

Abstract
Genetic tools and mutagenesis strategies based on transposable elements are currently under development with a vision to link primary DNA sequence information to gene functions in vertebrate models. By virtue of their inherent capacity to insert into DNA, transposons can be developed into powerful tools for chromosomal manipulations. Transposon-based forward mutagenesis screens have numerous advantages including high throughput, easy identification of mutated alleles and providing insight into genetic networks and pathways based on phenotypes. For example, the Sleeping Beauty transposon has become highly instrumental to induce tumors in experimental animals in a tissue-specific manner with the aim of uncovering the genetic basis of diverse cancers. Here, we describe a battery of mutagenic cassettes that can be applied in conjunction with transposon vectors to mutagenize genes, and highlight versatile experimental strategies for the generation of engineered chromosomes for loss-of-function as well as gain-of-function mutagenesis for functional gene annotation in vertebrate models, including zebrafish, mice and rats.
Keywords: genetic screens, insertional mutagenesis, forward genetics, animal models, stem cells, genomics
Forward genetic screens by insertional mutagenesis to annotate gene function
Diverse methods have been employed to address the challenge of functional annotation of every gene and elaborate genetic networks and pathways. Mutational analysis proved to be one of the most direct ways to interrogate gene functions. There are versatile strategies for generating mutant phenotypes. For example, reverse genetics approaches rely on targeting and disrupting genes of interest, preferably in pluripotent embryonic stem cells (ESCs), thereby creating engineered alleles that allow for the functional dissection of genes of interest on the cellular and organismal levels. In other words, reverse genetics is based on a candidate gene approach, in which the investigator addresses the functional consequences of modifying the expression of a given gene. Thus, this gene-by-gene approach does not facilitate gene discoveries related to a particular pathway of interest on a genome-wide scale. A shorter route to address this problem is designing genetic screens, in which phenotypes of interest are identified first and then followed by the identification of causative gene mutations. Indeed, forward genetic approaches aim to obtain mutant phenotypes by introducing loss-of-function or gain-of-function mutations in genomes of model organisms in a random and genome-wide fashion. In that manner, mutagenesis can be applied not only to decipher the functions of individual, mutated genes but also to provide insight into genetic pathways and networks.
Mutating genes by insertion of discrete pieces of foreign DNA has at least two advantages over other strategies of mutagenesis. First, insertion of foreign DNA is typically associated with a more profound mutagenic load than that of a point mutation, thereby generating more robust phenotypes. Second, the inserted DNA fragment can serve as a unique molecular tag that allows rapid, usually PCR-based, identification of the mutated allele. Insertional mutagenesis using engineered transposons can be one of the most productive and versatile approaches toward disrupting and manipulating genes on a genome-wide scale.
Transposon basics
DNA transposons are mobile genetic elements with the ability to change their positions within the genome via a “cut-and-paste” mechanism called transposition. During the transposition process, the transposon is removed from its original genomic location and is inserted somewhere else in the genome by the transposon-encoded transposase enzyme (Fig. 1). Because transposition involves insertion of the transposon DNA into a new locus, the process is inherently mutagenic. The potential of a transposon insertion to result in any alteration of gene function depends on whether the transposon lands in or near a gene. There are several ways by which a transposon can influence gene expression [1]. For example, the transposon may disrupt the coding region of a gene (Fig. 2A,B). Typically, this would lead to loss of function of that particular allele, although dominant negative effects may also arise, depending on where exactly within the gene the insertion occurred. Intronic insertions may be spliced out without having an effect on gene expression, but may ectopically drive transcription of a sense or antisense transcript from the transposon’s promoter (Fig. 2C,D). Cryptic splice sites present in the transposon sequence may result in the generation of truncated transcripts (Fig. 2E). Transposon insertions at the 5′-transcriptional regulatory region of a gene may introduce an alternative transcription start site or may override the transcriptional program of the gene’s endogenous promoter (Fig. 2F). Finally, transposon insertion in the 3′-UTR of a gene may introduce an alternative polyadenylation site (Fig. 2G). Mutagenesis caused by transposition resulting in unstable pigmentation phenotypes in maize kernels was the very clue for Barbara McClintock’s groundbreaking discovery of transposable elements [2].
Figure 1. Mechanism of cut-and-paste transposition.
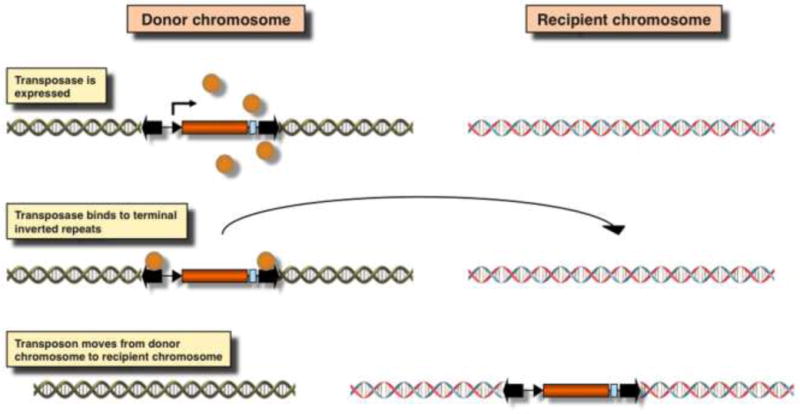
The transposon is a self-contained mobile genetic element containing a transposase coding sequence (orange box) flanked by terminal inverted repeats (TIRs; thick black arrows on the left and right). Transcriptional regulatory elements, including a promoter (small arrow) and polyA site (light blue box) regulate the expression of the transposase (orange spheres). Transposase molecules bind to the TIRs and catalyze the movement of the transposase to a new chromosomal location.
Figure 2. Possible mutagenic consequences of transposon integration in or close to a transcription unit.
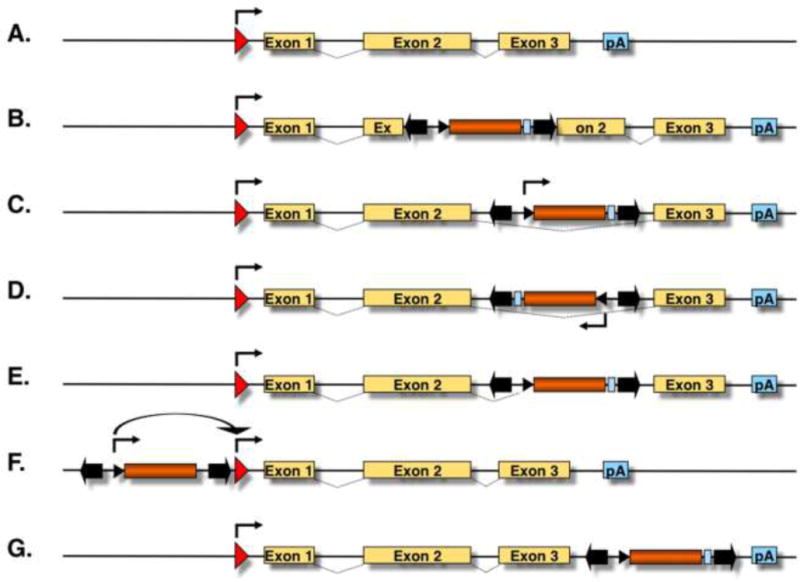
(A) The figure depicts a hypothetical transcription unit with a promoter (red arrow), three exons (yellow boxes) and a polyA signal (pA, light blue box). Splicing is indicated by dashed lines between the exons. (B) Transposition into an exon disrupts the coding sequence of the gene. Transposition into an intron may result in expression of a sense (C) or antisense (D) transcript from the transposon’s promoter. (E) Cryptic splice sites present in the transposon sequence may result in the generation of truncated transcripts. (F) Transposon insertions at the 5′-transcriptional regulatory region of a gene may introduce an alternative transcription start site or may override the transcriptional program of the gene’s endogenous promoter. (G) Transposon insertion in the 3′-UTR of a gene may introduce an alternative pA.
In nature, transposons exist as single units containing the transposase gene flanked by terminal inverted repeats (TIRs) that carry binding sites for transposase (Fig. 1). The underlying biochemical basis for using any transposon as a vector for genetic engineering is the trans-complementarity of the two functional components of transposition (i. e., the TIRs and the transposase). Because the transposase source can be physically separated from the TIRs, bi-component transposon systems can be engineered, in which virtually any DNA sequence of interest can be placed between the transposon TIRs and mobilized by conditionally expressing the transposase (Fig. 3A) from i) a transiently transfected expression plasmid, ii) transiently transfected mRNA and iii) a genomically located expression cassette. This feature makes transposons natural and easily controllable DNA delivery vehicles that can be used as tools for versatile applications ranging from somatic and germline transgenesis to functional genomics and gene therapy [3–5].
Figure 3. Mutagenic cassettes.
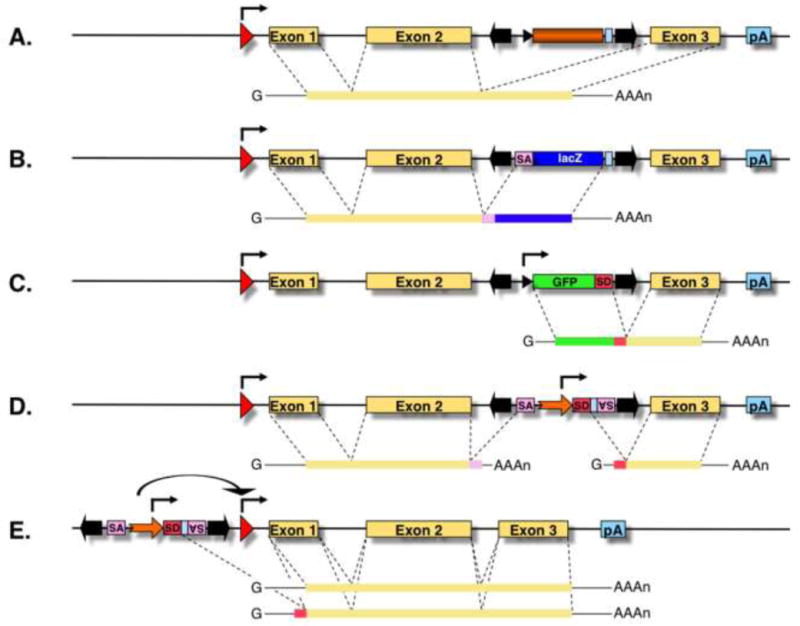
(A) A hypothetical transcription unit is depicted with an upstream promoter (red arrow), three exons (yellow boxes) and a polyadenylation signal (pA). An intronic transposon insertion is typically not mutagenic, because the transposon is spliced out from the primary RNA transcript together with the targeted intron sequences, thereby resulting in normal gene expression. (B) Gene trapping cassettes contain a splice acceptor (SA) followed by a reporter gene and a pA. The SA truncates the transcript, and expression of the reporter follows the expression pattern of the trapped gene. (C) Poly(A) traps contain a promoter followed by a reporter gene and a splice donor (SD) site, but they lack a pA signal. Therefore, reporter gene expression depends on splicing to downstream exon/s of a Pol II transcription unit containing a pA. (D) The oncogene trap contains SA signals in both orientations and a bidirectional pA signal to disrupt transcription, as well as a strong, viral enhancer/promoter (thick orange arrow) that drives transcription towards the outside of an inserted transposon, and thereby overexpresses a gene product. In case this transposon lands in the 5′ transcriptional regulatory region of a gene (E), a full-length transcript might be overexpressed.
Transposons have been successfully used in plants [6–9] as well as in invertebrate animal models, including C. elegans [10–13] and Drosophila [14–18] for transgenesis and insertional mutagenesis. In vertebrate animals, the molecular reconstruction of the Sleeping Beauty (SB) transposon system, a Tc1/mariner-type transposon found in several fish genomes [19], opened up new possibilities for genome engineering. This newly reactivated element allowed highly efficient transposition-mediated gene transfer in major vertebrate model species without the potential risk of cross-mobilization of endogenous transposon copies in host genomes. This is because the genomes of major models lack endogenous transposon sequences with sufficient sequence similarity that would be required for mobilization by an exogenously supplied SB transposase. SB has been successfully used as a tool for genetic modifications of a wide variety of vertebrate cell lines and species including humans (reviewed in [5, 20, 21]). During the past decade, other elements have been shown to catalyze efficient transposition in vertebrate model organisms. For example, the insect element piggyBac (PB) [22–24] catalyzes efficient transposition in mammalian cells. Moreover, the Tol2 element, a naturally occurring active transposon isolated from the medaka fish [25, 26], has been shown to be active in various vertebrate species [27]. The basic criteria for the applicability of a transposon in any given model organism are i) a sufficient level of transpositional activity in the given species, and ii) target site selection properties of the transposon, which are discussed in the next section.
Integration site preference
Target site preferences of different transposons range from essentially random to selective with respect to the actual DNA sequences, at which integrations occur, but invariably non-random at the genome-wide scale. For example, on the level of primary DNA sequence, the Tol2 element does not appear to exhibit a pronounced preference for any sequence for insertion [28]. In contrast, the Harbinger3_DR transposon from zebrafish is highly specialized to integrate into the palindromic AAACACCWGGTCTTT consensus sequence [29], the piggyBac transposon targets the sequence TTAA [30, 31], TcBuster and SPIN elements target palindromic 8-bp target sites with a conserved central TA [32], whereas all Tc1/mariner transposons, including SB [19], Frog Prince [33], Minos [34] and Hsmar1 [35], target their integration into TA dinucleotides. In the case of SB, this preference was studied in detail, and palindromic TA repeats were found to be preferred sites for integration [36]. However, computational analyses revealed that target selection is determined primarily on the level of DNA structure, not by specific base-pair interactions. For example, protein-induced deformability was shown to be associated with preferred SB insertion sites, whereas PB and Tol2 integration sites lack such consistent, clear-cut structural patterns [37, 38]. On the genomic scale, SB transposons exhibit a close-to-random integration profile with a slight bias towards integration into expressed genes and their upstream regulatory sequences in cultured mammalian cell lines and primary human cells [28, 32, 39–43]. This is in marked contrast to target site distributions of several other transposons including Tol2 [28, 41, 44], TcBuster [32], SPIN [32] and PB [28, 32, 41, 44] that all show significant difference from random insertion with respect to favored integration into genes and near chromatin marks characteristic of active transcription units (e.g., H3K27 acetylation and H3K4 monomethylation) and disfavored integration near marks characteristic of inactive chromatin (e.g., H3K27 trimethylation). The PB transposon, in particular, has been shown to favor open chromatin, expressed genes and TSSs (±5kb) associated with DNaseI hypersensitive sites, H3K4Me3 marks and Pol II-bound regions in mouse and human cells [23, 28, 44–49]. This control of integration at the chromatin level is poorly understood. One possible explanation for this can be the interaction of the transposase with chromatin-associated factors. Indeed, it has been recently shown that PB is targeted to TSSs through an interaction of the PB transposase with BET domain proteins [43], similar to the mechanism shown to be responsible for the enrichment of MLV integrations into TSSs [50]. Taken together, the preferences of particular elements to integrate into intragenic versus intergenic chromosomal regions, and preferences for integration sites within genes are substantially different.
Integration site preference can greatly influence the utility of transposon vectors for different applications. Indeed, the insertional biases associated with vector systems represent the main limitation to full genome coverage with individual transposon-based vectors. Thus, in this respect, the utility of transposons for mutagenesis is greatly enhanced by the availability of multiple, alternative vector systems with distinct preferences for insertion, such as SB, Tol2 and PB. Indeed, the propensity of Tol2 and PB to insert close to transcriptional start sites of genes might be particularly advantageous for enhancer trapping [51, 52] or for the overexpression of entire genes [53], whereas SB is expected to generate a wider range of mutant alleles.
Transposons and functional genomics
Mutagenic cassettes
As discussed above, transposons are natural mutagens. There are two main features that are typically incorporated in a transposon vector for mutagenesis purposes: a mutagenic cassette that enhances a loss-of-function or gain-of-function effect on gene expression and a reporter cassette that is conditionally expressed upon transposon insertion into a gene (Key Figure, Fig. 3).
There are several types of mutagenic cassettes that can be efficiently combined with transposon-based gene delivery for insertional mutagenesis, all designed to enhance the mutagenic effect of a transposon that landed in or near a gene (Fig. 3A). 5′ gene trap cassettes include splice acceptors (SA) and polyadenylation (pA) sequences so that transcription of genes can be disrupted upon vector insertion into introns (Fig. 3B) [54]. Frequently, such cassettes are also equipped with a reporter gene (usually, a fluorescent protein, β-galactosidase or antibiotic resistance), whose expression is dependent on the correct splicing between exons of the trapped gene and the SA site carried by the transposon vector [55, 56]. Commonly, such gene traps have been used for ESC mutagenesis [57, 58], and were also employed in combination with the SB delivery system in several studies for mutagenizing ESCs [59] as well as the germlines of experimental animals [60–68]. In addition, the efficiency of gene trapping can be further improved by inserting an internal ribosome entry site (IRES) sequence in front of the reporter gene, which allows for the expression of the reporter cassette irrespective of the reading frame of the disrupted gene [69]. Trapping and discovery of low expressing genes can be further facilitated by using transcriptional transactivation systems, in which an initial, low level of the gene trap reporter signal is amplified by trans-activation of a second reporter and hence made detectable. For example, a conditionally expressed tTA (tetracycline controllable) transcriptional activator has been included in an SB-based IRES-gene trap vector to amplify the trap signal by activating transcription of a reporter in zebrafish embryos and mice [64, 70]. Gene trapping was combined with a coat color marker in order to provide a method for fast and noninvasive identification of new transposition events and homozygous rats [68]. Because gene trap vectors depend on the transcriptional activities of endogenous promoters/enhancers to drive the expression of their reporter cassettes, they can only report insertions into genes that are actively transcribed in the tissue of interest in which mutagenesis is done.
Another type of cassette that can be incorporated into transposons is called a polyA trap that is equipped with an internal promoter, reporter cassette and splice donor (SD) site, but lacks a pA signal (Fig. 3C). If such cassette lands in a transcription unit in the right orientation, the RNA transcript initiated by the internal promoter splices to the endogenous, downstream exons and is processed and polyadenylated. Therefore, polyA traps can be used for trapping genes regardless of their transcriptional activity, but are only expected to be sufficiently mutagenic when combined with gene-breaking trap cassettes [21, 55, 67, 71]. As for the gene traps, efficiency of polyA trapping vectors can be improved by incorporating an IRES sequence, which suppresses nonsense-mediated RNA decay (NMD) of chimeric transcripts, between the reporter cassette and the SD site, thereby removing the bias in preferentially detecting those vector integrations that occurred in the last introns of the trapped genes [72].
A specific combination of gene- and polyA trap features was developed to discover proto-oncogenes as well as novel tumor suppressor genes in mice. This “oncogene trap” contains SA and pA signals in both orientations in order to disrupt transcription of endogenous genes (Fig. 3D), which is useful for knocking out tumor suppressor genes (TSGs). In addition, oncogene trap cassettes also include strong viral enhancers/promoters that can drive transcription outwards from the vector, and an SD site, at which the transcript is spliced to downstream exons, thereby leading to overexpression of a full-length or truncated protein product of the trapped gene (Fig. 3D,E) [73, 74]. Finally, the strong enhancer element inside the transposon may transactivate the promoter of an oncogene, thereby resulting in overexpression of an unfused transcript (Fig. 3E).
Genetic screens in cultured cells
In cell culture systems, mutagenic transposons can be introduced into the genome by transfection of plasmid DNA harboring the transposon system components or by establishing a transgenic master cell line containing a genomically integrated transposon that is mobilized to secondary locations by transient transposase expression (Fig. 4). Transfection-based, “plasmid-to-genome” delivery (Fig. 4A) yields insertion profiles characteristic to the different transposon systems, as described above; however, careful titration of the amount of the donor and transposase plasmid is required to provide the appropriate copy number per cell of the transposon. Intra-genomic, “genome-to-genome” mobilization (Fig. 4B) enables tight control over copy number, but local hopping poses some limitation with respect to the fraction of the genome that can be efficiently mutagenized [45, 59] (described in more detail below).
Figure 4. Experimental mobilzation of transposons.
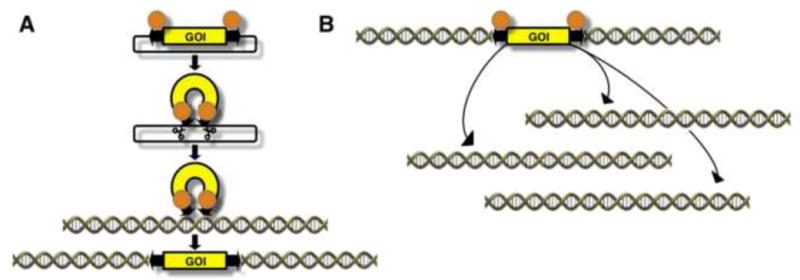
(A) Plasmid-to-genome mobilization. The transposon is typically mobilized out of transfected plasmids upon transient expression of the transposase. (B) Intra-genomic mobilization. Upon transposase expression, the genomically located transposon will be excised from the donor site and re-integrate at a different genomic location.
Pluripotent ESCs are attractive models for in vitro mutagenesis, because i) it is possible to perform preselection of modified cell clones before generating mutant animals, ii) they can differentiate into many cell types including the germline, and iii) they are amenable to sophisticated genetic manipulations, including transposon mutagenesis. However, insertional mutagenesis in somatic cells is challenged by the diploid genome. Both copies of a gene are nearly always required to be inactivated to evoke a phenotypic change, but the probability of generating bi-allelic mutations of a single locus by two independent “hits” is extremely low. Moreover, long generation times hamper the efficient production of homozygous knockouts in mammals. The recent discovery of haploid mammalian ESC lines can aid recessive genetic screens in mammalian cells, and can serve as an efficient experimental system way to generate homozygous, heritable genetic modifications [75–77]. Importantly, haploid ESCs isolated from androgenetic embryos have been demonstrated to support the generation of germline-modified mice via intracytoplasmic injection into oocytes [77, 78]. Moreover, haploid ESCs can serve as a convenient tool to generate homozygous mutants by transposon mutagenesis on a genome-wide scale for functional gene studies [79].
PB transposons carrying mutagenic SA sites in both orientations have been used for generating large pools of mutagenized mouse haploid ESCs by the “plasmid-to-genome” delivery approach [80]. Libraries containing up to 20,000 mutant cell clones containing, on average, a single transposon insertion per cell were selected, and exposed to drugs including 6-thioguanine and the Poly (ADP-Ribose) Polymerase (PARP) inhibitor olaparib, allowing the isolation of loss-of-function mutants of different mismatch repair genes and several mutant alleles of PARP superfamily genes, respectively [80]. Thus, haploid ESCs enable the isolation of knockout phenotypes in a single round of mutagenesis in a scalable manner. In another application, mutagenized haploid ESCs were screened for drug sensitivity phenotypes [81]. In this study ~100,000 mutant cell clones were generated by a gene trap PB transposon carrying an SA-IRES-neo cassette and a random barcode sequence, whose recovery by next generation sequencing enables quantitation of the number of cells bearing each barcode, and hence each transposon insertion. These mutant cell pools were then exposed to 13 different drugs followed by an assessment of barcode abundance in order to identify mutations that conferred sensitivity to inhibitors of topoisomerases, PARP and HSP90 [81]. The feasibility of introducing large numbers of mutagenic transposon insertions has also been demonstrated in haploid ESCs established from rats by gene trapping with PB vectors carrying an antibiotic selection marker [82].
In order to pass a mutation through the germline and to establish a homozygous knockout animal, mutagenized and clonally expanded ESCs need to be introduced into a mouse embryo by blastocyst injection, followed by two rounds of breeding: the first to pass the genotype through the germline and the second to bring the mutant allele to homozygosity (Fig. 5). In addition to the application of haploid ESCs described above, another technology that allows for obtaining a homozygous knockout (and thus a phenotype) in a more straightforward manner is mutagenesis in spermatogonial stem cells (SSCs). SSCs can be cultured in vitro similar to ESCs, and an approach to insertional mutagenesis in rat SSCs was recently established by using SB gene trap transposons [83–85]. A fundamental difference to ESC-based technologies is that mutagenized SSCs can be directly transplanted to repopulate the testes of sterilized, wild-type recipient male rats, thereby bypassing the step of generating a chimeric animal. Thus, the SSC genome is passed on to transgenic offspring upon a single cross of the recipient males with wild-type females (Fig. 5). By using this approach, several mutant alleles have been tagged including genes implicated in metabolic disorders, cancer, neurological and behavioral disorders, fertility, developmental defects, cardiovascular and kidney diseases, gastrointestinal disorders, bone defects and the immune system (http://www4.utsouthwestern.edu/hamralab/ko_disease.htm). Since several aspects of physiology in rats has evolved more closely to humans than that of mice, transposon mutagenesis in SSCs has the potential to develop powerful genomic tools for the rat, offering the opportunity to create a bridge between physiology and genomics.
Figure 5. Generation of knock-out animals by insertional mutagenesis in embryonic and spermatogonial stem cells.
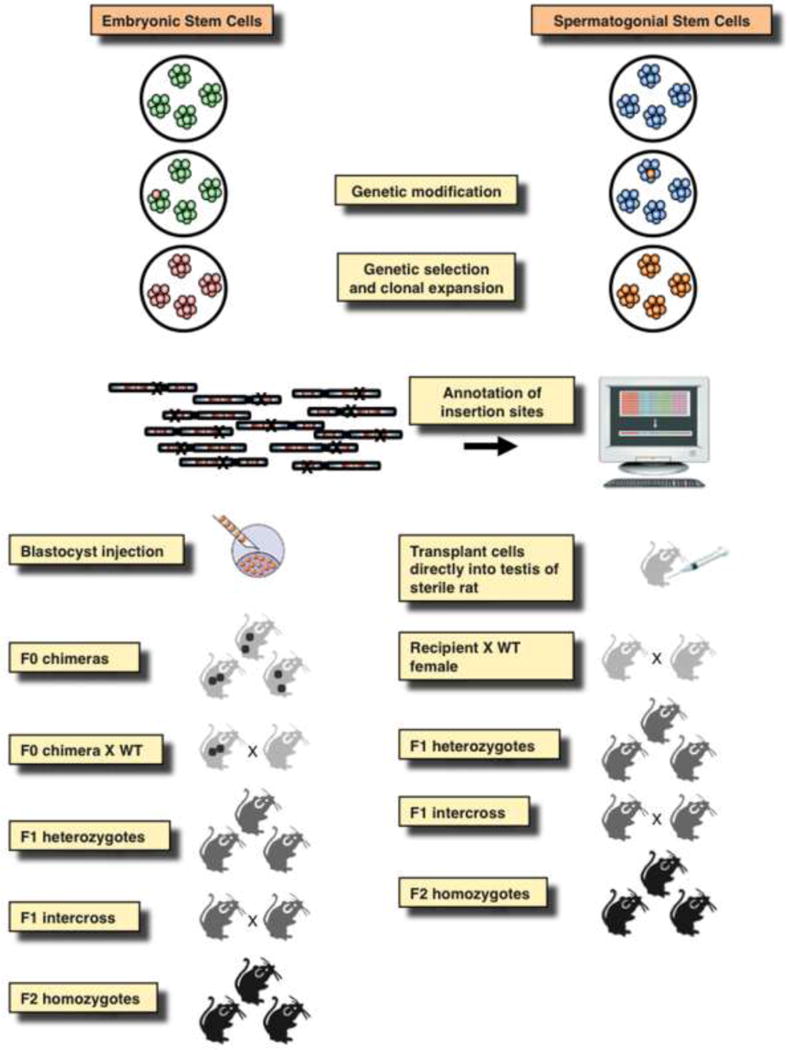
Cultured embryonic stem cells (ESCs) or spermatogonial stem cell (SSCs) are transfected with mutagenic transposon and transposase constructs that will lead to thousands of transposon insertions covering all chromosomes. Those cells, in which insertions occurred in genes can be selected based on activation of a reporter, and the insertion sites can be mapped. Clonally derived ESCs are transplanted into mouse embryos that will develop into chimeric animals that need to be crossed with wild-type (WT) animals to derive F1 heterozygotes. An F1 intercross will yield F2 homozygotes, in which a phenotype can be studied. SSC clones or polyclonal insertion libraries can be directly transplanted into the testes of sterile males, in which the spermatogonial step cells will undergo spermatogenesis. These transplanted males are crossed with WT females to pass the insertions through the germline and generate transgenic/knock-out animals.
Functional genomics in zebrafish using Tol2
The Tol2 transposon system has been used to create transgenic animals in zebrafish [86], other fish species including tilapia, killifish and cichlid fish [87–89], mouse [90], chicken [91] and frog [92]. Also, Tol2 vectors have been used for transfection of foreign DNAs into mammalian cells including ESCs, iPSCs and human lymphocytes [86, 93, 94]. In addition, Tol2 vectors with a fluorescent marker or a transcription activator such as rtTA have been introduced to chick and mouse embryos by electroporation to label and analyze specific cell types in vivo [95, 96]. More recently, these led to the development of the Tol2kit [97], pTransgenesis [98], and MAGIC systems [99].
The most significant contribution of Tol2 has been to zebrafish research. Although transposons of the Tc1/mariner family, namely Tc3 [100], mariner [101] and SB [102], have been shown to transpose in zebrafish and transmit to next generations through the germ line, the transgenic efficiencies with these transposons were not sufficiently high to develop robust technologies in zebrafish. This situation dramatically changed when Tol2 was shown to transpose very efficiently in the zebrafish germ lineage [103]. To successfully perform genetic screens such as gene trapping and enhancer trapping, it is required to create hundreds and thousands of transposon insertions efficiently in the genome since only small portions of insertions can give rise to transgenic fish with expression patterns of interest (Fig. 6). With the high transposition efficiency of Tol2, such screens became possible for the first time in zebrafish [51, 103]. Since then, Tol2-based transgenesis, including transfer of large DNA fragments such as BACs [104], became standard in the zebrafish research field. To date, numerous transgenic fish that express fluorescent markers or modified versions of the yeast transcription factor Gal4 have been generated and accelerated studies of developmental biology, organogenesis and neuroscience [105–107]. First, these transgenic fish are valuable to visualize specific cell types (Fig. 7). Since the body of a zebrafish larva is transparent through all developmental stages, such transgenic fish have been used to visualize the vascular system [108], the central and peripheral nervous systems [109, 110], the regenerating processes of the sensory system and fin [111, 112], the cellular process of muscle wound repair [113], and proteolytic cleavage of the extracellular domain of a membrane protein (Neuregulin) in the motor neurons [114], among others. Also, the transgenic fish were applied to image the activity of specific neuronal populations or endothelial cells by targeted expression of a calcium indicator GCaMP [115, 116]. In addition, gene trap vectors similar to those shown in Fig. 3B were applied to analyze and visualize the subcellular localization of hundreds of proteins [117, 118]. Second, with the Gal4-UAS system, these transgenic fish were used to modify functions of specific cells and tissues. Gal4-expressing specific cell types can be ablated by targeted expression of the nitroreductase gene and application of a prodrug which is converted into a toxic compound by the activity of nitroreductase [105]. Also, inhibition of specific neuronal populations have been performed by targeted expression of a neurotoxin gene [107, 119]. Third, when a transposon insertion landed within a gene, in some cases the function of the gene was disrupted and the mutant phenotype was analyzed [120, 121]. In sum, both Tol2 transposon-mediated forward genetic approaches, including gene trapping, as well as reverse genetic approaches, including a functional analysis of a cloned regulatory element, a gene or cDNA, have greatly contributed to the development of the zebrafish research.
Figure 6. A scheme for gene and enhancer trapping for the Gal4-UAS system.
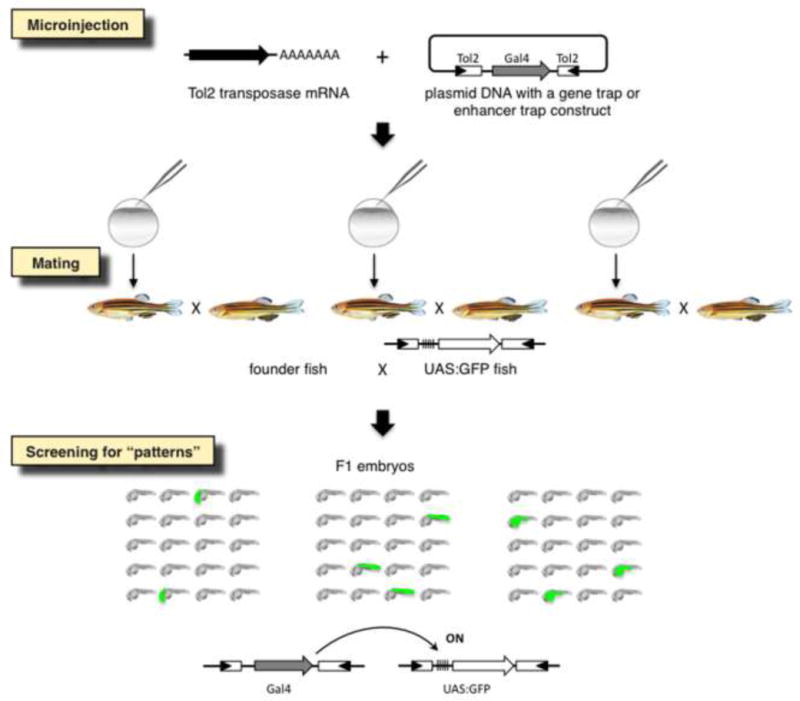
A gene trap or an enhancer trap construct containing gal4 is injected into fertilized eggs with the transposase mRNA. Injected founder fish are raised and mated with UAS:GFP reporter fish. When gal4 is expressed under the control of an endogenous promoter/enhancer, GFP expression is induced in the F1 embryos via the Gal4-UAS system. Double transgenic F1 embryos express GFP in spatially and temporally restricted fashions may be picked up.
Figure 7. Transgenic fish with specific gal4/gfp expression patterns.
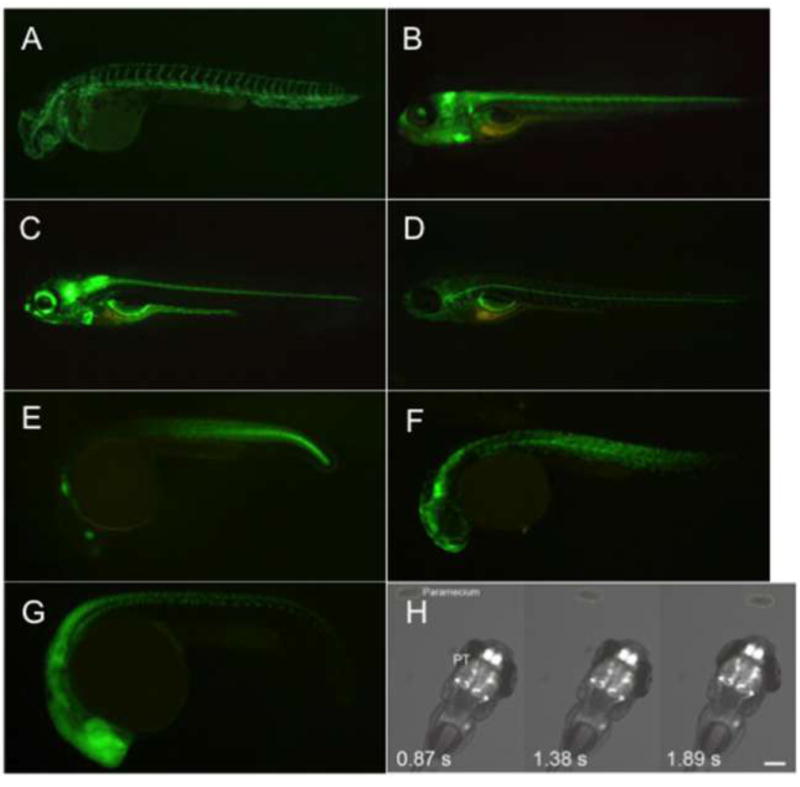
(A) Blood vessels in gSAIzGFF478A embryos at 3 days post-fertilization (dpf) [108]. (B) Central nervous system (cerebellum) in SAGFF128A embryo at 5 dpf [109]. (C) Central and enteric nervous system in SAGFF234A at 5 dpf [110]. (D) Lateral line glial cells in gSAGFF202A at 5 dpf [111]. (E) Caudal trunk (and the wound epidermis) in HGn21A embryo at 1 dpf [112]. (F) Fgf7b-positive muscle cells in gSAIzGFFD164A at 1 dpf [113]. (G) Central nervous system and motor neurons in the spinal cord in SAIGFF213A at 1 dpf [114]. (H) Calcium imaging of the pretectal neurons while a larva recognizes a paramecia (prey) [115].
Gain-of-function and loss-of-function screens for cancer in mice
Cut-and-paste DNA transposons, including SB and PB, have been used to introduce random insertion mutations in tissues of mice for the purpose of inducing cancer and identifying cancer genes [122] (Supplementary Table 1). These studies were initially motivated by older research showing that some retroviruses could establish chronic infections in animals and induce the formation of cancer via insertional mutagenesis [123]. These so-called “slow transforming retroviruses” (retroviruses that cause tumors after a latency period of 3–9 months [124]) can induce mammary tumors, leukemia, lymphoma and certain other types of cancer in sensitive species. The integration of the provirus causes the specific genetic damage that induces these cancers. Integration of a provirus can cause the activation of endogenous oncogenes, and indeed this is the most common type of specific genetic damage observed in these cases. In other rarer instances, the integrated provirus disrupts expression of endogenous TSGs. In the vast majority of infected cells, proviral integration has no consequence for the infected cell, while in rare cells proviral activation of an endogenous proto-oncogene or inactivation of a TSG begins the process of cancer development. The cancer develops via subclone selection of progressively more malignant cells that have additional cooperating mutations, some of which are also due to proviral insertional mutagenesis. Recurrent proviral insertions in the same genomic regions are observed in such cancers because they are selected for from among millions of insertions that cause no phenotype. These recurrent proviral insertions are called “common insertion sites” or “common integration sites” (CIS). The study of slow transforming retroviruses helped identify some of the first cancer genes such as Myc and Myb, but had some limitations as experimental tools. First, these viruses and the cancer they induce were found and not created. Second, slow transforming retroviruses were limited to induce only certain forms of cancer, including mammary tumors [by Mouse Mammary Tumor Viruses (MMTV)], or hematopoietic cancers [by Murine Leukemia Viruses (MuLV)]. Thus, when SB became the first cut-and-paste DNA transposon available for use in vertebrate models scientists quickly attempted to use it to create models of a wide variety of cancers induced by insertional mutagenesis. As with the slow transforming retroviruses, cancer models induced by transposon mutagenesis allow identification of specific cancer genes at CIS.
MuLV and MMTV have enhancers, promoters and SDs that can activate nearby or downstream endogenous proto-oncogenes upon insertion. They also have SAs and pA sites that can disrupt endogenous TSGs. Thus, such sequences were incorporated into SB and PB vectors mobilized body-wide or tissue-specifically for cancer gene identification using these transposons. The vectors for somatic cell transposon mutagenesis have included a strong promoter (CAGS or the U3 and R region of the Murine Stem Cell Virus long terminal repeat) and SD and two SAs (one in each orientation) and a bidirectional pA site [122]. In this way, transposon insertion can cause activation of endogenous proto-oncogenes or inactivation of TSGs (Fig. 3D, E).
SB and/or PB somatic cell transposon mutagenesis has been used to discover genes and genetic pathways that, when altered, can cause cancer in many different settings (Supplementary Table 1). Using tissue-specific expression of the transposases, generally using Cre-LoxP regulated transgenes, it has been possible to induce or accelerate the development of a variety of types of cancer from tissues representing all three germ layers including carcinomas, sarcomas, tumors of neuroepithelial origin, and various hematopoietic malignancies including B cell, T cell and myeloid leukemias [122]. These studies have recovered recurrent mutations in well-known cancer driver genes as well as novel genes. They have revealed drivers that act in tissue-specific manners or are more ubiquitously altered. The effects of various endogenous and exogenous factors on the spectrum of mutations that are selected for has also been determined. This includes the effects of Trp53 and other background gene mutations, liver damage from Hepatitis B virus gene expression, and sex on liver tumor development [125–129]. Ongoing unpublished studies will reveal the effects of fatty liver disease and liver fibrosis on the selective pressures for liver tumor development in mice.
In addition to getting information on how the context of tumor development (the identify of the initiating mutation, sex, and nature of pre-existing tissue damage) affects the gene mutations that are selected for, SB transposon mutagenesis has revealed candidate genes and pathways that influence the chance of metastatic disease [130] or the evolution of treatment resistance [131]. These are exciting new directions for cancer transposon mutagenesis. It would be highly desirable to use this system to understand the molecular and genetic basis of specific cancer traits, rather than just drivers of cancer initiation and primary tumor progression. So, SB mutagenesis should be married to new metastatic, genetically engineered mouse models and approaches for treating these models with the myriad therapies that human patients receive. The outcome will be unprecedented insight into what genetic changes can allow tumor cells to survive the selective pressures of the process of metastasis and development of treatment resistance. With this understanding may come methods to reverse the effects of these genetic adaptations.
Other advances in transposon mutagenesis for cancer are being made. New methods for recovery of the transposon insertions have allowed more scientists to recover more of the insertions that are present, and to determine their relative abundance within the tumor more accurately. Shear-splink and similar methods result in a more accurate association between the number of times a specific insertion is read and the true abundance of that insertional mutation within the tumor clone [132]. Thus, “trunk” mutations that occurred early in tumor development can be distinguished from “branch” mutations that may have caused progression. Single cell transposon insertion site recovery allows even better resolution for studies like these [133]. RNA sequencing-based transcriptomics is being applied to SB transposon-based models, and this reveals the tumor gene expression profile as well as the identity of genes altered by transposon insertion [134]. The transposon is designed to create fusion transcripts, initiated within the transposon and splicing from the SD to downstream exons, or initiated by cellular promoters and splicing to one of the transposon SAs (Fig. 3D, E). RNA sequencing powerfully allows one to associate specific genetic alterations with phenotypes that can be inferred from tumor gene expression profile, such as degree of cell cycle activation, white blood cell infiltration and others. Other advances in somatic cell transposon mutagenesis might be made by alterations to the design of the transposon so that it could reveal new classes of cancer genes. For example, transposon vectors designed to influence the local epigenome or result in local mutagenesis could be imagined. Other alterations to the transposase transgene could improve somatic cell transposon mutagenesis studies. For example, it would be helpful to be able to turn transposition on and then off again in mice. SB-induced or -accelerated tumors resulting from a “pulse” of transposase expression and transposon insertional mutagenesis may have far fewer transposon insertions and would thus be easier to interpret from a genetic perspective. Moreover, a “silent” empty transposon with no promoter, SAs or pA sites could be mobilized at a single point in time for lineage tracing. Thus, any cells that shared identical transposon insertions can be inferred to be descendants of a single cell that was present at the time of the pulse of transposase expression. New innovations such as these, and screens for more specific cancer-related traits, will make somatic cell transposon mutagenesis studies for cancer more and more valuable.
Concluding Remarks and Future Perspectives
Transposon-based technologies have enormous potential to develop powerful genomic tools with the vision of creating a bridge between physiology and genetics. As described in this article, it is now routine to create libraries of gene knockouts and to thereby establish new animal models of human disease for therapeutic and pharmaceutical intervention. The use of ESCs and SSCs allows for the generation of complex insertion libraries that can be screened for phenotypes either in vitro or in vivo upon transplantation into animals. Mutagenic transposon vectors can also be directly introduced into animal models, either by microinjection of the transposon components into early embryos or by mobilizing chromosomally resident transposons upon crossing of transgenic animal stocks encoding the different components of the transposon systems. One recent addition to the transposon toolbox is the Helraiser transposon that has been resurrected from ancient Helitron elements in the bat genome [135]. The fundamental difference to all other transposons that were described in this article is that Helitron elements transpose through a “copy-and-paste” reaction, in which the donor element does not excise, but it generates progeny that integrates elsewhere in the genome. Since the progeny can also act as donor in subsequent rounds of transposition, one could theoretically achieve very high copy numbers in experimental settings (either in cultured cells or in animals) starting from a single donor element that will amplify in the genome over time.
Other, alternative methods for gain-of-function or loss-of-function screens include the use of cDNA or short hairpin RNA (shRNA) libraries and the CRISPR/Cas9 system, a novel powerful tool for genome editing [136]. Cas9 derivatives lacking endonuclease activity have been engineered to promote transcriptional repression [137] or activation [138] of target genes when coexpressed with gRNAs. One major advantage of transposon-based insertional mutagenesis is, owing to the genetic elements within the transposons that can both intercept and promote transcription, the ability to generate compound phenotypes that depend on a given combination of collaborating loss-of-function and gain-of-function mutations in the same cell. In contrast, shRNA, cDNA and CRISPR/Cas9 screens allow for identification of either loss-of-function or gain-of-function, but not both at the same time [139]. Another bias of shRNA and CRISPR/Cas9 screens is that shRNAs and gRNAs are designed to target specific sequences. Thus, these screens are inherently biased by design.
Considering saturation of the genome and the range of phenotypes that can be generated by transposon-based screens, an important technical consideration is integration bias, which is governed by at least two factors: i) the intrinsic selectivity of each transposon system that can result in overlapping yet different genome-wide profiles of insertion and ii) the different patterns of insertion depending on whether transposition is “plasmid-to-genome” or “genome-to-genome”. Since transposons tend to have different integration preferences on both a local as well as on a genome-wide scale, applying multiple transposon systems in a complementary fashion may extend the range of genes that can be mutagenized by transposon insertions. Indeed, cancer screens based on SB (that displays an almost random integration profile in both mouse and human cells [39, 43]) might more frequently recover TSGs, wheres screens with PB [140, 141] (that displays a preference towards integration into TSSs [43]) might more frequently recover oncogenes.
Target site selection properties of transposons can be markedly different when transposition is launched from a chromosomal site, and are governed by „local hopping”. Local hopping describes a phenomenon of chromosomal transposition, in which transposons have a preference for landing into cis-linked sites in the vicinity of the donor locus [142], and seems to be a shared feature of transposons [45, 55, 60–62]. Local hopping can be a limiting factor in mutagenesis using chromosomally resident transposons, because i) it limits the chromosomal regions accessible to a transposon jumping out of a given chromosomal site, and ii) the donor chromosome containing the parental transposon concatemer has to be excluded from downstream bioinformatic analysis of integration sites. Thus, to probe all chromosomes by chromosomally located transposon donors, more than one transposon mouse strain has to be used [140]. Nonetheless, local hopping enables saturation of chromosomal regions with insertional mutations [59, 65].
In vivo transposon mutagenesis in the context of cancer screens requires up to four transgenic alleles (i.e., the transposon, the transposase, a Cre recombinase allele and a cancer-predisposing allele) to accelerate tumorigenesis in a tissue-specific manner in a sensitized background. Generating and maintaining compound mutant mouse strains is time consuming and costly, prompting alternative ways of utilizing the transposon systems. Molyneux and colleagues transduced immortalized primary human bone mesenchymal cells with a lentivirus harboring the elements of a mutagenic SB transposon, and, when injected into mice, the transplanted cells produced myxofibrosarcomas [143]. Thus, there is precedent for in vitro cell-based mutagenesis followed by in vivo phenotyping, but cancer screens fully based on in vitro mutagenesis coupled to a cell-based functional readout have thus far not been reported.
In cancer cells, loss of mRNA and protein expression can occur without any obvious genetic alteration in corresponding protein-coding regions. Only 2% of the mammalian genome encodes protein-coding genes; however, the non-coding portion of the genome, both transcribed (e.g., microRNAs, long non-coding RNAs) and non-transcribed (e.g., enhancers), plays crucial roles in physiology and pathology. However, transposon-based cancer mutagenesis screens predominantly have relied on mutagenizing protein-coding genes [53], and have barely scratched the surface of the non-coding genome. Advanced vector designs specifically tailored to tap into the non-coding sequence space are therefore expected to significantly extend our abilities to discover and annotate novel pathways that play a role in tumorigenic transformation (See Outstanding Questions).
Supplementary Material
TRENDS BOX.
Transposons are mobile genetic elements with the unique ability to change their position in the genome.
Because transposon integration can occur at vast numbers of locations in the genome, transposition is inherently mutagenic.
An entire array of sophisticated mutagenic and reporter cassettes have been established. Both loss-of-function and gain-of-function phenotypes can be induced by insertions of transposons carrying such cassettes.
Transposon-based functional genomics approaches have uncovered genes and genetic pathways in many areas of physiology and pathophysiology, including embryonic development, drug sensitivity and resistance and neurobiology.
Transposon forward mutagenesis has mainly been done in mice, on a variety of cancer-predisposing genetic or environmental backgrounds, for many types of cancer, and for cancer drug resistance mechanisms.
OUTSTANDING QUESTIONS BOX.
Can gene tagging/mutagenesis screens be efficiently conducted in all cell/tissue types? Are there limitations?
Can technologies be devised to obtain control over transposon copy number? Can transposon copy number be fine tuned? In all screens excess copies of the insertional mutagen can complicate assignment of genotype-phenotype linkage.
Could cancer screens, including phenotyping be done in vitro? How restrictive would such screens be with respect to biologically relevant conclusions?
One of the next challenges is to link transposon-induced tumor genotype to specific cancer phenotypes such as metastasis and treatment resistance.
GLOSSARY BOX
- reverse genetics
addressing the function of a gene by mutagenizing the gene and assessing its phenotypic consequence
- forward genetics
identifying mutant phenotypes in screens and then linking these phenotypes to mutations in particular genes
- loss-of-function
typically a knockout the gene is not able to carry out its normal task in the cell
- gain-of-function
overexpression of either the gene product or a variant of it that generates a dominant phenotype
- ectopic
the gene is expressed at the wrong place and/or at the wrong time
- cryptic splice sites
DNA sequences that resemble canonical splice sites and can be recognized by the splicing machinery
- conditional
expression of a gene that can be regulated at the spatio/temporal level
- protein-induced deformability
the ability of DNA to change shape when bound by a protein
- internal ribosome entry site
an RNA sequence that allows for translation independent of a cap at the 5′ end of mRNAs
- nonsense-mediated RNA decay
a surveillance mechanism that eliminates mRNAs that contain premature stop codons
- tumor suppressor gene
a gene that protects the cell from cancer
- oncogene
a gene that can cause cancer
- local hopping
a preference for transposition into cis-linked sites in the vicinity of the donor locus
- androgenetic
an embryo that contains chromosomes only from the male parent
- barcode
a short nucleotide sequence that uniquelly labels a DNA or RNA molecule
- Gal4-UAS system
A binary transcriptional regulatory system from yeast to activate gene expression in a heterologous species. Gal4 is a transcriptional activator and UAS (upstream activating sequence) is its binding site. A gene placed downstream of the UAS is activated in cells expressing Gal4
- driver mutation
a mutation of a gene that is causally involved in oncogenesis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feschotte C. Transposable elements and the evolution of regulatory networks. Nature reviews. Genetics. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mc CB. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci U S A. 1950;36:344–355. doi: 10.1073/pnas.36.6.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munoz-Lopez M, Garcia-Perez JL. DNA transposons: nature and applications in genomics. Current genomics. 2010;11:115–128. doi: 10.2174/138920210790886871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivics Z, Izsvak Z. The expanding universe of transposon technologies for gene and cell engineering. Mob DNA. 2010;1:25. doi: 10.1186/1759-8753-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivics Z, et al. Transposon-mediated genome manipulation in vertebrates. Nat Methods. 2009;6:415–422. doi: 10.1038/nmeth.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tadege M, et al. Insertional mutagenesis: a Swiss Army knife for functional genomics of Medicago truncatula. Trends in plant science. 2005;10:229–235. doi: 10.1016/j.tplants.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Brutnell TP. Transposon tagging in maize. Functional & integrative genomics. 2002;2:4–12. doi: 10.1007/s10142-001-0044-0. [DOI] [PubMed] [Google Scholar]
- 8.Ostergaard L, Yanofsky MF. Establishing gene function by mutagenesis in Arabidopsis thaliana. The Plant journal: for cell and molecular biology. 2004;39:682–696. doi: 10.1111/j.1365-313X.2004.02149.x. [DOI] [PubMed] [Google Scholar]
- 9.Parinov S, Sundaresan V. Functional genomics in Arabidopsis: large-scale insertional mutagenesis complements the genome sequencing project. Current opinion in biotechnology. 2000;11:157–161. doi: 10.1016/s0958-1669(00)00075-6. [DOI] [PubMed] [Google Scholar]
- 10.Rushforth AM, et al. Site-selected insertion of the transposon Tc1 into a Caenorhabditis elegans myosin light chain gene. Mol Cell Biol. 1993;13:902–910. doi: 10.1128/mcb.13.2.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zwaal RR, et al. Target-selected gene inactivation in Caenorhabditis elegans by using a frozen transposon insertion mutant bank. Proc Natl Acad Sci U S A. 1993;90:7431–7435. doi: 10.1073/pnas.90.16.7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bessereau JL, et al. Mobilization of a Drosophila transposon in the Caenorhabditis elegans germ line. Nature. 2001;413:70–74. doi: 10.1038/35092567. [DOI] [PubMed] [Google Scholar]
- 13.Bessereau JL. Insertional mutagenesis in C. elegans using the Drosophila transposon Mos1: a method for the rapid identification of mutated genes. Methods Mol Biol. 2006;351:59–73. doi: 10.1385/1-59745-151-7:59. [DOI] [PubMed] [Google Scholar]
- 14.Cooley L, et al. Insertional mutagenesis of the Drosophila genome with single P elements. Science. 1988;239:1121–1128. doi: 10.1126/science.2830671. [DOI] [PubMed] [Google Scholar]
- 15.Thibault ST, et al. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–287. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- 16.Venken KJ, Bellen HJ. Chemical mutagens, transposons, and transgenes to interrogate gene function in Drosophila melanogaster. Methods. 2014;68:15–28. doi: 10.1016/j.ymeth.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hummel T, Klambt C. P-element mutagenesis. Methods Mol Biol. 2008;420:97–117. doi: 10.1007/978-1-59745-583-1_6. [DOI] [PubMed] [Google Scholar]
- 18.Spradling AC, et al. Gene disruptions using P transposable elements: an integral component of the Drosophila genome project. Proc Natl Acad Sci U S A. 1995;92:10824–10830. doi: 10.1073/pnas.92.24.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivics Z, et al. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 20.Miskey C, et al. DNA transposons in vertebrate functional genomics. Cell Mol Life Sci. 2005;62:629–641. doi: 10.1007/s00018-004-4232-7. [DOI] [PubMed] [Google Scholar]
- 21.Mates L, et al. Technology transfer from worms and flies to vertebrates: transposition-based genome manipulations and their future perspectives. Genome Biol. 2007;8(Suppl 1):S1. doi: 10.1186/gb-2007-8-s1-s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding S, et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Wilson MH, et al. PiggyBac Transposon-mediated Gene Transfer in Human Cells. Mol Ther. 2007;15:139–145. doi: 10.1038/sj.mt.6300028. [DOI] [PubMed] [Google Scholar]
- 24.Yusa K. piggyBac Transposon. Microbiology spectrum. 2015;3 doi: 10.1128/microbiolspec.MDNA3-0028-2014. MDNA3-0028-2014. [DOI] [PubMed] [Google Scholar]
- 25.Koga A, et al. Transposable element in fish. Nature. 1996;383:30. doi: 10.1038/383030a0. [DOI] [PubMed] [Google Scholar]
- 26.Kawakami K, et al. Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proc Natl Acad Sci U S A. 2000;97:11403–11408. doi: 10.1073/pnas.97.21.11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawakami K. Tol2: a versatile gene transfer vector in vertebrates. Genome Biol. 2007;8(Suppl 1):S7. doi: 10.1186/gb-2007-8-s1-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grabundzija I, et al. Comparative analysis of transposable element vector systems in human cells. Mol Ther. 2010;18:1200–1209. doi: 10.1038/mt.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinzelle L, et al. Transposition of a reconstructed Harbinger element in human cells and functional homology with two transposon-derived cellular genes. Proc Natl Acad Sci U S A. 2008;105:4715–4720. doi: 10.1073/pnas.0707746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser MJ, et al. Transposon-mediated mutagenesis of a baculovirus. Virology. 1985;145:356–361. doi: 10.1016/0042-6822(85)90172-2. [DOI] [PubMed] [Google Scholar]
- 31.Fraser MJ, et al. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol Biol. 1996;5:141–151. doi: 10.1111/j.1365-2583.1996.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 32.Li X, et al. A resurrected mammalian hAT transposable element and a closely related insect element are highly active in human cell culture. Proc Natl Acad Sci U S A. 2013;110:E478–487. doi: 10.1073/pnas.1121543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miskey C, et al. The Frog Prince: a reconstructed transposon from Rana pipiens with high transpositional activity in vertebrate cells. Nucleic Acids Res. 2003;31:6873–6881. doi: 10.1093/nar/gkg910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franz G, Savakis C. Minos, a new transposable element from Drosophila hydei, is a member of the Tc1-like family of transposons. Nucleic Acids Res. 1991;19:6646. doi: 10.1093/nar/19.23.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miskey C, et al. The Ancient Mariner Sails Again: Transposition of the Human Hsmar1 Element by a Reconstructed Transposase and Activities of the SETMAR Protein on Transposon Ends. Mol Cell Biol. 2007 doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vigdal TJ, et al. Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. Journal of molecular biology. 2002;323:441–452. doi: 10.1016/s0022-2836(02)00991-9. [DOI] [PubMed] [Google Scholar]
- 37.Geurts AM, et al. Structure-based prediction of insertion-site preferences of transposons into chromosomes. Nucleic Acids Res. 2006;34:2803–2811. doi: 10.1093/nar/gkl301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hackett CS, et al. Predicting preferential DNA vector insertion sites: implications for functional genomics and gene therapy. Genome Biol. 2007;8(Suppl 1):S12. doi: 10.1186/gb-2007-8-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yant SR, et al. High-resolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol. 2005;25:2085–2094. doi: 10.1128/MCB.25.6.2085-2094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moldt B, et al. Comparative genomic integration profiling of Sleeping Beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells. Mol Ther. 2011;19:1499–1510. doi: 10.1038/mt.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ammar I, et al. Retargeting transposon insertions by the adeno-associated virus Rep protein. Nucleic Acids Res. 2012;40:6693–6712. doi: 10.1093/nar/gks317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voigt K, et al. Retargeting sleeping beauty transposon insertions by engineered zinc finger DNA-binding domains. Mol Ther. 2012;20:1852–1862. doi: 10.1038/mt.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gogol-Doring A, et al. Genome-wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the piggyBac Transposon in Primary Human CD4(+) T Cells. Mol Ther. 2016;24:592–606. doi: 10.1038/mt.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang X, et al. Gene transfer efficiency and genome-wide integration profiling of Sleeping Beauty, Tol2, and piggyBac transposons in human primary T cells. Mol Ther. 2010;18:1803–1813. doi: 10.1038/mt.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang Q, et al. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis. 2009;47:404–408. doi: 10.1002/dvg.20508. [DOI] [PubMed] [Google Scholar]
- 46.Wang H, et al. “Calling cards” for DNA-binding proteins in mammalian cells. Genetics. 2012;190:941–949. doi: 10.1534/genetics.111.137315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li MA, et al. The piggyBac transposon displays local and distant reintegration preferences and can cause mutations at noncanonical integration sites. Mol Cell Biol. 2013;33:1317–1330. doi: 10.1128/MCB.00670-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Jong J, et al. Chromatin landscapes of retroviral and transposon integration profiles. PLoS genetics. 2014;10:e1004250. doi: 10.1371/journal.pgen.1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galvan DL, et al. Genome-wide mapping of PiggyBac transposon integrations in primary human T cells. J Immunother. 2009;32:837–844. doi: 10.1097/CJI.0b013e3181b2914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Rijck J, et al. The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell reports. 2013;5:886–894. doi: 10.1016/j.celrep.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parinov S, et al. Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Dev Dyn. 2004;231:449–459. doi: 10.1002/dvdy.20157. [DOI] [PubMed] [Google Scholar]
- 52.Kondrychyn I, et al. Genome-wide analysis of Tol2 transposon reintegration in zebrafish. BMC Genomics. 2009;10:418. doi: 10.1186/1471-2164-10-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeNicola GM, et al. The utility of transposon mutagenesis for cancer studies in the era of genome editing. Genome Biol. 2015;16:229. doi: 10.1186/s13059-015-0794-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansen GM, et al. Large-scale gene trapping in C57BL/6N mouse embryonic stem cells. Genome Res. 2008;18:1670–1679. doi: 10.1101/gr.078352.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horie K, et al. Characterization of Sleeping Beauty transposition and its application to genetic screening in mice. Mol Cell Biol. 2003;23:9189–9207. doi: 10.1128/MCB.23.24.9189-9207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stanford WL, et al. Gene-trap mutagenesis: past, present and beyond. Nature reviews Genetics. 2001;2:756–768. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- 57.Joyner AL, et al. The gene trap approach in embryonic stem cells: the potential for genetic screens in mice. Ciba Found Symp. 1992;165:277–288. doi: 10.1002/9780470514221.ch16. discussion 288–297. [DOI] [PubMed] [Google Scholar]
- 58.Skarnes WC, et al. A gene trap approach in mouse embryonic stem cells: the lacZ reported is activated by splicing, reflects endogenous gene expression, and is mutagenic in mice. Genes Dev. 1992;6:903–918. doi: 10.1101/gad.6.6.903. [DOI] [PubMed] [Google Scholar]
- 59.Kokubu C, et al. A transposon-based chromosomal engineering method to survey a large cis-regulatory landscape in mice. Nat Genet. 2009;41:946–952. doi: 10.1038/ng.397. [DOI] [PubMed] [Google Scholar]
- 60.Dupuy AJ, et al. Transposition and gene disruption in the male germline of the mouse. Genesis. 2001;30:82–88. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- 61.Fischer SE, et al. Regulated transposition of a fish transposon in the mouse germ line. Proc Natl Acad Sci U S A. 2001;98:6759–6764. doi: 10.1073/pnas.121569298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carlson CM, et al. Transposon mutagenesis of the mouse germline. Genetics. 2003;165:243–256. doi: 10.1093/genetics/165.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roberg-Perez K, et al. MTID: a database of Sleeping Beauty transposon insertions in mice. Nucleic Acids Res. 2003;31:78–81. doi: 10.1093/nar/gkg045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geurts AM, et al. Conditional gene expression in the mouse using a Sleeping Beauty gene-trap transposon. BMC Biotechnol. 2006;6:30. doi: 10.1186/1472-6750-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keng VW, et al. Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat Methods. 2005;2:763–769. doi: 10.1038/nmeth795. [DOI] [PubMed] [Google Scholar]
- 66.Horie K, et al. Efficient chromosomal transposition of a Tc1/mariner- like transposon Sleeping Beauty in mice. Proc Natl Acad Sci U S A. 2001;98:9191–9196. doi: 10.1073/pnas.161071798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kitada K, et al. Transposon-tagged mutagenesis in the rat. Nat Methods. 2007 doi: 10.1038/nmeth1002. [DOI] [PubMed] [Google Scholar]
- 68.Lu B, et al. Generation of rat mutants using a coat color-tagged Sleeping Beauty transposon system. Mamm Genome. 2007;18:338–346. doi: 10.1007/s00335-007-9025-5. [DOI] [PubMed] [Google Scholar]
- 69.Bonaldo P, et al. Efficient gene trap screening for novel developmental genes using IRES beta geo vector and in vitro preselection. Exp Cell Res. 1998;244:125–136. doi: 10.1006/excr.1998.4208. [DOI] [PubMed] [Google Scholar]
- 70.Clark KJ, et al. Transposon vectors for gene-trap insertional mutagenesis in vertebrates. Genesis. 2004;39:225–233. doi: 10.1002/gene.20049. [DOI] [PubMed] [Google Scholar]
- 71.Sivasubbu S, et al. Gene-breaking transposon mutagenesis reveals an essential role for histone H2afza in zebrafish larval development. Mech Dev. 2006;123:513–529. doi: 10.1016/j.mod.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 72.Shigeoka T, et al. Suppression of nonsense-mediated mRNA decay permits unbiased gene trapping in mouse embryonic stem cells. Nucleic Acids Res. 2005;33:e20. doi: 10.1093/nar/gni022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dupuy AJ, et al. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer research. 2009;69:8150–8156. doi: 10.1158/0008-5472.CAN-09-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collier LS, et al. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 75.Elling U, et al. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell stem cell. 2011;9:563–574. doi: 10.1016/j.stem.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leeb M, Wutz A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011;479:131–134. doi: 10.1038/nature10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li W, et al. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature. 2012;490:407–411. doi: 10.1038/nature11435. [DOI] [PubMed] [Google Scholar]
- 78.Yang H, et al. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell. 2012;149:605–617. doi: 10.1016/j.cell.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 79.Pettitt SJ, et al. piggyBac transposon-based insertional mutagenesis in mouse haploid embryonic stem cells. Methods Mol Biol. 2015;1239:15–28. doi: 10.1007/978-1-4939-1862-1_2. [DOI] [PubMed] [Google Scholar]
- 80.Pettitt SJ, et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PloS one. 2013;8:e61520. doi: 10.1371/journal.pone.0061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pettitt SJ, et al. Genome-wide barcoded transposon screen for cancer drug sensitivity in haploid mouse embryonic stem cells. Scientific data. 2017;4:170020. doi: 10.1038/sdata.2017.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li W, et al. Genetic modification and screening in rat using haploid embryonic stem cells. Cell stem cell. 2014;14:404–414. doi: 10.1016/j.stem.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 83.Ivics Z, et al. Sleeping Beauty transposon mutagenesis of the rat genome in spermatogonial stem cells. Methods. 2011;53:356–365. doi: 10.1016/j.ymeth.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Izsvak Z, et al. Generating knockout rats by transposon mutagenesis in spermatogonial stem cells. Nat Methods. 2010;7:443–445. doi: 10.1038/nmeth.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ivics Z, et al. Sleeping Beauty transposon mutagenesis in rat spermatogonial stem cells. Nature protocols. 2011;6:1521–1535. doi: 10.1038/nprot.2011.378. [DOI] [PubMed] [Google Scholar]
- 86.Kawakami K, Noda T. Transposition of the Tol2 element, an Ac-like element from the Japanese medaka fish Oryzias latipes, in mouse embryonic stem cells. Genetics. 2004;166:895–899. doi: 10.1534/genetics.166.2.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fujimura K, Kocher TD. Tol2-mediated transgenesis in tilapia (Oreochromis niloticus) Aquaculture. 2011;319:342–346. doi: 10.1016/j.aquaculture.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Juntti SA, et al. Tol2-mediated generation of a transgenic haplochromine cichlid, Astatotilapia burtoni. PloS one. 2013;8:e77647. doi: 10.1371/journal.pone.0077647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Valenzano DR, et al. Transposon-Mediated Transgenesis in the Short-Lived African Killifish Nothobranchius furzeri, a Vertebrate Model for Aging. G3 (Bethesda) 2011;1:531–538. doi: 10.1534/g3.111.001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sumiyama K, et al. A simple and highly efficient transgenesis method in mice with the Tol2 transposon system and cytoplasmic microinjection. Genomics. 2010;95:306–311. doi: 10.1016/j.ygeno.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 91.Macdonald J, et al. Efficient genetic modification and germ-line transmission of primordial germ cells using piggyBac and Tol2 transposons. Proc Natl Acad Sci U S A. 2012;109:E1466–1472. doi: 10.1073/pnas.1118715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hamlet MR, et al. Tol2 transposon-mediated transgenesis in Xenopus tropicalis. Genesis. 2006;44:438–445. doi: 10.1002/dvg.20234. [DOI] [PubMed] [Google Scholar]
- 93.Hikichi T, et al. Transcription factors interfering with dedifferentiation induce cell type-specific transcriptional profiles. Proc Natl Acad Sci U S A. 2013;110:6412–6417. doi: 10.1073/pnas.1220200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsukahara T, et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther. 2015;22:209–215. doi: 10.1038/gt.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sato Y, et al. Stable integration and conditional expression of electroporated transgenes in chicken embryos. Dev Biol. 2007;305:616–624. doi: 10.1016/j.ydbio.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 96.Yoshida A, et al. Simultaneous expression of different transgenes in neurons and glia by combining in utero electroporation with the Tol2 transposon-mediated gene transfer system. Genes Cells. 2010;15:501–512. doi: 10.1111/j.1365-2443.2010.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kwan KM, et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 98.Love NR, et al. pTransgenesis: a cross-species, modular transgenesis resource. Development. 2011;138:5451–5458. doi: 10.1242/dev.066498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Loulier K, et al. Multiplex cell and lineage tracking with combinatorial labels. Neuron. 2014;81:505–520. doi: 10.1016/j.neuron.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 100.Raz E, et al. Transposition of the nematode Caenorhabditis elegans Tc3 element in the zebrafish Danio rerio. Curr Biol. 1998;8:82–88. doi: 10.1016/s0960-9822(98)70038-7. [DOI] [PubMed] [Google Scholar]
- 101.Fadool JM, et al. Transposition of the mariner element from Drosophila mauritiana in zebrafish. Proc Natl Acad Sci U S A. 1998;95:5182–5186. doi: 10.1073/pnas.95.9.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davidson AE, et al. Efficient gene delivery and gene expression in zebrafish using the Sleeping Beauty transposon. Dev Biol. 2003;263:191–202. doi: 10.1016/j.ydbio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 103.Kawakami K, et al. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev Cell. 2004;7:133–144. doi: 10.1016/j.devcel.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 104.Suster ML, et al. Transposon-mediated BAC transgenesis in zebrafish and mice. BMC Genomics. 2009;10:477. doi: 10.1186/1471-2164-10-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Davison JM, et al. Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Dev Biol. 2007;304:811–824. doi: 10.1016/j.ydbio.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scott EK, et al. Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nat Methods. 2007;4:323–326. doi: 10.1038/nmeth1033. [DOI] [PubMed] [Google Scholar]
- 107.Asakawa K, et al. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci U S A. 2008;105:1255–1260. doi: 10.1073/pnas.0704963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Venero Galanternik M, et al. A novel perivascular cell population in the zebrafish brain. Elife. 2017;6 doi: 10.7554/eLife.24369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Takeuchi M, et al. Establishment of Gal4 transgenic zebrafish lines for analysis of development of cerebellar neural circuitry. Dev Biol. 2015;397:1–17. doi: 10.1016/j.ydbio.2014.09.030. [DOI] [PubMed] [Google Scholar]
- 110.Heanue TA, et al. A Novel Zebrafish ret Heterozygous Model of Hirschsprung Disease Identifies a Functional Role for mapk10 as a Modifier of Enteric Nervous System Phenotype Severity. PLoS genetics. 2016;12:e1006439. doi: 10.1371/journal.pgen.1006439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xiao Y, et al. High-resolution live imaging reveals axon-glia interactions during peripheral nerve injury and repair in zebrafish. Dis Model Mech. 2015;8:553–564. doi: 10.1242/dmm.018184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shibata E, et al. Fgf signalling controls diverse aspects of fin regeneration. Development. 2016;143:2920–2929. doi: 10.1242/dev.140699. [DOI] [PubMed] [Google Scholar]
- 113.Pipalia TG, et al. Cellular dynamics of regeneration reveals role of two distinct Pax7 stem cell populations in larval zebrafish muscle repair. Dis Model Mech. 2016;9:671–684. doi: 10.1242/dmm.022251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kamezaki A, et al. Visualization of Neuregulin 1 ectodomain shedding reveals its local processing in vitro and in vivo. Sci Rep. 2016;6:28873. doi: 10.1038/srep28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Muto A, et al. Activation of the hypothalamic feeding centre upon visual prey detection. Nat Commun. 2017;8:15029. doi: 10.1038/ncomms15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yokota Y, et al. Endothelial Ca 2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. Elife. 2015;4 doi: 10.7554/eLife.08817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Clark KJ, et al. In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat Methods. 2011;8:506–515. doi: 10.1038/nmeth.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Trinh le A, et al. A versatile gene trap to visualize and interrogate the function of the vertebrate proteome. Genes Dev. 2011;25:2306–2320. doi: 10.1101/gad.174037.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sternberg JR, et al. Optimization of a Neurotoxin to Investigate the Contribution of Excitatory Interneurons to Speed Modulation In Vivo. Curr Biol. 2016;26:2319–2328. doi: 10.1016/j.cub.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 120.Nagayoshi S, et al. Insertional mutagenesis by the Tol2 transposon-mediated enhancer trap approach generated mutations in two developmental genes: tcf7 and synembryn-like. Development. 2008;135:159–169. doi: 10.1242/dev.009050. [DOI] [PubMed] [Google Scholar]
- 121.Rodriguez-Mari A, et al. Sex reversal in zebrafish fancl mutants is caused by Tp53-mediated germ cell apoptosis. PLoS genetics. 2010;6:e1001034. doi: 10.1371/journal.pgen.1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Moriarity BS, Largaespada DA. Sleeping Beauty transposon insertional mutagenesis based mouse models for cancer gene discovery. Current opinion in genetics & development. 2015;30:66–72. doi: 10.1016/j.gde.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jonkers J, Berns A. Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochimica et biophysica acta. 1996;1287:29–57. doi: 10.1016/0304-419x(95)00020-g. [DOI] [PubMed] [Google Scholar]
- 124.van Lohuizen M, Berns A. Tumorigenesis by slow-transforming retroviruses–an update. Biochimica et biophysica acta. 1990;1032:213–235. doi: 10.1016/0304-419x(90)90005-l. [DOI] [PubMed] [Google Scholar]
- 125.Keng VW, et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nature biotechnology. 2009;27:264–274. doi: 10.1038/nbt.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.O’Donnell KA, et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A. 2012;109:E1377–1386. doi: 10.1073/pnas.1115433109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Keng VW, et al. Sex bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology. 2013;57:120–130. doi: 10.1002/hep.26004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Riordan JD, et al. Identification of rtl1, a retrotransposon-derived imprinted gene, as a novel driver of hepatocarcinogenesis. PLoS genetics. 2013;9:e1003441. doi: 10.1371/journal.pgen.1003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bard-Chapeau EA, et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat Genet. 2014;46:24–32. doi: 10.1038/ng.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moriarity BS, et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat Genet. 2015;47:615–624. doi: 10.1038/ng.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perna D, et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci U S A. 2015;112:E536–545. doi: 10.1073/pnas.1418163112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Koudijs MJ, et al. High-throughput semiquantitative analysis of insertional mutations in heterogeneous tumors. Genome Res. 2011;21:2181–2189. doi: 10.1101/gr.112763.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mann KM, et al. Analyzing tumor heterogeneity and driver genes in single myeloid leukemia cells with SBCapSeq. Nature biotechnology. 2016;34:962–972. doi: 10.1038/nbt.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Temiz NA, et al. RNA sequencing of Sleeping Beauty transposon-induced tumors detects transposon-RNA fusions in forward genetic cancer screens. Genome Res. 2016;26:119–129. doi: 10.1101/gr.188649.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Grabundzija I, et al. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nat Commun. 2016;7:10716. doi: 10.1038/ncomms10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rad R, et al. PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science. 2010;330:1104–1107. doi: 10.1126/science.1193004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rad R, et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat Genet. 2015;47:47–56. doi: 10.1038/ng.3164. [DOI] [PubMed] [Google Scholar]
- 142.Ivics Z, Izsvak Z. Sleeping Beauty Transposition. Microbiology spectrum. 2015;3 doi: 10.1128/microbiolspec.MDNA3-0042-2014. MDNA3-0042-2014. [DOI] [PubMed] [Google Scholar]
- 143.Molyneux SD, et al. Human somatic cell mutagenesis creates genetically tractable sarcomas. Nat Genet. 2014;46:964–972. doi: 10.1038/ng.3065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.