

Abstract
p97 is an essential ATPase associated with various cellular activities (AAA+) that functions as a segregase in diverse cellular processes, including the maintenance of proteostasis. p97 interacts with different cofactors that target it to distinct pathways; an important example is the deubiquitinase ataxin3, which collaborates with p97 in endoplasmic reticulum-associated degradation. However, the molecular details of this interaction have been unclear. Here, we characterized the binding of ataxin3 to p97, showing that ataxin3 binds with low-micromolar affinity to both wild-type p97 and mutants linked to degenerative disorders known as multisystem proteinopathy 1 (MSP1); we further showed that the stoichiometry of binding is one ataxin3 molecule per p97 hexamer. We mapped the binding determinants on each protein, demonstrating that ataxin3's p97/VCP-binding motif interacts with the inter-lobe cleft in the N-domain of p97. We also probed the nucleotide dependence of this interaction, confirming that ataxin3 and p97 associate in the presence of ATP and in the absence of nucleotide, but not in the presence of ADP. Our experiments suggest that an ADP-driven downward movement of the p97 N-terminal domain dislodges ataxin3 by inducing a steric clash between the D1-domain and ataxin3's C terminus. In contrast, MSP1 mutants of p97 bind ataxin3 irrespective of their nucleotide state, indicating a failure by these mutants to translate ADP binding into a movement of the N-terminal domain. Our model provides a mechanistic explanation for how nucleotides regulate the p97–ataxin3 interaction and why atypical cofactor binding is observed with MSP1 mutants.
Keywords: ATPases associated with diverse cellular activities (AAA), conformational change, electron microscopy (EM), proteostasis, surface plasmon resonance (SPR), ataxin3, cofactor binding, multisystem proteinopathy 1 (MSP1), nucleotide regulation, p97/VCP
Introduction
The protein p97, also known as valosin-containing protein (VCP),2 is an essential ATPase associated with various cellular activities (type II AAA+ family) (1). p97 forms a hexamer, with each protomer comprising an N-terminal domain, D1- and D2-ATPase domains, and an unstructured C-terminal region (Fig. 1A). The D1- and D2-domains form two coaxially-stacked rings, with the N-domains arranged around the periphery of the D1 ring (2, 3). ATP hydrolysis in the D1- and D2-domains is harnessed to perform mechanical work, allowing p97 to function as a segregase in processes as varied as cell-cycle regulation, transcriptional activation, DNA-damage repair, and membrane fusion (4, 5). One of p97's most significant cellular roles is to assist in maintaining proteostasis, and the protein contributes to both proteasomal degradation and autophagy (6–10). As would be expected for a key component of regulatory and degradation pathways, p97 dysfunction is associated with numerous degenerative disorders (11, 12). Missense mutations in p97 cause a set of heterogeneous disorders collectively termed multisystem proteinopathy type 1 (MSP1), which include inclusion body myopathy, Paget disease of the bone and frontotemporal dementia, and amyotrophic lateral sclerosis (13–15). In cell culture, inhibiting or knocking down p97 induces endoplasmic reticulum (ER) stress and triggers the unfolded protein response, with a concomitant buildup of polyubiquitinated substrates (16–21). p97 protects against the toxic effects of aggregation-prone proteins and localizes to cytosolic aggregates (22–24). It extracts misfolded and toxic substrates from such aggregates, as well as from macromolecular complexes and cellular membranes, in quality-control processes associated with chromatin and mitochondria (25–30).
Figure 1.
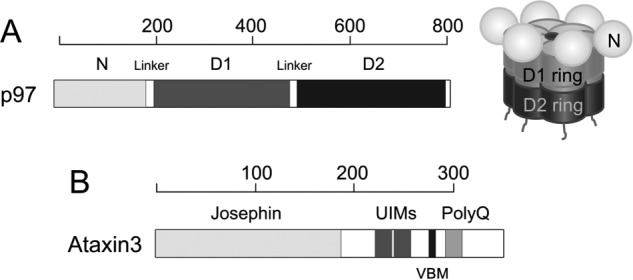
Schematic representation of p97 and ataxin3. A, structure of p97. At left is shown the domain organization of the p97 protomer. Each protomer comprises an N-terminal domain shown in light gray, D1- and D2-ATPase domains shown in dark gray and black, respectively, and an unstructured C-terminal region. At right is a schematic of the assembled hexamer, using the same shading. The D1- and D2-domains form two coaxially-stacked rings around a central pore, with the N-domains arranged along the periphery of the D1 ring. B, domain organization of ataxin3 showing the Josephin domain, two ubiquitin-interacting motifs (UIMs), the p97/VCP-binding motif (VBM), and the polyglutamine (polyQ) repeat region. The linkers, UIMs, VBM, and polyQ regions are not drawn to scale. The scales above each domain representation show length in amino acids.
Involvement of p97 in diverse cellular processes is coordinated by a multitude of cofactors that regulate its activity. These cofactors are roughly categorized into substrate recruiters, which direct p97 to specific pathways, and substrate processors, which are typically enzymes that fine-tune p97 function (31). The assembly of p97–cofactor complexes drives the subcellular localization of p97, its specificity for substrates, and the ultimate fate of these substrates (32). An important substrate-processing cofactor is ataxin3, a member of the Machado-Joseph disease class of deubiquitinating enzymes (DUBs) (33). Ataxin3 is intimately involved in the clearance of misfolded proteins, playing significant roles in the ubiquitin–proteasome system (34–36) and the aggregation–autophagy system (37–40). Through its ubiquitin-related activities, it suppresses neurodegeneration and cytotoxicity from various polyglutamine-disease proteins (41–43). In particular, ataxin3 collaborates with p97 in the process of ER-associated degradation (ERAD) (44), in which proteins that are misfolded or otherwise marked for destruction are removed from the ER and targeted to proteasomes. In addition, ataxin3 and p97 cooperate outside ERAD in other processes involving protein degradation, such as the control of protein levels involved in the DNA damage response (45). In general, ataxin3 seems to facilitate degradation of proteins targeted for destruction. For example, ataxin3 knock-out animals feature increased levels of polyubiquitinated proteins and increased levels of the stress response (46–48). Interestingly, a mutant form of ataxin3 containing an expanded polyglutamine tract gives rise to aggregates of misfolded protein and causes the neurodegenerative disorder spinocerebellar ataxia type-3 (SCA3, also known as Machado-Joseph disease (49–54)). Therefore, it appears that ataxin3 can both support normal protein degradation and (in its mutant form) disrupt this same process.
The p97–ataxin3 interaction was first observed via immunoprecipitation from cell extracts (24, 35, 55). Subsequent studies revealed that p97 and ataxin3 are found together in multiprotein ERAD complexes at the ER membrane and that the proteins also interact in vitro (24, 36, 54–56). Overexpression of a catalytically-inactive form of ataxin3 strongly inhibits the processing of ERAD substrates, whereas overexpression of the wild-type enzyme has a less pronounced but still inhibitory effect, possibly due to a dominant-negative effect (36, 56). The functionality of this interaction has been demonstrated in worms, where p97 and ataxin3 operate synergistically to promote degradation of key substrates (57). The p97–ataxin3 interaction is perturbed by mutations in p97 associated with MSP-1, as significantly elevated amounts of ataxin3 are associated with the mutant p97, as compared with wild-type protein (58).
The molecular details of how p97 interacts with cofactors such as ataxin3 are only now being elucidated. It is becoming clear that different cofactors assemble into complexes with differing stoichiometries (59–61). It is also apparent that cofactor binding is influenced by major conformational changes within p97, which are driven by nucleotide binding and hydrolysis (3, 62, 63). Most p97 cofactors interact with the protein's N-domain (64), which is significant because the N-domain can undergo large “up-down” motions relative to the D1 ring (3). These conformational changes are largely induced by the nucleotide bound in the D1-domain, with ATP favoring the “up” position, whereas ADP induces a “down” position. The up-down equilibrium is disrupted in MSP1 mutants (65, 66), in which all six N-domains preferentially adopt the up state, regardless of the nucleotide present; this behavior correlates with decreased ADP binding to the D1-domain in these mutants (67, 68). Even though the MSP1 mutations affect nucleotide binding, they are not located at nucleotide-binding sites, nor at cofactor-binding interfaces. Instead, they are mostly found in the N-domain, with some occurring in the D1-domain and the N-D1 linker (12), suggesting that they might affect cofactor binding by perturbing the N-domain conformation and/or inter-domain communication.
To understand the precise interplay between ataxin3, p97, and p97's ATPase cycle, a detailed analysis of binding is required. In this study, we characterized the p97–ataxin3 interaction for wild-type p97 and three common MSP1 mutants, localizing the determinants of binding for each partner and revealing that the dependence of binding upon nucleotide state is starkly different for the wild-type and mutant p97 proteins. We suggest a model in which ataxin3 binding is controlled by a nucleotide-induced shift of the N-domain conformation, which leads to steric displacement of ataxin3 in the ADP-bound state. This model has potential consequences for protein quality-control pathways in both normal and disease states and may prove applicable to other p97 cofactors.
Results
p97 interaction with ataxin3
It has previously been demonstrated that GST-fused ataxin3 can pull down p97 from cell lysates, as well as from a solution of purified p97 (24, 56). To quantitatively characterize this interaction, we used surface plasmon resonance (SPR), focusing first on the interaction between the full-length proteins. A C-terminal 1D4 epitope (69) was used to immobilize the p97 hexamer in an oriented manner on a sensor chip. Ataxin3 was found to bind with an equilibrium dissociation constant (KD) of 3.7 ± 0.9 μm (Fig. 2A). A similar KD value (2.2 ± 0.2 μm) was derived using a different type of sensor chip and immobilization chemistry, indicating that the binding observed is not tag-specific (see supplemental Fig. S1, A and B). Because most p97 cofactors interact with the protein's N-terminal domain, we next tested the binding of ataxin3 to the p97 N-domain fragment, obtaining a KD of 6.4 ± 1.5 μm (Fig. 2B). This is comparable with the affinity observed with full-length p97 and suggests that the N-domain is sufficient for ataxin3 binding. We then verified these results using isothermal titration calorimetry (ITC), an orthogonal technique that should be free of any artifacts and surface effects associated with SPR. The calorimetry experiments yielded KD values of 8.4 ± 0.1 and 4.5 ± 0.2 μm for ataxin3 binding to full-length p97 and the p97 N-domain, respectively (Fig. 2, C and D), in good agreement with the SPR data. In addition, we derived stoichiometries of 1.38 ± 0.01 and 1.1 ± 0.2 ataxin3 molecules bound per full-length p97 hexamer and N-domain, respectively. The binding data are summarized in Table 1. Thus, we were able to confirm a direct interaction between p97 and ataxin3, with low-micromolar affinity, centered on p97's N-terminal domain.
Figure 2.

p97 interacts directly with ataxin3. A and B, ataxin3 binding to p97 as measured by SPR. A, full-length ataxin3 binding to full-length p97; B, full-length ataxin3 binding to the p97 N-domain. The upper panels show normalized equilibrium binding responses, fit to a one-site binding model; the lower panels show representative sensorgrams. The dashed lines represent the response range used to determine the equilibrium fit (for both A and B, n ≥ 3 for each concentration). C and D, ataxin3 binding to p97 as measured by ITC. C, full-length p97; D, the p97 N-domain. The upper panels show the raw data for injection of full-length ataxin3 into a cell containing either full-length p97 or the p97 N-domain, and the bottom panels show the integrated heat data as a function of the p97/ataxin3 mole ratio (closed circles). The solid lines represent the best fit of a one-site model to the data.
Table 1.
Equilibrium dissociation constant (KD) values for the interaction between p97 and ataxin3 fragments
| p97 constructs | Ataxin3 constructs | KD |
|---|---|---|
| μm | ||
| Full-length (1–806) | Full-length (1–345) | 3.7 ± 0.9a; 8.4 ± 0.1b |
| Full-length (1–806) | Ataxin3ΔC (1–292) | 4.3 ± 0.6a |
| p97 N-domain (1–187) | Full-length (1–345) | 6.4 ± 1.5a; 4.5 ± 0.2b |
| p97 N-domain (1–187) | Ataxin3ΔC (1–292) | 2.8 ± 0.1b |
| p97 N-domain (1–187) | Ataxin3ΔN (220–345) | 4.0 ± 0.4b |
a Values were calculated using the equilibrium binding response from three or more SPR experiments for each interacting pair. Data were fit to a one-site specific binding model using GraphPad Prism 7.0.
b Values were calculated from three or more ITC experiments for each interacting pair. Data were fit to a one-site independent binding model using NanoAnalyze 3.5.0.
Electron microscopy (EM) analysis of the p97–ataxin3 complex
As a homohexamer, p97 can potentially bind up to six cofactor molecules. Our ITC experiments suggest that the binding stoichiometry of ataxin3 to the full-length p97 hexamer is near 1:1. To confirm the stoichiometry, we analyzed the complex by negative-stain EM. The proteins were incubated together and passed through gel filtration, and the complex-containing fractions were negatively stained and imaged. Purified p97 alone was imaged as a control. Our preliminary analyses of the complex fractions revealed an additional blurry density at the periphery of the p97 hexamer that was absent in the control (supplemental Fig. S2, A and B). The blurred appearance could result from dissociation of the complex (supplemental Fig. S3A) or a flexible association between the two proteins. Additionally, only around 5% of the total particles selected showed evidence for complex formation, indicating a significant amount of unbound p97 was present. Therefore, to stabilize and further enrich for the p97–ataxin3 complex, we cross-linked the proteins using glutaraldehyde, followed by affinity chromatography to remove unbound p97 and a final round of gel filtration (supplemental Fig. S3B). The fractions containing cross-linked p97–ataxin3 were stained with 1% uranyl formate and imaged. Around 7,100 particles were selected from individual micrographs (Fig. 3A) and sorted into 50 classes. Representative 2D class averages are shown in Fig. 3B, in which a single distinct projection is visible at one vertex of the p97 hexamer, indicated by the white arrows. We confirmed that this additional density corresponds to ataxin3 by imaging Nanogold-labeled ataxin3 in complex with p97 (supplemental Fig. S5). Together with the ITC data, our EM results clearly show that one ataxin3 molecule binds to the p97 hexamer.
Figure 3.
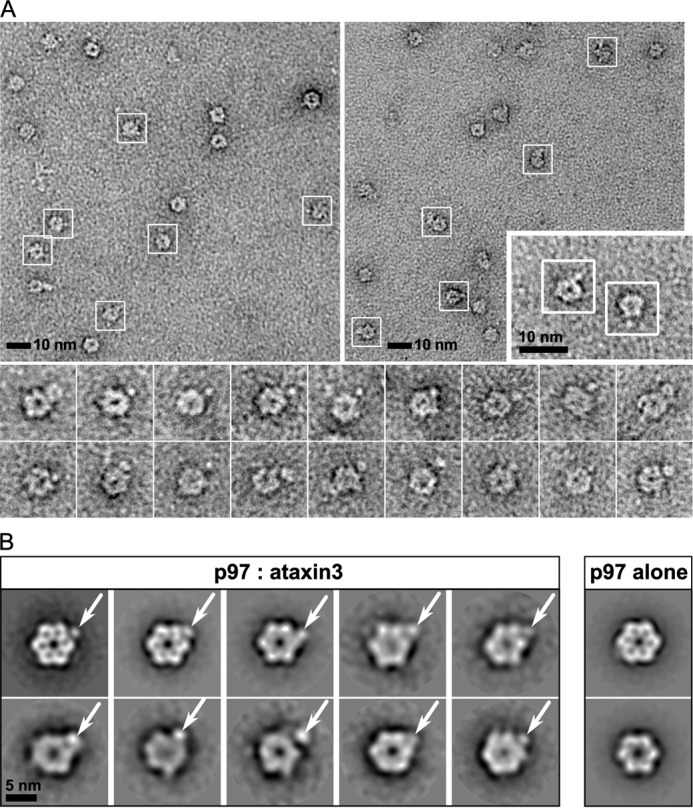
Visualization of the p97–ataxin3 complex by negative-stain EM. A, electron micrographs taken from different portions of the grid, showing negatively-stained hexamers of p97 cross-linked to ataxin3; white boxes show particles identified as complexes. The inset (lower right) shows a 2× magnified view of two p97–ataxin3 complexes from a third micrograph, and the bottom panels show additional raw images of the complex viewed down the 6-fold axis. B, representative 2D class averages derived from ∼7,100 particles for the p97–ataxin3 complex (left) and p97 alone (right). White arrows indicate the bound ataxin3 molecule.
Analysis of the p97–ataxin3 binding interface
We next turned to identifying the portions of the ataxin3 and p97 proteins responsible for their interaction. Ataxin3 has a highly-conserved N-terminal Josephin domain that is a cysteine protease with DUB activity; the protein also contains a C-terminal polyglutamine region, two ubiquitin-interacting motifs (UIMs), and an arginine/lysine-rich p97/VCP-binding motif (VBM) (Fig. 1B). The VBM has previously been identified as crucial for the p97–ataxin3 interaction (24) and is well-conserved across different species (Fig. 4A). This motif corresponds to residues 282–285 and has the sequence RKRR. To determine whether this minimal VBM is sufficient for interaction with p97, we mutated these residues to ANAA (56) and assessed the proteins' interaction by SPR, which revealed that binding was completely abolished for the mutant (Fig. 4D). We also tested the binding of various deletion constructs of ataxin3 (Fig. 4, B and C). N-terminal fragments corresponding to the Josephin domain alone or the Josephin domain plus the UIMs failed to bind to p97. However, an ataxin3 N-terminal fragment truncated after the VBM (ataxin3(1–292), denoted hereafter as ataxin3ΔC) bound p97 with the same affinity as full-length ataxin3 (Fig. 4D). These results were supported by ITC experiments that showed full-length ataxin3, ataxin3ΔC, and an ataxin3(220–345) C-terminal fragment (ataxin3ΔN) all bind the p97 N-domain with equal affinities (Fig. 4, E and F; Table 1). Therefore, we conclude that ataxin3's VBM sequence is necessary and sufficient to mediate binding with p97.
Figure 4.

Ataxin3 VBM is necessary and sufficient for interaction with p97. A, alignment of the VBMs from different ataxin3 homologs showing conservation across species. B, SDS-polyacrylamide gel of the different purified ataxin3 constructs used, stained with Coomassie Brilliant Blue. C, schematic representation of the ataxin3 constructs shown in B. D, left, representative SPR responses for ataxin3 constructs binding to immobilized full-length p97 (for all constructs, analyte concentration = 10 μm); right, normalized equilibrium binding responses for the same constructs, fit to a one-site binding model (n ≥ 3 for each concentration). The dashed line in the left panel represents the response range used to determine the equilibrium fit. E, and F, ITC raw and fitted data for the binding of the p97 N-domain to ataxin3 deletion constructs. E, injection of ataxin3ΔC into the p97 N-domain; F, injection of ataxin3ΔN into the p97 N-domain.
To map the region of p97 that interacts with ataxin3, we first noted in two recent crystal structures that peptides containing either a VBM motif or a related p97/VCP-interacting motif (VIM) bind in the same site, within a cleft separating the two lobes of the p97 N-terminal domain (70–72). Other interacting partners also bind in or near the same site; these include the ubiquitin regulatory X (UBX) and UBXL domains, whose binding sites overlap the VBM/VIM cleft, despite their possessing very different structures (60, 73–75). To test whether ataxin3 utilizes the same binding cleft, we performed competition pulldown assays, using a VIM-containing peptide from the ubiquitin ligase gp78 (71). This peptide prevented His6-tagged ataxin3 from pulling down the p97 fragment containing N and D1 domains (Fig. 5C), suggesting competition for the same site. We then mutated residues lining this binding cleft (Fig. 5A), and we tested the mutants' abilities to bind ataxin3 using SPR. Changing residues Tyr143 and Leu72 to alanine completely abolished binding, whereas the R53A and G54W mutations both partially reduced the interaction (Fig. 5, B and D). These results collectively demonstrate that the VBM of ataxin3 binds in the same inter-lobe cleft of the N-domain that is utilized by many other p97 cofactors.
Figure 5.

Inter-lobe cleft of the p97 N-domain forms the ataxin3-binding site. A, schematic representation of a p97 protomer showing the approximate positions of mutated residues (black arrows) in the N-domain. B, surface representation of the p97 N-domain (PDB entry 3TIW) showing the binding cleft that separates the two lobes and the residues lining the cleft that were mutated for binding experiments. Residues in red are the most crucial for ataxin3 interaction, because their mutation completely abolishes binding. Mutating the residues in orange and yellow partially reduces binding; thus, these residues contribute moderately to the interaction. C, competition pulldown assays involving His6-tagged ataxin3 and the p97 N- and D1-domain constructs, incubated in the presence or absence of a VIM-containing peptide from gp78. D, representative SPR responses for 10 μm ataxin3 passed over immobilized wild-type or mutant p97 (n ≥ 3 for each). The dashed line represents the time for which the response values were compared.
Effect of nucleotides on the p97–ataxin3 interaction
In the p97 ATPase cycles, nucleotide hydrolysis is accompanied by major conformational changes in the hexamer (2, 76); hence, it is reasonable to suspect that ataxin3 binding would be sensitive to the nucleotide-binding state, and indeed, evidence is available to support this idea (24). To quantify this effect, we measured the binding of ataxin3 to p97 in the presence of various nucleotides. As shown in Fig. 6A, p97 binds ataxin3 in the presence of ATP, ATPγS, and AMP-PNP, as well as in the absence of added nucleotide. In contrast, ADP significantly inhibits the interaction, with an IC50 of 5.5 ± 1.1 μm (Fig. 6, A and B).
Figure 6.

ADP inhibits the p97–ataxin3 interaction by binding to the D1-domain. A, left, SPR responses for 10 μm ataxin3 binding to immobilized full-length p97, in the presence of different nucleotides (1 mm), or in the absence of added nucleotide; right, normalized equilibrium responses, with and without 1 mm of the indicated nucleotide. The dashed line represents the response range used to determine the equilibrium fit. B, inhibition of ataxin3 binding to immobilized full-length p97 by ADP. Ataxin3 concentration was 10 μm; the IC50 value was calculated from the nonlinear least-squares regression fit, represented by the solid line. C, ribbon representation of the side view of full-length hexameric p97 bound to ADP (PDB entry 5FTK); for clarity's sake, only three of the six protomers are shown. Two protomers are colored light blue and light gray, and the third is colored green, cyan, and deep blue (denoting the N-, D1-, and D2-domains, respectively). ADP is shown in red (ball and stick form), and two conserved Walker A residues crucial for ADP binding are shown in yellow (Lys251 in the D1-domain and Lys524 in the D2-domain). D, normalized equilibrium SPR responses for ataxin3 passed over immobilized wild-type p97 and the Walker mutants. E–H, effect of ADP on ataxin3 binding to immobilized p97. E, wild-type p97; F, D1 mutant; G, D2 mutant; and H, D1/D2 double mutant. For each p97 construct, ataxin3 binding is shown in the presence of 0, 10, and 100 μm ADP. For all SPR experiments shown, n ≥ 3 for each concentration; equilibrium binding curves are fit to single-site binding models.
There are two nucleotide-binding sites per p97 monomer, one each in the D1- and D2-domains (Fig. 6C); the D1-domain binds ADP ∼10-fold more tightly than the D2-domain (18, 65, 77, 78). In p97 preparations, a fraction of the D1-domains is typically occupied by ADP that is carried through the purification (77). Thus, even when no nucleotide is added, some ADP is likely present; added nucleotide is expected to saturate unoccupied sites and compete for the pre-occupied sites. To determine which site ADP acts through to disrupt the p97–ataxin3 interaction, we generated mutations in the Walker A motifs that selectively perturb nucleotide binding in each domain, introducing the K251A mutation in the D1-domain and the K524A mutation in the D2-domain. The mutant proteins form normal hexamers and exhibit decreased ATPase activity, especially the D2 mutants (supplemental Fig. S6), which is consistent with previous findings (77). We used SPR to test the interaction of these mutants with ataxin3 in the presence of 0, 10, and 100 μm ADP (Fig. 6, D–H). In the absence of ADP, all the mutants bound ataxin3 with the same affinity as wild-type p97 (Fig. 6D). When ADP was included in the binding assay, the mutant in which the D2-domain was disrupted behaved like wild-type p97, displaying an ADP-dependent inhibition of ataxin3 binding (Fig. 6, E and G). However, mutants in which the D1-domain was disrupted (K251A and K251A/K524A) were still able to interact with ataxin3, even in the presence of 100 μm ADP (Fig. 6, F and H). Thus, ADP's inhibition of the p97–ataxin3 interaction appears to be mediated largely through nucleotide binding to the D1-domain.
Conformationally-locked form of p97 cannot bind ataxin3
We next probed the mechanism by which ADP binding inhibits the p97–ataxin3 interaction. Recent cryo-EM structures of p97 reveal that when ADP occupies the D1-domain, the N-domain is positioned along the side of the hexamer, coplanar to the D1 ring (3); this conformation is referred to as the down state (65). In contrast, in the presence of ATP, the N-domain moves above the hexamer, to adopt the up state. To test whether this conformational change affects ataxin3 binding, we generated the R155C/N387C mutant of p97. These two residues form a disulfide bond under oxidizing conditions, which tethers the N-domain to the D1-domain, mimicking the down state, even in the absence of ADP (Fig. 7A) (62). In the disulfide-locked form, this mutant has very low ATPase activity; activity can be restored by addition of reducing agent (supplemental Fig. S8). Under oxidizing conditions, ataxin3 did not interact with the R155C/N387C double mutant; however, under reducing conditions (+DTT), the interaction was restored (Fig. 7B). The interaction of wild-type p97 with ataxin3 served as a control for any nonspecific effects of the reducing agent (Fig. 7C). These results suggest that the down-state conformation does not allow for ataxin3 binding and that ADP inhibits the p97–ataxin3 interaction by driving the N-domain into the down state.
Figure 7.

Conformationally-locked form of p97 cannot bind ataxin3. A, ribbon representations of the side view of a p97 protomer (PDB entries left, 5FTK and right, 5FTM), with the N-, D1-, and D2-domains shown in green, cyan, and deep blue respectively, and the two linkers in red. The yellow circles mark the positions of the R155C and N387C mutations that form a disulfide bond under oxidizing conditions (− DTT), locking the N-domain in the down state (left). When the disulfide bond is reduced (+ DTT), the N-domain is flexible and free to move to the up state (right), as indicated by the gray arrow. B and C, top panels show normalized SPR equilibrium responses fit to one-site binding models, and bottom two panels show binding to immobilized p97 with and without 7 mm DTT. B, R155C/N387C double mutant; C, wild-type p97 (n ≥ 3 for each concentration). The gray dashed lines in the sensorgrams represent the response range used to determine the fit.
Ataxin3 binding is sterically hindered in the down-state conformation of p97
Crystal structures are available for the p97 N-domain bound to VBM and VIM peptides (Protein Data Bank ID codes 5EPP and 3TIW respectively) (70, 71). When these structures are superimposed on the structure of the full-length p97 hexamer in the down state, the peptides are oriented with their C termini projecting directly toward the D1 ring of the hexamer (Fig. 8A). In ataxin3, there are ∼60 residues located downstream of the VBM (Fig. 4C); hence, if the ataxin3 VBM adopts the same binding pose as that observed in 5EPP and 3TIW, these 60 residues at the protein's C-terminal end would sterically clash against the D1 ring when the p97 hexamer is in the down-state conformation (Fig. 8B). This suggests an explanation for why ataxin3 fails to bind when p97 is in the ADP (down state) form. To test this hypothesis, we assessed the binding properties of the C-terminal truncation ataxin3ΔC. Unlike the full-length protein, ataxin3ΔC was still able to bind to p97 in the presence of ADP, and it could also bind to the R155C/N387C double mutant under both oxidizing and reducing conditions (Fig. 8, C and D; supplemental Fig. S9A). These results are consistent with a steric clash between the ataxin3 C-terminal region and p97's D1 ring in the down-state conformation, providing a structural rationale for the effects of ADP upon the p97–ataxin3 interaction.
Figure 8.

Ataxin3 binding is sterically hindered in the down-state conformation of p97. A, superposition of the structure of the p97 N-domain bound to the gp78 VIM peptide (PDB entry 3TIW), and the structure of one protomer of full-length p97 in the ADP-bound form (PDB entry 5FTK). The N-, D1-, and D2-domains are shown in green, cyan, and deep blue. respectively, the gp78 VIM peptide in orange, and ADP in red. The N- and C-terminal ends of the peptide are labeled in orange. B, diagram depicting the predicted steric clash of ataxin3's C terminus against the D1-domain in the down-state conformation, and unobstructed binding in the up state. The orientation and color scheme for the p97 subunit are as in A, and ataxin3 is shown in orange. C, and D, binding of full-length ataxin3 and ataxin3ΔC to immobilized full-length p97, with and without 1 mm ADP. Left panels show normalized SPR equilibrium responses fit to one-site binding models, and center and right panels show representative sensorgrams (n ≥ 3 for each concentration). The gray dashed lines in the sensorgrams represent the response ranges used to determine the equilibrium fits.
ADP does not affect the binding of p97 MSP1 mutants to ataxin3
MSP1 mutations in p97 alter the equilibrium between the down and up conformations of the N-domain (62, 65, 79). These mutations also increase p97's ATPase activity (62, 80) and lower the ADP affinity for the D1-domain (65, 79). Moreover, the binding of MSP1 mutants to cofactors is perturbed relative to that of the wild-type protein, as seen both in cells (58, 80, 81) and when using purified proteins (58, 68). Because many of these cofactors bind in the same inter-lobe cleft in the N-domain that is used by ataxin3, we sought to determine whether the interaction with ataxin3 is altered for MSP1 mutants.
The pathogenic MSP1 mutations in p97 are principally located in the N-domain, the N-D1 linker, and the D1-domain. We chose to study one mutation from each of these three regions, namely R155H, L198W, and A232E (Fig. 9, A and B). The Arg155 mutation is the most prevalent and widely analyzed, and A232E has been linked to early-onset and more severe myopathy and dementia (82). L198W was initially detected in MSP1 tissue samples containing rimmed vacuoles and cytoplasmic inclusions (15), and its effect on N-domain dynamics has been investigated by NMR (66). None of these mutations are at nucleotide-binding sites or in the binding cleft for cofactors (Fig. 9B). All three mutant proteins had unaltered oligomeric states and elevated ATPase activities as compared with wild-type p97 (supplemental Fig. S7) (62, 67). We immobilized them on an SPR sensor chip, using the same oriented attachment strategy as for wild-type p97, and we measured their interaction with ataxin3 with and without ADP. In the absence of ADP, all three mutants bound ataxin3 with affinities similar to that of wild-type p97 (Fig. 9C). However, in the presence of ADP the mutants still interacted with ataxin3, unlike the wild-type protein (Fig. 9D). ADP did not inhibit the interaction with ataxin3, even at concentrations well above the reported KD values for the binding of ADP to the D1-domain (18, 65, 77, 78). In wild-type p97, binding of ADP to the D1-domain triggers the down-state conformation, disrupting the ataxin3 interaction; thus, our results indicate that in the MSP1 mutants, ADP binding fails to induce the down state, and therefore it does not inhibit the ataxin3 interaction. To test this, we introduced the L198W and A232E mutations into the disulfide-locked R155C/N387C variant of p97. Both the L198W and A232E versions of the R155C/N387C mutant bound to ataxin3, even under oxidizing conditions (supplemental Fig. S10, A and B), suggesting that Cys155 and Cys387 are never in close enough proximity to form a disulfide bond and lock the protein in the down state. In line with previous findings (65, 66, 83), it appears that the mutants rarely adopt the down state, favoring instead a conformation placing the N-domains in the up position.
Figure 9.

ADP does not inhibit ataxin3 binding to p97 MSP1 mutants. A, schematic representation of p97 showing the approximate positions of the three MSP1-related mutations (black arrows). B, ribbon representations of the side view of a p97 protomer (PDB entry 5FTM), with the N-, D1-, and D2-domains shown in green, cyan, and deep blue, respectively, and the two linkers in red. The purple circles mark the positions of the R155H, L198W and A232E MSP1 mutations in the N-domain, N-D1 linker, and D1-domain respectively. The yellow diamonds represent the approximate D1 and D2 nucleotide-binding sites, and the light pink ellipse highlights the cofactor binding cleft. The view on the left is rotated 60° anti-clockwise around the in-plane vertical axis to obtain the view on the right. C and D, normalized equilibrium SPR responses are shown for ataxin3 binding to immobilized wild-type p97 and each of the MSP1 mutants. C shows ataxin3 binding to wild-type and mutant p97 in the absence of ADP. D shows the effects of 0, 10, 100, and 1000 μm ADP on ataxin3 binding to wild-type p97, and the three MSP1 mutants R155H, L198W, and A232E (n ≥ 3 for each concentration).
Discussion
Using both SPR and ITC, we have shown that ataxin3 binds p97 with an affinity in the low-micromolar range. This is comparable with affinities reported for other p97 cofactors, which typically range from ∼0.5 to 20 μm for intact proteins and isolated VIMs and from ∼50 to 70 μm for isolated VBMs (supplemental Table S3). Our affinity measurements are also consistent with ITC studies that measured the binding of the ataxin3 VBM peptide to the p97 N-domain, reporting a KD of 15.6 μm (84). In contrast, our measurements are not consistent with a previous report in which SPR was used to derive a substantially higher affinity estimate for the p97–ataxin3 interaction (KD ∼1 nm) (85). However, as we have shown in this work, the p97–ataxin3 system presents challenges when using SPR to measure binding affinities, and if mass transport and rebinding events are not accounted for appropriately, the affinity and binding kinetics will not be calculated correctly (see supplemental data, SPR experimental details, and supplemental Figs. S12 and S13). The experiments described in Ref. 85 feature high ligand immobilization density, low analyte flow rates, and short dissociation times, all conditions likely to promote surface-induced artifacts, which are reflected in poor global fits to the kinetic data. Hence, the apparent disagreement between our affinity measurements and those reported in the earlier paper (85) may simply reflect differences in data analysis and is not a genuine difference in the behavior of the proteins.
We have also shown that ataxin3 associates with p97 in a stoichiometry of one ataxin3 molecule per p97 hexamer. Binding stoichiometries vary considerably for different p97 cofactors; for example, the p97 hexamer binds to a single Ufd1 (ubiquitin-fusion degradation protein 1)/Npl4 (nuclear localization protein 4) heterodimer and a single molecule of the deubiquitinating enzyme Otu1 (ovarian tumor domain-containing protein 1), but three molecules of p47 (59, 60, 86). Binding of only a single ataxin3 molecule per hexamer may leave room for the binding of other cofactors, or alternatively, binding of one ataxin3 molecule may prevent interaction with other cofactors. For example, overexpression experiments in cell culture suggest that the binding of ataxin3 and Ufd1 to p97 is mutually exclusive (56). It is currently still unclear how the binding of a single ataxin3 cofactor to the p97 hexamer is sensed by the remaining five protomers, but it seems reasonable to speculate that the sensing mechanism would entail conformational changes involving the N-terminal domains.
Our experiments do reveal how conformational changes control the nucleotide-dependent recognition of p97 by ataxin3. p97 binds many different cofactors, some of which have antagonistic functions; therefore, cofactor binding must be carefully controlled. This control may be exerted through different mechanisms, such as post-translational modification (87) or up-regulation of cofactor levels (11). In the case of ataxin3, binding is clearly regulated by the nucleotide-binding state of the p97 hexamer, as demonstrated by pull-down experiments using cell lysates and recombinant proteins (24). A similar, albeit less pronounced, nucleotide-based control of binding has also been reported for the cofactor p47 (68). We have probed nucleotide control of ataxin3 binding to p97 using purified proteins, confirming that the two proteins associate in the presence of ATP or the absence of nucleotide but not in the presence of ADP. We propose a mechanism to explain this behavior, drawing upon recent cryo-EM studies that reveal that different nucleotides give rise to distinct p97 conformations in which the N-terminal domain moves relative to the D1 ring (3). Taken together with our data, these experiments suggest that an ADP-driven downward movement of the N-terminal domain dislodges ataxin3 by causing a steric clash between the D1-domain and ataxin3's C terminus. Because ataxin3 uses a binding site on the N-terminal domain that is also used by other p97 cofactors, this steric dislodgement mechanism may also occur with other cofactors, depending upon their geometry of binding.
This model helps to explain how recognition of ataxin3 is altered in MSP1 mutants of p97. In cells, MSP1 mutants bind significantly elevated amounts of ataxin3 as compared with wild-type p97 (58). We show that the mutant proteins' affinities for ataxin3 are unchanged; however, whereas ADP abolishes ataxin3 binding to wild-type p97, it does not inhibit ataxin3 binding for the mutants. Hence, the elevated ataxin3 binding observed in cells can be explained by a failure of the mutant p97 proteins to release ataxin3, even after ATP has been hydrolyzed to ADP. This atypical ataxin3 binding reflects defective inter-domain communication in the MSP1 mutants, which makes them unable to transduce the ADP-bound signal from the D1- to the N-domain. The failure to release ataxin3, even in the presence of ADP, could perturb normal processing of ubiquitinated substrates, possibly by preventing other cofactors from binding; for example, MSP1 mutants that bind elevated levels of ataxin3 bind reduced amounts of the ubiquitin ligase E4B (58). Additionally, an increased residence time on p97 might disrupt ataxin3's normal patterns of subcellular localization, because residues within ataxin3's VBM form a functional nuclear localization sequence (88). Binding to p97 may mask this sequence and reduce nuclear levels of ataxin3. Notably, ataxin3 is not the only cofactor for which binding is perturbed in the MSP1 mutants; nucleotide control of p47 binding is also altered in the mutants (68), which suggests that the ADP-driven conformational changes may be a general mechanism for regulating cofactor binding.
Ataxin3 is thought to coordinate with p97 to maintain the flux of proteins through ERAD (44); however, its specific role has not been elucidated at the molecular level. In ERAD, p97 acts as a segregase, coupling ATP hydrolysis with the removal of ubiquitinated substrates from the ER. The importance of ubiquitination is quite clear: substrates must be ubiquitinated in order for p97 to be recruited to the site of protein dislocation (89), and non-ubiquitinated substrates are not efficiently unfolded and translocated by p97 (90, 91). Furthermore, if the substrate is to be targeted to the proteasome, it should remain ubiquitinated once released from p97. However, the function of deubiquitination in ERAD is less clear, and thus the part played by ataxin3 has not been immediately obvious. One possibility is that ataxin3 edits non-Lys-48 ubiquitin linkages that will not be efficiently recognized by the proteasome; this is consistent with the report that the enzyme recognizes mixed-linkage ubiquitin chains (92). Another possibility is that ataxin3 serves to trim excessively long ubiquitin chains to a minimum length necessary for proteasomal degradation. Such an activity would allow for recycling of ubiquitin molecules; it might also ease the burden on p97, because it appears that ubiquitin chains attached to substrates must be threaded through the central pore of the p97 hexamer, along with the substrate (91). Such a trimming activity for ataxin3 would be consistent with its observed substrate specificity, which is skewed toward longer ubiquitin chains. In fact, ataxin3 is quite inefficient at cleaving tetra-ubiquitin and smaller chains (93), which coincides with the minimum size required for efficient proteasome targeting (94). Thus, one possible model is that p97 is first recruited to a ubiquitinated substrate by the cofactors Ufd1/Npl4, after which ataxin3 binds (possibly triggering release of Ufd1/Npl4). The ataxin3 then can trim excessive ubiquitin chains and/or remove incorrect linkages as the substrate is being translocated. Our EM results suggest that the linkage between ataxin3 and p97 is a flexible one, which could allow for continuous processing of ubiquitin chains even as the substrate is being threaded through the pore. Once the p97 catalytic cycle is finished and ATP is hydrolyzed to ADP in the D1 ring, ataxin3 would be released. Given that the D1 ring only needs to undergo one or a few hydrolysis cycles in order for an entire substrate to be processed (91), ataxin3 may be able to fully complete its task during a single ATP/ADP cycle. It is also possible that ataxin3, once dislodged from p97, can re-engage with ubiquitinated substrates that have been released from p97, possibly facilitating their hand-off to shuttling factors like Rad23 and Dsk2 for delivery to the proteasome (95, 96).
Experimental procedures
Reagents
Enzymes for cloning were purchased from New England Biolabs (Ipswich, MA). PCR primers were purchased from Integrated DNA Technologies Inc. (Coralville, IA), and sequencing of constructs was performed by Genewiz (South Plainfield, NJ). All chemicals were from Sigma unless otherwise stated, and media components were purchased from Thermo Fisher Scientific. Chromatography columns were obtained from GE Healthcare, and all surface plasmon resonance sensor chips and chemicals were purchased from Bio-Rad. The Rho1D4 monoclonal antibody was obtained from the University of British Columbia (Vancouver, Canada), and the 5 nm Ni-NTA-Nanogold® was purchased from Nanoprobes Inc. (Yaphank, NY).
Cloning and mutagenesis
Human p97 and ataxin3 proteins were cloned into the pETHSUL vector (97), to generate N-terminal hexahistidine (His6)-tagged SUMO fusion constructs, under T7 promoter control. p97 was amplified by PCR from cDNA (Open Biosystems; clone ID 6502535). Ataxin3 was amplified from clones kindly provided by Randall Pittman and Henry Paulson; our ataxin3 sequence corresponds to that given in Kawaguchi et al. (52), except that the 26-residue polyglutamine tract corresponding to residues 292–317 was replaced by 11 glutamines. Constructs produced using the pETHSUL vector included one extra glycine at the N terminus (not included in the numbering scheme). The following constructs were used in this work: full-length p97 (amino acids (aa) 1–806); p97 N-domain (aa 1–187); p97 ND1 (aa 1–480); full-length ataxin3 (aa 1–345); ataxin3 Josephin domain (aa 1–190); ataxin3 Josephin + UIMs (aa 1–263); ataxin3ΔC (aa 1–292); and ataxin3ΔN (aa 220–345). The pProEX-HTb vector (Invitrogen) was also used to generate a full-length, N-terminal His6-tagged ataxin3 construct (aa 1–361). The ataxin3 mutant 282ANAA285 and all p97 mutants (Y143A, L72A, R53A, G54W, K251A, K524A, K251A/K524A, R155C/N387C, R155H, L198W, A232E, L198W/R155C/N387C, and A232E/R155C/N387C) were generated by one-step PCR-mediated site-directed mutagenesis (98). The C-terminal 1D4 epitope was introduced into the full-length p97 pETHSUL construct by one-step insertion mutagenesis (98). The C-terminal His6-tagged full-length p97 was cloned as a SUMO-p97 fusion insert into the pETCH vector (97) by XbaI/XmaI digestion and ligation. Primer sequences and additional details are provided in the supplemental Tables S1 and S2. Cloning, mutagenesis, and plasmid amplification were performed using the Escherichia coli Mach1 strain (Invitrogen). The human gp78 peptide (622VTLRRRMLAAAAERRLQKQ640) was purchased from Biomatik (Wilmington, DE).
Protein expression and purification
Detailed information about the preparation of various proteins is given in the supplemental data. Briefly, all proteins were expressed in E. coli Rosetta (DE3) cells (Novagen) by induction with 1 mm isopropyl β-d-thiogalactopyranoside at an A600 of 0.5–0.8, followed by overnight incubation at 24 °C. Proteins were then purified by subtractive metal affinity chromatography on a HiTrap IMAC HP column (97), followed by size-exclusion chromatography (SEC) on a HiPrep Sephacryl S-300 HR column (supplemental Fig. S11). The ataxin3 constructs required an additional anion-exchange chromatography step using a HiTrap-Sepharose Q HP column. All proteins were concentrated to 8–10 mg/ml using an Amicon® stirred cell (Merck Millipore), shock-frozen in liquid nitrogen, and stored at −80 °C.
SPR
All SPR experiments were performed on a Bio-Rad ProteOn XPR36 at 25 °C using ProteOn GLC sensor chips, unless otherwise stated. The full-length p97 hexamer was captured on the chip in an oriented manner, by employing the Rho1D4 epitope–antibody system (69, 99). The Rho1D4 monoclonal antibody was first immobilized to a density of ∼3,500 response units (RU) on the chip using amine coupling by injecting 0.1–0.5 mg/ml antibody for 7 min, using PBS (Alfa Aesar, Haverhill, MA), pH 7.4, and 0.005% Tween® 20 (VWR, Radnor, PA) as the running buffer. 1D4-tagged full-length wild-type p97 and mutants were captured as ligands, to densities of 100–150 RU, on the antibody-coated surfaces. Various concentrations of analytes, including ataxin3 and ataxin3 fragments with and without nucleotides, were passed over the chip at 150 μl/min, for the maximum possible contact time of 163 s. All interaction experiments were performed in a running buffer containing 25 mm Tris, pH 8.0, 200 mm KCl, 5 mm MgCl2, 0.1 mm tris(2-carboxyethyl)phosphine (TCEP) (Gold Biotechnology Inc., Olivette, MO), 0.005% Tween® 20. Some experiments involving the p97 R155C/N387C variant included 7 mm DTT instead of TCEP, as indicated in the text. The surface was regenerated completely with three injections of freshly prepared 10 mm NaOH and 1% N-octyl-β-d-glucopyranoside (Anatrace, Maumee, OH), at 100 μl/min for 18 s, before the next round of 1D4-tagged ligand capture. Additionally, full-length ataxin3 was used as an analyte for the p97 N-domain, which was directly immobilized on the sensor chip by amine coupling (500 RU), and for the His6-tagged full-length p97 hexamer, which was captured on a ProteOn HTG sensor chip (200–300 RU) (supplemental Fig. S1). Sensorgrams were processed and double-referenced, and the data were fit to an equilibrium binding model using GraphPad Prism 7.0 to derive the binding affinity. Detailed information on all SPR experiments and data analyses are available in the supplemental data section.
ITC
ITC measurements were conducted at 25 °C on a Nano ITC calorimeter (TA Instruments). Proteins were extensively dialyzed against 20 mm sodium phosphate, pH 7.0, 200 mm KCl, 5 mm β-mercaptoethanol (βME), and degassed for 15 min before each experiment. Full-length ataxin3, ataxin3ΔC, or ataxin3ΔN (400–855 μm) was placed in the syringe and titrated as ligands into the sample cell containing 45 μm full-length p97 or 60–80 μm p97 N-domain. The first technical injection of 0.4 μl was followed by 25 injections of 2 μl into the sample cell (300 μl), using 250 rpm stirring and a 200-s delay between successive injections. At least three titration experiments were performed for each ligand, and the data were corrected for heats of dilution by injecting the same ligand concentration into a matched buffer. Corrected data were analyzed with a one-site binding model and nonlinear least-squares fitting to derive the KD value, using the NanoAnalyze 3.5.0 software package.
In vitro-binding assay
For competition pulldown assays, His6-tagged ataxin3 was immobilized on nickel-Sepharose Fast Flow beads (GE Healthcare) by mixing 50 μl of a 50% slurry with 100 μl of His6–ataxin3 (5 mg/ml), for 5 min at room temperature, and washing off unbound protein. The beads were then incubated at room temperature for 5 min with 80 μm p97 ND1 or p97 ND1 pre-incubated with 800 μm of the competing gp78 peptide (4 °C for 20 min, followed by 5 min at room temperature). Nickel-Sepharose Fast Flow beads alone were used as a control. All assays were performed in 50 mm Tris, pH 8.0, 250 mm KCl, 5% glycerol, 5 mm MgCl2, and 5 mm βME. The beads were centrifuged at 5,000 × g for 30 s in a 0.2-μm Nanosep® MF centrifugal filter (ODM02C34; PALL Laboratory) and additionally washed four times with 400 μl of buffer. Bound proteins were eluted in 100 μl of buffer containing 300 mm imidazole after a 5-min incubation at room temperature and analyzed by 12% (v/v) SDS-PAGE.
ATPase assays
The ATPase activities of wild-type and mutant p97 proteins (supplemental Figs. S6A, S7, and S8) were measured spectrophotometrically using an NADH-coupled system (100). The decrease in absorbance at 340 nm associated with oxidation of NADH was measured at 0.5-min intervals over 40 min on a DU800 spectrophotometer (Beckman Coulter). The rate of ATP hydrolysis was calculated from the change in A340/min. The assay was performed at room temperature, in standard buffer (25 mm Tris, pH 8.0, 200 mm KCl, 5 mm MgCl2), containing 2 mm ATP, 3 mm phosphoenolpyruvate, pyruvate kinase (20 units/ml), lactate dehydrogenase (20 units/ml), NADH (200 μg/ml), and 5 μm protein, in a final volume of 85 μl.
Electron microscopy
The complex of full-length ataxin3 + p97 was prepared by incubating 5 μm purified p97 hexamer (∼3 mg/ml) with an 11-fold molar excess of ataxin3 at 4 °C for 15 min. The complex was further purified by SEC on a HiPrep Sephacryl S-300 HR column in 25 mm Tris, pH 8.0, 200 mm KCl, 5 mm MgCl2, 0.5 mm DTT (supplemental Figs. S3B and S4). The fraction corresponding to the complex peak was diluted 20-fold (final concentration ∼25 μg/ml) to achieve sufficient dispersion of individual particles and applied to freshly glow-discharged 400 square mesh copper EM grids (Electron Microscopy Sciences, Hatfield, PA), coated with Formvar/carbon film. The grids were stained with 2% uranyl acetate, and excess liquid was blotted away with filter paper. Micrographs were collected on an FEI Tecnai T12 (120 kV, LaB6 filament) microscope, equipped with a Gatan UltraScan1000 CCD camera (2k × 2k pixels), using an indicated magnification of 52,000×, a defocus range between −0.7 and −1.5 μm, and an electron dose of 20 e−/Å2. A total of 20,256 particles were selected with the EMAN Boxer (101). Two-dimensional (2D) classification generated in the RELION software (102, 103) resulted in 50 class-average images.
For the chemical cross-linking, a mixture of 5 μm p97 hexamer and 55 μm His6-tagged ataxin3 was incubated with EM-grade glutaraldehyde (0.05% final concentration) at 4 °C for 10 min, in 20 mm HEPES, pH 8.0, 150 mm KCl, 2.5 mm MgCl2. The N-terminally His6-tagged ataxin3 construct was used because the VBM is near ataxin3's C terminus (see Fig. 4). The cross-linking reaction was terminated with Tris, pH 8.0 (25 mm final concentration). Unbound p97 was then removed by immobilized metal-affinity chromatography, which retained the His6-ataxin3-p97 complex. After elution, unbound His6-ataxin3 was removed by centrifugation at 5,000 × g for 15 min using a 100-kDa cutoff Amicon® Ultra-15 centrifugal filter (Merck Millipore). After a final round of gel filtration as described above, the complex fraction was diluted 25-fold (final concentration ∼10 μg/ml), loaded onto grids, and stained with pH-neutralized 1% uranyl formate. 7,171 images were collected and processed as above, to give 50 class-average images.
For the Nanogold-labeling, the His6-ataxin3-p97 complex (prepared as described above) was incubated with a 5-fold molar excess of 5-nm diameter Ni-NTA-Nanogold particles at 4 °C for 10 min. Excess Nanogold was removed by centrifugation at 3,000 × g for 15 min using a 10-kDa cutoff Amicon® Ultra-0.5 centrifugal filter (Merck Millipore). The mixture was further purified by gel filtration using a Yarra SEC-3000 column (Phenomenex, Torrance, CA), in 25 mm Tris, pH 7.5, 200 mm KCl, 5 mm MgCl2, 0.5 mm DTT, and the complex fraction was directly applied to grids (∼3 μg/ml), stained with 2% uranyl formate, and imaged. See supplemental Fig. S3B for sample preparation workflow and SECs for all EM experiments.
Author contributions
M. V. R. and P. J. L. designed the experiments. D. R. W. and M. V. R. performed the negative-stain electron microscopy, and M. V. R. conducted the remainder of the experiments. S. C. contributed reagents, provided access to SPR instrumentation, and helped design some of the research. M. V. R. and P. J. L. analyzed the data and wrote the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge insightful comments from Dr. Dale Haines and extensive discussions and encouragement throughout the course of this work from Dr. Tara Davis. We thank Drs. Alexander Mazin and Olga Mazina for sharing reagents for the ATPase assays.
This work was supported in part by National Institutes of Health Grant R01 NS065140. Research in this publication includes work carried out at the Sidney Kimmel Cancer Center X-ray Crystallography and Molecular Interaction Facility at Thomas Jefferson University, which is supported in part by NCI Cancer Center Support Grant P30 CA56036. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S13 and Tables S1–S3.
- VCP
- valosin-containing protein
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- DUB
- deubiquitinase
- SPR
- surface plasmon resonance
- ITC
- isothermal titration calorimetry
- UIM
- ubiquitin-interacting motif
- VBM
- p97/VCP-binding motif
- VIM
- p97/VCP-interacting motif
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- AMP-PNP
- adenylyl-imidodiphosphate
- aa
- amino acid
- SEC
- size-exclusion chromatography
- RU
- response units
- Ni-NTA
- nickel-nitrilotriacetic acid
- RT
- room temperature
- TCEP
- tris(2-carboxyethyl)phosphine
- βME
- β-mercaptoethanol
- PDB
- Protein Data Bank.
References
- 1. Ogura T., and Wilkinson A. J. (2001) AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6, 575–597 [DOI] [PubMed] [Google Scholar]
- 2. DeLaBarre B., and Brunger A. T. (2003) Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat. Struct. Biol. 10, 856–863 [DOI] [PubMed] [Google Scholar]
- 3. Banerjee S., Bartesaghi A., Merk A., Rao P., Bulfer S. L., Yan Y., Green N., Mroczkowski B., Neitz R. J., Wipf P., Falconieri V., Deshaies R. J., Milne J. L., Huryn D., Arkin M., and Subramaniam S. (2016) 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xia D., Tang W. K., and Ye Y. (2016) Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 583, 64–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meyer H., and Weihl C. C. (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 127, 3877–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meyer H., Bug M., and Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 7. Baek G. H., Cheng H., Choe V., Bao X., Shao J., Luo S., and Rao H. (2013) Cdc48: a Swiss army knife of cell biology. J. Amino Acids 2013, 183421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bug M., and Meyer H. (2012) Expanding into new markets–VCP/p97 in endocytosis and autophagy. J. Struct. Biol. 179, 78–82 [DOI] [PubMed] [Google Scholar]
- 9. Dargemont C., and Ossareh-Nazari B. (2012) Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim. Biophys. Acta 1823, 138–144 [DOI] [PubMed] [Google Scholar]
- 10. Liebl M. P., and Hoppe T. (2016) It's all about talking: two-way communication between proteasomal and lysosomal degradation pathways via ubiquitin. Am. J. Physiol. Cell Physiol. 311, C166–C178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chapman E., Fry A. N., and Kang M. (2011) The complexities of p97 function in health and disease. Mol. BioSystems 7, 700–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang W. K., and Xia D. (2016) Mutations in the human AAA+ chaperone p97 and related diseases. Front. Mol. Biosci. 3, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim H. J., Kim N. C., Wang Y. D., Scarborough E. A., Moore J., Diaz Z., MacLea K. S., Freibaum B., Li S., Molliex A., Kanagaraj A. P., Carter R., Boylan K. B., Wojtas A. M., Rademakers R., et al. (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P., and Kimonis V. E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381 [DOI] [PubMed] [Google Scholar]
- 15. Watts G. D., Thomasova D., Ramdeen S. K., Fulchiero E. C., Mehta S. G., Drachman D. A., Weihl C. C., Jamrozik Z., Kwiecinski H., Kaminska A., and Kimonis V. E. (2007) Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin. Genet. 72, 420–426 [DOI] [PubMed] [Google Scholar]
- 16. Locke M., Toth J. I., and Petroski M. D. (2014) Lys11- and Lys48-linked ubiquitin chains interact with p97 during endoplasmic-reticulum-associated degradation. Biochem. J. 459, 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chou T. F., Brown S. J., Minond D., Nordin B. E., Li K., Jones A. C., Chase P., Porubsky P. R., Stoltz B. M., Schoenen F. J., Patricelli M. P., Hodder P., Rosen H., and Deshaies R. J. (2011) Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. U.S.A. 108, 4834–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Magnaghi P., D'Alessio R., Valsasina B., Avanzi N., Rizzi S., Asa D., Gasparri F., Cozzi L., Cucchi U., Orrenius C., Polucci P., Ballinari D., Perrera C., Leone A., Cervi G., et al. (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 9, 548–556 [DOI] [PubMed] [Google Scholar]
- 19. Wójcik C., Rowicka M., Kudlicki A., Nowis D., McConnell E., Kujawa M., and DeMartino G. N. (2006) Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 17, 4606–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wójcik C., Yano M., and DeMartino G. N. (2004) RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J. Cell Sci. 117, 281–292 [DOI] [PubMed] [Google Scholar]
- 21. Jarosch E., Taxis C., Volkwein C., Bordallo J., Finley D., Wolf D. H., and Sommer T. (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 4, 134–139 [DOI] [PubMed] [Google Scholar]
- 22. Nishikori S., Yamanaka K., Sakurai T., Esaki M., and Ogura T. (2008) p97 Homologs from Caenorhabditis elegans, CDC-48.1 and CDC-48.2, suppress the aggregate formation of huntingtin exon1 containing expanded polyQ repeat. Genes Cells 13, 827–838 [DOI] [PubMed] [Google Scholar]
- 23. Song C., Wang Q., and Li C. C. (2007) Characterization of the aggregation-prevention activity of p97/valosin-containing protein. Biochemistry 46, 14889–14898 [DOI] [PubMed] [Google Scholar]
- 24. Boeddrich A., Gaumer S., Haacke A., Tzvetkov N., Albrecht M., Evert B. O., Müller E. C., Lurz R., Breuer P., Schugardt N., Plassmann S., Xu K., Warrick J. M., Suopanki J., Wüllner U., et al. (2006) An arginine/lysine-rich motif is crucial for VCP/p97-mediated modulation of ataxin-3 fibrillogenesis. EMBO J. 25, 1547–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu S., Peng G., Wang Y., Fang S., and Karbowski M. (2011) The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 22, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi T., Manno A., and Kakizuka A. (2007) Involvement of valosin-containing protein (VCP)/p97 in the formation and clearance of abnormal protein aggregates. Genes Cells 12, 889–901 [DOI] [PubMed] [Google Scholar]
- 27. van den Boom J., Wolf M., Weimann L., Schulze N., Li F., Kaschani F., Riemer A., Zierhut C., Kaiser M., Iliakis G., Funabiki H., and Meyer H. (2016) VCP/p97 extracts sterically trapped Ku70/80 rings from DNA in double-strand break repair. Mol. Cell 64, 189–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ballar P., Pabuccuoglu A., and Kose F. A. (2011) Different p97/VCP complexes function in retrotranslocation step of mammalian ER-associated degradation (ERAD). Int. J. Biochem. Cell Biol. 43, 613–621 [DOI] [PubMed] [Google Scholar]
- 29. Christianson J. C., and Ye Y. (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Franz A., Ackermann L., and Hoppe T. (2016) Ring of change: CDC48/p97 drives protein dynamics at chromatin. Front. Genet. 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jentsch S., and Rumpf S. (2007) Cdc48 (p97): a “molecular gearbox” in the ubiquitin pathway? Trends Biochem. Sci. 32, 6–11 [DOI] [PubMed] [Google Scholar]
- 32. Buchberger A., Schindelin H., and Hänzelmann P. (2015) Control of p97 function by cofactor binding. FEBS Lett. 589, 2578–2589 [DOI] [PubMed] [Google Scholar]
- 33. Li X., Liu H., Fischhaber P. L., and Tang T. S. (2015) Toward therapeutic targets for SCA3: insight into the role of Machado-Joseph disease protein ataxin-3 in misfolded proteins clearance. Prog. Neurobiol. 132, 34–58 [DOI] [PubMed] [Google Scholar]
- 34. Burnett B., Li F., and Pittman R. N. (2003) The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 12, 3195–3205 [DOI] [PubMed] [Google Scholar]
- 35. Doss-Pepe E. W., Stenroos E. S., Johnson W. G., and Madura K. (2003) Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol. Cell. Biol. 23, 6469–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Q., Li L., and Ye Y. (2006) Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 174, 963–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Onofre I., Mendonça N., Lopes S., Nobre R., de Melo J. B., Carreira I. M., Januário C., Gonçalves A. F., and de Almeida L. P. (2016) Fibroblasts of Machado Joseph disease patients reveal autophagy impairment. Sci. Rep. 6, 28220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nascimento-Ferreira I., Santos-Ferreira T., Sousa-Ferreira L., Auregan G., Onofre I., Alves S., Dufour N., Colomer Gould V. F., Koeppen A., Déglon N., and Pereira de Almeida L. (2011) Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 134, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 39. Menzies F. M., Huebener J., Renna M., Bonin M., Riess O., and Rubinsztein D. C. (2010) Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 133, 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashkenazi A., Bento C. F., Ricketts T., Vicinanza M., Siddiqi F., Pavel M., Squitieri F., Hardenberg M. C., Imarisio S., Menzies F. M., and Rubinsztein D. C. (2017) Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sutton J. R., Blount J. R., Libohova K., Tsou W. L., Joshi G. S., Paulson H. L., Costa M. D. C., Scaglione K. M., and Todi S. V. (2017) Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of spinocerebellar ataxia type 3. Hum. Mol. Genet. 26, 1419–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsou W. L., Ouyang M., Hosking R. R., Sutton J. R., Blount J. R., Burr A. A., and Todi S. V. (2015) The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 82, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Warrick J. M., Morabito L. M., Bilen J., Gordesky-Gold B., Faust L. Z., Paulson H. L., and Bonini N. M. (2005) Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell 18, 37–48 [DOI] [PubMed] [Google Scholar]
- 44. Liu Y., and Ye Y. (2012) Roles of p97-associated deubiquitinases in protein quality control at the endoplasmic reticulum. Curr. Protein Pept. Sci. 13, 436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ackermann L., Schell M., Pokrzywa W., Kevei É., Gartner A., Schumacher B., and Hoppe T. (2016) E4 ligase-specific ubiquitination hubs coordinate DNA double-strand-break repair and apoptosis. Nat. Struct. Mol. Biol. 23, 995–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schmitt I., Linden M., Khazneh H., Evert B. O., Breuer P., Klockgether T., and Wuellner U. (2007) Inactivation of the mouse Atxn3 (ataxin-3) gene increases protein ubiquitination. Biochem. Biophys. Res. Commun. 362, 734–739 [DOI] [PubMed] [Google Scholar]
- 47. Hübener J., Vauti F., Funke C., Wolburg H., Ye Y., Schmidt T., Wolburg-Buchholz K., Schmitt I., Gardyan A., Driessen S., Arnold H. H., Nguyen H. P., and Riess O. (2011) N-terminal ataxin-3 causes neurological symptoms with inclusions, endoplasmic reticulum stress and ribosomal dislocation. Brain 134, 1925–1942 [DOI] [PubMed] [Google Scholar]
- 48. Rodrigues A. J., Neves-Carvalho A., Teixeira-Castro A., Rokka A., Corthals G., Logarinho E., and Maciel P. (2011) Absence of ataxin-3 leads to enhanced stress response in C. elegans. PLoS One 6, e18512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramani B., Harris G. M., Huang R., Seki T., Murphy G. G., Costa Mdo C., Fischer S., Saunders T. L., Xia G., McEachin R. C., and Paulson H. L. (2015) A knockin mouse model of spinocerebellar ataxia type 3 exhibits prominent aggregate pathology and aberrant splicing of the disease gene transcript. Hum. Mol. Genet. 24, 1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boy J., Schmidt T., Schumann U., Grasshoff U., Unser S., Holzmann C., Schmitt I., Karl T., Laccone F., Wolburg H., Ibrahim S., and Riess O. (2010) A transgenic mouse model of spinocerebellar ataxia type 3 resembling late disease onset and gender-specific instability of CAG repeats. Neurobiol. Dis. 37, 284–293 [DOI] [PubMed] [Google Scholar]
- 51. Goti D., Katzen S. M., Mez J., Kurtis N., Kiluk J., Ben-Haïem L., Jenkins N. A., Copeland N. G., Kakizuka A., Sharp A. H., Ross C. A., Mouton P. R., and Colomer V. (2004) A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J. Neurosci. 24, 10266–10279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., and Akiguchi I. (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 8, 221–228 [DOI] [PubMed] [Google Scholar]
- 53. Chai Y., Koppenhafer S. L., Shoesmith S. J., Perez M. K., and Paulson H. L. (1999) Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum. Mol. Genet. 8, 673–682 [DOI] [PubMed] [Google Scholar]
- 54. Yang H., Li J. J., Liu S., Zhao J., Jiang Y. J., Song A. X., and Hu H. Y. (2014) Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: implication in a loss-of-function pathology. Sci. Rep. 4, 6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hirabayashi M., Inoue K., Tanaka K., Nakadate K., Ohsawa Y., Kamei Y., Popiel A. H., Sinohara A., Iwamatsu A., Kimura Y., Uchiyama Y., Hori S., and Kakizuka A. (2001) VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 8, 977–984 [DOI] [PubMed] [Google Scholar]
- 56. Zhong X., and Pittman R. N. (2006) Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum. Mol. Genet. 15, 2409–2420 [DOI] [PubMed] [Google Scholar]
- 57. Kuhlbrodt K., Janiesch P. C., Kevei É., Segref A., Barikbin R., and Hoppe T. (2011) The Machado-Joseph disease deubiquitylase ATX-3 couples longevity and proteostasis. Nat. Cell Biol. 13, 273–281 [DOI] [PubMed] [Google Scholar]
- 58. Fernández-Sáiz V., and Buchberger A. (2010) Imbalances in p97 co-factor interactions in human proteinopathy. EMBO Rep. 11, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pye V. E., Beuron F., Keetch C. A., McKeown C., Robinson C. V., Meyer H. H., Zhang X., and Freemont P. S. (2007) Structural insights into the p97-Ufd1-Npl4 complex. Proc. Natl. Acad. Sci. U.S.A. 104, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim S. J., Cho J., Song E. J., Kim S. J., Kim H. M., Lee K. E., Suh S. W., and Kim E. E. (2014) Structural basis for ovarian tumor domain-containing protein 1 (OTU1) binding to p97/valosin-containing protein (VCP). J. Biol. Chem. 289, 12264–12274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hänzelmann P., and Schindelin H. (2016) Characterization of an additional binding surface on the p97 N-terminal domain involved in bipartite cofactor interactions. Structure 24, 140–147 [DOI] [PubMed] [Google Scholar]
- 62. Niwa H., Ewens C. A., Tsang C., Yeung H. O., Zhang X., and Freemont P. S. (2012) The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J. Biol. Chem. 287, 8561–8570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hänzelmann P., and Schindelin H. (2016) Structural basis of ATP hydrolysis and intersubunit signaling in the AAA+ ATPase p97. Structure 24, 127–139 [DOI] [PubMed] [Google Scholar]
- 64. Yeung H. O., Kloppsteck P., Niwa H., Isaacson R. L., Matthews S., Zhang X., and Freemont P. S. (2008) Insights into adaptor binding to the AAA protein p97. Biochem. Soc. Trans. 36, 62–67 [DOI] [PubMed] [Google Scholar]
- 65. Tang W. K., Li D., Li C. C., Esser L., Dai R., Guo L., and Xia D. (2010) A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 29, 2217–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schuetz A. K., and Kay L. E. (2016) A dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 5, e20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang X., Gui L., Zhang X., Bulfer S. L., Sanghez V., Wong D. E., Lee Y., Lehmann L., Lee J. S., Shih P. Y., Lin H. J., Iacovino M., Weihl C. C., Arkin M. R., Wang Y., and Chou T. F. (2015) Altered cofactor regulation with disease-associated p97/VCP mutations. Proc. Natl. Acad. Sci. U.S.A. 112, E1705–E1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bulfer S. L., Chou T. F., and Arkin M. R. (2016) p97 disease mutations modulate nucleotide-induced conformation to alter protein-protein interactions. ACS Chem. Biol. 11, 2112–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Molday L. L., and Molday R. S. (2014) 1D4: a versatile epitope tag for the purification and characterization of expressed membrane and soluble proteins. Methods Mol. Biol. 1177, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lim J. J., Lee Y., Ly T. T., Kang J. Y., Lee J. G., An J. Y., Youn H. S., Park K. R., Kim T. G., Yang J. K., Jun Y., and Eom S. H. (2016) Structural insights into the interaction of p97 N-terminal domain and VBM in rhomboid protease, RHBDL4. Biochem. J. 473, 2863–2880 [DOI] [PubMed] [Google Scholar]
- 71. Hänzelmann P., and Schindelin H. (2011) The structural and functional basis of the p97/valosin-containing protein (VCP)-interacting motif (VIM): mutually exclusive binding of cofactors to the N-terminal domain of p97. J. Biol. Chem. 286, 38679–38690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stapf C., Cartwright E., Bycroft M., Hofmann K., and Buchberger A. (2011) The general definition of the p97/valosin-containing protein (VCP)-interacting motif (VIM) delineates a new family of p97 cofactors. J. Biol. Chem. 286, 38670–38678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Buchberger A., Howard M. J., Proctor M., and Bycroft M. (2001) The UBX domain: a widespread ubiquitin-like module. J. Mol. Biol. 307, 17–24 [DOI] [PubMed] [Google Scholar]
- 74. Dreveny I., Kondo H., Uchiyama K., Shaw A., Zhang X., and Freemont P. S. (2004) Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J. 23, 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hänzelmann P., Buchberger A., and Schindelin H. (2011) Hierarchical binding of cofactors to the AAA ATPase p97. Structure 19, 833–843 [DOI] [PubMed] [Google Scholar]
- 76. Rouiller I., Butel V. M., Latterich M., Milligan R. A., and Wilson-Kubalek E. M. (2000) A major conformational change in p97 AAA ATPase upon ATP binding. Mol. Cell 6, 1485–1490 [DOI] [PubMed] [Google Scholar]
- 77. Briggs L. C., Baldwin G. S., Miyata N., Kondo H., Zhang X., and Freemont P. S. (2008) Analysis of nucleotide binding to P97 reveals the properties of a tandem AAA hexameric ATPase. J. Biol. Chem. 283, 13745–13752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chou T. F., Bulfer S. L., Weihl C. C., Li K., Lis L. G., Walters M. A., Schoenen F. J., Lin H. J., Deshaies R. J., and Arkin M. R. (2014) Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J. Mol. Biol. 426, 2886–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tang W. K., and Xia D. (2013) Altered intersubunit communication is the molecular basis for functional defects of pathogenic p97 mutants. J. Biol. Chem. 288, 36624–36635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Manno A., Noguchi M., Fukushi J., Motohashi Y., and Kakizuka A. (2010) Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells 15, 911–922 [DOI] [PubMed] [Google Scholar]
- 81. Erzurumlu Y., Kose F. A., Gozen O., Gozuacik D., Toth E. A., and Ballar P. (2013) A unique IBMPFD-related P97/VCP mutation with differential binding pattern and subcellular localization. Int. J. Biochem. Cell Biol. 45, 773–782 [DOI] [PubMed] [Google Scholar]
- 82. Wojcik C. (2011) in Muscle Aging: Inclusion-Body Myositis and Myopathies (Askanas V., and Engel W. K., eds) pp. 206–218, Wiley-Blackwell, Hoboken, NJ [Google Scholar]
- 83. Tang W. K., and Xia D. (2012) Structural and functional deviations in disease-associated p97 mutants. J. Struct. Biol. 179, 83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu S., Fu Q. S., Zhao J., and Hu H. Y. (2013) Structural and mechanistic insights into the arginine/lysine-rich peptide motifs that interact with P97/VCP. Biochim. Biophys. Acta 1834, 2672–2678 [DOI] [PubMed] [Google Scholar]
- 85. Almeida B., Abreu I. A., Matos C. A., Fraga J. S., Fernandes S., Macedo M. G., Gutiérrez-Gallego R., Pereira P. J., Carvalho A. L., and Macedo-Ribeiro S. (2015) SUMOylation of the brain-predominant Ataxin-3 isoform modulates its interaction with p97. Biochim. Biophys. Acta 1852, 1950–1959 [DOI] [PubMed] [Google Scholar]
- 86. Kondo H., Rabouille C., Newman R., Levine T. P., Pappin D., Freemont P., and Warren G. (1997) p47 is a cofactor for p97-mediated membrane fusion. Nature 388, 75–78 [DOI] [PubMed] [Google Scholar]
- 87. Ewens C. A., Kloppsteck P., Förster A., Zhang X., and Freemont P. S. (2010) Structural and functional implications of phosphorylation and acetylation in the regulation of the AAA+ protein p97. Biochem. Cell Biol. 88, 41–48 [DOI] [PubMed] [Google Scholar]
- 88. Macedo-Ribeiro S., Cortes L., Maciel P., and Carvalho A. L. (2009) Nucleocytoplasmic shuttling activity of ataxin-3. PLoS One 4, e5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ye Y., Meyer H. H., and Rapoport T. A. (2003) Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blythe E. E., Olson K. C., Chau V., and Deshaies R. J. (2017) Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP*NPLOC4*UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. U.S.A. 114, E4380–E4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bodnar N. O., and Rapoport T. A. (2017) Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell 169, 722–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Winborn B. J., Travis S. M., Todi S. V., Scaglione K. M., Xu P., Williams A. J., Cohen R. E., Peng J., and Paulson H. L. (2008) The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 283, 26436–26443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Weeks S. D., Grasty K. C., Hernandez-Cuebas L., and Loll P. J. (2011) Crystal structure of a Josephin-ubiquitin complex: evolutionary restraints on ataxin-3 deubiquitinating activity. J. Biol. Chem. 286, 4555–4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Thrower J. S., Hoffman L., Rechsteiner M., and Pickart C. M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Raasi S., Orlov I., Fleming K. G., and Pickart C. M. (2004) Binding of polyubiquitin chains to ubiquitin-associated (UBA) domains of HHR23A. J. Mol. Biol. 341, 1367–1379 [DOI] [PubMed] [Google Scholar]
- 96. Richly H., Rape M., Braun S., Rumpf S., Hoege C., and Jentsch S. (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120, 73–84 [DOI] [PubMed] [Google Scholar]
- 97. Weeks S. D., Drinker M., and Loll P. J. (2007) Ligation independent cloning vectors for expression of SUMO fusions. Protein Exp. Purif. 53, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Locatelli-Hoops S. C., Gorshkova I., Gawrisch K., and Yeliseev A. A. (2013) Expression, surface immobilization, and characterization of functional recombinant cannabinoid receptor CB2. Biochim. Biophys. Acta 1834, 2045–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nørby J. G. (1988) Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 156, 116–119 [DOI] [PubMed] [Google Scholar]
- 101. Ludtke S. J., Baldwin P. R., and Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 102. Scheres S. H. (2012) RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Scheres S. H. (2012) A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.