

Abstract
Exogenous fatty acids provide substrates for energy production and biogenesis of the cytoplasmic membrane, but they also enhance cellular signaling during cancer cell proliferation. However, it remains controversial whether dietary fatty acids are correlated with tumor progression. In this study, we demonstrate that increased Src kinase activity is associated with high-fat diet–accelerated progression of prostate tumors and that Src kinases mediate this pathological process. Moreover, in the in vivo prostate regeneration assay, host SCID mice carrying Src(Y529F)-transduced regeneration tissues were fed a low-fat diet or a high-fat diet and treated with vehicle or dasatinib. The high-fat diet not only accelerated Src-induced prostate tumorigenesis in mice but also compromised the inhibitory effect of the anticancer drug dasatinib on Src kinase oncogenic potential in vivo. We further show that myristoylation of Src kinase is essential to facilitate Src-induced and high-fat diet–accelerated tumor progression. Mechanistically, metabolism of exogenous myristic acid increased the biosynthesis of myristoyl CoA and myristoylated Src and promoted Src kinase–mediated oncogenic signaling in human cells. Of the fatty acids tested, only exogenous myristic acid contributed to increased intracellular myristoyl CoA levels. Our results suggest that targeting Src kinase myristoylation, which is required for Src kinase association at the cellular membrane, blocks dietary fat–accelerated tumorigenesis in vivo. Our findings uncover the molecular basis of how the metabolism of myristic acid stimulates high-fat diet–mediated prostate tumor progression.
Keywords: diet, fatty acid, prostate cancer, protein myristoylation, Src
Introduction
Src kinase is a member of the Src family kinases (SFKs),2 a group of non-receptor tyrosine kinases that play important roles in prostate cancer initiation and progression (1). More than 90% of castration-resistant prostate tumors carry elevated Src kinase activity (2). Src kinase is a pleiotropic activator in numerous signal transduction pathways, including the MAPK, PI3K–Akt, integrin–FAK, and STAT3 pathways, consequently leading to cell survival, proliferation, migration, tumor adhesion, and angiogenesis (3). Particularly, overexpression of Src kinase promotes castration resistance in prostate cancer progression (4, 5).
Fatty acids (FAs) are involved in multiple biological processes, such as lipid synthesis, protein acylation, energy production, and transcriptional regulation (6, 7). Similar to the “Warburg effect” of glucose metabolism in cancer cells, dysregulation of lipid metabolism facilitates tumor initiation and progression (8, 9). FAs are derived from endogenous de novo synthesis and intracellular lipid metabolism as well as exogenous fatty acids from dietary uptake. Numerous epidemiological studies suggest that the dietary saturated FAs myristic acid (MA) and palmitic acid (PA) significantly increase the risk of prostate cancer–specific mortality among patients diagnosed with localized disease (10, 11). Additionally, the incorporation of exogenous free FAs into the cytoplasmic membrane promotes cancer invasion by reduction of cell–cell contact and an increase in surface adhesion (12). However, the molecular mechanisms explaining these effects of dietary FAs in promoting prostate cancer progression remains unknown.
Fatty acylation modulates SFK activity and oncogenic potential. All SFK members have an SH4 domain that mediates membrane association via myristoylation and, depending on the SFK, palmitoylation as well as SH3 and SH2 domains that mediate inter- and intramolecular interactions and the SH1 domain for tyrosine kinase catalytic activity (13). Src kinase is modified by myristoylation solely at the N terminus, which determines its membrane association and kinase activity (13, 14). Given the essential role of myristoylation in Src kinase, we demonstrate here that Src kinase activity mediates high-fat diet–accelerated tumor progression. Metabolism of exogenous myristate led to an increase of myristoyl-CoA, myristoylated Src kinase, and Src-mediated oncogenic signaling. These results offer a molecular mechanism for the association between a high-fat diet and cancer progression in advanced prostate cancer, suggesting diet as a potential intervention for reducing the risk of prostate cancer progression.
Results
Elevation of Src kinase activity is associated with high-fat diet–accelerated prostate tumors
The expression and activity of Src kinase is frequently elevated in advanced stages of prostate cancer (2). To examine whether dietary fat accelerates Src-mediated prostate tumor progression, we selected PC-3 cancer cells, which showed the highest expression level of pSrc among the tested cancer cell lines for the dietary study (supplemental Fig. S1, A and B). Host mice carrying PC-3 prostate cancer xenografts were placed on different fat diets. Diets were chosen that differed in the percentage of kilocalories derived from fat: 10% (low-fat diet, LFD) and 45% and 60% (HFD) (supplemental Table S1). As expected, increases in ectopic fat accumulation in hepatocytes and increases in adipocyte size in white adipose tissue were observed with increasing dietary fat in the host mice (Fig. 1, A and B). The 45% and 60% fat diets significantly promoted the growth of xenografts in comparison with the 10% fat diet (Fig. 1, C and D). Analysis of PC-3 xenografts grown under high dietary fat (45% and 60% fat diet) showed significantly elevated expression levels of pSrc and pErk in comparison with those in LFD xenografts (Fig. 1E), suggesting a correlation of high-fat diet with elevated Src kinase activity.
Figure 1.

Elevated Src kinase activity is associated with high-fat diet–accelerated prostate tumor growth. PC-3 human prostate cancer cells (3 × 105 cells/xenograft) were subcutaneously injected into both flanks of SCID mice (n = 9). The SCID mice were fed a 10%, 45%, or 60% fat diet for 8 weeks. A and B, representative images from H&E staining of white adipose and liver tissue from host SCID mice on a 10%, 45%, or 60% fat diet. The accumulation of ectopic fat in hepatocytes and the size of adipocytes increased in mice on the 45% and 60% fat diets compared with 10% fat diet. Scale bars = 200 μm. C, images of representative tumor-bearing mice and isolated tumors. D, the size and weight of tumors were measured. E, expression levels of total Src, pSrc(Tyr-416), Erk, pErk, and GAPDH in three representative PC-3 xenografts from 10%, 45%, and 60% fat diets. The expression levels of pSrc/total Src and pErk/total Erk were quantified. #, p < 0.05; ##, p < 0.01; ###, p < 0.005.
Src kinase mediates high-fat diet–accelerated PC-3 xenografts
To examine whether high-fat diet–accelerated tumor growth depends on Src kinase activity, a lentiviral vector to knock down Src expression was created. The lentivector is a bi-cistron vector in which shRNA-Src is driven by the H1 promoter and RFP is driven by the CMV promoter (supplemental Fig. S2, A and B). PC-3 cells were transduced with shRNA-control or shRNA-Src by lentiviral infection and implanted into SCID mice. The xenograft tumors were RFP-positive, indicating successful gene transduction. The tumors expressing shRNA-control were red because of a higher titer (with RFP marker) and/or larger in size. The tan color visualized in the tumors expressing shRNA-Src might be due to a lower titer of viral infection and/or smaller size (Fig. 2A).
Figure 2.

Src kinase mediates accelerated prostate tumor growth by dietary fat. A, PC-3 cells were transduced with shRNA-Control or shRNA-Src and subcutaneously injected into both flanks of SCID mice (n = 6/group). The mice were placed on a 10% or 45% fat diet for 8 weeks. Representative images of mice and of excised xenografts are shown (scale bar = 8 mm). B, the size and weight of xenografts are represented as mean ± S.E. (n = 6/group). Statistics were performed using two-way analysis of variance by the SAS system. C–F, RFP fluorescence and IHC staining of total Src kinase, pSrc(Tyr-416), and pErk1/2 of PC-3 xenografts from A (scale bars = 200 μm). G and H, the expression levels of pSrc and pErk in E and F were analyzed by image analysis software. #, p < 0.05; ##, p < 0.01; ###, p < 0.005; N.S., not significant.
Knockdown of Src kinase significantly reduced the size of tumors (Fig. 2, A and B) in both LFD and HFD groups. More importantly, the growth of shSrc xenografts did not respond to an increase in dietary fat (Fig. 2, A and B). Similarly, although the expression level of Ki67 was elevated in tumors grown under an HFD in comparison with those under an LFD, the expression level was inhibited in tumors expressing shRNA-Src with both diets (supplemental Fig. S3A). RFP fluorescence (a marker for lentiviral infection) and total Src staining confirmed Src knockdown efficiency in the PC-3 xenografts (Fig. 2, C and D). The levels of pSrc and pErk were elevated in the shRNA-control HFD group compared with that in the LFD group, but there was no difference when comparing the shSrc groups between the LFD and HFD groups (Fig. 2, E–H). Additionally, tumors derived from the HFD were also associated with elevated expression levels of CD34 (supplemental Fig. S3B) and inflammatory factors such as IL1-β and TNF-α (supplemental Fig. S3, C and D), and the expression levels were suppressed in tumors expressing shRNA-Src (supplemental Fig. S3, B–D). This result indicates that Src kinase mediates high-fat diet–accelerated tumor progression.
High fat diet–accelerated Src kinase–induced prostate tumors
To further examine whether dietary fat accelerates Src-induced prostate tumor progression, a cancer model established previously using the constitutively active mutant Src(Y529F) was utilized to recapitulate activated Src (15). Host mice carrying Src(Y529F)-induced prostate tumors were placed on the LFD or HFD (supplemental Fig. S4A and Fig. 3A). Both groups of mice showed no significant difference in the total amount of calories consumed (16, 17) (Fig. 3B). Host mice showed increases in liver weight, white adipose tissue, and body weight with no change in kidney, heart, or lung weight (supplemental Fig. S4, B and C). Hepatocytes accumulated more ectopic fat, and the size of adipocytes in white adipose tissue increased in the HFD group (supplemental Fig. S4D). Strikingly, the weight of regenerated tumor tissue derived from the HFD group was 3-fold greater than that from the LFD group (Fig. 3, C and D). Growth of tumors with both diets was significantly suppressed by dasatinib (Fig. 3, C and D), suggesting that dietary fat acceleration of tumor growth was Src kinase–dependent. Src(Y529F)-induced tumors showed sheets of tumorigenic cells, as reported previously (4) (Fig. 3E). Although total Src levels were similar, the level of pSrc and its target pErk were highly elevated (Fig. 3, E and F) together with down-regulation of cytokeratin 8 (CK8) with the HFD (Fig. 3E), which, as suggested previously, indicates Src-mediated epithelial–mesenchymal transition (4). Collectively, the results suggest that an increase in dietary fat promotes Src-induced tumorigenesis.
Figure 3.

Dietary fat accelerates Src-induced prostate tumor progression. A, schematic of the in vivo prostate regeneration assay. Host SCID mice carrying Src(Y529F)-transduced regeneration tissues were placed on a 10% or 60% fat diet for 8 weeks and treated with vehicle or dasatinib from week 5 to week 8 (n = 4/group). UGSM, urogenital sinus mesenchyme. B, total calorie intake was not significantly different between the diet groups. C, representative images of regenerated prostate tumors. The dashed lines show the regenerated prostate grafts on the kidney (scale bar = 4 mm). D, prostate tumor weight from A represented as mean ± S.E. (n = 4/group). Asterisks indicate unpaired, two-tailed t test. E, representative H&E (panoramic view, scale bars = 400 μm) and IHC staining (selected tumorigenic region, scale bars = 100 μm) of total Src, pSrc(Tyr-416), and pErk1/2 and co-staining of CK5, a basal epithelial cell marker, CK8, a luminal epithelial cell marker, and DAPI of tumors from C. F, the expression levels of pSrc and pErk were analyzed in IHC samples based on image analysis. #, p < 0.05; ##, p < 0.01; N.S., not significant.
Myristoylation is required for Src kinase–mediated and dietary fat–accelerated prostate tumorigenesis
Myristoylation is essential for Src kinase activity (13). We examined whether loss of myristoylation inhibits Src-driven high-fat diet–accelerated tumor progression. Src(Y529F) tumor size increased with increasing fat percentage in the diets (Fig. 4A). Src(Y529F)-transformed tumors were composed of poorly differentiated carcinoma cells and lost glandular structures in regenerated tissues derived from all diets. In contrast, the size of the regenerated tissues derived from Src(Y529F/G2A) groups was significantly smaller than Src(Y529F) tumors (Fig. 4A). Grafts derived from Src(Y529F/G2A) contained normal tubule structure and did not change with increasing dietary fat (Fig. 4, B and C). Although Src expression levels were equivalent in Src(Y529F) and Src(Y529F/G2A) regenerated prostate tissue grafts (Fig. 4D), the expression levels of pSrc and pErk were much higher in Src(Y529F) tumors from the 45% and 60% fat diet (Fig. 4, E and F, and supplemental Fig. S5) but below the detection limit in the Src(Y529F/G2A) group (Fig. 4, E and F). Dasatinib treatment of Src(Y529F) tumors decreased pSrc and pErk levels with the 10% fat diet; however, the inhibitory effect of dasatinib on Src kinase activity was compromised in the high-fat diet groups (Fig. 4, E and F, and supplemental Fig. S5). These results demonstrate that loss of myristoylation inhibits the tumorigenic potential of Src-mediated and HFD-accelerated tumorigenesis.
Figure 4.

Loss of myristoylation inhibits Src-mediated HFD-accelerated tumor progression. A, the in vivo prostate regeneration assay was performed with Src(Y529F) and Src(Y529F/G2A) under 10%, 45%, and 60% fat diets with vehicle or dasatinib treatment (75 mg/kg/day weeks 5 to 8), a similar experimental setting as described in Fig. 1. The dashed lines represent the regenerated prostate tumors grown on the kidney (scale bars = 4 mm). B–F, H&E panoramic view (B, scale bars = 300 μm) and selected region (C, scale bars = 100 μm) and IHC staining of total Src kinase, pSrc(Tyr-416), and pErk1/2 of regenerated prostate tissue (D–F, scale bars = 100 μm) in A. See also supplemental Fig. S5.
Metabolism of exogenous MA elevates the levels of myristoyl-CoA
Because myristoylation is essential for modification of Src kinase and its kinase activity, we examined the conversion of FAs to myristoyl-CoA. Exogenous fatty acids change the composition of acyl-CoAs in cells (18). Although the amount of intracellular myristoyl-CoA was markedly increased after addition of MA in the tested cell lines, other fatty acids, such as decanoic acid (C10:0, DA), lauric acid (C12:0, LA), and PA (C16:0) had limited effects on the amount of myristoyl-CoA (Fig. 5, A and B). The levels of myristoyl-CoA were maintained after 24 h but decreased with longer incubation (supplemental Fig. S6). Exogenous LA also led to detectable increases in myristoyl-CoA (Fig. 5, A and B). The data suggest that exogenous MA could be efficiently converted into the corresponding myristoyl-CoA, but a small portion of shorter FAs could undergo metabolic conversion to contribute to the myristoyl-CoA pool in vitro.
Figure 5.

Exogenous MA or an HFD increases intracellular myristoyl-CoA in cells or xenograft tumors. A, 293T+TRE/Src(Y529F) cells were treated with DMSO, DA (C10:0), LA (C12:0), MA (C14:0), or PA (C16:0) for 2 or 24 h (three repeats in each group). Con, control. B, PC-3 or DU145 cells were treated similarly for 24 h. The levels of myristoyl-CoA were analyzed by LC/MS-MS, and the levels in the control were set as 1 (three repeats in each group). C, the levels of myristoyl-CoA in PC-3 xenograft tumors from Fig. 1 were analyzed by LC/MS-MS. The amount of myristoyl-CoA was standardized to the amount of total protein in the tumors. Sixteen tumors per group were analyzed. #, p < 0.05; ##, p < 0.01. See also supplemental Fig. S6.
The composition of individual FAs varied between dietary fats. For example, the amount of MA increased ∼10-fold (10% versus 45% fat), respectively (supplemental Table S2). We further examined whether an HFD could substantially increase the levels of myristoyl-CoA in xenograft tumors. Indeed, the levels of myristoyl-CoA in tumors derived from an HFD (45% and 60% fat) were significantly elevated in comparison with those from an LFD (10% fat) (Fig. 5C), suggesting that exogenous myristoyl-CoA provides an increased amount of substrate.
Myristoyl-CoA derived from exogenous MA increases the incorporation of MA into Src kinase
Src kinase is myristoylated at the N terminus (Gly-2), and mutation of Gly-2 to Ala blocks myristoylation (14). To determine whether exogenous MA could lead to myristoyl-Src, the analog MA-azide and click chemistry were used (Fig. 6A). The click chemistry reaction allowed detection of myristoylated proteins in SYF1 (Src−/−Yes−/−Fyn−/−) cells (control). Of note, the band at 60 kDa in the first lane is not myristoylated Src in SYF1 cells (Fig. 6B). However, a 60 kDa band that reacted with streptavidin increased only in the Src(WT) lane compared with Src(G2A) or control cells (Fig. 6B). Immunoprecipitation of Src confirmed that myristoylation occurred only for Src(WT) but not for the Src(G2A) mutant (Fig. 6C). Additionally, the density of the myristoylated 60-kDa protein band increased with increasing exogenous MA-azide in SYF1+Src(WT) cells (Fig. 6D) or PC-3 cells (Fig. 6E) but decreased with shRNA-Src (Fig. 6E) or by increasing the concentration of unlabeled MA (supplemental Fig. S7A). Additionally, only high levels of LA or PA competed with the incorporation of MA-azide into Src kinase (supplemental Fig. S7, B and C), most likely because of metabolic conversion to myristoyl-CoA (Fig. 5). Collectively, these data validate the technique for analyzing Src kinase myristoylation and suggest a positive correlation between the levels of myristoyl-Src and increasing MA.
Figure 6.

Exogenous MA elevates myristoylated Src kinase and promotes Src oncogenic signaling. A, schematic of click chemistry to detect the myristoylation of Src kinase. Cells were grown with myristic acid-azide overnight. NMT1/2 catalyzes the cellular acylation of Src kinase. As a result, myristic acid-azide was processed intracellularly and incorporated into de novo synthesized Src kinase (Step 1). Myristic acid-azide–modified Src kinase in the cell lysate was reacted with a biotin-conjugated alkyne in vitro in a click reaction (Step 2). The biotin-myristoylated Src was immunoprecipitated and visualized by immunoblotting using streptavidin-HRP (Step 3). Total Src was visualized by immunoblotting. B, SYF1 (Src−/−Yes−/−Fyn−/−) cells expressing Src(WT), Src(G2A), or control were grown in medium containing 100 μm myristic acid-azide. Myristoylated proteins were detected by a click chemistry reaction. C, identification of myristoylated Src(WT) from click chemistry reactions by immunoprecipitation (IP, using a Src antibody) and immunoblotting with streptavidin-HRP. D, SYF1 cells expressing Src(WT) were cultured with 0, 20, and 100 μm of myristic acid-azide. Myristoylated proteins and the expression of Src were detected by click chemistry reactions (left) and immunoblotting (right). WB, Western blot. E, PC-3 cells expressing control or shRNA-Src were cultured with 0, 20, or 60 μm myristic acid-azide. The arrowhead indicates the band of Src kinase. F and G, 293T cells expressing doxycycline-inducible Src(Y529F) (293T+TRE/Src(Y529F)) were grown in DMEM with 2% BSA and treated with MA (F) with/without Dox (1 μg/ml) or dasatinib (10 nm) for 24 h. PC-3 cells were grown in F-12K medium with 2% BSA and treated with 0, 100, 300, 600, or 1200 μm MA overnight (G). The levels of total Src, pSrc(Tyr-416), total FAK, pFAK (Tyr-925), total Erk, and pErk1/2 were determined by immunoblotting. The data represent three independent experiments.
Exogenous MA enhances Src kinase–mediated oncogenic signaling
To identify whether exogenous MA possesses the ability to increase Src-mediated signaling, a doxycycline (Dox)-inducible Src(Y529F) expression system was utilized (supplemental Fig. S8). Based on the physiological concentration of MA in the blood of healthy men 8 h after a meal (1 g of fat/kg of body weight), 200 and 400 μm MA were examined (19). Although 200 and 400 μm LA did not enhance pSrc, pErk, or pFAK (data not shown), the levels of pErk and pFAK increased with Dox induction of pSrc in an MA dose–dependent manner (Fig. 6F). This effect was inhibited by dasatinib, suggesting that MA-induced MAPK and FAK signaling is dependent on Src activity. MA (400 μm) also induced Src-independent pErk expression (Fig. 6F). Similarly, elevated expression levels of pSrc and pErk correlated with an increase of MA in PC-3 cancer cells (Fig. 6G). Collectively, these results indicate that MA promotes Src-mediated MAPK and FAK signaling.
Exogenous myristate elevates the expression levels of Src kinase at the membrane fraction
Biosynthesis of myristoyl-Src is important for enhancing its kinase activity. The incorporation of myristoyl-CoA into Src kinase is catalyzed by N-myristoyltransferase (NMT1) (20). We examined whether overexpression of NMT1 altered the expression levels of endogenous Src kinase at the membrane fraction. Overexpression of NMT1 did not change total Src expression, nor did it change Src expression levels in the membrane fraction (supplemental Fig. S9), suggesting that the endogenous levels of NMT1 might be sufficient to catalyze the biosynthesis process.
We further examined whether elevation of myristate correlates with an elevation of the expression levels of Src in the membrane fraction. As shown in Fig. 7A, first and second lanes), a fraction of Src was located at the cytosol in SYF1+Src(WT) cells. Although exogenous myristate did not alter Src expression levels in the total lysate, it significantly increased the levels of Src in the membrane fraction, with a slight decrease in the cytosol fraction (Fig. 7A). Of note, because the cytosol fraction contained a large volume of lysate and only a limited amount of the cytosol protein was able to be loaded in the gel, this subtle change might reflect a larger amount of Src kinase decreased in the total cytosol fraction.
Figure 7.

Exogenous myristate elevates the expression levels of Src kinase at the membrane fraction. A, SYF1 cells expressing Src(WT) were grown in medium containing 400 μm myristic acid. The expression levels of Src kinase in the cytosol (Cyt) and membrane (Mem) fraction and total lysate were analyzed by immunoblotting. The amount of Src expression in the cytosol fraction treated with DMSO was set as 1. B, SYF1 cells expressing Src(WT) were transduced with shRNA-NMT1 by lentiviral infection with increased amounts of lentivirus. SYF1 and SYF1 cells expressing Src(WT) and different expression levels of NMT1 were grown in medium containing 60 μm myristic acid-azide. Myristoylated proteins were detected by a click chemistry reaction. Additionally, the levels of total Src, NMT1, and GAPDH were determined by immunoblotting. The amount of myristoyl-Src expression in cells without shNMT1 was set as 1. The data represent three independent experiments.
We further analyzed whether down-regulation of NMT1 expression inhibits myristoylation of Src kinase. In comparison with SYF1 cells, Src expression and myristoyl-Src were visible at 60 kDa in SYF1 cells expressing Src(WT). Indeed, knockdown of NMT1 significantly inhibited myristoylated Src in an NMT1 dose–dependent manner (detected by streptavidin-HRP) but did not change the expression levels of Src kinase (Fig. 7B). The data suggest that a portion of Src kinase is in the non-myristoylated state when the NMT:Src (or other proteins) ratio is stoichiometrically imbalanced. Exogenous myristate might potentially increase myristoyl-Src under endogenous levels of NMT1.
Discussion
Our study reveals a biological mechanism for how the metabolism of dietary fat promotes Src kinase–mediated MAPK signaling and prostate tumorigenesis. We have demonstrated the potential co-existence of both non-myristoylated and myristoylated Src kinase in cells. The expression levels of Src kinase are elevated in numerous cancers, including advanced prostate cancer (2). Aberrant expression of Src might lead to the accumulation of non-myristoylated Src in cancer cells. Our data show an increase in the biosynthesis of myristoyl-CoA from exogenous MA. As a result, exogenous MA might promote the formation of myristoylated Src, subsequently enhancing Src kinase activity (Fig. 8). Myristoylation of Src kinase is essential for its membrane localization, oncogenic signaling, and HFD-accelerated activity and tumor progression. This is supported by evidence that an HFD causes c-Src partitioning and activation in detergent-resistant membranes and inhibits insulin signaling (21). Lipid rafts are the major platform where proteins can associate and initiate oncogenic signaling (22).
Figure 8.
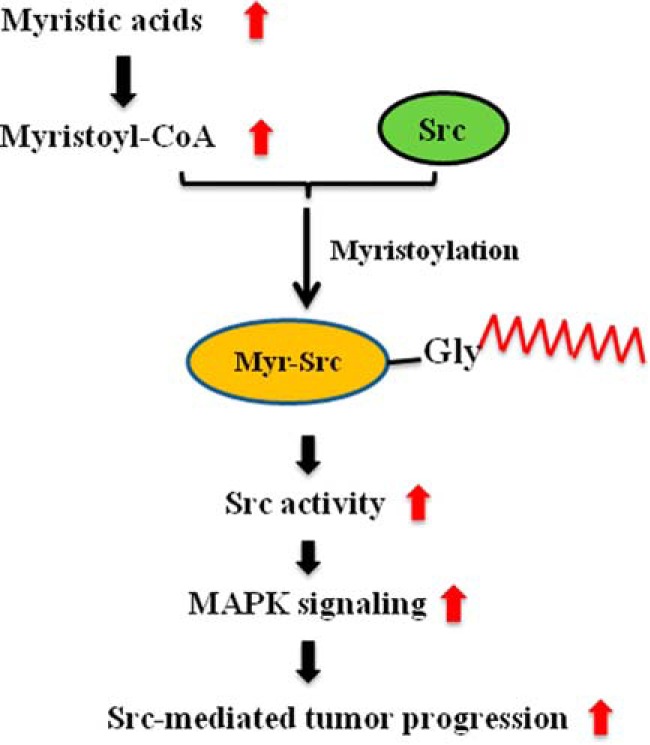
Mechanism of Src-mediated prostate tumor progression by exogenous myristic acid. Exogenous myristic acid is converted into the corresponding myristoyl-CoA. Myristoyl-CoA will be incorporated into the de novo synthesized Src kinase, which is essential for its kinase activity, by N-myristoyltransferases. The elevation of Src myristoylation enhances Src oncogenic signaling, including the phosphorylation of MAPK and FAK, thereby accelerating Src-mediated tumor progression.
Myristoylation determines dephosphorylation at Tyr-529, which is required for the regulation of Src kinase activity (23). Myristate might enhance Src kinase activity intramolecularly. Based on structural analyses, it has been proposed that the Src myristoyl group binds to the myristate binding pocket in the SH1 catalytic domain, which helps the unphosphorylated Tyr-529 site lock into the SH2 domain of Src kinase and maintain Src kinase in the inactive state. It is possible that exogenous myristate might compete with the myristate-binding pocket, which promotes phosphorylation of Tyr-416 in the kinase active site (24). Further studies will be needed to understand how myristate affects the activity of Src kinase on an intramolecular level. Non-myristoylated Src is stabilized through down-regulation of ubiquitination and degradation (14). Stabilization might act as a dominant negative factor to inhibit Src kinase activity. Our study and others indicate that myristoylation of Src kinase is a favorable target for inhibiting its mediated tumorigenesis (14).
Our study demonstrates that loss of myristoylation inhibits Src kinase–mediated tumor progression under dietary fat. This suggests that targeting Src myristoylation may be an effective approach for inhibiting Src-mediated tumor progression. Two mammalian isozymes of NMT, NMT1 and NMT2, catalyze the transfer of the myristoyl group from myristoyl-CoA to Gly-2 in Src kinase (25). Our data show that ablation of NMT1 inhibits the expression levels of myristoylated Src. This result is in agreement with a report in which ablation of NMT1 led to loss of activation of Src and its down-stream target FAK as well as the MAPK pathway, subsequently down-regulating cell proliferation (20). NMT activity is also suggested to regulate Src membrane anchoring and/or trafficking in the Golgi apparatus during monocytic differentiation (26). We have demonstrated that an increase in exogenous myristate increases the expression levels of Src kinase in the membrane fraction, most likely through NMT1-catalyzed synthesis of myristoylated Src kinase. Further confirmation of myristoylated Src in the membrane fraction is required to test this hypothesis. Inhibition of NMTs, suppressing myristoylation of oncogenic proteins, has been considered as a chemotherapeutic approach for targeting cancer (27). Compounds targeting NMT activity have been reported to effectively target cancer cells (28, 29).
Several drugs, including dasatinib, have been developed, targeting the Src kinase catalytic domain at the ATP binding site, and show promising efficacy in suppression of tumor proliferation and metastasis in prostate cancer and beneficial effects in reducing tumor size and numbers in preclinical and phase I and II studies (30). Our study shows that dasatinib efficiently inhibits Src-mediated tumor growth in vivo. However, surprisingly, dietary fat significantly compromises the inhibition of Src-mediated tumorigenesis by dasatinib in vivo and reduced dasatinib efficacy in vitro. Dasatinib together with docetaxel shows limited improvement when treating metastatic castration-resistant prostate cancer in a clinical trial (31). Documentation of the differential consumption of dietary fat might be helpful to further understand the limited success of dasatinib in the trial.
Our study highlights the association of exogenous saturated FAs with Src-mediated tumor progression. High intake of saturated fat increases the risk of biochemical failure with localized prostate cancer treated with prostatectomy (32, 33). However, because of the lack of a molecular mechanistic understanding, correlation of the risk of prostate cancer with dietary FAs has been inconsistent. For example, Epstein et al. (10) and Crowe et al. (11) reported that dietary myristate is correlated with a risk of health deterioration of prostate cancer patients. However, Crowe et al. (34) suggest no correlation of prostate cancer risk with six FAs in the blood, including MA. Further stratification of the investigated clinical cases based on the expression and activity of Src kinase will be helpful to correlate cancer risk with dietary fat.
Our data indicate that exogenous MA elevates myristoylation of Src and promotes Src-mediated tumor progression. It should be noted that exogenous MA could also have pleiotropic effects on the pathological process of tumorigenic cells. For example, exogenous MA could increase the β-oxidation process and lipogenesis, including biosynthesis of triglycerides and phospholipids (6). Additionally, a high-fat diet could also promote inflammatory factors such as IL1-β and TNF-α, among others. Src family kinases are essential nodes to mediate inflammatory factors (35, 36). Elevated myristoylation promotes the activity of Src family kinases and accelerates the inflammatory response in cancer progression. Our study supports epidemiological observation, highlighting dietary choice as an important factor in prostate cancer progression. It is anticipated that dietary MA could accelerate the growth of tumors that express high levels of activated Src kinase in other cancer types.
Experimental procedures
Plasmid constructs and lentivirus production
Lentiviral vectors were constructed using the FUCRW parental vector, as reported previously (4). Loss of the myristoylation site Src(G2A) or Src(Y529F/G2A) was created by PCR using the forward primer F-G2A and the reverse primer R-G2A (supplemental Table S3). The PCR products were cloned into the XbaI and EcoRI sites in the FUCRW vector. Additionally, doxycycline-inducible lentiviral vectors for the regulation of Src(Y529F) and Src(Y529F/G2A) expression were created by a similar strategy using pTK380 as a parental vector, designated TRE/Src(Y529F).
To knock down the human Src gene, four pairs of primers were commercially synthesized and annealed together (supplemental Table S3). The annealed products were ligated into the psiRNA-W (H1.4) vector at the BbsI site. The fragment containing the PacI site was released and subcloned into the FUCRW lentiviral vector. As a result, Src-shRNAs regulated by an H1 promoter were created in a lentiviral vector. Lentivirus production and infection were performed as described previously (37). All procedures followed the safety guidelines and regulations of the University of Georgia.
Prostate regeneration assay and PC-3 xenografts
C57BL/6J and CB.17SCID/SCID (SCID) mice were purchased from Taconic (Hudson, NY). For the prostate regeneration assay, primary prostate cells were isolated from 8- to 12-week-old male C57BL/6J mice and infected with lentivirus according to an experimental setup. Infected cells (2–3 × 105 cells/graft) were combined with urogenital sinus mesenchyme (2–3 × 105 cells/graft) together with 25 μl of collagen type I (adjusted to pH 7.0) (4). After overnight incubation, the grafts were implanted under the kidney capsule in SCID mice by survival surgery. After 3 days of implantation, host SCID mice carrying oncogene-transduced grafts were placed on a normal chow diet or switched from a normal chow diet to a 10% (catalog no. D12450J), 45% (catalog no. D08091803), or 60% (catalog no. D12492) fat diet designed by Research Diets (New Brunswick, NJ). All three diets contained the same amount of total calories, including 20% kcal from protein, and the composition of each diet is shown in supplemental Table S1. The identities of fatty acids in the diets are shown in supplemental Table S2. The amount of food consumed by the mice under the 10% or 60% diet was recorded, and the total amount calories consumed were calculated.
SCID mice carrying the Src(Y529F) transduced grafts were also administrated dasatinib (BioVision, Milpitas, CA) or vehicle by gavage (75 mg/kg body weight) per day for 4 weeks. Dasatinib, an Src family kinase inhibitor, was dissolved in 80 mm citrate buffer (pH 3.0) with 5% DMSO. Body weight was measured once a week. All animals were sacrificed 8–12 weeks after the grafts were implanted. The grafts, liver, and fat mass were collected and measured and then formalin-fixed and paraffin-embedded for histology and immunohistochemistry analysis.
For xenograft experiments, PC-3 cells transduced with Src-shRNA or control shRNA by lentiviral infection were grown in DMEM with 10% FBS. 3 × 105 cells were mixed with 50 μl of collagen type I (pH 7.0) (BD Biosciences) and inoculated subcutaneously in both lateral flanks of SCID mice. Host mice carrying PC-3 xenografts were placed on 10%, 45%, or 60% fat diets for 2 months. Tumors were measured (length × width) with Vernier calipers once a week. After 8 weeks, the animals were sacrificed, and the tumors were harvested and weighed.
All animals were maintained and used according to the surgical and experimental procedures of Protocol A2013 03-008. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Georgia.
Cell culture
Human cancer cell lines, including LNCaP, 22Rv1, PC3, DU145, VCaP, HT-29, and HCT116, were purchased from the ATCC and grown in ATCC-recommended media. SYF1 (Src−/−Yes−/−Fyn−/−) mouse fibroblast cells transduced with Src(WT) or Src mutants or 293T cells carrying the reverse tetracycline activator (rtTA, a gift from the laboratory of Dr. Kathrin Plath, University of California, Los Angeles) transduced with the tetracycline-responsive element (TRE)/Src(Y529F) by lentiviral infection were grown in DMEM with 10% FBS. To exclude uninfected cells, RFP-positive cells were isolated by cell sorting (BD Biosciences). The expression of Src kinase was induced by doxycycline in TRE/Src(Y529F)-infected cells.
Protein lysate preparation and antibodies
To examine how individual fatty acids affected Src-mediated signaling, Src-expressing cells were serum-starved for 6–24 h and then cultured with 2% fatty acid–free BSA with or without fatty acid overnight. For total protein expression analysis, protein lysate was extracted using radioimmune precipitation assay lysis buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm EDTA, 10% glycerol, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, protease inhibitor mixture, and phosphatase inhibitor mixtures 1 and 2. Src kinase and downstream signaling were analyzed by immunoblotting.
Src, pSrc(Tyr-416), pErk1/2, Fak, pFak(Tyr-925), GAPDH, and γ-tubulin antibodies were purchased from Cell Signaling Technology (Beverly, MA). Androgen receptor and Erk2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Protein fractionation
The fractionation protocol for the cytosol and the membrane fraction was described previously (38). Briefly, NIH/3T3 and SYF1 cells expressing Src(WT) or Src(G2A) were transduced with NMT1 or shRNA NMT1 by lentiviral infection and cultured in DMEM with 10% FBS. After lysing with TNE lysis buffer (50 mm Tris, 150 mm NaCl, 2 mm EDTA (pH 7.4), protease inhibitor mixture, and phosphatase inhibitor cocktails), the protein extracts were homogenized using a 25-gauge needle syringe (20 strokes) and centrifuged at 14,000 rpm for 20 min. The supernatant was collected as the cytosolic fraction. To extract the membrane fraction, pellets were rinsed twice with TNE lysis buffer and resuspended in TNE lysis buffer containing 60 mm β-octylglucoside. Samples were incubated on ice for 30 min and centrifuged at 14,000 rpm for 20 min at 4 °C. The supernatants were collected as the membrane fraction. Src total, Caveolin-1, NMT1, and GAPDH proteins were analyzed by immunoblotting with specific antibodies.
Immunohistochemistry and quantification of protein expression
Illuminated and fluorescent images were taken with dissecting and fluorescence microscopes. The primary antibodies and dilutions for immunohistochemistry (IHC) analysis were performed as described previously (4). Formalin-fixed/paraffin-embedded grafts and tissues were sectioned at 4-μm thickness and mounted on positively charged microscope slides. Sections were stained with H&E, and IHC analysis was performed as follows: primary antibodies and dilutions for Src (1:250), pSrc (Tyr-416) (1:50), androgen receptor (1:200), and pErk1/2 (1:50) were used.
The expression levels of pSrc and pErk in IHC samples were analyzed by the ImageJ software. Images of each sample were taken under the same exposure time, light intensity, and magnification. Images were taken at a low magnification (×10) to cover all of the tumor area. An image (.jpg file) was loaded into the software for image analysis. Images were split into three channels, including “red,” “green,” and “blue,” under the function of “Image” and subfunction of “color.” The image in the blue channel was processed under the function of Image, subfunction of “Adjust,” and subsubfunction of “Threshold.” A threshold was selected until all positive pixels were selected. Additionally, the threshold should not contain any oversaturated pixels. The same threshold was applied for all images. The intensity of the selected pixels was reported under the function of “Analyze” and subfunction of “Measure.” The expression levels are defined in “%Area,” which represents the percentage of an area with positive IHC staining in an image. A representative image was selected to represent each treatment in the experiment.
For quantification of protein expression in Western blots, bands in the film were uploaded to ImageJ, and the bands of interest were selected. Because the film had different exposure times, the bands with unsaturated signals were used for the analysis. The area of a peak was given by the software.
Detection of Src kinase myristoylation by click chemistry
SYF1 cells expressing Src(WT) or Src(G2A) were grown in DMEM with 2% fatty acid–free BSA containing 20 or 100 μm myristic acid-azide for 24 h. Similarly, PC-3 cells expressing control or shRNA-Src were cultured in DMEM with 2% fatty acid–free BSA containing 0, 20, or 60 μm myristic acid-azide for 24 h. Cells were washed twice with PBS and lysed on ice with M-PER buffer (Thermo Scientific) containing protease inhibitors and phosphatase inhibitors. Cell lysates were centrifuged at 14,000 rpm for 20 min, and the supernatants were collected. Protein concentration was determined using the Bio-RadDC protein assay kit. The click chemistry reaction was accomplished with 40 μg of protein lysates, 100 μl of Click-iT reaction buffer containing 40 μM alkyne-biotin, 10 μl of CuSO4, 10 μl of additive 1 solution, and 20 μl of additive 2 solution according to the instructions of the manufacturer (Life Technologies). After 30 min of incubation at room temperature, the samples were boiled for 5 min and resolved by SDS-PAGE gel. Streptavidin-HRP was used for detecting azide-labeled myristoylated Src kinase using immunoblotting.
Acyl-CoA analysis
293T cells carrying doxycycline-inducible Src kinase, PC-3 cells, and DU145 cells were cultured in DMEM with 2% BSA for treatment with various fatty acids. Cell culture medium was removed when cells reached 80% confluence. The method for extraction and determination of acyl-CoAs from biological samples were described previously (18). Cells were washed with PBS twice and incubated with 2 ml methanol and 15 μl of 25 μg/ml pentadecanoyl-CoA in methanol (internal standard) at −80 °C for 15 min. When xenograft tumors were used, each tumor sample with a similar wet weight was homogenized with 2 ml of methanol and 15 μl of 25 μg/ml pentadecanoyl CoA on ice. The cell or tissue lysate was collected and centrifuged at 15,000 × g at 5 °C for 5 min. The supernatant was transferred to a glass tube, mixed with 1 ml of acetonitrile, and evaporated in a vacuum concentrator at 55 °C for 2 h. The sample was reconstituted with 120 μl of methanol, briefly vortexed, and centrifuged, and 100 μl of the supernatant was transferred into an autosampler vial for injection.
Various acyl-CoAs were purchased from Avanti Polar Lipids (Alabaster, AL). Ammonium hydroxide, ammonium acetate, LC/MS-grade acetonitrile, methanol, and water were purchased from Sigma-Aldrich (St. Louis, MO).
An Agilent 1100 binary pump HPLC system (Santa Clara, CA) interfaced to a Waters Micromass Quattro Micro triple quadrupole mass spectrometer with an electrospray ionization (ESI) (+) source (Milford, MA) was used for LC/MS-MS analysis. Masslynx 4.0 software by Waters (Beverly, MA) was applied for instrument control and data processing. A Labconco CentriVap Complete Vacuum Concentrator (Kansas City, MO) was used to evaporate samples. The instrumentation settings for the LC/MS-MS measurements can be found in the Supplemental Experimental procedures (18).
Statistical analysis
Prism software was used to carry out statistical analysis. The data were presented as mean ± S.E. and analyzed using Student's t test. All t tests were performed at the two-sided 0.05 level for significance. The SAS system was used to analyze the effect of two factors, factor 1 with diet levels and factor 2 with/without Src knockdown, in a two-way analysis of variance: #, p < 0.05; ##, p < 0.01; ###, p < 0.005; N.S., not significant.
Author contributions
S. K. and H. C. designed the experiments and wrote the manuscript. S. K. performed the majority of the experiments. X. Y. and M. G. B. designed and measured the acyl-CoA. Q. L. helped with maxi-prep of lentiviral vectors and preparation of some lentiviruses in some experiments. M. W. performed immunohistochemical staining. Z. B. initiated the protein fractionation experiments and edited the manuscript. L. C. managed the mouse colony, performed diet maintenance, and helped with data collection for in vivo experiments.
Supplementary Material
Acknowledgments
We thank Dr. Lorene Leiter from Research Diets for guidance regarding rodent diet design, Dr. George Zheng for guidance with click chemistry experiments, Dr. Lily Wang for help with statistical analysis, and Dr. Dexi Liu for critical reading of the manuscript.
This work was supported by National Institutes of Health Grant R01CA172495 and Department of Defense Grant W81XWH-15-1-0507 (to H. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S9, Tables S1–S3, and Experimental procedures.
- SFK
- Src family kinase
- FA
- fatty acid
- MA
- myristic acid
- PA
- palmitic acid
- SH
- Src homology
- LFD
- low-fat diet
- HFD
- high-fat diet
- DA
- decanoic acid
- LA
- lauric acid
- DOX
- doxycycline
- NMT
- N-myristoyltransferase
- TRE
- tetracycline-responsive element
- IHC
- immunohistochemistry
- RFP
- red fluorescent protein
- FAK
- focal adhesion kinase.
References
- 1. Thomas S. M., and Brugge J. S. (1997) Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13, 513–609 [DOI] [PubMed] [Google Scholar]
- 2. Guo Z., Dai B., Jiang T., Xu K., Xie Y., Kim O., Nesheiwat I., Kong X., Melamed J., Handratta V. D., Njar V. C., Brodie A. M., Yu L. R., Veenstra T. D., Chen H., and Qiu Y. (2006) Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10, 309–319 [DOI] [PubMed] [Google Scholar]
- 3. Yeatman T. J. (2004) A renaissance for SRC. Nat. Rev. Cancer 4, 470–480 [DOI] [PubMed] [Google Scholar]
- 4. Cai H., Babic I., Wei X., Huang J., and Witte O. N. (2011) Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer Res. 71, 862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Y., Sawyers C. L., and Scher H. I. (2008) Targeting the androgen receptor pathway in prostate cancer. Curr. Opin. Pharmacol. 8, 440–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grevengoed T. J., Klett E. L., and Coleman R. A. (2014) Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 34, 1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Currie E., Schulze A., Zechner R., Walther T. C., and Farese R. V. Jr. (2013) Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Menendez J. A., and Lupu R. (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 [DOI] [PubMed] [Google Scholar]
- 9. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 10. Epstein M. M., Kasperzyk J. L., Mucci L. A., Giovannucci E., Price A., Wolk A., Håkansson N., Fall K., Andersson S. O., and Andrén O. (2012) Dietary fatty acid intake and prostate cancer survival in Orebro County, Sweden. Am. J. Epidemiol. 176, 240–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crowe F. L., Allen N. E., Appleby P. N., Overvad K., Aardestrup I. V., Johnsen N. F., Tjønneland A., Linseisen J., Kaaks R., Boeing H., Kröger J., Trichopoulou A., Zavitsanou A., Trichopoulos D., Sacerdote C., et al. (2008) Fatty acid composition of plasma phospholipids and risk of prostate cancer in a case-control analysis nested within the European Prospective Investigation into Cancer and Nutrition. Am. J. Clin. Nutr. 88, 1353–1363 [DOI] [PubMed] [Google Scholar]
- 12. Le T. T., Huff T. B., and Cheng J. X. (2009) Coherent anti-Stokes Raman scattering imaging of lipids in cancer metastasis. BMC Cancer 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Resh M. D. (1994) Myristylation and palmitylation of Src family members: the fats of the matter. Cell 76, 411–413 [DOI] [PubMed] [Google Scholar]
- 14. Patwardhan P., and Resh M. D. (2010) Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell. Biol. 30, 4094–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai H., Smith D. A., Memarzadeh S., Lowell C. A., Cooper J. A., and Witte O. N. (2011) Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 6579–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Higuchi T., Mizuno A., Narita K., Ichimaru T., and Murata T. (2012) Leptin resistance does not induce hyperphagia in the rat. J. Physiol. Sci. 62, 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bullen J. W. Jr, Ziotopoulou M., Ungsunan L., Misra J., Alevizos I., Kokkotou E., Maratos-Flier E., Stephanopoulos G., and Mantzoros C. S. (2004) Short-term resistance to diet-induced obesity in A/J mice is not associated with regulation of hypothalamic neuropeptides. Am. J. Physiol. Endocrinol. Metab. 287, E662–E670 [DOI] [PubMed] [Google Scholar]
- 18. Yang X., Ma Y., Li N., Cai H., and Bartlett M. G. (2017) Development of a method for the determination of Acyl-CoA compounds by liquid chromatography mass spectrometry to probe the metabolism of fatty acids. Anal. Chem. 89, 813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tholstrup T., Sandström B., Bysted A., and Hølmer G. (2001) Effect of 6 dietary fatty acids on the postprandial lipid profile, plasma fatty acids, lipoprotein lipase, and cholesterol ester transfer activities in healthy young men. Am. J. Clin. Nutr. 73, 198–208 [DOI] [PubMed] [Google Scholar]
- 20. Ducker C. E., Upson J. J., French K. J., and Smith C. D. (2005) Two N-myristoyltransferase isozymes play unique roles in protein myristoylation, proliferation, and apoptosis. Mol. Cancer Res. 3, 463–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holzer R. G., Park E. J., Li N., Tran H., Chen M., Choi C., Solinas G., and Karin M. (2011) Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147, 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mollinedo F., and Gajate C. (2015) Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 57, 130–146 [DOI] [PubMed] [Google Scholar]
- 23. Bagrodia S., Taylor S. J., and Shalloway D. (1993) Myristylation is required for Tyr-527 dephosphorylation and activation of pp60c-Src in mitosis. Mol. Cell Biol. 13, 1464–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cowan-Jacob S. W., Fendrich G., Manley P. W., Jahnke W., Fabbro D., Liebetanz J., and Meyer T. (2005) The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 13, 861–871 [DOI] [PubMed] [Google Scholar]
- 25. Farazi T. A., Waksman G., and Gordon J. I. (2001) The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 276, 39501–39504 [DOI] [PubMed] [Google Scholar]
- 26. Shrivastav A., Varma S., Lawman Z., Yang S. H., Ritchie S. A., Bonham K., Singh S. M., Saxena A., and Sharma R. K. (2008) Requirement of N-myristoyltransferase 1 in the development of monocytic lineage. J. Immunol. 180, 1019–1028 [DOI] [PubMed] [Google Scholar]
- 27. Felsted R. L., Glover C. J., and Hartman K. (1995) Protein N-myristoylation as a chemotherapeutic target for cancer. J. Natl. Cancer Inst. 87, 1571–1573 [DOI] [PubMed] [Google Scholar]
- 28. French K. J., Zhuang Y., Schrecengost R. S., Copper J. E., Xia Z., and Smith C. D. (2004) Cyclohexyl-octahydro-pyrrolo[1,2-a]pyrazine-based inhibitors of human N-myristoyltransferase-1. J. Pharmacol. Exp. Ther. 309, 340–347 [DOI] [PubMed] [Google Scholar]
- 29. Thinon E., Serwa R. A., Broncel M., Brannigan J. A., Brassat U., Wright M. H., Heal W. P., Wilkinson A. J., Mann D. J., and Tate E. W. (2014) Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat. Commun. 5, 4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aleshin A., and Finn R. S. (2010) SRC: a century of science brought to the clinic. Neoplasia 12, 599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Araujo J. C., Trudel G. C., Saad F., Armstrong A. J., Yu E. Y., Bellmunt J., Wilding G., McCaffrey J., Serrano S. V., Matveev V. B., Efstathiou E., Oudard S., Morris M. J., Sizer B., Goebell P. J., et al. (2013) Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol. 14, 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strom S. S., Yamamura Y., Forman M. R., Pettaway C. A., Barrera S. L., and DiGiovanni J. (2008) Saturated fat intake predicts biochemical failure after prostatectomy. Int. J. Cancer 122, 2581–2585 [DOI] [PubMed] [Google Scholar]
- 33. Whittemore A. S., Kolonel L. N., Wu A. H., John E. M., Gallagher R. P., Howe G. R., Burch J. D., Hankin J., Dreon D. M., and West D. W. (1995) Prostate cancer in relation to diet, physical activity, and body size in blacks, whites, and Asians in the United States and Canada. J. Natl. Cancer Inst. 87, 652–661 [DOI] [PubMed] [Google Scholar]
- 34. Crowe F. L., Appleby P. N., Travis R. C., Barnett M., Brasky T. M., Bueno-de-Mesquita H. B., Chajes V., Chavarro J. E., Chirlaque M. D., English D. R., Gibson R. A., Giles G. G., Goodman G. E., Henning S. M., Kaaks R., et al. (2014) Circulating fatty acids and prostate cancer risk: individual participant meta-analysis of prospective studies. J. Natl. Cancer Inst. 106, dju240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu S. T., Pham H., Pandol S. J., and Ptasznik A. (2013) Src as the link between inflammation and cancer. Front. Physiol. 4, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kovács M., Németh T., Jakus Z., Sitaru C., Simon E., Futosi K., Botz B., Helyes Z., Lowell C. A., and Mócsai A. (2014) The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J. Exp. Med. 211, 1993–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xin L., Ide H., Kim Y., Dubey P., and Witte O. N. (2003) In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc. Natl. Acad. Sci. U.S.A. 100, 11896–11903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adam R. M., Yang W., Di Vizio D., Mukhopadhyay N. K., and Steen H. (2008) Rapid preparation of nuclei-depleted detergent-resistant membrane fractions suitable for proteomics analysis. BMC Cell Biol. 9, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.