

Abstract
The ability to detect and respond to oxidative stress is crucial to the survival of living organisms. In cells, sensing of increased levels of reactive oxygen species (ROS) activates many defensive mechanisms that limit or repair damage to cell components. The ROS-signaling responses necessary for cell survival under oxidative stress conditions remain incompletely understood, especially for the translational machinery. Here, we found that drug treatments or a genetic deficiency in the thioredoxin system that increase levels of endogenous hydrogen peroxide in the yeast Saccharomyces cerevisiae promote site-specific endonucleolytic cleavage in 25S ribosomal RNA (rRNA) adjacent to the c loop of the expansion segment 7 (ES7), a putative regulatory region located on the surface of the 60S ribosomal subunit. Our data also show that ES7c is cleaved at early stages of the gene expression program that enables cells to successfully counteract oxidative stress and is not a prerequisite or consequence of apoptosis. Moreover, the 60S subunits containing ES7c-cleaved rRNA cofractionate with intact subunits in sucrose gradients and repopulate polysomes after a short starvation-induced translational block, indicating their active role in translation. These results demonstrate that ES7c cleavage in rRNA is an early and sensitive marker of increased ROS levels in yeast cells and suggest that changes in ribosomes may be involved in the adaptive response to oxidative stress.
Keywords: oxidation-reduction (redox), oxidative stress, reactive oxygen species (ROS), redox signaling, ribosomal ribonucleic acid (rRNA) (ribosomal RNA), ribosome, RNA degradation, RNA structure, Saccharomyces cerevisiae, stress response
Introduction
Cells of the budding yeast Saccharomyces cerevisiae regularly encounter stressful conditions in their environment, including fluctuations in temperature, pH, nutrient availability, and exposure to toxic compounds. It is not surprising that these unicellular eukaryotes have evolved a wide range of sophisticated survival mechanisms to combat adverse effects of stress and adapt to new conditions. One of the best-studied environmental stressors is the exposure to elevated levels of reactive oxygen species (ROS),2 a condition also known as oxidative stress. In addition to extracellular environment, ROS can be also generated from intracellular sources, such as mitochondria and the ER (reviewed in Ref. 1). ROS are highly reactive chemical products that include superoxide anions (O2˙̄), H2O2, and the hydroxyl radicals (OH•), formed upon incomplete reduction of oxygen. ROS-induced damage to cellular components including DNA, lipids, and proteins contributes to a variety of pathologies and aging (2, 3). To fight deleterious effects of ROS, cells possess an arsenal of enzymatic and non-enzymatic defense systems (4, 5). Enzymatic components for scavenging ROS and maintaining the proper redox state include thioredoxin-dependent peroxiredoxins, superoxide dismutases, glutathione peroxidases, and catalases. These enzymes often have specialized functions within the cell that differ by the target substrate on which they act, cellular compartment where they function, mode of expression, and mechanism of catalysis (4).
Aside from causing cell damage, ROS also play roles beneficial for the organism. For example, at low concentration, ROS can provide protection from invading pathogens, participate in tissue repair, and control gene expression (discussed in Ref. 6). Moreover, the accumulated body of evidence indicates that ROS, especially H2O2, can serve as intracellular messengers to regulate various physiological processes (7). At low doses, hydrogen peroxide specifically reacts with sensor molecules in the cell in a process termed redox signaling (8). In its role of a signal transducer, H2O2 regulates a range of cellular responses that help cells to adapt to the continuously changing environment (9).
Although cellular H2O2-sensing pathways are still not completely understood, one well-characterized mechanism in both prokaryotes and eukaryotes relies on transcription factors that regulate expression of antioxidant genes (9). For example, H2O2-mediated oxidation promotes formation of an intramolecular disulfide bond between two Cys residues of the yeast transcription factor Yap1 and its cofactor Gpx3 (10, 11). This interaction results in oxidized Yap1 that retains nuclear localization (12) and up-regulates transcription of a large number of stress-responsive genes (reviewed in Ref. 13). Among other proteins identified as H2O2-signal sensors are components of the antioxidant machinery (thioredoxins, peroxiredoxins), glycolytic enzymes, structural proteins (actin, myosin), and protein folding and degradation factors (heat shock proteins and components of the proteasome).
Translation, the process of protein synthesis that occurs in every living cell, is affected by elevated levels of ROS on many levels. Multiple studies have described oxidation of protein factors and enzymes involved in the initiation, elongation, and termination of translation (14–17), as well as oxidation-induced misacylation of aminoacyl-tRNA synthetases (18–20). These modifications may result in the inhibition of protein synthesis or a decrease of translational fidelity. Other examples include oxidation of mRNAs resulting in ribosome stalling and production of misfolded proteins prompted to aggregation (21, 22); alterations of post-transcriptional tRNA modifications leading to selective translation of codon-biased mRNAs for stress response proteins (23, 24); and fragmentation of mature tRNAs generating tRNA fragments (reviewed in Ref. 25). It is thought that tRNA fragments can reprogram translation (26–29), thereby fine-tuning protein synthesis during stress conditions. In contrast to tRNAs, the question of how ROS may affect the abundant ribosomal RNAs (rRNAs) has received limited experimental attention. Ribosomal RNAs (25S, 18S, 5.8S, and 5S rRNAs in yeast) constitute the structural and functional core of the ribosome (30) and are essential for ribosome function in translation. Previous studies found that high-level oxidative stress capable of inducing cell apoptosis results in extensive fragmentation and subsequent degradation of 25S and 5.8S rRNAs (31). However, little is known about how these complex macromolecules are affected by disruptions in redox homeostasis that are not accompanied by cell lethality.
In this study, we demonstrate that treatment of yeast cells with sublethal doses of oxidants causes cleavage in expansion segment 7 (ES7) of 25S rRNA. ES7 is an rRNA extension that is universally present in eukaryotic ribosomes and predicted to possess a regulatory function (32, 33), although its precise role remains unknown. Remarkably, our data indicate that ES7 undergoes hydrolysis following even slight perturbations in redox balance. Moreover, ES7c cleavage is not a part of cell death but coincides with early events in the gene expression program that leads to the successful adaptation to oxidative stress. This is the first study to describe a direct involvement of an rRNA molecule in the oxidative stress response.
Results
DTT treatment induces endonucleolytic cleavage within the ES7c region of 25S rRNA
The initial question we were addressing in our studies was how cells deal with ribosomes stalled in translation because of aberrations/misfolding of nascent peptide chains. We subjected cells to various conditions known to cause nascent chain misfolding, including high temperature (38 and 40 °C), treatments with ethanol, aminoglycosides (hygromycin B and G418), and proteotoxic derivatives of proline (l-azetidine-2-carboxylic acid) and arginine (canavanine) (34). Notably, none of these conditions affected the integrity of rRNAs, as assessed by hybridization analysis of RNA extracted from the stressed cells (Fig. 1A). Surprisingly, treatment of cells with DTT, an agent known to reduce cysteine thiols and cause protein misfolding (34), resulted in the appearance of a distinct rRNA-derived fragment (Fig. 1A) detected with the rRNA-specific probe y540 complementary to the 5′ end of 25S rRNA (Fig. 1B). This effect of DTT was dose-dependent and affected 25S rRNA but not 18S rRNA (Fig. 1A), suggesting that it is specific to the large ribosomal subunit.
Figure 1.

25S rRNA undergoes endonucleolytic cleavage in the ES7c region upon treatment with DTT. A, wild-type BY4741 cells were treated for 4 h with different stressors in synthetic medium (canavanine (Can) and l-azetidine-2-carboxylic acid (AZC) treatments, first three lanes) or YPDA medium (treatments with DTT, hygromycin B (HygB), geneticin (G418), ethanol (EtOH), and elevated temperatures). − indicates that no drugs or elevated temperature were applied to the culture. RNA was extracted and analyzed by Northern hybridizations with 32P-labeled 25S rRNA probe y540 and 18S rRNA probe y500. For the hybridization with probe y540, two exposures are shown. The asterisk denotes a fragment derived from 25S rRNA. The positions of full-length rRNAs are indicated on the right. B, schematic representation of the hybridization sites for 25S rRNA oligonucleotide probes y503 and y540 relative to the position for the observed endonucleolytic cleavage. C, mid-log tsa1Δ culture and its isogenic wild-type control (BY4741) were adjusted to the same cell density; 2 × 106 cells were serially diluted (1:5), plated onto YPDA agar plates with or without 8 mm DTT, and incubated at 30 °C for 3 days. D, overnight cultures of wild-type and tsa1Δ cells were diluted with YPDA to an A600 of ∼0.3 and grown for 2 h. DTT was added to the final concentration of 20 mm. Total RNA was extracted at the indicated time points and analyzed by hybridization with 25S and 18S rRNA probes. E, cells were processed as described in D, except that DTT was added (+) or not (−) for 2 h. RNA was analyzed with two 25S rRNA probes, y540 and y503. Methylene blue (MB) staining of one of the membranes is shown, and the positions of full-length 25S rRNA and major cleavage fragments are indicated. F, RNA from E was used for primer extensions to map the ES7c cleavage site. Primer extension reactions were separated on a 6% polyacrylamide/urea gel next to a sequencing reaction. G, the secondary structure of the ES7 region, adapted from the RiboVision website (apollo.chemistry.gatech.edu/RiboVision/; please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site).
To investigate the rRNA-destabilizing effect of DTT further, we performed a time course analysis using a strain lacking TSA1, a gene that provides tolerance to DTT exposure (35). The tsa1Δ mutant, previously identified in a genetic screen for increased DTT sensitivity, was confirmed to be highly sensitive to this drug (Fig. 1C) and showed increased generation of the 25S rRNA fragment over time (Fig. 1D). Next, we used experimental conditions upon which 25S rRNA was excessively cleaved in tsa1Δ, but not in wild-type cells (2 h of treatment with 20 mm DTT; Fig. 1D) to map the cleavage site in 25S rRNA. First, we hybridized RNA prepared from DTT-treated and untreated wild-type and tsa1Δ cells with radiolabeled 25S rRNA probes y540 and y503 (Fig. 1B). The 5′ rRNA probe y540 detected a 5′ shorter fragment (∼600 bases) of 25S rRNA (Fig. 1, D and E), whereas probe y503, which anneals ∼1500 nucleotides downstream of y540 (Fig. 1B), detected a larger 3′ fragment (∼2.8 kb) in DTT-treated tsa1Δ cells (Fig. 1E). This result indicates that in response to DTT treatment, 25S rRNA undergoes endonucleolytic cleavage in the region located between the y540 and y503 hybridization sites (Fig. 1B). Using primer extensions on 25S rRNA from wild-type and tsa1Δ cells, we detected novel reverse transcriptase stops appearing after 20 mm DTT treatment at positions corresponding to cleavage after nucleotides 610 and 611 (Fig. 1F). This site is located within ES7c (Fig. 1G), one of the largest rRNA extensions present in the eukaryotic ribosome (36, 37). Hereafter, we will refer to the identified cleavage of 25S rRNA within the ES7 region as “ES7c cleavage.”
Although the precise role of ES7 in ribosome function is unknown, structural studies have revealed its high accessibility on the 60S subunit surface and predicted its involvement in recruitment of various protein factors (33). In support of the structural analysis data, recent in vitro pulldown assays identified 36 proteins that interact with the ES7 region, including aminoacyl-tRNA synthetases, protein quality control factors, and chaperones (32), supporting the idea that ES7 functions as a regulatory part in the ribosome.
ES7c cleavage is not part of the unfolded protein response
Stability of rRNA in cells subjected to multiple proteotoxic stress conditions (Fig. 1A) indicated that ES7c cleavage was not triggered by nascent chains misfolding and that DTT induces 25S rRNA cleavage by a different mechanism. Given that DTT causes ER stress and activates the unfolded protein response (UPR) (38), we asked whether cleavage of 25S rRNA was a consequence of an activated UPR. To address this possibility, we examined the effects of deleting the ER-stress transmitter gene IRE1, critical for mounting the UPR (39), using tsa1Δ cells, which showed efficient ES7c cleavage formation in response to DTT (Fig. 1, D and E). To confirm that the UPR was suppressed in the generated ire1Δ strains, we monitored the formation of the spliced form of the HAC1 gene, HAC1i, which gives rise to the transcriptionally competent Hac1 factor necessary for transcription of UPR-responsive genes (40). As expected, HAC1i formation was completely blocked in ire1Δ and tsa1Δire1Δ strains after ER stress induced by treatment with DTT or another classic ER stress inducer, tunicamycin (Tm; Fig. 2, bottom panel). An ES7c cleavage fragment formation induced by DTT still occurred in the tsa1Δire1Δ cells, but it was not observed after Tm treatment in either of the strains (Fig. 2, top panel). Taken together, these data indicate that cleavage of 25S rRNA in the ES7c region is not part of the UPR.
Figure 2.
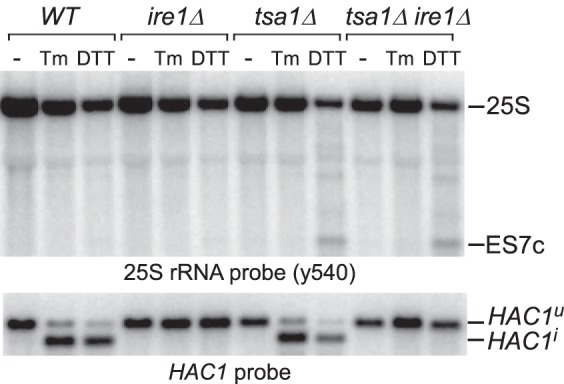
ES7c cleavage is not part of the UPR. Overnight cultures from the indicated strains were diluted with YPDA to an A600 of ∼0.3, grown for 2 h, and treated for 2 h with 20 mm DTT or 1 μg/ml Tm or left untreated (−). RNA was extracted, resolved on a gel in duplicate, and hybridized with the 25S rRNA probe y540 (top panel) and a probe that recognizes spliced (HAC1i) and unspliced (HAC1u) forms of HAC1 (bottom panel).
DTT treatment induces ES7c cleavage by increasing H2O2 levels
Having established that ES7c cleavage does not occur because of DTT acting as an activator of proteotoxic stress or the UPR, we next sought to investigate whether it may be related to the property of DTT to generate hydrogen peroxide as a by-product of its oxidation (41–46). The ability of DTT and other thiols to act as a source of ROS in cells depends on multiple environmental and cellular factors including thiol concentration and the availability of metal ions, chelators, and activity of radical scavengers in the cell (43). This side of DTT was investigated primarily in mammalian cell culture studies (42, 43, 46), and the extent of this potential effect of DTT in yeast cells was unclear.
To address the hypothesis that ROS production mediates the effects of DTT treatment on RNA integrity, we first measured H2O2 levels in yeast cultures treated with DTT using Amplex Red hydrogen peroxide assays. Because H2O2 readily diffuses through the plasma membrane and exists in the intra/extracellular equilibrium, Amplex Red measurements of extracellular H2O2 reflect its presence inside the cell (43, 47). Indeed, we found that treatment of wild-type cells with DTT resulted in a significant increase of H2O2 over time when compared with control untreated cells (Fig. 3A).
Figure 3.

ES7c cleavage occurs in response to H2O2. A, mid-log wild-type cells grown in YPDA were treated with 20 mm DTT for 2 or 4 h or left untreated (n/t). The relative amounts of H2O2 present in treated and untreated cells were determined using an Amplex Red assay: an equal number of cells (1 × 106) from each culture was collected, washed, and incubated in 500 μl of the assay buffer for 30 min at 30 °C to allow H2O2 release; 100 μl of the supernatant was used per reaction. The data show mean relative fluorescence units (RFU) in triplicate samples; the error bars represent S.D. The differences between the treated and untreated samples were significant. *, p < 0.01, unpaired two-tail t test. B, overnight tsa1Δ cultures were diluted with YPDA to an A600 of ∼0.2 and grown for 3 h. One half of the culture was pretreated with 10 μm ebselen for 1 h, whereas the other was left untreated (−). 20 mm DTT was added to both cultures, and the cells were harvested at the indicated time points. RNA extracted from cells was analyzed by Northern hybridizations with the indicated rRNA probes. The hybridization signal corresponding to the ES7c-cleaved rRNA fragment was converted to phosphorimaging units and normalized to the 18S rRNA signal in the same lane. The data on the graph show the mean values from three independent experiments; the error bars represent S.D. C, wild-type cells were diluted with YPDA to an A600 of ∼0.2, grown for 4 h, and treated for 2 h with 20 mm DTT, 25 μm menadione, 6 μm plumbagin, or left untreated (−). Treatment with 0.2 mm inorganic H2O2 was for 30 min. RNA was analyzed by Northern hybridization as in Fig. 1A.
To confirm a functional connection between DTT-induced H2O2 production and DTT-induced ES7c cleavage, we used a dual approach. First, we examined ES7c cleavage in response to DTT in cells pretreated with the hydrogen peroxide scavenger ebselen (48). Because of ebselen sensitivity to light, this assay was conducted in a tsa1Δ strain, which demonstrates fast kinetics of ES7c cleavage in response to DTT (Fig. 1D). Hybridization analysis of rRNA revealed that the presence of ebselen in media before and during incubation of tsa1Δ with DTT significantly decreased ES7c-fragment formation (Fig. 3B). This is consistent with the idea that DTT causes ES7c cleavage by promoting generation of H2O2. As a second approach, we tested rRNAs extracted from wild-type cells treated with various drugs known to increase cellular H2O2 levels. Similar to DTT, treatments with redox-cycling agents (menadione and plumbagin) or inorganic hydrogen peroxide promoted formation of ES7c-cleaved fragments (Fig. 3C). The combination of these data strongly supported the possibility that production of hydrogen peroxide mediates 25S rRNA cleavage in the ES7c region in response to DTT.
ES7c cleavage represents an early response to low-level oxidative stress and is not a consequence of yeast apoptosis
Menadione and hydrogen peroxide are two oxidants often used as extracellular stressors of yeast cells because of their good stability in medium and high cell wall/membrane permeability. Interestingly, these drugs can trigger different cellular responses depending on the drug concentration and duration of treatment (49). Previous studies have shown that rRNA fragmentation can occur in response to activation of an apoptotic program in yeast cells (31). Although much harsher conditions were used in the previous studies to induce apoptosis (e.g. 600 μm menadione for 200 min) (31), we asked whether treatment of cells with the lower oxidant doses that induced ES7c cleavage (Fig. 3C) might also be inducing the yeast death program.
First, we tested whether deletion of genes involved in yeast apoptosis might prevent or decrease the formation of the ES7c cleavage. To this end, we analyzed strains deficient for the mitochondrial cell death effector gene AIF1 (50) and metacaspase gene YCA1 (51). As in the wild-type control, treatment of aif1Δ and yca1Δ cells with low concentrations of menadione and hydrogen peroxide led to clear ES7c fragment formation (Fig. 4A).
Figure 4.

ES7c cleavage is unrelated to apoptosis. A, overnight cultures from the indicated yeast strains were diluted with YPDA to an A600 of ∼0.2, grown for 4 h, and treated with 25 or 50 μm menadione for 2 h, 0.2 mm H2O2 for 30 min or left untreated (−). RNA was extracted and analyzed by Northern hybridization with the indicated 32P-labeled probes. B, wild-type yeast cells were processed as described in A. The cells were converted to spheroplasts, stained with annexin V–FITC, and analyzed by flow cytometry. The percentage of annexin-positive cells was determined using FlowJo software and is indicated on the right side of each panel.
Next, we tested the possibility that ES7c cleavage of 25S rRNA might be associated with an early apoptotic response occurring upstream of Yca1 and Aif1. Annexin V binding to phosphatidylserine exposed on the surface of apoptotic cells is an established marker for early apoptotic cells (52). We assessed the percentage of annexin V-positive cells in yeast cultures that were subjected to various drug treatments (Fig. 4B). We observed no increase in the amount of annexin V-positive cells upon treatment with low concentrations of menadione (25 and 50 μm) or 0.2 mm H2O2 (Fig. 4B). In contrast, incubation with apoptosis-inducing 2 mm H2O2 (49) caused a 3-fold increase in the annexin V-positive cells population (Fig. 4B).
Our initial data showed that 25S rRNA undergoes ES7c cleavage upon mild oxidant treatment (Fig. 3C). Time course analysis demonstrated formation of detectable levels of the ES7c-cleaved rRNA after only 15 min of incubation with 50 μm menadione (Fig. 5A). It was therefore possible that the endonucleolytic cleavage within the ES7c region might occur at the very beginning of the apoptotic program, prior to other detectable apoptotic events, or it could reflect cell death by a non-apoptotic mechanism. To address these possibilities, we directly assessed cell viability after treatments with different doses of menadione by performing clonogenic assays. The number of viable cells that formed colonies after treatment with 25 and 50 μm menadione was not reduced when compared with untreated cells (Fig. 5B), indicating that ES7c fragment formation under these conditions (Fig. 5C, second and third lanes) was not a marker for impending cell death. By comparison, 100 μm menadione resulted in a significant decline in the number of colonies, with only ∼37% cells surviving (Fig. 5B). Consistent with this result, exposing cells to 100 μm menadione strongly affected culture growth, whereas only modest growth delay was observed at 50 μm, and there was virtually no detectable effect at 25 μm (Fig. 5D). Taken together, results presented on Fig. 5 (B–D) illustrate concentration-dependent effects of oxidants on cell growth and viability. For brevity, we will refer to the oxidant concentrations that do not reduce cell viability as “low dose” as opposed to concentrations that inhibit growth and induce cell death.
Figure 5.

ES7c cleavage occurs rapidly in response to oxidant doses that do not reduce cell viability. A, overnight culture of wild-type cells was diluted with YPDA to A600∼0.2 and grown for 4 h, and 50 μm menadione was added. RNA was extracted at the indicated time points and analyzed by Northern hybridization with rRNA probes. ES7c-cleaved fragment was quantified as described in Fig. 3B. The data show mean values of three independent experiments; the error bars represent S.D. B, cells grown as in A were treated with 25, 50, or 100 μm menadione for 2 h. To compare total cell numbers in each culture, the cells were counted using a hemocytometer (8 counts/culture; left panel). For the clonogenic assay (right panel), equal volumes of each culture were plated onto YPDA agar plates (5 replicates/culture). The plates were incubated at 30 °C for 3 days, and colonies formed on each plate were counted. The 100 μm menadione treatment was the only statistically significant condition (p < 0.0001, unpaired, two-tail t test) compared with the no-treatment (N/T) control, other treatments had no significant differences in the number of colony-forming units (cfu) relative to the no-treatment culture. C, rRNA cleavage in cells treated with menadione as in B. The ratio of the hybridization signals of the full-length 25S rRNA and the ES7c-cleaved fragment is indicated for each lane. The graph shows relative levels of ES7c-cleaved fragment normalized to 18S rRNA quantified in three biological replicates as described for Fig. 3B; the error bars represent S.D. D, growth curves of wild-type BY4741 cells in YPDA containing different menadione concentrations. Triplicate cultures in a 96-well plate were incubated at 30 °C with shaking. A600 measurements were recorded using a microplate reader; the data show the mean value at each time point. E, polysome traces for the wild-type strain treated with the indicated concentrations of menadione for 2 h. F, Northern analysis of rRNA from wild-type cells treated for 2 h with various oxidants at the indicated concentrations.
Sucrose gradient sedimentation analysis (Fig. 5E) revealed that the inhibitory effects of 100 μm menadione on culture growth were accompanied by a severe inhibition of translation, as indicated by increased 40S, 60S, and monosome 80S fractions and a dramatic decrease in polysomes (Fig. 5E, bottom right). In contrast, treatment of cells with 25 μm menadione had no detectable effect on the polysome profile when compared with untreated cells (Fig. 5E, upper panels), whereas 50 μm menadione treatment resulted in a moderate translation inhibition (Fig. 5E, bottom left). The dose-dependent effect of menadione on translation correlated well with the effects of this drug on culture growth and viability (Fig. 5, B and D).
To better understand how ES7c cleavage is related to the extensive fragmentation of 25S rRNA observed during apoptosis (31), we assessed effects of different concentrations of menadione (25, 50, and 100 μm) on 25S rRNA. Hybridization of rRNA showed that the formation of an ES7c fragment was dose-dependent and that treatment with a higher dose (100 μm) of menadione led to generation of additional rRNA degradation products and increased smear in hybridizations with an rRNA-specific probe (Fig. 5, C and F). The detrimental effect of 100 μm menadione on cell viability (Fig. 5, B and D) is consistent with the activation of an apopotic program, accompanied by extensive RNA breakdown in the cell (31). Similar to menadione's effect on 25S rRNA, exposure of growing wild-type cells to low concentrations of inorganic H2O2 or plumbagin also resulted in 25S rRNA cleavage primarily in the ES7c region (Fig. 3C), whereas prolonged treatment with high doses of these oxidants caused extensive fragmentation of 25S rRNA (Fig. 5F). Thus, our results revealed that the duration and strength of oxidative stress dictate the intensity of its effects on ribosomes. Based on these data, we conclude that endonucleolytic cleavage of ES7c can be initiated by oxidants at levels that are below the threshold needed for triggering cell death.
ES7c cleavage coincides with activation of early responders to redox imbalance
Our time course analysis (Fig. 5A), oxidant titration assays (Figs. 3C and 5, C and F), sucrose gradient sedimentation analysis (Fig. 5E), and cell viability data (Fig. 5, B and D) suggest that the ES7c cleavage is an early effect of ROS on ribosomes that occurs when cells have not lost viability or the capacity to combat ROS. Multiple studies have shown that at low levels, H2O2 can function as a signaling molecule that promotes the oxidative stress response, a series of defensive mechanisms that serve to restrict the buildup of intracellular reactive oxygen species (discussed in Refs. 7, 9, and 53). As part of this response, various ROS-detoxifying enzymes are employed including dismutases, catalases, peroxidases, and enzymes of the thioredoxin system (4). The oxidative stress response is a dynamic process that develops through multiple stages, with different genes undergoing up- and down-regulation (54). Gene expression studies also revealed differential gene expression patterns dependent on the type, strength, and duration of the toxic stimuli (16, 54–56), indicating that this response can be tailored to a particular stress-inducing situation.
To understand at which stage of the oxidative stress response the ES7c cleavage might be initiated, we monitored the expression patterns of transcriptionally regulated early (TRX2 and TRR1) and late (CTT1 and CTA1) genes in cells following treatment with 0.2 mm H2O2 for 30 min. For comparison, we used a longer, 2-h treatment with 2 mm H2O2 that results in severe oxidative stress and apoptosis (Fig. 4B) (31). Consistent with data from gene array studies (54, 55), our qRT-PCR analysis showed a significant increase in the amount of all the tested mRNAs at the high dose of H2O2 (Fig. 6A, dark bars), whereas the short treatment with the low concentration predominantly induced expression of early “redox-control” genes TRX2 and TRR1 (54) but not catalases CTT1 or CTA1 (Fig. 6A, light bars). To obtain a more detailed picture of the dynamics of differential gene expression, we examined transcript levels of TRX2, CTT1, and TSA1 (a downstream target of Trx2) over time in cultures treated with 0.2 mm H2O2. The initial transcriptional activation of TRX2 was observed at 30 min of the H2O2 treatment, significantly declined within the next 30 min, and remained only slightly elevated in the following 7 h of the time course (Fig. 6B, top panel). Expression of TSA1 was slightly delayed compared with TRX2, peaking at 1 h (Fig. 6B, middle panel), whereas CTT1 peaked significantly later during the time course, with maximum up-regulation observed at 4–8 h (Fig. 6B, bottom panel). In agreement with previously published studies (54), induction of all these mRNAs by H2O2 was abolished (Fig. 6B) in yap1Δ cells lacking the transcriptional activator Yap1, a key element of oxidative stress response (discussed in Ref. 57).
Figure 6.

ES7c cleavage coincides with activation of the thioredoxin system during low-dose oxidative stress. A, wild-type overnight culture was diluted with YPDA to an A600 of ∼0.2, grown for 4 h and treated with 0.2 mm H2O2 for 30 min or 2 mm H2O2 for 2 h. RNA was extracted from the cells, and the levels of TRX2, TRR1, CTT1, and CTA1 mRNAs were analyzed by qRT-PCR. Transcript levels relative to the ACT1 mRNA are shown as mean values normalized to no-treatment conditions (dashed horizontal line, y = 1) in triplicate samples; the error bars show S.D. B, overnight cultures of wild-type and yap1Δ strains were analyzed as in A after treatment with 0.2 mm H2O2 for the indicated times. C, Northern hybridizations of the rRNA from cultures shown in B. D, ES7c-cleaved fragments from samples treated as in B and C were quantified as described in Fig. 3B. The data show mean values of three independent experiments; the error bars represent S.D.
To relate the expression of the oxidative stress-responsive genes to ES7c cleavage formation, we examined RNA from the same cells that were used for the RT-qPCR analysis (Fig. 6B) by Northern hybridizations with a 25S rRNA-specific probe. Consistent with the results shown above (Fig. 3C), an ES7c cleavage-derived rRNA fragment was readily evident at 30 min of 0.2 mm H2O2 treatment of wild-type cells (Fig. 6, C and D, second lane), at the time when the thioredoxin TRX2 mRNA is maximally induced (Fig. 6B). Remarkably, ES7c fragment levels decreased upon longer treatment with the low H2O2 concentration in wild-type cells (Fig. 6, C and D, left panels), but not in yap1Δ cells (Fig. 6, C and D, right panels), suggesting that the cells exposed to low doses of H2O2 undergo adaptation within the first hour, and this requires Yap1-dependent gene expression.
We next examined rRNA in strains deleted for thioredoxin genes to evaluate the functional connection between the thioredoxin system and ES7c cleavage. We reasoned that diminishing the protective ability of these major redox regulators should accelerate redox imbalance, and this should increase ES7c cleavage. In agreement with this prediction, deletion of TRX1, TRX2, or their transcriptional regulator YAP1 resulted in elevated amounts of the ES7c fragment upon treatment with 0.2 mm H2O2 (Fig. 7A) or 25 μm menadione (Fig. 7B). Thus, ES7c cleavage caused by a low concentration of H2O2 present in the medium coincides with the activation of the thioredoxin system, which is considered one of the early responders to redox imbalance in the cell (54).
Figure 7.

ES7c cleavage of rRNA in oxidative stress response-deficient cells can occur with and without drug treatment. A and B, quantification of the ES7c-cleaved fragment levels in wild-type and the indicated mutant strains treated with 0.2 mm H2O2 for 30 min (A) or with 25 μm menadione for 2 h (B). Representative Northern blots are shown; graphs show mean values from three independent experiments; the error bars represent S.D. Quantification was done as in Fig. 3B. C, representative growth curves of wild-type and tsa1Δ cells. Overnight cultures were diluted with YPDA to an A600 of ∼0.2 and grown for 4 h; the cells were collected, washed, resuspended in synthetic complete (SC) medium to an A600 of ∼0.1 and grown at 30 °C with shaking in a 96-well plate (200 μl/well) for 20 h. A600 was recorded every 5 min. D, increased production of H2O2 in post-diauxic tsa1Δ cells. Equal numbers of cells (9 × 106) grown as described in C for 20 h were collected, washed, and incubated in 500 μl of the Amplex Red assay buffer for 30 min at 30 °C to allow H2O2 diffusion into the buffer, and 100 μl was assayed with Amplex Red as described in Fig. 3A. The data show the mean values of relative fluorescence units (RFU) in three biological replicates; the error bars represent S.D. E, ES7c cleavage occurs in tsa1Δ cells upon natural accumulation of ROS in postdiauxic cells. rRNA extracted from cells grown as in C and D was analyzed by Northern hybridizations with the indicated probes. The same image is presented as a heat map to better visualize the ES7c-cleavage product.
ES7c cleavage occurs is response to endogenously generated ROS
In the experiments shown above, ES7c cleavage was observed after chemically inducing redox imbalance in cells. However, ROS are also produced as part of normal metabolism, raising the question of whether this rRNA cleavage can occur under native growth conditions. Increased levels of ROS generated as by-products of aerobic metabolism (discussed in Ref. 58) require up-regulation of numerous antioxidant genes during the shift from fermentative growth to respiration, including genes of the thioredoxin-dependent ROS scavenging system (55, 59–61). With this in mind, we examined 25S rRNA in wild-type and tsa1Δ strains that were grown in synthetic complete medium for 20 h until they reached post-diauxic phase (Fig. 7C). As expected from the critical ROS scavenging function of Tsa1, significantly higher levels of H2O2 were present in post-diauxic tsa1Δ cells in comparison with wild-type cells, as measured by an Amplex Red assay (Fig. 7D). Northern hybridization analysis revealed accumulation of an ES7c cleavage product in post-diauxic tsa1Δ cells but not in the matching wild-type culture (Fig. 7E). These results indicate that cleavage of 25S rRNA in the ES7c region is part of a natural process that can be triggered by ROS generated endogenously.
ES7c-cleaved ribosomes are active during translation
To begin addressing the functional role of the ES7c cleavage, we performed polysome analysis in tsa1Δ cells using DTT treatment conditions that generate readily detectable 25S cleavage products (Fig. 1D). Consistent with previous reports (62), DTT treatment decreased the amount of polysomes and led to a pronounced ribosome accumulation in the 80S (monosome) fraction (Fig. 8A, left panel). Northern hybridization of RNA extracted from the gradient fractions showed ribosomes containing ES7c-cleaved 25S rRNA in polysome fractions (Fig. 8A, left panel), suggesting that this cleavage may occur during translation.
Figure 8.

ES7c cleaved ribosomes are translationally competent. A, sucrose gradient analysis of ribosomes extracted from tsa1Δ cells treated for 2 h with 20 mm DTT alone (left panel) or in combination with 50 μg/ml CHX (right panel). In the middle panel, lysate was incubated with EDTA for 10 min on ice prior to loading on the gradient. Cell lysates were centrifuged through a 15–45% sucrose gradient and fractionated with the continuous measurement of absorbance at 254 nm to visualize ribosomal peaks. RNA isolated from individual fractions was separated on a gel and visualized by SYBR gold staining. RNA was then transferred onto nylon membranes and analyzed by Northern hybridization. The asterisk indicates the 3′ fragment derived from 25S rRNA upon ES7c cleavage. B, ribosome redistribution after a starvation-induced translation arrest. An overnight culture of wild-type BY4741 cells was diluted with YPDA to an A600 of ∼0.3; the cells were grown for 2 h and shifted to dextrose-free medium for 15 min, and a portion of the culture was withdrawn for the gradient analysis (left panel). Menadione was added to the remaining culture to a final concentration of 50 μm for 2 h to induce ES7c cleavage (middle panel). The remaining cells were pelleted, washed and resuspended in fresh YPDA, grown for another 2 h, and harvested (right panel). The lysates were analyzed by sucrose gradients as in A.
Next, we treated cell lysates with EDTA prior to loading onto gradients to split ribosomes into 40S and 60S subunits. The ES7c-cleavage fragment and full-length 25S rRNA were found together in the same 60S subunit peak after the EDTA treatment (Fig. 8A, middle panel), indicating that ES7c-cleaved 60S particles retain their overall structure and resist breakdown even after stripping of Mg2+ ions. To further evaluate the possibility of cleavage in translating ribosomes, we included elongation inhibitor cycloheximide (CHX) during DTT treatment of tsa1Δ cells. CHX suppressed DTT-induced redistribution of ribosomes to the 80S monosome peak (Fig. 8A, right panel) and effectively prevented the ES7c-cleaved fragment from redistribution from polysome-containing fractions to the 80S fraction.
Taken together, the above results suggest that ES7c cleavage can occur in intact ribosomes engaged in translation. However, the presence of ES7c-cleaved 60S subunits in polysomal fractions does not prove by itself that cleaved subunits remain part of an actively translating pool. To test for translational competency, we first repressed translation in cells by a short incubation in dextrose-free media. As shown in previous studies (63), this reduces the amount of actively translated ribosomes and causes a shift from polysomes into the 80S fractions (Fig. 8B, left panel). We next treated the dextrose-depleted cells with menadione to induce ES7c cleavage (Fig. 8B, middle panel). When these cells were washed off the drug and resuspended in dextrose-containing medium, translation quickly recovered, as evidenced by redistribution of ribosomes from the 80S peak to polysomes (Fig. 8B, right panel). Remarkably, ES7c-cleaved 25S rRNA moved to polysome fractions like the intact 25S (Fig. 8B, right panel), indicating that 60S subunits cleaved in the ES7c region were capable of reengaging 40S subunits and forming translationally competent complexes.
ES7c cleavage is induced during the hormetic response to oxidative stress
The appearance of ES7c-cleaved 60S subunits among the actively translated polysomes could reflect functions beneficial for cells under oxidative stress conditions. The ability of cells to mount an adaptive response by sensing increasing concentrations of ROS, such as H2O2, allows cells to survive conditions of severe oxidative stress, a phenomenon known as oxidative hormesis (64). Several lines of evidence are consistent with the possibility that ES7c cleavage might be part of such an adaptive pathway: ES7c cleavage of 25S rRNA occurs rapidly in response to redox disbalance (Figs. 5A and 6C); it can be triggered by low, sublethal ROS levels, such as by failure to completely neutralize ROS produced endogenously (Fig. 7, C–E); and it is not a part of the cell death program (Figs. 4 and 5). To further address the potential adaptive function of ES7c cleavage, we examined whether the low doses of oxidants that induce ES7c cleavage in our experiments are capable of increasing survival of cells subjected to severe oxidative stress conditions. Consistent with previous studies, 2 mm H2O2 severely inhibited cell growth, but pretreatment with either 0.2 mm H2O2 or 25 μm menadione markedly improved growth when cells were later exposed to the high dose of H2O2 (Fig. 9A). Both drugs functioned as hormetic agents within the same pathway, because menadione cross-protected cells against H2O2. The observed growth inhibition in the culture treated with 2 mm H2O2 was primarily due to a prolonged lag period following the treatment rather than a change in doubling time after the cells resumed growth (Fig. 9B). Presumably, cells exposed to high-intensity oxidative stress require time to repair damage (compare bars 1 and 4 in Fig. 9B), whereas hormetic adaptation greatly shortens the recovery process (compare bars 4–6 in Fig. 9B). Low-dose oxidant pretreatment also alleviated the loss of viability observed in cells exposed to 2 mm H2O2 (Fig. 9C).
Figure 9.

Low doses of oxidants that induce ES7c cleavage promote cell resistance to severe oxidative stress. An overnight culture of wild-type BY4741 cells was diluted with YPDA to an A600 of ∼0.2 and grown for 4 h. The cells were then pretreated or not with low doses of menadione (25 μm for 2 h, K3) or H2O2 (0.2 mm for 30 min). Each culture was divided into two, and 2 mm H2O2 was added for 200 min to one part to induce acute oxidative stress, after which cells were washed with fresh YPDA and analyzed. A, representative growth curves after the indicated treatments. B, lag time and doubling time of the cultures. All treatments were done in triplicate; the error bars represent S.D. C, viability of cells tested by serial dilution (1:5) of 2 × 106 cells from each culture; the cells were incubated on a YPDA agar plate at 30 °C for 3 days. D, 25S rRNA degradation analyzed by Northern hybridization. During high-intensity oxidative stress treatment, both 120- and 200-min time points were analyzed. Hybridization signals corresponding to full-length 25S rRNA and faster-migrating degradation products (dp) in each sample are shown as percentages of the total signal per lane. The asterisk indicates the ES7c-cleaved rRNA fragment. E, working model for rRNA cleavage as a sensor of H2O2 during adaptation to oxidative stress. During normal growth, ROS produced by metabolic cellular processes are neutralized via the basal capacity of cellular ROS scavenger systems (sector 1). Slight increases in H2O2 levels (H2O2↑) trigger expression of enzymes from the redox-control group (including thioredoxin Trx2 and peroxiredoxin Tsa1). During this time, a fraction of ribosomes becomes cleaved in the ES7 region (sector 2). In addition to inactivating ROS, this adaptation period also prepares cells for harsher oxidative stress conditions, with ES7c-cleaved ribosomes potentially playing a role in this process. If the functional capacity of the ROS-protecting systems is exceeded and ROS continue to accumulate (H2O2↑↑), the cells begin to experience severe oxidative stress, which is accompanied by expression of catalase Ctt1, further increase in ES7c-cleaved ribosomes, and appearance of other rRNA cleavages (sector 3). Finally, when ROS reach levels incompatible with cell viability (H2O2↑↑↑), an apoptotic program is activated, during which ribosomes become extensively degraded (sector 4).
To determine whether the adaptive response induced by pretreatment with low-dose oxidants would protect rRNA in yeast ribosomes from the excessive breakdown associated with high doses of oxidants (Fig. 5F), cells were incubated with 0.2 mm H2O2, 25 μm menadione or left untreated and then exposed to 2 mm H2O2 and collected after 120 and 200 min of the high-dose treatment. Northern analysis of the rRNA showed that the severe oxidative stress resulted in intensive 25S rRNA fragmentation (Fig. 9D, lanes 3 and 4), indicative of active apoptosis (31). Applying low-dose H2O2 or menadione prior to oxidative stress reduced 25S rRNA fragmentation (Fig. 9D, lanes 5, 6, 8, and 9), consistent with a hormetic effect. Thus, pretreatment with low doses of oxidants not only rescues cell viability but also confers protection on the translation machinery from a destructive rRNAs degradation. Induction of ES7c cleavage by the hormetic doses of oxidants provides a strong indication that this change in ribosomes occurs as part of the adaptive oxidative stress response in budding yeast.
Discussion
Analysis of ribosomes in yeast cells subjected to various conditions that stress the translation machinery revealed that treatments with low doses of oxidants can cause site-specific cleavage of the large subunit rRNA adjacent to the c loop of ES7 (ES7c; Fig. 1G). This cleavage was observed to occur rapidly in cells exposed to diverse chemicals capable of disturbing the redox balance, including classic oxidants, such as inorganic hydrogen peroxide, and those that generate H2O2 indirectly, such as DTT. Our analyses have also shown that the effect of oxidants on ES7c is unlikely to be a consequence of proteotoxic stress, nor it is part of the UPR. The ES7c cleavage affects a limited number of ribosomes and does not correlate with a loss of cell viability, differing in this respect from the extensive rRNA breakdown previously shown to take place during apoptosis (31). By studying the time course of the ES7c cleavage, we found that it occurs in unison with the activation of the thioredoxin system (Fig. 9E), an early mechanism required for cell adaptation to acute oxidative stress (54). Importantly, we also found that in cells with a defective thioredoxin system, ES7c can be cleaved even during natural accumulation of ROS under growth conditions requiring respiration, indicating that this is a physiologically relevant process and not an artifact of drug treatment. These results are strongly consistent with the model in which the ES7 cleavage in yeast ribosomes is part of an adaptive oxidative stress response (Fig. 9E).
What could be the functional consequences of breaking rRNA in a ribosomal subunit? Several types of changes in ribosomes have been described in bacteria subjected to harsh environments. For example, cleavages in rRNA observed during transition to stationary phase were proposed to provide a fast way of repressing protein synthesis (65). In yeast, only a limited number of ribosomes is cleaved if the oxidant dose is survivable (Fig. 5, A–C), arguing against the scenario in which the ES7c cleavage simply serves to reduce global protein synthesis. Another possibility might be that cleavages in rRNA are not indiscriminate but rather target ribosomes damaged by ROS to remove them from the actively translating pool. Indeed, H2O2 was shown to cross-link r-proteins to rRNA (66) and oxidize cysteine residues in r-proteins (67). A precedent for rRNA cleavage by the RNase YbeY in defective 70S ribosomes during stress has been described in bacteria (68). Given that ES7c-cleaved yeast ribosomes appear to be translationally competent (Fig. 8), we consider it unlikely that cleavage of 25S rRNA within the ES7c region functions only to promote the disposal of dysfunctional ribosomes.
An intriguing role has been proposed for rRNA cleavage by the stress-induced bacterial endoribonuclease MazF (69, 70). It was found that instead of rendering ribosomes inactive, MazF-induced cleavage of 16S rRNA at the decoding center of 30S subunits generates a subpopulation of “specialized” ribosomes that selectively translate a small number of proteins required for survival during stress (69). Targeting the small ribosomal subunits by MazF was therefore suggested to function as a stress-response mechanism (70).
In our study, we found that yeast ribosomes cleaved in the ES7c region retain their translational competency, consistent with a non-degradative role of this cleavage. Based on the sedimentation properties of ES7c-cleaved 60S subunits (Fig. 8) and the fact that ES7c cleavage can be triggered by mild stress that preconditions cells to high-intensity oxidative treatment (Fig. 9), we currently favor the model in which this 25S rRNA cleavage alters the translational behavior of a subset of ribosomes to adapt the translation machinery to an ROS-rich environment. It remains to be determined what specific functions of the ribosome are affected by the cleavage. The low abundance and lack of biochemical features suitable for selective purification of ES7c-cleaved ribosomes currently hinder their analysis. Potential effects of rRNA cleavage in 60S subunits could involve interactions of nascent polypeptide chains with ribosome-associated chaperones, altered elongation speed of ribosomes, or their ability to overcome translational stalls. More work would be necessary to address these possibilities.
ES7 is a eukaryotic extension of the rRNA helix 25 that localizes on the surface of the 60S ribosomal subunit (30, 71). This region is 210 nucleotides long in S. cerevisiae, lacks base modifications (72), and folds into an evolutionarily conserved tripartite stem-loop structure (Fig. 1G). Structural studies have predicted a regulatory function for ES7 because of its flexibility and accessibility (33, 36), whereas recent biochemical evidence suggests that ES7 might be a docking area for multiple proteins (32). Thus, changes in ES7 conformation and/or its interactions with proteins during oxidative stress could conceivably result in the opening of this part of 25S rRNA, providing access to a nuclease. Given the rapid kinetics of ES7c cleavage when yeast cells are subjected to ROS-inducing conditions, a useful practical application of our findings is that loss of integrity of this rRNA loop can serve as an early and sensitive biomarker of oxidative stress.
Experimental procedures
Yeast media, drugs, and treatment conditions
Wild-type BY4741 (MATa his3–1 leu2–0 met15–0 ura3–0) and its derivative KanMX6 deletion strains (tsa1Δ, ire1Δ, aif1Δ, yca1Δ, yap1Δ, trx1Δ, trx2Δ) were obtained from Open Biosystems. The tsa1Δ ire1Δ strain was generated by homology-based replacement of the TSA1 gene with a LEU2 cassette in the ire1Δ strain by standard PCR-based techniques (73). We used standard recipes for YPDA (1% yeast extract, 2% peptone, 2% dextrose, 10 mg/liter adenine) and synthetic drop-out medium. Unless indicated otherwise, overnight yeast cultures were diluted with fresh YPDA an A600 of ∼0.2 and grown for an additional 2–4 h at 30 °C before each experiment.
The cells were treated with DTT (Sigma), H2O2 (Sigma), menadione (Enzo), plumbagin (Acros Organics), tunicamycin (Calbiochem), and ebselen (TCI) as indicated in the text. To induce proteotoxic stress, YPDA-grown day cultures were shifted to 38 or 40 °C for 2 h or incubated at 30 °C for 2 h in the presence of the following drugs: 50 μg/ml hygromycin B (Calbiochem), 40 μg/ml G418 (Thermo Fisher), or 6% ethanol. For treatments with 2 μg/ml canavanine (TCI) or 0.3 mm l-azetidine-2-carboxylic acid (TCI), day cultures were grown in synthetic medium without Arg for 2 h.
RNA extraction, Northern blotting, and signal quantification
Total RNA was isolated from cells using one-step extraction with formamide-EDTA as described in Ref. 74. RNA was separated on 1.2% agarose gels containing 1.3% formaldehyde (75), transferred to nylon membranes (Hybond N; GE Biosciences), and visualized by methylene blue staining. Individual rRNA species were detected by Northern hybridizations using 32P-labeled oligonucleotide probes as described (76). We used the following probes: y500 against 18S rRNA (5′-AGAATTTCACCTCTGACAATTG), y503 (5′-ACCCACGTCCAACTGCTGT), and y540 against 25S rRNA (5′-TCCTACCTGATTTGAGGTCAAAC).
To visualize spliced (HAC1i) and unspliced (HAC1u) HAC1 mRNA, membranes were hybridized in the Church buffer (77) at 50 °C with a probe against the first HAC1 exon, which recognizes both HAC1u and HAC1i (38). The probe was generated by PCR using genomic DNA as a template and primers 5′-AGCGGATCCATGGAAATGACTGATTTTGAAC (forward) and 5′-TCTTGATCACTGTAGTTTCCTGGTCATC (reverse).
All hybridizations were analyzed using a Typhoon 9200 PhosphorImager and ImageQuant software (GE Biosciences). For quantification, volume of the hybridization signal corresponding to the fragment of interest was converted to phosphorimaging units, and the background (average image background) was subtracted. To correct for unequal loading between lanes, the data were normalized to the 18S rRNA signal in the same samples.
Sucrose gradient analysis
Sucrose gradient centrifugation analysis was performed as described in Ref. 78. Briefly, the cells were treated for 5 min with 100 μg/ml CHX prior to harvesting, pelleted by centrifugation, washed, and lysed by glass bead shearing in buffer A (100 mm NaCl, 3 mm MgCl2, 10 mm Tris-HCl, pH 7.4, 100 μg/ml CHX, 200 μg/ml heparin, and 100 μm PMSF). Lysates were clarified by centrifugation, and an aliquot corresponding to 50 A260 units was loaded onto 11 ml, 15–45% (w/v) sucrose gradients prepared in 70 mm NH4Cl, 4 mm MgCl2, and 10 mm Tris-HCl, pH 7.4. Gradients were centrifuged at 188,000 × g at 4 °C for 4 h 15 min (Beckman SW41Ti rotor, 36,000 rpm) and fractionated using a Beckman fraction recovery system connected to an EM-1 UV monitor (Bio-Rad). A260 traces were digitally recorded using Windaq data acquisition software. RNA was extracted from each fraction by phenol/chloroform extraction followed by isopropanol precipitation and analyzed by Northern hybridizations (75).
RT-qPCR
A total of 1.5 μg of RNA was treated with 2 units of DNase (Thermo Fisher) in 100 μl for 15 min at 37 °C. RNA was purified by phenol/chloroform extraction, precipitated with isopropanol, annealed with an oligo(dT)18 primer (0.4 μm) at 65 °C for 5 min, and reverse-transcribed using the ReverseAid enzyme (Thermo Fisher) for 1.5 h at 42 °C. Reaction products were diluted to 150 μl, and 5 μl was used as a template for real-time qPCR using the SYBR FAST Universal qPCR Master Mix (KAPA Biosystems) on a Mastercycler ep realplex (Eppendorf) with an initial step at 95 °C for 2 min and cycles at 95 °C for 15 s, 55 °C for 15 s, and 68 °C for 20 s. All samples were analyzed in triplicate, and ACT1 was used to normalize the mRNA expression levels.
Primer extension
A total of 3 μg of RNA from WT and tsa1Δ cells treated with 20 mm DTT for 2 h was separated on a guanidine thiocyanate-containing gel, and RNA was extracted from the ∼2.8-kb fragment as described in Ref. 79. RNA was annealed with 2 pmol of the 32P-labeled primer Prex1 (5′-GACTCCTTGGTCCGTGTTTC) for 5 min at 65 °C and used for primer extensions as described (80). Reaction products were purified by phenol/chloroform extraction, precipitated with ethanol, dissolved in 10 μl of loading buffer (95% formamide, 10 mm EDTA, 0.1% xylene cyanol, and 0.1% bromphenol blue, at final pH 11), and analyzed on a 6% polyacrylamide/urea gel. The sequencing reactions were performed with SequiTherm EXCEL II (Epicenter) using 250 ng of the pJD694 plasmid containing rDNA (81) as a template and the Prex1 primer.
Amplex Red assay
We used an Amplex Red Assay kit (Thermo Fisher) as described in Ref. 47 with a few minor modifications. Briefly, the same numbers of mid-log phase cells (as specified in figure legends) were collected, washed with PBS, and resuspended in 0.5 ml of the reaction buffer supplied with the kit. The cells were incubated for 30 min in a 30 °C shaker and centrifuged, and 100 μl of supernatant was taken into an Amplex Red assay reaction that was performed in triplicate according to the manufacturer's protocol. Oxidation of Amplex Red by horseradish peroxidase requires H2O2 (from the sample) and produces resorufin, whose fluorescence was measured at 590 nm using a Synergy HT microplate reader (BioTek).
Cell viability and growth assays
A culture of wild-type BY4741 cells was diluted with YPDA to an A600 of ∼0.2, grown for 4 h, and diluted again to an A600 of ∼0.2. The cells were either left untreated or treated with 25, 50, or 100 μm menadione for 2 h at 30 °C with shaking. The total cell number in each culture was determined by counting multiple aliquots using a hemocytometer. Equal culture volumes were plated onto YPDA agar plates (five replicates for each culture). Colonies were counted after 2–3 days at 30 °C, and both the colony number and total cell count were analyzed using GraphPad Prism 5.
For growth assays, yeast cultures were adjusted to an A600 of ∼0.1 with appropriate media, 200 μl/well was inoculated into 96-well plates in three replicates, and the cultures were grown for 20–24 h at 30 °C with shaking. A600 measurements were taken every 5 min and automatically recorded using a BioTek Synergy HT microplate reader. The lag-phase time and Vmax were determined using the instrument's Gen5 software; the doubling time was defined as Ln(2)/Vmax as described in Ref. 82.
FITC–annexin V staining and flow cytometry
To determine the number of apoptotic cells, an overnight culture was diluted with YPDA to an A600 of ∼0.2, grown for additional 4 h, and treated with oxidants. The cells were collected by centrifugation, washed, and converted to spheroplasts using a protocol modified from that described in Ref. 83. We used Zymolase T-100 (Sunrise Science Products) at the final concentration of 30 μg/ml in spheroplasting buffer (0.8 m d-sorbitol, 10 mm β-mercaptoethanol, 20 mm Tris-HCl, pH 7.4) for 15 min at 30 °C. Efficiency of spheroplasting was monitored microscopically. Spheroplasts were washed twice with PBS, pH 7.0, and once in the 1× binding buffer containing 0.6 m d-sorbitol, supplied with the Alexa Fluor 488 annexin V apoptosis kit (Thermo Fisher). Spheroplasts (2 × 106 cells) were treated with FITC–annexin V and propidium iodide according to the manufacturer's recommendations. Annexin V–FITC–stained spheroplasts were analyzed using a BD Accuri C6 flow cytometer and FlowJo software (version 10).
Author contributions
N. S., D. G. P., and D. S. conceived and designed the experiments. D. S., J. A. Z., E. G., and N. S. performed the experiments. N. S., D. G. P., and D. S. analyzed the data. N. S. and D. G. P. wrote the manuscript.
Acknowledgments
We are thankful to Jonathan Dinman for the pJD694 plasmid. We are grateful to Randy Strich, Arnab Ghosh, Russell Sapio, and Leonid Anikin for valuable technical advice and constructive comments on the manuscript. Special thanks to Jonathan Harbin for technical assistance.
This work was supported by National Institutes of Health Grant R01GM114308 (to N. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ROS
- reactive oxygen species
- rRNA
- ribosomal RNA
- ES
- expansion segment
- UPR
- unfolded protein response
- Tm
- tunicamycin
- CHX
- cycloheximide
- qPCR
- quantitative PCR.
References
- 1. Strich R. (2015) Programmed cell death initiation and execution in budding yeast. Genetics 200, 1003–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cook J. A., Gius D., Wink D. A., Krishna M. C., Russo A., and Mitchell J. B. (2004) Oxidative stress, redox, and the tumor microenvironment. Semin. Radiat. Oncol. 14, 259–266 [DOI] [PubMed] [Google Scholar]
- 3. Lowell B. B., and Shulman G. I. (2005) Mitochondrial dysfunction and type 2 diabetes. Science 307, 384–387 [DOI] [PubMed] [Google Scholar]
- 4. Jamieson D. J. (1998) Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast 14, 1511–1527 [DOI] [PubMed] [Google Scholar]
- 5. Klaunig J. E., and Kamendulis L. M. (2004) The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 44, 239–267 [DOI] [PubMed] [Google Scholar]
- 6. Bhattacharyya A., Chattopadhyay R., Mitra S., and Crowe S. E. (2014) Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 94, 329–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Autréaux B., and Toledano M. B. (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8, 813–824 [DOI] [PubMed] [Google Scholar]
- 8. Schieber M., and Chandel N. S. (2014) ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marinho H. S., Real C., Cyrne L., Soares H., and Antunes F. (2014) Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2, 535–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Delaunay A., Isnard A. D., and Toledano M. B. (2000) H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J. 19, 5157–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delaunay A., Pflieger D., Barrault M. B., Vinh J., and Toledano M. B. (2002) A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111, 471–481 [DOI] [PubMed] [Google Scholar]
- 12. Kuge S., Toda T., Iizuka N., and Nomoto A. (1998) Crm1 (XpoI) dependent nuclear export of the budding yeast transcription factor yAP-1 is sensitive to oxidative stress. Genes Cells 3, 521–532 [DOI] [PubMed] [Google Scholar]
- 13. Temple M. D., Perrone G. G., and Dawes I. W. (2005) Complex cellular responses to reactive oxygen species. Trends Cell Biol. 15, 319–326 [DOI] [PubMed] [Google Scholar]
- 14. Doronina V. A., Staniforth G. L., Speldewinde S. H., Tuite M. F., and Grant C. M. (2015) Oxidative stress conditions increase the frequency of de novo formation of the yeast [PSI+] prion. Mol. Microbiol. 96, 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagano T., Yutthanasirikul R., Hihara Y., Hisabori T., Kanamori T., Takeuchi N., Ueda T., and Nishiyama Y. (2015) Oxidation of translation factor EF-G transiently retards the translational elongation cycle in Escherichia coli. J. Biochem. 158, 165–172 [DOI] [PubMed] [Google Scholar]
- 16. Shenton D., Smirnova J. B., Selley J. N., Carroll K., Hubbard S. J., Pavitt G. D., Ashe M. P., and Grant C. M. (2006) Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J. Biol. Chem. 281, 29011–29021 [DOI] [PubMed] [Google Scholar]
- 17. Sideri T. C., Koloteva-Levine N., Tuite M. F., and Grant C. M. (2011) Methionine oxidation of Sup35 protein induces formation of the [PSI+] prion in a yeast peroxiredoxin mutant. J. Biol. Chem. 286, 38924–38931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baxter C. J., Redestig H., Schauer N., Repsilber D., Patil K. R., Nielsen J., Selbig J., Liu J., Fernie A. R., and Sweetlove L. J. (2007) The metabolic response of heterotrophic Arabidopsis cells to oxidative stress. Plant Physiol. 143, 312–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ling J., and Söll D. (2010) Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc. Natl. Acad. Sci. U.S.A. 107, 4028–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Netzer N., Goodenbour J. M., David A., Dittmar K. A., Jones R. B., Schneider J. R., Boone D., Eves E. M., Rosner M. R., Gibbs J. S., Embry A., Dolan B., Das S., Hickman H. D., Berglund P., et al. (2009) Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 462, 522–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simms C. L., Hudson B. H., Mosior J. W., Rangwala A. S., and Zaher H. S. (2014) An active role for the ribosome in determining the fate of oxidized mRNA. Cell Rep. 9, 1256–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanaka M., Chock P. B., and Stadtman E. R. (2007) Oxidized messenger RNA induces translation errors. Proc. Natl. Acad. Sci. U.S.A. 104, 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan C. T., Pang Y. L., Deng W., Babu I. R., Dyavaiah M., Begley T. J., and Dedon P. C. (2012) Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gu C., Begley T. J., and Dedon P. C. (2014) tRNA modifications regulate translation during cellular stress. FEBS Lett. 588, 4287–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raina M., and Ibba M. (2014) tRNAs as regulators of biological processes. Front. Genet. 5, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gebetsberger J., Zywicki M., Künzi A., and Polacek N. (2012) tRNA-derived fragments target the ribosome and function as regulatory non-coding RNA in Haloferax volcanii. Archaea 2012, 260909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ivanov P., Emara M. M., Villen J., Gygi S. P., and Anderson P. (2011) Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 43, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ivanov P., O'Day E., Emara M. M., Wagner G., Lieberman J., and Anderson P. (2014) G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc. Natl. Acad. Sci. U.S.A. 111, 18201–18206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang S., Sun L., and Kragler F. (2009) The phloem-delivered RNA pool contains small noncoding RNAs and interferes with translation. Plant Physiol. 150, 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yusupova G., and Yusupov M. (2014) High-resolution structure of the eukaryotic 80S ribosome. Annu. Rev. Biochem. 83, 467–486 [DOI] [PubMed] [Google Scholar]
- 31. Mroczek S., and Kufel J. (2008) Apoptotic signals induce specific degradation of ribosomal RNA in yeast. Nucleic Acids Res. 36, 2874–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gómez Ramos L. M., Smeekens J. M., Kovacs N. A., Bowman J. C., Wartell R. M., Wu R., and Williams L. D. (2016) Yeast rRNA expansion segments: folding and function. J. Mol. Biol. 428, 4048–4059 [DOI] [PubMed] [Google Scholar]
- 33. Melnikov S., Ben-Shem A., Garreau de Loubresse N., Jenner L., Yusupova G., and Yusupov M. (2012) One core, two shells: bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 19, 560–567 [DOI] [PubMed] [Google Scholar]
- 34. Zhao Y., Macgurn J. A., Liu M., and Emr S. (2013) The ART-Rsp5 ubiquitin ligase network comprises a plasma membrane quality control system that protects yeast cells from proteotoxic stress. eLife. 2, e00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rand J. D., and Grant C. M. (2006) The thioredoxin system protects ribosomes against stress-induced aggregation. Mol. Biol. Cell 17, 387–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Armache J.-P., Jarasch A., Anger A. M., Villa E., Becker T., Bhushan S., Jossinet F., Habeck M., Dindar G., Franckenberg S., Marquez V., Mielke T., Thomm M., Berninghausen O., Beatrix B., et al. (2010) Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-A resolution. Proc. Natl. Acad. Sci. U.S.A. 107, 19748–19753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taylor D. J., Devkota B., Huang A. D., Topf M., Narayanan E., Sali A., Harvey S. C., and Frank J. (2009) Comprehensive molecular structure of the eukaryotic ribosome. Structure 17, 1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Back S. H., Schröder M., Lee K., Zhang K., and Kaufman R. J. (2005) ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 35, 395–416 [DOI] [PubMed] [Google Scholar]
- 39. Sidrauski C., and Walter P. (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 [DOI] [PubMed] [Google Scholar]
- 40. Cox J. S., and Walter P. (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404 [DOI] [PubMed] [Google Scholar]
- 41. Kachur A. V., Held K. D., Koch C. J., and Biaglow J. E. (1997) Mechanism of production of hydroxyl radicals in the copper-catalyzed oxidation of dithiothreitol. Radiat. Res. 147, 409–415 [PubMed] [Google Scholar]
- 42. Amrolia P., Sullivan S. G., Stern A., and Munday R. (1989) Toxicity of aromatic thiols in the human red blood cell. J. Appl. Toxicol. 9, 113–118 [DOI] [PubMed] [Google Scholar]
- 43. Held K. D., Sylvester F. C., Hopcia K. L., and Biaglow J. E. (1996) Role of Fenton chemistry in thiol-induced toxicity and apoptosis. Radiat. Res. 145, 542–553 [PubMed] [Google Scholar]
- 44. Misra H. P. (1974) Generation of superoxide free radical during the autoxidation of thiols. J. Biol. Chem. 249, 2151–2155 [PubMed] [Google Scholar]
- 45. Munday R. (1989) Toxicity of thiols and disulphides: involvement of free-radical species. Free Radic. Biol. Med. 7, 659–673 [DOI] [PubMed] [Google Scholar]
- 46. Tartier L., McCarey Y. L., Biaglow J. E., Kochevar I. E., and Held K. D. (2000) Apoptosis induced by dithiothreitol in HL-60 cells shows early activation of caspase 3 and is independent of mitochondria. Cell Death Differ. 7, 1002–1010 [DOI] [PubMed] [Google Scholar]
- 47. Karthikeyan G., Lewis L. K., and Resnick M. A. (2002) The mitochondrial protein frataxin prevents nuclear damage. Hum. Mol. Genet. 11, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 48. Satheeshkumar K., and Mugesh G. (2011) Synthesis and antioxidant activity of peptide-based ebselen analogues. Chemistry 17, 4849–4857 [DOI] [PubMed] [Google Scholar]
- 49. Jin C., Strich R., and Cooper K. F. (2014) Slt2p phosphorylation induces cyclin C nuclear-to-cytoplasmic translocation in response to oxidative stress. Mol. Biol. Cell 25, 1396–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wissing S., Ludovico P., Herker E., Büttner S., Engelhardt S. M., Decker T., Link A., Proksch A., Rodrigues F., Corte-Real M., Fröhlich K.-U., Manns J., Candé C., Sigrist S. J., Kroemer G., and Madeo F. (2004) An AIF orthologue regulates apoptosis in yeast. J. Cell Biol. 166, 969–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Madeo F., Herker E., Maldener C., Wissing S., Lächelt S., Herlan M., Fehr M., Lauber K., Sigrist S. J., Wesselborg S., and Fröhlich K. U. (2002) A caspase-related protease regulates apoptosis in yeast. Mol. Cell 9, 911–917 [DOI] [PubMed] [Google Scholar]
- 52. Fadok V. A., Voelker D. R., Campbell P. A., Cohen J. J., Bratton D. L., and Henson P. M. (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216 [PubMed] [Google Scholar]
- 53. Winterbourn C. C., and Hampton M. B. (2008) Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 45, 549–561 [DOI] [PubMed] [Google Scholar]
- 54. Lucau-Danila A., Lelandais G., Kozovska Z., Tanty V., Delaveau T., Devaux F., and Jacq C. (2005) Early expression of yeast genes affected by chemical stress. Mol. Cell Biol. 25, 1860–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gasch A. P., Spellman P. T., Kao C. M., Carmel-Harel O., Eisen M. B., Storz G., Botstein D., and Brown P. O. (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11, 4241–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koerkamp M. G., Rep M., Bussemaker H. J., Hardy G. P., Mul A., Piekarska K., Szigyarto C. A., De Mattos J. M., and Tabak H. F. (2002) Dissection of transient oxidative stress response in Saccharomyces cerevisiae by using DNA microarrays. Mol. Biol. Cell 13, 2783–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coleman S. T., Epping E. A., Steggerda S. M., and Moye-Rowley W. S. (1999) Yap1p activates gene transcription in an oxidant-specific fashion. Mol. Cell Biol. 19, 8302–8313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Farrugia G., and Balzan R. (2012) Oxidative stress and programmed cell death in yeast. Front. Oncol. 2, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DeRisi J. L., Iyer V. R., and Brown P. O. (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680–686 [DOI] [PubMed] [Google Scholar]
- 60. Garrido E. O., and Grant C. M. (2002) Role of thioredoxins in the response of Saccharomyces cerevisiae to oxidative stress induced by hydroperoxides. Mol. Microbiol. 43, 993–1003 [DOI] [PubMed] [Google Scholar]
- 61. Park S. G., Cha M. K., Jeong W., and Kim I. H. (2000) Distinct physiological functions of thiol peroxidase isoenzymes in Saccharomyces cerevisiae. J. Biol. Chem. 275, 5723–5732 [DOI] [PubMed] [Google Scholar]
- 62. Trotter E. W., Rand J. D., Vickerstaff J., and Grant C. M. (2008) The yeast Tsa1 peroxiredoxin is a ribosome-associated antioxidant. Biochem. J. 412, 73–80 [DOI] [PubMed] [Google Scholar]
- 63. van den Elzen A. M., Schuller A., Green R., and Séraphin B. (2014) Dom34-Hbs1 mediated dissociation of inactive 80S ribosomes promotes restart of translation after stress. EMBO J. 33, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ludovico P., and Burhans W. C. (2014) Reactive oxygen species, ageing and the hormesis police. FEMS Yeast Res. 14, 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luidalepp H., Berger S., Joss O., Tenson T., and Polacek N. (2016) Ribosome shut-down by 16S rRNA fragmentation in stationary-phase Escherichia coli. J. Mol. Biol. 428, 2237–2247 [DOI] [PubMed] [Google Scholar]
- 66. Mirzaei H., and Regnier F. (2006) Protein-RNA cross-linking in the ribosomes of yeast under oxidative stress. J. Proteome Res. 5, 3249–3259 [DOI] [PubMed] [Google Scholar]
- 67. Brandes N., Reichmann D., Tienson H., Leichert L. I., and Jakob U. (2011) Using quantitative redox proteomics to dissect the yeast redoxome. J. Biol. Chem. 286, 41893–41903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jacob A. I., Köhrer C., Davies B. W., RajBhandary U. L., and Walker G. C. (2013) Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell 49, 427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Amitai S., Kolodkin-Gal I., Hananya-Meltabashi M., Sacher A., and Engelberg-Kulka H. (2009) Escherichia coli MazF leads to the simultaneous selective synthesis of both “death proteins” and “survival proteins.” PLoS Genet. 5, e1000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vesper O., Amitai S., Belitsky M., Byrgazov K., Kaberdina A. C., Engelberg-Kulka H., and Moll I. (2011) Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 147, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nilsson J., Sengupta J., Gursky R., Nissen P., and Frank J. (2007) Comparison of fungal 80 S ribosomes by cryo-EM reveals diversity in structure and conformation of rRNA expansion segments. J. Mol. Biol. 369, 429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Piekna-Przybylska D., Decatur W. A., and Fournier M. J. (2008) The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res. 36, D178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Longtine M. S., McKenzie A. 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., and Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 74. Shedlovskiy D., Shcherbik N., and Pestov D. G. (2017) One-step hot formamide extraction of RNA from Saccharomyces cerevisiae. RNA Biol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mansour F. H., and Pestov D. G. (2013) Separation of long RNA by agarose-formaldehyde gel electrophoresis. Anal. Biochem. 441, 18–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pestov D. G., Lapik Y. R., and Lau L. F. (2008) Assays for ribosomal RNA processing and ribosome assembly. Curr. Protoc. Cell Biol. Chapter 22, Unit 22.11 [DOI] [PubMed] [Google Scholar]
- 77. Nilsen T. W. (2015) An alternative method for processing Northern blots after capillary transfer. Cold Spring Harb. Protoc. 2015, 314–318 [DOI] [PubMed] [Google Scholar]
- 78. Shcherbik N., Chernova T. A., Chernoff Y. O., and Pestov D. G. (2016) Distinct types of translation termination generate substrates for ribosome-associated quality control. Nucleic Acids Res. 44, 6840–6852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang M., Parshin A. V., Shcherbik N., and Pestov D. G. (2015) Reduced expression of the mouse ribosomal protein Rpl17 alters the diversity of mature ribosomes by enhancing production of shortened 5.8S rRNA. RNA 21, 1240–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kent T., Lapik Y. R., and Pestov D. G. (2009) The 5′ external transcribed spacer in mouse ribosomal RNA contains two cleavage sites. RNA 15, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rakauskaite R., and Dinman J. D. (2006) An arc of unpaired “hinge bases” facilitates information exchange among functional centers of the ribosome. Mol. Cell Biol. 26, 8992–9002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Toussaint M., Levasseur G., Gervais-Bird J., Wellinger R. J., Elela S. A., and Conconi A. (2006) A high-throughput method to measure the sensitivity of yeast cells to genotoxic agents in liquid cultures. Mutat. Res. 606, 92–105 [DOI] [PubMed] [Google Scholar]
- 83. Lee J. Y., Jun D. Y., Park J. E., Kwon G. H., Kim J.-S., and Kim Y. H. (2017) Pro-apoptotic role of the human YPEL5 gene identified by functional complementation of a yeast moh1Δ mutation. J. Microbiol. Biotechnol. 27, 633–643 [DOI] [PubMed] [Google Scholar]