

Abstract
Expansion of adipose tissue in response to a positive energy balance underlies obesity and occurs through both hypertrophy of existing cells and increased differentiation of adipocyte precursors (hyperplasia). To better understand the nutrient signals that promote adipocyte differentiation, we investigated the role of glucose availability in regulating adipocyte differentiation and maturation. 3T3-L1 preadipocytes were grown and differentiated in medium containing a standard differentiation hormone mixture and either 4 or 25 mm glucose. Adipocyte maturation at day 9 post-differentiation was determined by key adipocyte markers, including glucose transporter 4 (GLUT4) and adiponectin expression and Oil Red O staining of neutral lipids. We found that adipocyte differentiation and maturation required a pulse of 25 mm glucose only during the first 3 days of differentiation. Importantly, fatty acids were unable to substitute for the 25 mm glucose pulse during this period. The 25 mm glucose pulse increased adiponectin and GLUT4 expression and accumulation of neutral lipids via distinct mechanisms. Adiponectin expression and other early markers of differentiation required an increase in the intracellular pool of total NAD/P. In contrast, GLUT4 protein expression was only partially restored by increased NAD/P levels. Furthermore, GLUT4 mRNA expression was mediated by glucose-dependent activation of GLUT4 gene transcription through the cis-acting GLUT4–liver X receptor element (LXRE) promoter element. In summary, this study supports the conclusion that high glucose promotes adipocyte differentiation via distinct metabolic pathways and independently of fatty acids. This may partly explain the mechanism underlying adipocyte hyperplasia that occurs much later than adipocyte hypertrophy in the development of obesity.
Keywords: adiponectin, glucose metabolism, glucose transporter type 4 (GLUT4), lipogenesis, NAD, adipocyte differentiation
Introduction
The prevalence of obesity and of obesity-related diseases such as type 2 diabetes mellitus, cardiovascular disease, and metabolic syndrome has dramatically increased in the past 20 to 30 years. Adipose tissue is thought to play an important role in the development of these diseases because of the changes in adipocyte function and adipokine secretion that occur with obesity. During the onset of obesity, expansion of the adipose mass occurs initially through cellular hypertrophy, during which the size of individual adipocytes is increased. As obesity progresses, continued expansion of the fat pad occurs through hyperplasia, during which the generation of new adipocytes is increased (1–3). In genetic models of obesity, rounds of adipocyte hyperplasia occur with a periodicity of ∼55 days (4). It is unclear what triggers the periodic increase in adipogenesis during the progression of obesity.
A primary function of adipose tissue is to increase uptake of both glucose and fatty acids from a meal in response to insulin signaling (5, 6). The insulin-responsive glucose transporter GLUT4 is responsible for the insulin-dependent glucose uptake in fat cells and plays a significant role in whole-body glucose homeostasis (7). GLUT4 is up-regulated as the adipocyte differentiates and matures as GLUT1, a non-insulin-responsive transporter, is down-regulated (8). Under obesogenic conditions, GLUT4 levels are markedly reduced in adipose tissue (9). Reduced GLUT4 expression and reduced insulin-dependent glucose uptake are markers of adipocyte dysfunction in hypertrophic adipose tissue (for a review, see Ref. 10).
A second important function of healthy adipocytes is the production and secretion of adipokines, such as adiponectin. Adiponectin acts on a variety of tissues, enhancing their insulin sensitivity through signaling pathways, such as AMP kinase, and transcriptional regulators, such as PPARα (11–14). Adiponectin expression increases 3 to 4 days post-differentiation and is considered to be a late marker of a mature adipocytes (15, 16). Similar to GLUT4, the expression of adiponectin is greatly reduced in obesity (16), thus serving as another biomarker of adipocyte dysfunction.
Hyperplastic expansion of adipose is preferable to hypertrophic expansion, as it is known that adipose hypertrophy is highly associated with metabolic disease (17). Although it is clear that hyperplastic adipose expansion occurs after a period of hypertrophic growth, the signals that promote adipogenesis in the expanding fat pad are unknown (3, 4, 17). We hypothesized that elevated levels of glucose in the extracellular fluid of Glut4-deficient, hypertrophic fat may be providing the nutrient requirement to support adipogenesis. Our hypothesis predicts that adipogenesis in vitro is regulated by glucose concentration in the culture medium. To test this, we used gain-of-function and loss-of-function approaches to determine how glucose regulates differentiation of 3T3-L1 adipocytes in vitro. Our data demonstrate that a brief pulse of high glucose is required early in differentiation for development of the cellular pools of NAD/P, and increased pentose pathway activity is required for development of mature adipocytes. Thus, these data support the hypothesis that adipocyte hyperplasia is regulated by extracellular glucose concentrations that may increase as a result of decreased insulin-dependent glucose uptake in obese, hypertrophic fat cells. Importantly, these findings demonstrate that extracellular signaling is not sufficient to promote adipogenesis. Differentiation requires both extracellular signaling and appropriate nutritional support (or nutrient excess) for production of cellular metabolites, such as NAD/P, that are required for differentiation and maturation to proceed.
Results
GLUT4 expression is glucose-dependent
Two defining features of healthy, mature adipose tissue adipose tissue are the ability to mediate Glut4-dependent glucose uptake and to secrete adiponectin (18, 19). Both of these functions rely on maximal expression of both GLUT4 and adiponectin in the mature adipocyte. To understand the metabolic conditions that result in maximal expression of these important adipocyte proteins, the expression pattern of GLUT4 and adiponectin protein was measured as a function of time after differentiation. The protocol (Fig. 1A) for differentiation began at day 0 with a growth-arrested plate of 3T3-L1 preadipocytes grown to confluence in 25 mm glucose-containing medium supplemented with 10% calf serum (standard conditions). To initiate differentiation, medium was changed at day 0 to 25 mm glucose medium supplemented with 10% fetal bovine serum, 175 nm insulin, 1 μm dexamethasone, and 0.5 mm isobutyl-1-methylxanthine. After 3 days of differentiation, the medium was changed to 25 mm glucose medium supplemented with only 10% fetal bovine serum to allow maturation of the adipocytes for the following 6 days. Cells were harvested each day for 9 days for measurement of GLUT4 and adiponectin protein (Fig. 1B). GLUT4 was first expressed at low levels on day 5 and reached maximal expression by day 9. Adiponectin expression was first detected at day 4, 1 day earlier than GLUT4.
Figure 1.
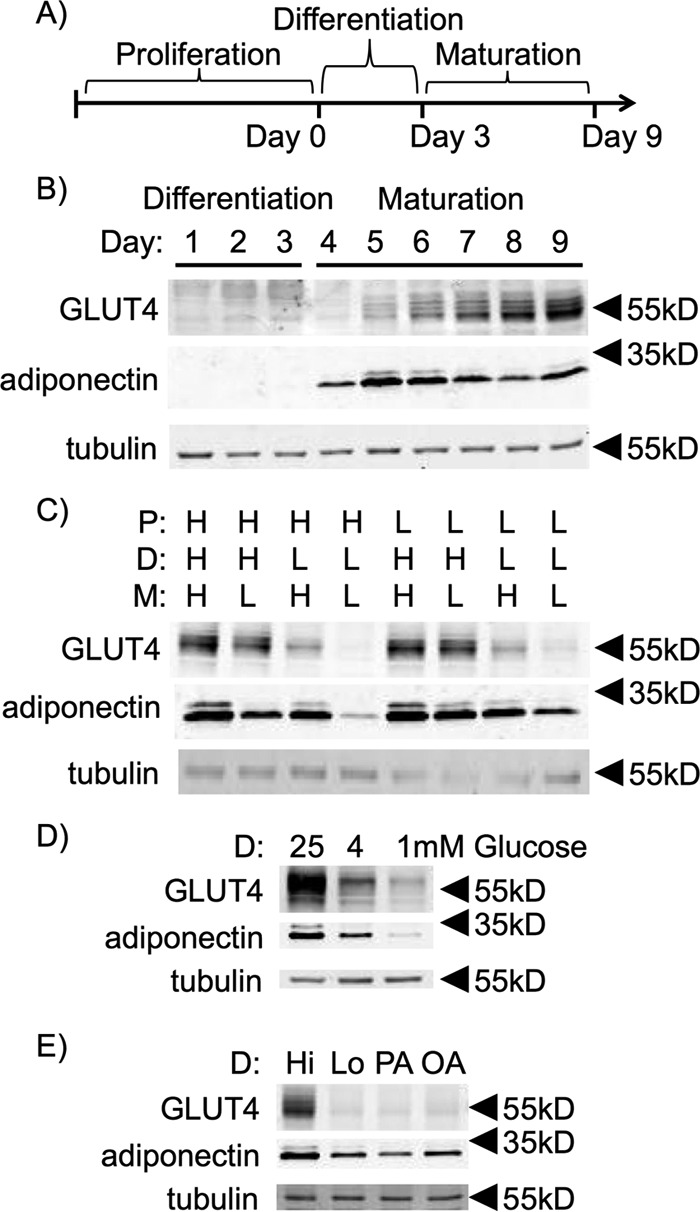
GLUT4 and adiponectin expression are glucose-dependent. A, schematic of the three major phases of 3T3-L1 adipocyte cell culture. B, 3T3-L1 adipocytes grown under standard medium conditions and harvested every day post-differentiation induction with dexamethasone, isobutylmethylxanthine, and insulin (DII). C, 3T3-L1 adipocytes cultured in either 4 mm glucose-containing (L) or 25 mm glucose-containing (H) medium during the different phases of adipocyte cell culture as indicated. D, preadipocytes isolated from the stromal vascular fraction of mouse subcutaneous adipose tissue were induced for differentiation and pulsed with varying concentrations of glucose. E, 3T3-L1 adipocytes were induced for differentiation and pulsed with 25 mm glucose (Hi), 4 mm glucose (Lo), 50 μm palmitic acid and 4 mm glucose (PA) or 50 μm oleic acid and 4 mm glucose (OA) and cultured in Lo glucose medium during the P and M phases of their cell culture. Protein from whole-cell lysates of day 9 adipocytes were separated via SDS-PAGE and immunoblotted for GLUT4, adiponectin, and tubulin. Data shown are representative of n = 3 independent experiments
3T3-L1 adipocytes have traditionally been grown and differentiated in DMEM containing 25 mm glucose, a supraphysiological concentration. We wanted to learn whether high levels of glucose were required for adipocyte differentiation and maturation to occur. To test this, cells were cultured in medium containing either 4 mm (Lo) or 25 mm (Hi) glucose during their proliferation (P)2 phase, differentiation (D) phase, and maturation (M) phase (as indicated in Fig. 1A). Maximal expression of GLUT4 at day 9 post-differentiation required Hi glucose during the 3-day D phase. 25 mm glucose in either the P or M phases was not required (Fig. 1C). In contrast, adiponectin expression was optimal when cells were pulsed with Hi glucose during either the D phase or M phase (Fig. 1C). These data suggest that the role for glucose in adipocyte development and gene expression may impact multiple signaling pathways during the developmental program. Glucose-dependent gene expression was also observed during differentiation of primary stromal vascular cells isolated from subcutaneous fat from mice (Fig. 1D), indicating that the glucose requirement was not unique to the transformed 3T3-L1 cells.
Because glucose is used for de novo lipogenesis in rodent adipose tissue (20), we determined whether fatty acids could compensate for Hi glucose during the D phase. Cells differentiated in Lo glucose medium were pulsed with either palmitic acid or oleic acid during the D phase (Fig. 1E). Neither fatty acid was able to compensate for Hi glucose in the D phase to promote differentiation. These data suggest that plasma carbohydrate levels are more important than plasma lipid levels for activating differentiation of adipocyte progenitors.
Next, we sought to determine whether direct addition of glucose metabolites during the D phase is sufficient for adipocyte maturation. To do this, we supplemented Lo glucose medium with equal carbon equivalents of intermediate glucose metabolites. Lo glucose, D phase medium was pulsed with 42 mm pyruvate (Pyr), 42 mm lactate (Lac), or 42 mm sodium acetate (Ac) (Fig. 2A). The pulsed cells were returned to Lo glucose medium during the M phase and harvested at day 9 post-differentiation. Pyr was able to rescue expression of adiponectin (Fig. 2A). In contrast, Pyr only partially rescued GLUT4 protein levels (Fig. 2A). Lactate and sodium acetate had no effect on either GLUT4 or adiponectin.
Figure 2.
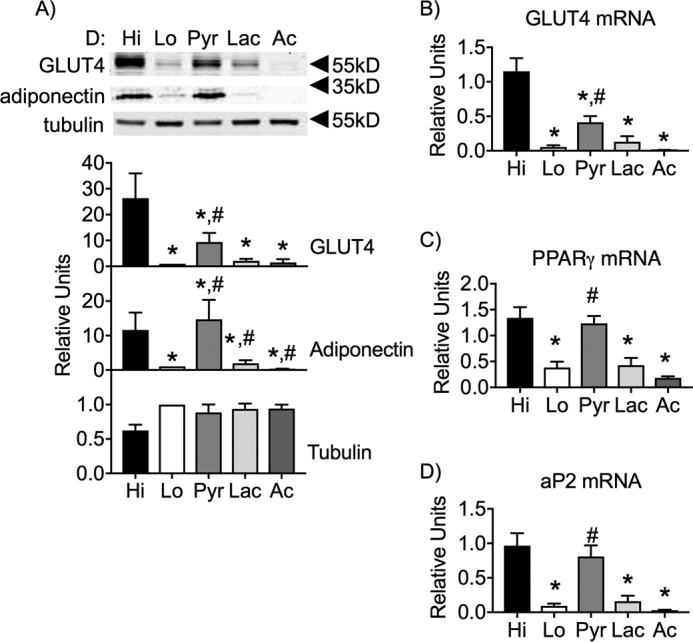
Pyruvate partially restores GLUT4 expression in the absence of Hi glucose and fully supports adiponectin expression. 3T3-L1 adipocytes were induced for differentiation and pulsed with Hi glucose, Lo glucose, 42 mm sodium pyruvate and 4 mm glucose (Pyr), 42 mm lactate and 4 mm glucose (Lac), or 42 mm sodium acetate and 4 mm glucose (Ac) and cultured in Lo glucose medium during the P and M phases of their cell culture. A, protein from whole-cell lysates of day 9 adipocytes were separated via SDS-PAGE and immunoblotted for GLUT4, adiponectin, and tubulin. B–D, mRNA isolated from day 9 adipocytes were analyzed via qRT-PCR for the relative expression of glut4, pparg, and ap2. 95% confidence intervals were used to determine statistical differences in immunoblot densitometry. One-way analysis of variance was used to identify significant differences (p < 0.05 for n = 3 independent experiments) in expression data. *, differences compared with Hi glucose cells; #, differences compared with Lo glucose cells.
To determine the extent to which Pyr was able to compensate for Hi glucose, we measured glut4 mRNA expression as well as mRNA for ap2 and pparγ, early markers of adipocyte differentiation (Fig. 2, B–D). Pyr fully supported the expression of the two early differentiation markers but not glut4 mRNA. These data suggest that there is a nutrient-dependent regulation of GLUT4 expression that is independent of other markers of adipocyte differentiation and maturation.
We next determine whether Lo glucose inhibited differentiation because of glucose starvation. We first measured the change in glucose concentration from day 0 through day 3 in D phase cells pulsed with Hi, Lo, or Pyr medium (Fig. 3A). The glucose concentration in Lo cells at day 3 was greater than 1 mm, indicating no glucose starvation. The magnitude in change of the glucose concentration was significantly greater in Hi glucose cells, indicating that a significant portion of glucose in the medium was consumed during the D phase. A Pyr pulse showed the least change in glucose concentration, consistent with the notion that glucose is partly used for production of pyruvate. We next determined whether maintaining glucose concentrations in cells pulsed with in Lo or Pyr during the D phase would restore differentiation. We found that glucose supplementation to maintain constant 4 mm glucose had no effect on differentiation in Lo and Pyr cells (Fig. 3B). These data suggested that a sustained glucose concentration of greater than 4 mm is required for differentiation. Finally, a glucose dose response revealed that at least 15 mm glucose was required during the D phase for full differentiation (Fig. 3C).
Figure 3.
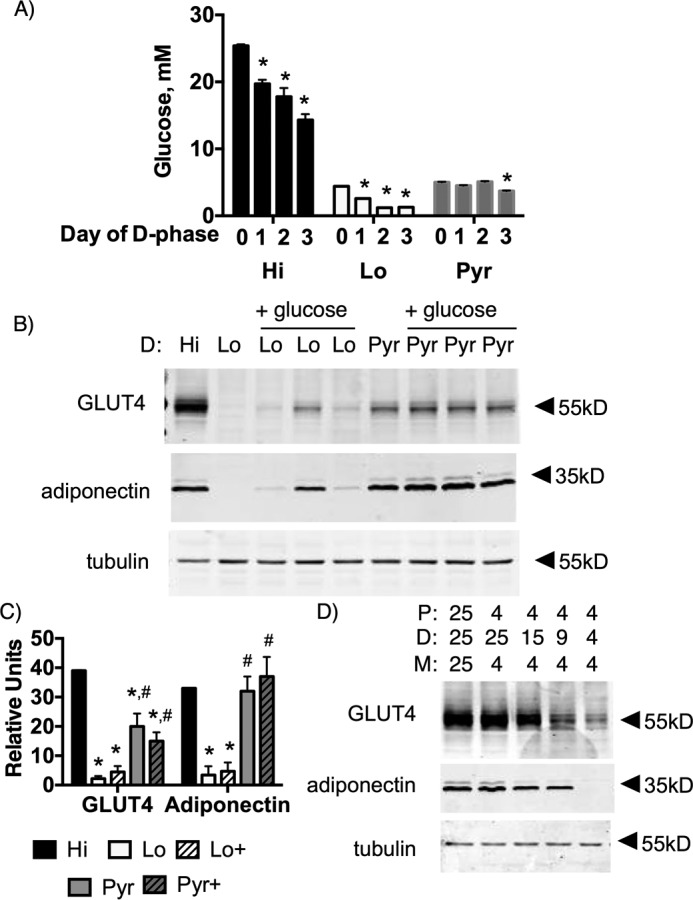
4 mm glucose does not inhibit differentiation as a result of glucose starvation. A, glucose concentration in medium on days 0, 1, 2, and 3 of the D phase. *, p < 0.001 for n = 4 independent experiments, a significant difference from day 0 glucose concentration. One-way analysis of variance was used to identify significant differences in glucose concentration. 3T3-L1 cells were grown in 4 mm glucose and pulsed with 4 mm glucose (Lo), 25 mm glucose (Hi) or 4 mm glucose + 42 mm pyruvate (Pyr) during the D phase. Glucose was added (+glucose) on day 2 to restore glucose levels at starting concentrations. B, a representative cohort. C, quantification of three replicates of the cohort. Data were analyzed by one-way analysis of variance (p < 0.05 for n = 3 independent cohort experiments. *, significant difference compared with Hi glucose cells; #, significant difference compared with Lo glucose cells. D, glucose pulse dose response during D phase. Protein from whole-cell lysates of day 9 adipocytes were separated via SDS-PAGE and immunoblotted for GLUT4, adiponectin, and tubulin in both B and D.
Low glucose exhibits reduced total NADH/P levels
We hypothesized that Hi glucose might be required for anabolic metabolism mediated by flux through the pentose phosphate pathway (PPP) for either NADPH regeneration, nucleotide biosynthesis via generation of phosphoribosyl pyrophosphate (PRPP), or both. To assess the importance of the PPP, cells treated during the D phase in Hi, Lo, or Pyr medium without or with 6-AN, a potent inhibitor of the PPP that targets 6-phosphogluconate dehydrogenase, were harvested at day 9 post-differentiation. 6-AN treatment during the D phase completely inhibited the ability of the 3T3-L1 preadipocytes to differentiate (Fig. 4A), indicating that the PPP is required during the D phase for differentiation and maturation of adipocytes.
Figure 4.
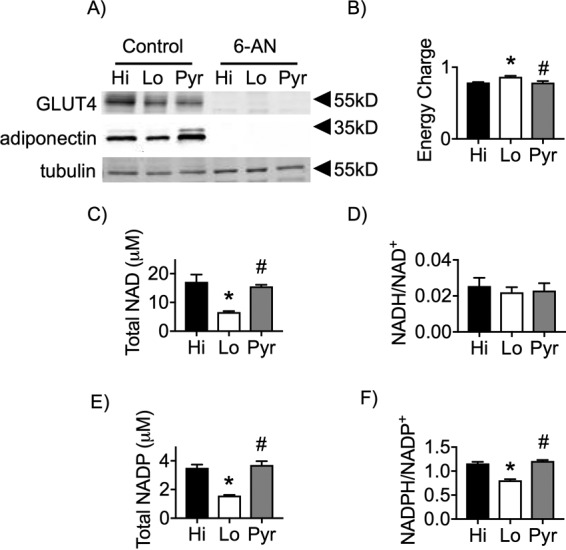
Lo glucose–differentiated cells exhibit a reduced NADH/P ratio. A, 3T3-L1 adipocytes were induced for differentiation and pulsed with Hi glucose, Lo glucose, or Pyr without or with 5 mm 6-AN and cultured in Lo glucose medium during the P and M phases of their cell culture. Protein from whole-cell lysates of day 9 adipocytes were separated via SDS-PAGE and immunoblotted for GLUT4, adiponectin, and tubulin. B–F, 3T3-L1 adipocytes were induced for differentiation and pulsed with Hi glucose, Lo glucose, or Pyr and cultured in Lo glucose medium during the P and M phases of their cell culture. Day 9 adipocytes were collected and lysed accordingly, and the supernatants were analyzed via HPLC purification and subsequent spectrofluorophotometry. One-way analysis of variance was used to identify significant differences (p < 0.05 for n = 3 independent experiments) in expression data. *, differences compared with Hi glucose cells; #, differences compared with Lo glucose cells.
The energy charge of the cells under each of the nutrient conditions was found to be within the normal, expected range (Fig. 4B), supporting that changes in ATP, ADP, and AMP levels are not mediating the differential effects of Lo versus Hi glucose. Because 6-AN is known to decrease cellular NAD levels (21), we next measured total NAD and NADP pools. Cells treated with Lo glucose during D phase exhibited a significant reduction in their total NAD and NADP pools, whereas Pyr maintained levels equivalent to that of cells differentiated under Hi conditions (Fig. 4, C and E). Despite differences in total NAD levels, there were no significant differences in the NADH/NAD+ ratio (Fig. 4D), consistent with the measures of energy charge. In contrast, the NADPH/NADP+ ratio was decreased in Lo glucose cells, suggesting that the capacity for reductive biochemistry is reduced.
Nicotinamide promotes adipocyte differentiation and adiponectin
To begin to determine why Lo glucose in the D phase led to lower total NAD/P levels, we evaluated several aspects of the of the NAD/P synthesis pathway (Fig. 5A). Expression of nadk mRNA, which encodes NAD kinase, which produces NADP from NAD+, was unaffected by nutrient conditions during the D phase (Fig. 5B). In contrast, expression of NAMPT protein, responsible for regeneration of NAD from NAM in the salvage pathway, was significantly reduced under both Lo and Pyr nutrient conditions (Fig. 5C). This suggested that these cells had a reduced ability to produce NAD through the salvage pathway (22). Similarly, the total pool of PRPP was significantly reduced under both Lo and Pyr conditions when measured on day 3 at the end of the D phase (Fig. 5D). Taken together, neither reduced NAMPT expression nor reduced PRPP levels were limiting for the synthesis of NAD/P in the maturing adipocytes pulsed with Pyr.
Figure 5.
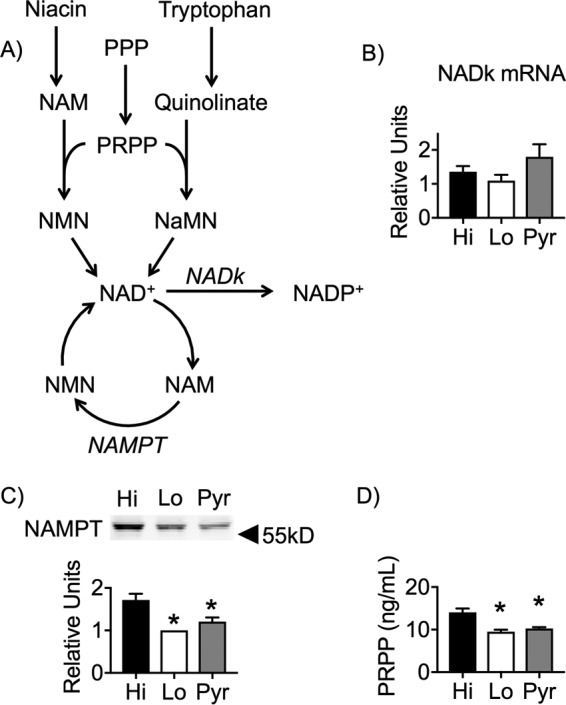
Reductions in both NAMPT and PRPP levels were not sufficient to impair NAD synthesis in Pyr cells. A, diagram of NAD/P synthesis metabolic pathways. 3T3-L1 adipocytes were induced for differentiation and pulsed with Hi glucose, Lo glucose, or Pyr and were cultured in Lo glucose medium during the P and M phases of their cell culture. B, mRNA isolated from day 9 adipocytes was analyzed via qRT-PCR for the relative expression of nadk. C, protein from whole-cell lysates of day 9 adipocytes were separated via SDS-PAGE and Western-blotted for NAMPT. D, day 9 adipocytes were collected and lysed accordingly, and the supernatants were analyzed via ELISA for levels of PRPP. 95% confidence intervals were used to determine statistical differences in immunoblot densitometry. One-way analysis of variance was used to identify significant differences (p < 0.05 for n = 3 independent experiments) in expression and ELISA data. *, differences compared with Hi glucose cells; #, differences compared with Lo glucose cells.
We next treated the cells with NAM, a bioavailable precursor for NAD synthesis, during the D phase to promote flux through the pathway and increase the total NAD pool (Fig. 6A). This successfully increased the levels of NAD in the Lo glucose cells at day 3 of differentiation. Interestingly, we discovered that NAM treatment of Lo glucose cells partially recovered GLUT4 expression and, more importantly, completely rescued adiponectin (Fig. 6B). Taken together, these data suggest that NAD is required for adipocyte differentiation and maturation, as determined by expression of adiponectin when, at day 3 of differentiation, total NAD levels matched the levels found under Hi glucose conditions. NAM was only partially able to restore glut4 mRNA and protein levels in Lo glucose cells to match those found in Pyr-treated cells but not Hi glucose (Fig. 6, B and C). Finally, NAM treatment was sufficient to restore pparγ and aP2 mRNA in Lo glucose cells, indicating that NAM was limiting for early markers of adipocyte differentiation (Fig. 6, D and E).
Figure 6.
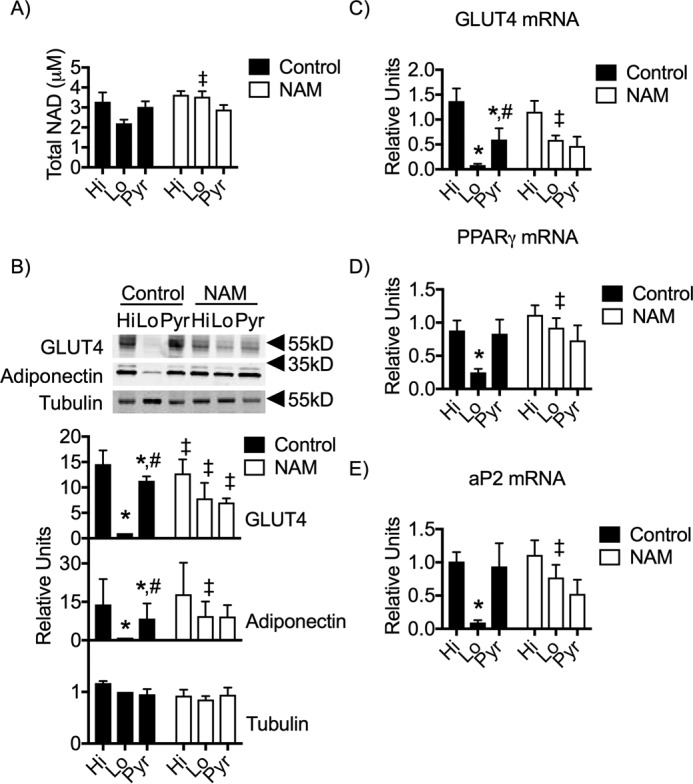
NAM supplementation partially rescues GLUT4 expression and fully supports adiponectin expression. 3T3-L1 adipocytes were induced for differentiation and pulsed with Hi glucose, Lo glucose, or Pyr without or with 15 mm NAM and cultured in Lo glucose medium during the P and M phases of their cell culture A, day 3 adipocytes were collected and lysed accordingly, and the supernatants were analyzed via ELISA for levels of total NAD. B, protein from whole-cell lysates of day 9 adipocytes were separated via SDS-PAGE and immunoblotted for GLUT4, adiponectin, and tubulin. C–E, mRNA isolated from day 9 adipocytes were analyzed via qRT-PCR for the relative expression of glut4, pparg, and ap2. 95% confidence intervals were used to determine statistical differences in immunoblot densitometry. One-way analysis of variance was used to identify significant differences in expression and ELISA data. *, differences compared with Hi glucose cells; #, differences compared with Lo glucose cells. ‡, difference comparing NAM treatment with the untreated control.
Loss of LXRE in the GLUT4 promotor blocks the glucose effect
Expression of GLUT4 mRNA and protein were only partially restored by either NAM or pyruvate pulses in the D phase, indicating an alternative glucose-dependent pathway leading to Glut4 expression. To determine whether the reduction of glut4 mRNA was due to a transcriptional mechanism, we performed an in vitro glut4 promoter/luciferase reporter (-895-hGLUT4-Luc) transcription assay in day 6 adipocytes grown in Lo glucose and pulsed with Hi glucose or Pyr during the D phase. As expected, luciferase activity was highest in cells with the Hi glucose pulse and intermediate in cells pulsed with Pyr (Fig. 7). Previous studies from our laboratory have shown that metabolic signals regulate glut4 promoter activity in 3T3-L1 adipocytes through the LXRE element in the Glut4 gene promoter (23). To test the role of the glut4 LXRE, we tested a mutant version of -895-hGLUT4-Luc that carried a loss-of-function mutation in the LXRE (ΔLXRE-Luc). The glucose-dependent activity of the mutated ΔLXRE-Luc probes was lost, and expression of the mutant probe remained at the basal level of activation. These data suggest that Hi glucose activates glut4 transcription through the LXRE.
Figure 7.
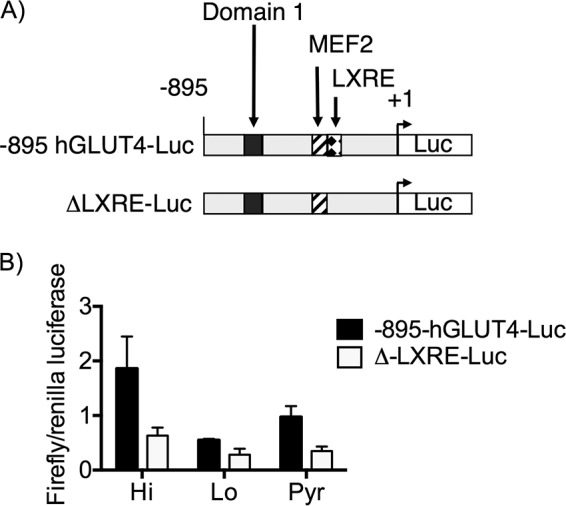
The LXRE region of the glut4 promoter is required for glucose-dependent differentiation. Shown is a schematic of the luciferase reporter gene under the regulator control of two different glut4 promotors, differing only by their inclusion of the LXRE region. 3T3-L1 adipocytes were induced for differentiation and pulsed with Hi glucose, Lo glucose, or Pyr and cultured in Lo glucose medium during the P and M phases of their cell culture. Day 6 adipocytes were collected and lysed accordingly, and the supernatants were analyzed for luciferase expression. 95% confidence intervals were used to determine statistical differences (p < 0.05 for n = 3 independent experiments. *, differences compared with Hi glucose cells; #, differences compared with Lo glucose cells with the same glut4 promoter region.
Differentiation in low glucose inhibits lipogenesis
A key function of an adipocyte is to convert and store excess nutrients in lipid droplets. Oil Red O staining of day 9 adipocytes showed that Lo glucose cells exhibited a significant reduction in their ability to accumulate neutral lipid (Fig. 8, A and B). Pulsing cells with high glucose resulted in an increased number of cells incorporating neutral lipid and clearly more neutral lipid per cell. Interestingly, Pyr-differentiated cells displayed an intermediate effect with numerous cells incorporating less neutral lipid per cell. To determine whether this Pyr effect is an artifact of partial adipocyte differentiation, we determined whether the enzymes and organelles required for lipogenesis were intact. Expression of fas and srebp1c mRNAs was significantly reduced in Lo glucose cells, whereas their expression was fully supported with a Pyr pulse (Fig. 8, C and D). Similarly, mitochondrial and peroxisomal levels, as determined by succinate dehydrogenase and peroxisomal catalase, were indistinguishable when cells were pulsed with either Hi glucose or Pyr (Fig. 8E). Although pyruvate supplementation of Lo glucose levels during differentiation was able to support the machinery needed for lipogenesis to proceed, total lipid accumulation was lower, consistent with the lower levels of GLUT4 expression. Collectively, these results support the notion that a brief exposure to high glucose is required, in combination with relevant hormonal signals, to initiate a program of adipocyte differentiation and maturation of new adipocytes.
Figure 8.
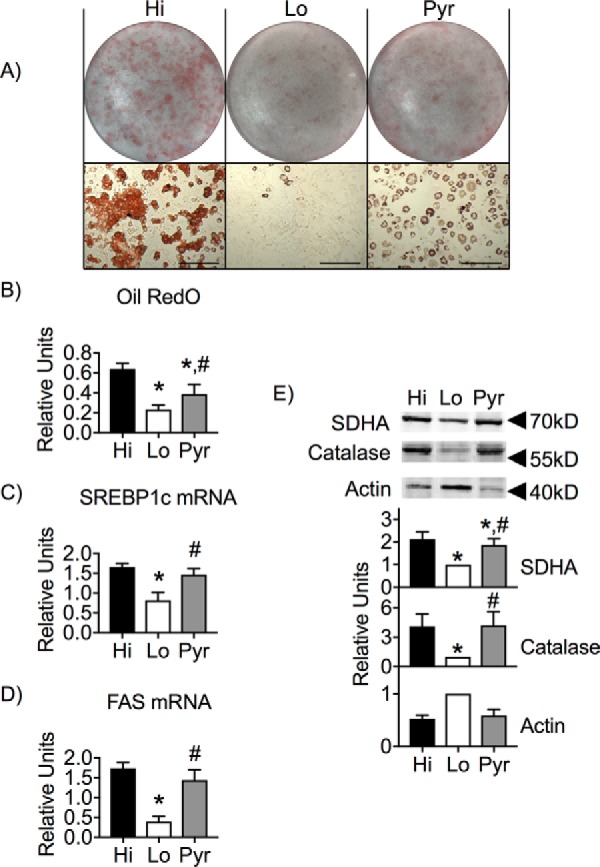
Neutral lipid accumulation is glucose-dependent. 3T3-L1 adipocytes were induced for differentiation and pulsed with Hi glucose, Lo glucose, or Pyr and cultured in Lo glucose medium during the P and M phases of their cell culture. A and B, day 9 adipocytes were fixed and stained with Oil Red O (A), which was then eluted for detection of absorbance at 500 nm (B). C and D, mRNA isolated from day 9 adipocytes were analyzed via qRT-PCR for the relative expression of srebp1c and fas. E, protein from whole-cell lysates of day 9 adipocytes were separated via SDS-PAGE and immunoblotted for succinate dehydrogenase, peroxisomal catalase, and actin. 95% confidence intervals were used to determine statistical differences in immunoblot densitometry and Oil Red O absorbance. One-way analysis of variance was used to identify significant differences (p < 0.03 for n = 3 independent experiments) in expression data. *, differences compared with Hi glucose cells; #, differences compared with Lo glucose cells.
Discussion
We have shown that a Hi glucose (25 mm) pulse early in differentiation plays a critical role in the regulation of adipocyte development in vitro. When differentiation is initiated, maturation of the adipocyte is no longer dependent on 25 mm glucose but, rather, proceeds normally at 4 mm glucose. We showed that there is a critical role for the PPP during the D phase of differentiation. This finding was supported by the observation that the NADPH/NADP+ ratio was significantly lower in cells pulsed with 4 mm glucose during the D phase. This strongly suggests that there is a burst of anabolic metabolism required during the D phase of adipocyte development. The burst of anabolic metabolism is driven by high levels of nutrients, in this case glucose, exactly as one would expect for expansion of adipocytes under chronic positive energy balance.
We looked specifically at several important functions of a differentiated and mature adipocyte that are regulated uniquely by the availability of glucose during the initial days of differentiation. The first is the expression of GLUT4, which is required in the mature adipocyte to properly clear glucose from the blood and store it in the form of triglycerides. The second function related to GLUT4 function is de novo lipogenesis and accumulation of neutral lipid in mature adipocytes. Both of these functions were dependent upon glucose availability during the D phase (days 0–3) of adipocyte development in vitro. Pulsing cells with pyruvate instead of high glucose (4 mm glucose and 42 mm pyruvate) only partially compensated for 25 mm glucose with intermediate promotion of GLUT4 expression and accumulation of neutral lipids at day 9 of development. Interestingly, 3T3-L1 adipocytes differentiated in medium containing only pyruvate as the primary carbon source (no glucose and 50 mm pyruvate) showed a complete lack in their ability to differentiate (data not shown). Therefore, the pyruvate supplementation is likely serving to spare what little glucose is available during the D phase, allowing it to be directed where it is needed for adipocyte differentiation to occur.
Another very important adipose function is the production of adipokines, such as adiponectin. This secreted protein functions primarily in sensitizing tissues to insulin stimulation and is important for whole-body glucose homeostasis (14). In contrast to the nutrient storage functions of adipose tissue, we found that adiponectin expression was fully compensated by supplementation of low-glucose medium with pyruvate. Furthermore, this was mechanistically linked to the redox reactions of the cell because supplementation of 4 mm glucose medium with NAM to increase NAD/P levels restored adiponectin expression.
Interestingly, Wellen et al. (24) reported a glucose dependence for differentiation of 3T3-L1 adipocytes. By knocking down ATP-citrate lyase, they demonstrated that glucose was required for production of cytosolic acetyl-CoA to be used for histone acetylation and full expression of GLUT4 mRNA as of day 4 of differentiation. Under these conditions, supplementation with sodium acetate was able to restore GLUT4 expression. In our studies, a pulse of sodium acetate was not able to compensate for 25 mm glucose for either GLUT4 or adiponectin expression in cells harvested after 9 days of differentiation and maturation. Thus, glucose is playing a role in additional pathways during differentiation.
GLUT4 and adiponectin regulation are both glucose-dependent, but their regulation by glucose is not by the same mechanism. Adiponectin expression and early markers of differentiation were rescued in Lo glucose by pulsing with NAM. Because we found that total NAD levels were already rising in 3T3-L1 adipocytes on day 3 of the pulse with 25 mm glucose or 42 mm pyruvate, it is possible that NAD+ was limiting in the Lo glucose cells, preventing differentiation from proceeding. These data are consistent with Shapiro et al. (25), who found that NAM was required for development of ppar-γ mRNA expression in umbilical cord–derived mesenchymal stem cells differentiation in vitro in 4 mm glucose.
GLUT4 expression was only partially restored by pulsing cells with NAM and/or Pyr. We showed that transcriptional activity of the glut4 promoter in vitro required a 25 mm glucose pulse and that a loss-of-function mutation in the glut4 LXRE was no longer responsive to glucose availability. It is unclear how a glucose-dependent signal is transduced through the glut4 LXRE. In 3T3-L1 adipocytes, the glut4 LXRE is partially occupied by LXR-α (26–28). Glucose has been shown to be a direct activator of LXR and LXR target genes in the liver (29). It is uncertain whether LXR isoforms serve as positive activators of the glut4-LXRE in adipose tissue. LXR-α/β knock-out mice have increased glut4 mRNA expression in the perigonadal fat pads (30). Thus, it is an open question what binds to the glut4-LXRE and how signaling through high glucose is transduced through this promoter element.
NAM supplementation increased the total pool of NAD available in Lo glucose–cultured cells. In addition to regulating the redox state of the cell, NAD is key to enzymatic function of sirtuins and PARPs, which convert NAD+ to NAM to either deacetylate or PARylate target proteins, respectively (22, 31). Because Sirt1 and Sirt2 have both been shown to inhibit adipogenesis, we do not favor that an increase in NAD+ required for increasing sirtuin activity is occurring during the D phase (32, 33). NAD+-dependent PARylation through PARP1 activity may play a specific role in adipogenesis; however, the literature supporting this role is conflicting. Whole-body knockout of PARP1 has revealed a drastic reduction of adipocyte differentiation, complete loss of PPARγ2, aP2, and adiponectin in vitro, and reduced adiposity in mice in vivo (34, 35). In contrast Luo et al. (36) showed that conditional knockdown of PARP1 activity in adipose tissue during the first 48 h of adipocyte differentiation might be a key regulator of the transcriptional program that underlies adipocyte differentiation. These papers highlight both a permissive role and an inhibitory role for NAD+ PARP1 activity. Our data suggested that NAD metabolism was permissive for adipogenesis, presumably through regulation of the NADP pool size and NADPH/NADP+ ratio. Because the signal for expansion of the adipose pad arises from nutrient excess, an allosteric mechanism for promoting differentiation is relevant. Further investigation is warranted to elucidate the precise mechanism through which glucose and NADP metabolism are mediating these effects.
Our findings may explain, in part, why adipocyte hyperplasia occurs later than adipocyte hypertrophy in the progression of obesity (3, 4, 17). Obesity leads to decreased expression of GLUT4 protein in adipose tissue (9). In the hypertrophic adipose pad, decreased GLUT4-dependent glucose uptake and higher plasma glucose levels may provide an optimal environment for increasing the rate of differentiation of adipocyte progenitors in the fat pad. Obese patients with impaired ability to increase adipose hyperplasia during the development of obesity may be at higher risk for developing metabolic disease (37). Our data begin to provide a mechanistic understanding of nutrient control of adipocyte differentiation. We demonstrate that carbohydrate metabolism, likely through pentose phosphate pathway intermediates and NADPH metabolism, plays a significant role in multiple pathways required for adipogenesis.
Materials and methods
Cell culture
3T3-L1 cells were cultured and differentiated as described previously, except the glucose concentration of the medium was varied as indicated (38). Preadipocytes were grown to confluence in 4 mm glucose medium containing 10% calf serum. Differentiation was stimulated by changing to medium containing 10% fetal bovine serum and a mixture of 175 nm insulin, 1 μm dexamethasone, and 0.5 mm isobutyl-1-methylxanthine. After 3 days in this differentiation medium, cells were then changed to medium containing 10% fetal bovine serum without the mixture and replenished again 3 days later. Unless otherwise indicated, cells were cultured in 4 mm glucose-containing medium on the first 7 days and the last 6 days of tissue culture, whereas the medium supplied during the middle 3-day D phase contained either 4 or 25 mm glucose, 4 mm glucose supplemented with 42 mm pyruvate, 4 mm glucose supplemented with 50 μm palmitic acid, or 4 mm glucose supplemented with 50 μm oleic acid. Where indicated, cells were treated with 15 mm nicotinamide or 5 mm 6-aminonicotinamide only during the 3-day D phase.
The adipose stromal fraction was isolated from inguinal fat pads from C57BL6 mice. The stromal vascular fraction was isolated by differential centrifugation following collagenase digestion of adipose pads as described previously (39). Stromal vascular cells were counted, plated on 35-mm dishes, and grown to confluence in F12/DMEM supplemented with 10% FBS. Two days post-confluence, cells were switched to differentiation medium as described for 3T3-L1 preadipocytes.
Whole-cell lysates and immunoblots
Whole-cell extracts were prepared in lysis buffer containing 20 mm HEPES, 1% Nonidet P-40, 2 mm EDTA, 10 mm sodium fluoride, 10 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 1 mm molybdate, protease inhibitor mixture (Complete Mini EDTA-free protease inhibitor mixture, Roche), 1 mm PMSF, and 50 mm DTT. Protein concentrations were determined using a Coomassie Plus (Bradford) assay kit (Pierce). Lysates were fractionated using SDS-PAGE, and proteins were transferred to Immobilon-FL polyvinylidene fluoride membranes (EMD Millipore Corp., Billerica, MA) Membranes were stained with anti-GLUT4 antibody (sc-1608, Santa Cruz Biotechnology), anti-adiponectin antibody (a gift from Philipp Scherer), anti-NAMPT antibody (A300–372A, Bethyl), anti-SDH antibody (mitochondrial biogenesis mixture, ab123545, Abcam), anti-peroxisomal catalase antibody (ab15834, Abcam), anti-actin antibody (mitochondrial biogenesis mixture, ab123545, Abcam), or anti-α/β-tubulin antibody (2148, Cell Signaling Technology) and visualized with appropriate secondary antibodies conjugated with Alexa Fluor 680 (Invitrogen). Fluorescence was quantified with a Li-Cor Odyssey imager (Li-Cor Biosciences). Protein expression data were normalized to tubulin as a loading control.
RNA isolation and qRT-PCR
mRNA was extracted using guanidinium isothiocyanate and purified by density equilibrium centrifugation using a CsCl gradient as described previously (40). Samples were stored as ethanol precipitates at −20 °C until further analysis. RNA was quantified by UV absorbance at 260, 280, and 320 nm. Messenger RNA levels of GLUT4, aP2, PPARγ, SREBP-1c, and FAS were quantified by quantitative real-time PCR (qRT-PCR). Primer sequences were as follows: mGLUT4, 5′-AAAAGTGCCTGAAACCAGAG-3′ (forward), 5′-TCACCTCCTGCTCTAAAAGG-3′ (reverse); maP2 5′-GCGTGGAATTCGATGAAATCA-3′ (forward), 5′-CCCGCCATCTAGGGTTATGA-3′ (reverse); mPPARγ 5′-GTGCCAGTTTCGATCCGTAGA-3′ (forward), 5′-GGCCAGCATC GTGTAGATGA-3′ (reverse); mSREBP-1c, 5′-GTGAGCCTGACAAGCAATCA-3′ (forward), 5′-GGTGCCTACAGAGCAAGAG-3′ (reverse); mFAS, 5′-GCTGCGGAAACTTCAGGAAAT-3′ (forward), 5′-AGAGACGTGTCACTCCTGGACTT-3′ (reverse). The relative mRNA levels were calculated using a standard curve developed from control 3T3-L1 adipocytes. All qRT-PCRs were completed using a modular thermal cycler platform composed of a C1000 TouchTM thermal cycler chassis (Bio-Rad) and a CFX96TM optical reaction module (Bio-Rad). Data from qRT-PCR was read using CFX ManagerTM software (Bio-Rad).
NAD/P and PRPP measurements
The total pool of NAD was estimated using a colorimetric quantification kit (ab65348, Abcam), and the total pool of PRPP was estimated using a solid-phase competitive ELISA (173862, USBiological). The ratios of NAD+/NADH and NADP+/NADPH were determined by HPLC purification and subsequent spectrofluorophotometry as described previously (41). Briefly, HPLC (Shimadzu LC-20A high precision binary gradient HPLC system) and fluorescence or UV-visible diode array spectrometry were used to resolve and detect NADH, NADPH, NAD+, and NADP+. The mobile phase consisted of 100 mm KH2PO4 and 1.0 mm tetrabutylammonium sulfate at pH 6.0 (buffer A) and CH3CN (buffer B) with a flow rate of 1.0 ml/min over an Eclipse Plus C18 column with 5-μm-diameter beads, 4.6 × 150 mm in length (Agilent). NADH and NADPH were resolved (100-μl injection) using the following stepwise gradients of buffer A/B: 100%/0% for 2.5 min, 95%/5% for 5 min, 85%/15% for 7.5 min, and 100%/0% for 10 min. NAD+ and NADP+ were resolved (100-μl injection) using the following stepwise gradients of buffer A/B: 100%/0% for 5 min, 85%/15% for 5 min, and 100/0% for 10 min. Concentrations of NADH and NADPH were detected by fluorescence (excitation, 340 nm; emission, 460 nm) and NAD+ and NADP+ by absorption at 254 nm and quantified based on the integrated area of standards.
Glucose assay
Glucose concentration was measured using the anthrone reagent as described previously (42). The glucose standard curve was generated using glucose-free medium supplemented with 10% FBS instead of water.
Luciferase reporter assays
3T3-L1 adipocytes were differentiated and electroporated as described previously (38). The luciferase constructs -895-hGLUT4-Luc and ΔLXRE-Luc have been described previously and were used where indicated (28, 38) Luciferase assays were performed 24 h after transfection using the Dual-Glo luciferase kit (Promega).
Oil Red O staining
Oil Red O staining was performed in cells that had been washed twice in PBS prior to fixation by 10% formalin for 30 min at room temperature. After fixation, the cells were washed twice with PBS and then incubated in 60% isopropanol for 5 min at room temperature. The cells were then stained with a filtered, 0.3% solution of Oil Red O in 60% isopropanol for 5 min at room temperature. The Oil Red O solution was removed, and cells were washed five times with water or until the rinse water ran clear. The Oil Red O stain was eluted with 2 ml of 99% isopropanol, and absorbance at 500 nm was recorded. The cells were washed twice with water and counterstained with 5 mg/ml solution of crystal violet in water for 30 min at room temperature. Crystal violet stain was removed, and cells were washed five times with water or until the rinse water ran clear. The stain was eluted in 2 ml of methanol. The absorbance at 540 nm was recorded.
Statistical analysis
Data are expressed as mean and standard error of the mean. Differences were reported through the use of either 95% confidence intervals for data normalized to control or one-way analysis of variance statistical tests, as indicated.
Author contributions
A. L. O. conceived and coordinated the study. A. L. O. and R. M. G. analyzed the data and wrote the paper. R. M. G. and B. A. G. acquired data for Figs. 1–5 and 8. R. M. J. and L. I. S. acquired data for Fig. 6. J. M. G. acquired data for Fig. 7. All authors approved the final version of manuscript.
This manuscript was supported in part by a grant from the Harold Hamm Diabetes Center. The authors declare that they have no conflicts of interest with the contents of this article.
- P
- proliferation
- D
- differentiation
- M
- maturation
- Pyr
- pyruvate
- Lac
- lactate
- Ac
- sodium acetate
- PPP
- pentose phosphate pathway
- PRPP
- phosphoribosyl pyrophosphate
- PARP
- Poly(ADP-ribose)polymerase
- qRT-PCR
- quantitative real-time PCR
- 6-AN
- 6-aminonicotinamide
- NAM
- nicotinamide
- NAMPT
- nicotinamide phosphoribosyltransferase.
References
- 1. Jo J., Gavrilova O., Pack S., Jou W., Mullen S., Sumner A. E., Cushman S. W., and Periwal V. (2009) Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comput. Biol. 5, e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sun K., Kusminski C. M., and Scherer P. E. (2011) Adipose tissue remodeling and obesity. J. Clin. Invest. 121, 2094–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Q. A., Tao C., Gupta R. K., and Scherer P. E. (2013) Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 19, 1338–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. MacKellar J., Cushman S. W., and Periwal V. (2010) Waves of adipose tissue growth in the genetically obese Zucker fatty rat. PLoS ONE 5, e8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosen E. D., and Spiegelman B. M. (2014) What we talk about when we talk about fat. Cell 156, 20–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rutkowski J. M., Stern J. H., and Scherer P. E. (2015) The cell biology of fat expansion. J. Cell Biol. 208, 501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leto D., and Saltiel A. R. (2012) Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 13, 383–396 [DOI] [PubMed] [Google Scholar]
- 8. Hauner H., Röhrig K., Spelleken M., Liu L. S., and Eckel J. (1998) Development of insulin-responsive glucose uptake and GLUT4 expression in differentiating human adipocyte precursor cells. Int. J. Obes. Relat. Metab. Disord. 22, 448–453 [DOI] [PubMed] [Google Scholar]
- 9. Atkinson B. J., Griesel B. A., King C. D., Josey M. A., and Olson A. L. (2013) Moderate GLUT4 overexpression improves insulin sensitivity and fasting triglyceridemia in high-fat diet-fed transgenic mice. Diabetes 62, 2249–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith U., and Kahn B. B. (2016) Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J. Internal Med. 280, 465–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tomas E., Tsao T. S., Saha A. K., Murrey H. E., Zhang C. C., Itani S. I., Lodish H. F., and Ruderman N. B. (2002) Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc. Natl. Acad. Sci. U.S.A. 99, 16309–16313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamauchi T., Kamon J., Minokoshi Y., Ito Y., Waki H., Uchida S., Yamashita S., Noda M., Kita S., Ueki K., Eto K., Akanuma Y., Froguel P., Foufelle F., Ferre P., et al. (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295 [DOI] [PubMed] [Google Scholar]
- 13. Yamauchi T., Kamon J., Waki H., Imai Y., Shimozawa N., Hioki K., Uchida S., Ito Y., Takakuwa K., Matsui J., Takata M., Eto K., Terauchi Y., Komeda K., Tsunoda M., et al. (2003) Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J. Biol. Chem. 278, 2461–2468 [DOI] [PubMed] [Google Scholar]
- 14. Ye R., and Scherer P. E. (2013) Adiponectin, driver or passenger on the road to insulin sensitivity? Mol. Metab. 2, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scherer P. E., Williams S., Fogliano M., Baldini G., and Lodish H. F. (1995) A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 270, 26746–26749 [DOI] [PubMed] [Google Scholar]
- 16. Hu E., Liang P., and Spiegelman B. M. (1996) AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 271, 10697–10703 [DOI] [PubMed] [Google Scholar]
- 17. Krotkiewski M., Björntorp P., Sjöström L., and Smith U. (1983) Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J. Clin. Invest. 72, 1150–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kadowaki T., Yamauchi T., Kubota N., Hara K., Ueki K., and Tobe K. (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116, 1784–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saltiel A. R., and Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 20. Letexier D., Pinteur C., Large V., Fréring V., and Beylot M. (2003) Comparison of the expression and activity of the lipogenic pathway in human and rat adipose tissue. J. Lipid Res. 44, 2127–2134 [DOI] [PubMed] [Google Scholar]
- 21. Budihardjo I. I., Walker D. L., Svingen P. A., Buckwalter C. A., Desnoyers S., Eckdahl S., Shah G. M., Poirier G. G., Reid J. M., Ames M. M., and Kaufmann S. H. (1998) 6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin. Clin. Cancer Res. 4, 117–130 [PubMed] [Google Scholar]
- 22. Revollo J. R., Grimm A. A., and Imai S. (2004) The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 279, 50754–50763 [DOI] [PubMed] [Google Scholar]
- 23. Griesel B. A., Weems J., Russell R. A., Abel E. D., Humphries K., and Olson A. L. (2010) Acute inhibition of fatty acid import inhibits GLUT4 transcription in adipose tissue, but not skeletal or cardiac muscle tissue, partly through liver X receptor (LXR) signaling. Diabetes 59, 800–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wellen K. E., Hatzivassiliou G., Sachdeva U. M., Bui T. V., Cross J. R., and Thompson C. B. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shapiro A. L., Boyle K. E., Dabelea D., Patinkin Z. W., De la Houssaye B., Ringham B. M., Glueck D. H., Barbour L. A., Norris J. M., and Friedman J. E. (2016) Nicotinamide promotes adipogenesis in umbilical cord-derived mesenchymal stem cells and is associated with neonatal adiposity: The Healthy Start BabyBUMP Project. PLoS ONE 11, e0159575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dalen K. T., Ulven S. M., Bamberg K., Gustafsson J. A., and Nebb H. I. (2003) Expression of the insulin-responsive glucose transporter GLUT4 in adipocytes is dependent on liver X receptor α. J. Biol. Chem. 278, 48283–48291 [DOI] [PubMed] [Google Scholar]
- 27. Laffitte B. A., Chao L. C., Li J., Walczak R., Hummasti S., Joseph S. B., Castrillo A., Wilpitz D. C., Mangelsdorf D. J., Collins J. L., Saez E., and Tontonoz P. (2003) Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc. Natl. Acad. Sci. U.S.A. 100, 5419–5424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weems J. C., Griesel B. A., and Olson A. L. (2012) Class II histone deacetylases downregulate GLUT4 transcription in response to increased cAMP signaling in cultured adipocytes and fasting mice. Diabetes 61, 1404–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitro N., Mak P. A., Vargas L., Godio C., Hampton E., Molteni V., Kreusch A., and Saez E. (2007) The nuclear receptor LXR is a glucose sensor. Nature 445, 219–223 [DOI] [PubMed] [Google Scholar]
- 30. Korach-André M., Archer A., Barros R. P., Parini P., and Gustafsson J. Å. (2011) Both liver-X receptor (LXR) isoforms control energy expenditure by regulating brown adipose tissue activity. Proc. Natl. Acad. Sci. U.S.A. 108, 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bai P., and Cantó C. (2012) The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab 16, 290–295 [DOI] [PubMed] [Google Scholar]
- 32. Picard F., Kurtev M., Chung N., Topark-Ngarm A., Senawong T., Machado De Oliveira R., Leid M., McBurney M. W., and Guarente L. (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 429, 771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jing E., Gesta S., and Kahn C. R. (2007) SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 6, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Erener S., Hesse M., Kostadinova R., and Hottiger M. O. (2012) Poly(ADP-ribose)polymerase-1 (PARP1) controls adipogenic gene expression and adipocyte function. Mol. Endocrinol. 26, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Devalaraja-Narashimha K., and Padanilam B. J. (2010) PARP1 deficiency exacerbates diet-induced obesity in mice. J. Endocrinol. 205, 243–252 [DOI] [PubMed] [Google Scholar]
- 36. Luo X., Ryu K. W., Kim D. S., Nandu T., Medina C. J., Gupte R., Gibson B. A., Soccio R. E., Yu Y., Gupta R. K., and Kraus W. L. (2017) PARP-1 Controls the adipogenic transcriptional program by PARylating C/EBPβ and modulating its transcriptional activity. Mol. Cell 65, 260–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gustafson B., Gogg S., Hedjazifar S., Jenndahl L., Hammarstedt A., and Smith U. (2009) Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Endocrinol. Metab. 297, E999–E1003 [DOI] [PubMed] [Google Scholar]
- 38. Eyster C. A., Duggins Q. S., and Olson A. L. (2005) Expression of constitutively active Akt/protein kinase B signals GLUT4 translocation in the absence of an intact actin cytoskeleton. J. Biol. Chem. 280, 17978–17985 [DOI] [PubMed] [Google Scholar]
- 39. Church C. D., Berry R., and Rodeheffer M. S. (2014) Isolation and study of adipocyte precursors. Methods Enzymol. 537, 31–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thai M. V., Guruswamy S., Cao K. T., Pessin J. E., and Olson A. L. (1998) Myocyte enhancer factor 2 (MEF2)-binding site is required for GLUT4 gene expression in transgenic mice: regulation of MEF2 DNA binding activity in insulin-deficient diabetes. J. Biol. Chem. 273, 14285–14292 [DOI] [PubMed] [Google Scholar]
- 41. Rindler P. M., Cacciola A., Kinter M., and Szweda L. I. (2016) Catalase-dependent H2O2 consumption by cardiac mitochondria and redox-mediated loss in insulin signaling. Am. J. Physiol. Heart Circ. Physiol. 311, H1091–H1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hassid W. Z., and Abraham S. (1975) Determination of glycogen with anthrone reagent. Methods Enzymol. 3, 34–37 [Google Scholar]