads were randomly sampled with replacement until the required coverage was reached. These resampled set of reads were processed in the same manner as the original samples using HaplotypR. For resampling only sequence files from HB3-3D7 mixtures were used that had a coverage of >3000 reads.
Additional files
Supporting information. Supplementary text, figures and tables. (DOCX 2111 kb)
List of 200 bp He windows of whole P. falciparum genome. (XLSX 96 kb)
List of haplotype calls. (XLSX 67 kb)
Acknowledgements
We are grateful to the study participants and their guardians and to the field and laboratory team of the PNG Institute of Medical Research, in particular Anna Rosanas-Urgell and Alice Ura. This publication uses data from the MalariaGEN Plasmodium falciparum Community Project as described in Genomic epidemiology of artemisinin resistant malaria, eLife, 2016 (DOI: 10.7554/eLife.08714).
Funding
This work was supported by the Swiss National Science Foundation [310030_159580] and the International Centers of Excellence in Malaria Research [U19 AI089686]. AL was partly funded by Novartis Foundation for Medical-Biological Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
Sequencing data have been deposited at the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRR5412316. A list with the haplotype calls can be found in Additional file 3. HaplotypR can be downloaded from https://github.com/lerch-a/HaplotypR.git.
Abbreviations
- CE
Capillary electrophoresis
- CPMP
PF3D7_0104100, conserved Plasmodium membrane protein
- CSP
Circumsporozoite protein
- He
Expected heterozygosity
- indel
Insertion and deletion
- MOI
Multiplicity of infection
- MSP
Merozoite surface proteins
- PCR
Polymerase chain reaction
- PNG
Papua New Guinea
- SNP
Single nucleotide polymorphism
- SRA
Sequence read archive
Authors’ contributions
Conceived and designed the experiments: IF, IM, AL, CK, SW. Performed the experiments: AL, CK, JHK, NH, CM, SW. Supervised field work and responsible for acquisition of samples: IB. Analysed the data: AL. Supervision: IF, IM, LOC. Writing - draft: AL, IF. Writing - review & editing: CK, NH, JHK, IM, LOC. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Ethical clearance was obtained from PNG Institute of Medical Research Institutional Review Board (IRB 07.20) and PNG Medical Advisory Committee (07.34). Informed written consent was obtained from all parents or guardians prior to recruitment of each child. No medical records were used for this study.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4260-y) contains supplementary material, which is available to authorized users.
Contributor Information
Anita Lerch, Email: anita.lerch@unibas.ch.
Cristian Koepfli, Email: ckoepfli@uci.edu.
Natalie E. Hofmann, Email: natalie.hofmann@swisstph.ch
Camilla Messerli, Email: cammess@gmail.com.
Stephen Wilcox, Email: swilcox@wehi.edu.au.
Johanna H. Kattenberg, Email: jhkattenberg@gmail.com
Inoni Betuela, Email: ibetuela@gmail.com.
Liam O’Connor, Email: ofinwit@gmail.com.
Ivo Mueller, Email: mueller@wehi.edu.au.
Ingrid Felger, Email: Ingrid.Felger@unibas.ch.
References
- 1.Mueller I, Schoepflin S, Smith T a, Benton KL, Bretscher MT, Lin E, et al. Force of infection is key to understanding the epidemiology of plasmodium falciparum malaria in Papua new Guinean children. Proc Natl Acad Sci. 2012;109:10030–10035. doi: 10.1073/pnas.1200841109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snounou G, Beck HP. The use of PCR genotyping in the assessment of recrudescence or reinfection after antimalarial drug treatment. Parasitol Today. 1998;14:462–467. doi: 10.1016/S0169-4758(98)01340-4. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization . Methods and techniques for clinical trials on antimalarial drug efficacy: genotyping to identify parasite populations. 2008. [Google Scholar]
- 4.Anderson TJ, Su XZ, Bockarie M, Lagog M, Day KP. Twelve microsatellite markers for characterization of plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119:113–125. doi: 10.1017/S0031182099004552. [DOI] [PubMed] [Google Scholar]
- 5.Falk N, Maire N, Sama W, Owusu-Agyei S, Smith T, Beck H-P, et al. Comparison of PCR-RFLP and Genescan-based genotyping for analyzing infection dynamics of plasmodium falciparum. Am J Trop Med Hyg. 2006;74:944–950. [PubMed] [Google Scholar]
- 6.Anderson TJC, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, et al. Microsatellite markers reveal a Spectrum of population structures in the malaria parasite plasmodium falciparum. Mol Biol Evol. 2000;17:1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- 7.Messerli C, Hofmann NE, Beck H-P, Felger I. Critical Evaluation of Molecular Monitoring in Malaria Drug Efficacy Trials and Pitfalls of Length-Polymorphic Markers. Antimicrob. Agents Chemother. 2017;61:AAC.01500–16. [DOI] [PMC free article] [PubMed]
- 8.Juliano JJ, Porter K, Mwapasa V, Sem R, Rogers WO, Ariey F, et al. Exposing malaria in-host diversity and estimating population diversity by capture-recapture using massively parallel pyrosequencing. Proc Natl Acad Sci U S A. 2010;107:20138–20143. doi: 10.1073/pnas.1007068107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neafsey DE, Juraska M, Bedford T, Benkeser D, Valim C, Griggs A, et al. Genetic diversity and protective efficacy of the RTS,S/AS01 malaria vaccine. N Engl J Med. 2015;373:2025–2037. doi: 10.1056/NEJMoa1505819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin JT, Hathaway NJ, Saunders DL, Lon C, Balasubramanian S, Kharabora O, et al. Using amplicon deep sequencing to detect genetic signatures of plasmodium vivax relapse. J Infect Dis. 2015;212:999–1008. doi: 10.1093/infdis/jiv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey J a, Mvalo T, Aragam N, Weiser M, Congdon S, Kamwendo D, et al. Use of massively parallel pyrosequencing to evaluate the diversity of and selection on Plasmodium falciparum csp T-cell epitopes in Lilongwe, Malawi. J Infect Dis. 2012;206:580–587. doi: 10.1093/infdis/jis329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedrich LR, Popovici J, Kim S, Dysoley L, Zimmerman PA, Menard D, et al. Complexity of infection and genetic diversity in Cambodian plasmodium vivax. PLoS Negl Trop Dis. 2016;10:e0004526. doi: 10.1371/journal.pntd.0004526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang H, Worby CJ, Yeka A, Nankabirwa J, Kamya MR, Staedke SG, et al. THE REAL McCOIL: a method for the concurrent estimation of the complexity of infection and SNP allele frequency for malaria parasites. PLoS Comput Biol. 2017;13:e1005348. doi: 10.1371/journal.pcbi.1005348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 15.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 16.Pallen MJ. Reply to updating benchtop sequencing performance comparison. Nat Biotechnol. 2013;31:296. doi: 10.1038/nbt.2531. [DOI] [PubMed] [Google Scholar]
- 17.MalariaGEN Plasmodium falciparum Community Project Genomic epidemiology of artemisinin resistant malaria. elife. 2016;5:1–29. doi: 10.7554/eLife.08714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esling P, Lejzerowicz F, Pawlowski J. Accurate multiplexing and filtering for high-throughput amplicon-sequencing. Nucleic Acids Res. 2015;43:2513–2524. doi: 10.1093/nar/gkv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301:1503–1508. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]
- 20.Mobegi V a, Duffy CW, Amambua-ngwa A, Loua KM, Laman E, Nwakanma DC, et al. Genome-wide analysis of selection on the malaria parasite plasmodium falciparum in west African populations of differing infection endemicity. Mol Biol Evol. 2014;31:1490–1499. doi: 10.1093/molbev/msu106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barry AE, Schultz L, Buckee CO, Reeder JC. Contrasting population structures of the genes encoding ten leading vaccine-candidate antigens of the human malaria parasite, plasmodium falciparum. PLoS One. 2009;4:e8497. doi: 10.1371/journal.pone.0008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahé F, Rognes T, Quince C, de Vargas C, Dunthorn M. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ. 2014;2:e593. doi: 10.7717/peerj.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahé F, Rognes T, Quince C, de Vargas C, Dunthorn M. Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ. 2015;3:e1420. [DOI] [PMC free article] [PubMed]
- 24.Mideo N, Bailey JA, Hathaway NJ, Ngasala B, Saunders DL, Lon C, et al. A deep sequencing tool for partitioning clearance rates following antimalarial treatment in polyclonal infections. Evol Med public Heal. 2016;2016:21–36. doi: 10.1093/emph/eov036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinozuka H, Forster JW. Use of the melting curve assay as a means for high-throughput quantification of Illumina sequencing libraries. PeerJ. 2016;4:e2281. doi: 10.7717/peerj.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris JK, Sahl JW, Castoe TA, Wagner BD, Pollock DD, Spear JR. Comparison of normalization methods for construction of large, multiplex amplicon pools for next-generation sequencing. Appl Environ Microbiol. 2010;76:3863–3868. doi: 10.1128/AEM.02585-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun. 2012;3:811. doi: 10.1038/ncomms1814. [DOI] [PubMed] [Google Scholar]
- 28.Rao PN, Uplekar S, Kayal S, Mallick PK, Bandyopadhyay N, Kale S, et al. A method for amplicon deep sequencing of drug resistance genes in plasmodium falciparum clinical isolates from India. J Clin Microbiol. 2016;54:JCM.00235-16. doi: 10.1128/JCM.00235-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levitt B, Obala A, Langdon S, Corcoran D, O’Meara WP, Taylor SM. Overlap extension Barcoding for the next generation sequencing and genotyping of plasmodium falciparum in individual patients in western Kenya. Sci Rep. 2017;7:41108. doi: 10.1038/srep41108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, et al. An inducible Lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations InVivo. Cell Rep. 2015;10:1422–1432. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Beerenwinkel N, Günthard HF, Roth V, Metzner KJ. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol. 2012;3:329. doi: 10.3389/fmicb.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betuela I, Rosanas-Urgell A, Kiniboro B, Stanisic DI, Samol L, de Lazzari E, et al. Relapses contribute significantly to the risk of plasmodium vivax infection and disease in Papua new Guinean children 1–5 years of age. J Infect Dis. 2012;206:1771–1780. doi: 10.1093/infdis/jis580. [DOI] [PubMed] [Google Scholar]
- 33.Kibbe WA. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:43–46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quail M a, Otto TD, Gu Y, Harris SR, Skelly TF, McQuillan JA, et al. Optimal enzymes for amplifying sequencing libraries. Nat Methods. 2012;9:10–11. doi: 10.1038/nmeth.1814. [DOI] [PubMed] [Google Scholar]
- 35.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ Prepr. 2016;4:e2409v1. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information. Supplementary text, figures and tables. (DOCX 2111 kb)
List of 200 bp He windows of whole P. falciparum genome. (XLSX 96 kb)
List of haplotype calls. (XLSX 67 kb)
Data Availability Statement
Sequencing data have been deposited at the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRR5412316. A list with the haplotype calls can be found in Additional file 3. HaplotypR can be downloaded from https://github.com/lerch-a/HaplotypR.git.