

Abstract
Interleukin-1 receptor–associated kinase 4 (IRAK4) plays a critical role in innate immune signaling by Toll-like receptors (TLRs), and loss of IRAK4 activity in mice and humans increases susceptibility to bacterial infections and causes defects in TLR and IL1 ligand sensing. However, the mechanism by which IRAK4 activity regulates the production of downstream inflammatory cytokines is unclear. Using transcriptomic and biochemical analyses of human monocytes treated with a highly potent and selective inhibitor of IRAK4, we show that IRAK4 kinase activity controls the activation of interferon regulatory factor 5 (IRF5), a transcription factor implicated in the pathogenesis of multiple autoimmune diseases. Following TLR7/8 stimulation by its agonist R848, chemical inhibition of IRAK4 abolished IRF5 translocation to the nucleus and thus prevented IRF5 binding to and activation of the promoters of inflammatory cytokines in human monocytes. We also found that IKKβ, an upstream IRF5 activator, is phosphorylated in response to the agonist-induced TLR signaling. Of note, IRAK4 inhibition blocked IKKβ phosphorylation but did not block the nuclear translocation of NFκB, which was surprising, given the canonical role of IKKβ in phosphorylating IκB to allow NFκB activation. Moreover, pharmacological inhibition of either IKKβ or the serine/threonine protein kinase TAK1 in monocytes blocked TLR-induced cytokine production and IRF5 translocation to the nucleus, but not nuclear translocation of NFκB. Taken together, our data suggest a mechanism by which IRAK4 activity regulates TAK1 and IKKβ activation, leading to the nuclear translocation of IRF5 and induction of inflammatory cytokines in human monocytes.
Keywords: cytokine, inflammation, interferon regulatory factor, IRF5, NFκB, Toll-like receptor, TLR, IRAK4, TAK1, IKKβ
Introduction
Interleukin-1 receptor–associated kinase 4 (IRAK4)7 is a critical component of the Toll-like receptor/interleukin-1 receptor (TLR/IL1R) signaling pathway that plays a major role in innate immune responses and inflammation. Ablation of IRAK4 expression in mouse and in humans causes susceptibility to bacterial infections and defects in the ability to sense TLR and IL1 ligands (1, 2). Mice expressing kinase-inactive IRAK4 are resistant to multiple models of autoimmune and inflammatory disease and inhibitors of IRAK4 are currently under investigation in the clinic as therapeutics for multiple autoimmune and inflammatory disease as well as cancers (3). TLRs recognize and bind bacterial, viral, and fungal pathogen ligands, and IL1R recognizes the inflammatory IL1 family cytokines. Both TLRs and the IL1 family of receptors have Toll/IL1 receptor domains that bind the adaptor protein MyD88, which then associates with IRAK4 and IRAK1/2 via death domain interactions. The IRAKs then activate the ubiquitin E3 ligase TRAF6. Activation of the pathway induces strong inflammatory responses through nuclear factor kappa B (NFκB), interferon regulatory factor (IRF), and activator protein-1 (AP-1) responsive genes (4). However, work by multiple investigators has shown that inactivation of IRAK4 kinase activity has minimal effects on the activation of the canonical inflammatory pathways of NFκB and the MAP kinases in mice and human cells (5–7). These data lead to the question of how IRAK4 kinase activity controls pro-inflammatory cytokine production. Studies in murine cells suggest that IRAK4 kinase activity may control the stability of cytokine mRNA (6) but the impact of IRAK4 kinase activity on transcription factors other than NFκB or AP-1 has not been examined.
Interferon regulatory factor family members regulate defenses against pathogens via their DNA-binding domain, which recognizes interferon-stimulated response elements (ISRE) found on many genes that encode type I interferons as well as pro-inflammatory cytokines and chemokines. Overexpression of interferon-induced genes is a characteristic of multiple autoimmune disorders, such as systemic lupus erythematosus (SLE) (8), psoriasis (9), dermatomyositis (10), and rheumatoid arthritis (RA) (11). The transcription factor IRF5 has been implicated in TLR/IL1R signaling and is activated in a MyD88-dependent manner to induce cytokines and interferon (12). In particular, IRF5 is known to be a critical mediator of TLR7 signaling that requires not only MyD88, but also IRAK1/4 and TRAF6 in this pathway. Activation requires phosphorylation, dimerization, nuclear translocation, and binding to promoters with IRF binding elements (13). IRF5 is known to cooperate with the NFκB subunit RelA to promote transcription of pro-inflammatory cytokines in myeloid cells (14). Several groups have recently provided evidence in human myeloid cells that phosphorylation of IRF5 by IKKβ results in IRF5 activation and nuclear translocation following TLR ligation (15, 16) and that this process involved TAK1 (17).
In humans, IRF5 polymorphisms are associated with autoimmune and inflammatory conditions such as inflammatory bowel disease (18), RA (19), and systemic lupus erythematosus (20). Furthermore, it has been shown that IRF5−/− mice are protected from development of lupus in the Fcgr2b−/− Yaa, Fcgr2b−/− (21), and pristane-induced (22) models of the disease. In addition, mice deficient in IRF5 have decreased pro-inflammatory cytokines, such as IL6 and TNF, in their serum. They are therefore more susceptible to viral infections. After stimulation with unmethylated DNA and lipopolysaccharide, IRF5-deficient mice are more resistant to lethal shock. However, activation of NFκB is comparable to wild type in these mice (23). This is similar to what is seen in immune cells from IRAK4 kinase–inactive knock-in mice. Given the importance of IRF5 in regulating TLR-mediated cytokine and type I interferon production as described above, we set out to investigate the role of IRAK4 kinase in regulating IRF5 activation. Our data, generated using a highly potent and selective IRAK4 inhibitor (24), led us to propose that activation of the transcription factor IRF5 via TAK1 and IKKβ is the central mechanism by which IRAK4 kinase inhibitors mediate their anti-inflammatory effects. Thus we uncovered a novel role of IRAK4 in activation of the TAK1-IKKβ-IRF5 axis that leads to the induction of cytokines and interferons following TLR7/8 stimulation.
Results
IRAK4 kinase is essential for inflammatory mRNA and cytokine production
To determine the effect of IRAK4 kinase inhibition on TLR-induced cytokines we employed a recently developed potent and selective IRAK4 inhibitor PF-06426779 (compound 38 in Ref. 24). This compound has an IC50 of 1 nm against full-length IRAK4 kinase, a cell-based IC50 of 12 nm, and excellent selectivity against the human kinome and is >100× selective for IRAK4 over IRAK1 (24). PF-06426779 was assessed in a KiNativTM panel (ActivX Biosciences Inc.) in hPBMC cell lysates at 10, 1, and 0.1 μm. Estimated IC50s were more than 1 μm for all kinases assayed except for IRAK4 (0.01–0.026 μm) and IRAK3 (0.73 μm) (supplemental Table 1).
Primary human monocytes from healthy donors were pretreated with 1 μm compound for 30 min and then stimulated with the TLR7/8 ligand R848. Robust induction of IL1, IL6, and TNF cytokine protein production occurred with R848 treatment alone, whereas R848-induced IL1, IL6, and TNF cytokine production was significantly reduced by the inhibitor (5). Furthermore, mRNA levels of IL1, IL6, and TNF were induced by R848 by at least 10-fold at 1 h and by at least 100-fold at 4 h in all donors (Fig. 1). IRAK4 kinase inhibition reduced R848-induced IL1 mRNA levels by an average of 1.7-fold at 1 h (p = 0.008) and 2.6-fold at 4 h (p < 0.001); IL6 mRNA by 2.3-fold at 1 h (p = 0.001) and 4.2-fold at 4 h (p < 0.001); and TNF mRNA by 3.5- and 3.6-fold at 1 h (p = 0.003) and 4 h (p = 0.012) (n = 8 donors, Student's t test, Fig. 1), respectively. We had previously determined that despite the ability of IRAK inhibitors to decrease cytokine production, there are minimal effects on the activation status of the NFκB pathway (5). Because we observed here that regulation of cytokine production occurred at the mRNA level, we chose to query the mechanism of cytokine inhibition through genome-wide transcriptome analysis.
Figure 1.

IRAK4 kinase is essential for inflammatory cytokine mRNA expression. Primary human monocytes were stimulated with R848 in the presence and/or absence of the IRAK4 kinase inhibitor. IL1, IL6, and TNF mRNA expression was measured 1 and 4 h after R848 stimulation (n = 8 donors).
Transcriptional analysis suggests IRAK4 kinase regulates IRF5-mediated transcripts
Our goal was to determine what genes, groups of genes, and pathways were affected when IRAK4 kinase is inhibited. To answer this question, human monocytes were isolated from nine healthy donors. They were treated with and without IRAK4 kinase inhibitor and with and without R848, and RNA was collected at 1 h post stimulation. Global transcriptional analysis using Affymetrix genome-wide U133 2.0 plus arrays (GEO accession no. GSE86234) identified 860 genes that were differentially regulated based on a Welch's t test (p < 0.01, -fold change ≥ 4.0) by R848 stimulation. Hierarchical clustering of the regulated genes identified three main patterns of expression (Fig. 2A). One group was induced by R848 but unaffected by IRAK4 kinase inhibition. Thus, this group contains genes that work through an IRAK4 kinase–independent pathway that may utilize the scaffold function of IRAK4. A second group was induced by R848 and reversed by IRAK4 kinase inhibition. This group contains genes that are regulated in an IRAK4 kinase–dependent manner. The final group contained genes that were inhibited by R848 but were not significantly affected by IRAK4 inhibition.
Figure 2.

IRAK4 kinase inhibition results in differential regulation of genes in monocytes. A, 860 probes that showed a significant difference in expression (n = 8 donors, Welch's p value <0.01, -fold difference >4.0) following treatment with R848 for 1 h were visualized in TIBCO Spotfire 3.3.2. Each row was z-score normalized and then subjected to hierarchical clustering. The middle bracketed cluster represents 203 genes that were down-regulated following treatment with IRAK4, the lower bracketed cluster represents 185 genes that remained elevated despite IRAK4 inhibitor treatment. Ingenuity Pathway Analysis was used to identify the transcriptional regulators significantly associated with these two clusters of genes and the top are presented here. B and C, ChIP-qPCR demonstrates that IRAK4 kinase controls IRF5 binding to promoters of target genes but not NFκB. Human monocytes were pretreated with DMSO or IRAK4 kinase inhibitor for 30 min followed by treatment with 1 μg/ml R848 or untreated for 30 min. Increased binding of both IRF5 (B) and NFκB (C) were seen upon R848 treatment as compared with untreated conditions. Binding of IRF5 to promoters of target genes CXCL10 and TNFa was reduced with IRAK4 kinase inhibition (B), whereas no reduction of NFκB binding was observed with IRAK4 kinase inhibition (C). Error bars are standard deviation from the mean.
To determine the role of IRAK4 kinase activity in TLR-induced cytokine production, we focused on the second group, containing IRAK4 kinase–dependent genes (Fig. 2A, middle bracket). Upstream analysis in Ingenuity Pathway Analysis (IPA) of the 203 genes in this cluster identified the IRF family of transcription factors (IRF1, 3, 5, 7, and 8) as being five of six of the top predicted transcriptional regulators of genes induced by R848 and reversed by IRAK4 kinase inhibition. On the other hand, the genes not significantly affected by IRAK4 inhibition tended to be regulated by non-IRF transcription factors such as NFκB, Jun, and NFAT (Fig. 2A). This demonstrates that IRAK4 kinase regulates a narrow selection of TLR-induced transcription factors, mainly within the IRF family.
Mice expressing kinase-inactive IRAK4 have normal NFκB and MAP kinase activation but are deficient in cytokine responses when challenged with TLR ligands (6, 7). Similarly, mice deficient in Irf5 expression have a nearly identical phenotype, exhibiting normal activation of NFκB and MAP kinases but deficiencies in the production of multiple inflammatory cytokines in response to TLR ligands (23). IRF5 is the only member of the IRF family of transcription factors reported to control all MyD88-dependent effects on cytokines and interferon (25). In addition, human monocyte expression of IRF5 is significantly higher as compared with other IRFs (IRF3, 7) (26). We therefore focused first on IRF5 and investigated the binding of IRF5 to the promoters of IRAK4 target genes. We chose two genes identified as highly induced by R848 and reversed by IRAK4 kinase inhibition from our profiling data that had both IRF5 and NFκB binding sites in their promoters, CXCL10 and TNF. ChIP-quantitative PCR (qPCR) (Active Motif, Carlsbad, CA) confirmed increased IRF5 binding in the upstream promoter of CXCL10 and TNF upon R848 treatment. Binding of IRF5 is reduced by IRAK4 kinase inhibition in both of these genes, suggesting IRF5 as a downstream target of IRAK4 activity (Fig. 2B). In contrast, although R848 increased NFκB binding to the promoters of CXCL10 and TNF, IRAK4 kinase inhibition did not significantly reduce NFκB binding to the promoters of these genes (Fig. 2C). Combined with previous data that IRAK4 kinase minimally affects phosphorylation of p65 (5), this suggests that IRAK4 kinase activity does not regulate NFκB activity. Therefore, regulation of cytokine production in myeloid cells by IRAK4 kinase activity occurs through the regulation of IRF5 but not NFκB.
IRAK4 kinase controls IRF5 nuclear translocation
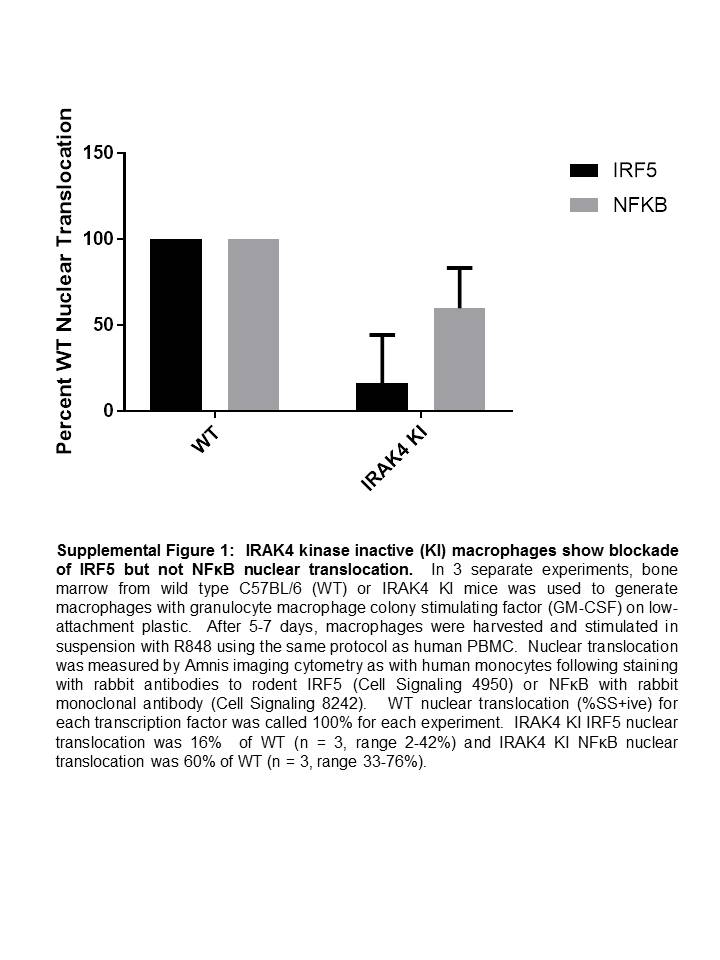
To determine the mechanism by which IRAK4 inhibition is blocking IRF5-mediated transcription, we investigated IRF5 translocation to the nucleus of primary human monocytes stimulated with R848. As measured by Amnis imaging cytometry in CD14+ human monocytes and depicted in Fig. 3, A–C, upon stimulation with R848, IRF5 translocates to the nucleus (86.7 ± 1.1 versus 10.2 ± 0.5% similarity score percent positive, p < 0.0001). This R848-induced IRF5 nuclear translocation was blocked by the IRAK4 inhibitor (Fig. 3C, 86.7 ± 1.1 versus 21.8 ± 2.9% similarity score percent positive, p < 0.0001). By contrast (Fig. 3C), NFκB p65 translocation was also induced by R848 (86.5 ± 3.6 versus 11.0 ± 0.9% similarity score percent positive, p = 0.0002), but was not significantly inhibited by the IRAK4 inhibitor (86.5 ± 3.6 versus 77.9 ± 8.4% positive, p = 0.2). To confirm that the lack of NFκB inhibition was not specific to the IRAK4 inhibitor or species, we performed the same experiment using bone marrow–derived macrophages from Irak4 knock-in mice, which confirmed that IRAK4 kinase activity was required for IRF5 but not NFκB p65 nuclear translocation (supplemental Fig. 1). Finally, an independent flow cytometry platform was used to measure NFκB pathway activation by R848 and inhibition by the IRAK4 inhibitor (Fig. 3D) using IκB degradation as a readout. In PBMC isolated from 59 healthy human volunteers, R848 induced IκB degradation, and this was minimally affected by pretreatment with the IRAK4 inhibitor. The median control IκB expression (L2DiffAF) was 10.48, whereas those for R848 stimulated and R848 stimulated in the presence of IRAK4 inhibitor were significantly lower, indicating IκB degradation at 8.31 and 8.85, respectively (n = 59, two-sided paired Wilcoxon exact form (“signed rank test”), p < 2.2 × 10–16). Because we determined that IRAK4 kinase activates transcription of inflammatory cytokines through IRF5, we next sought to investigate the mechanism by which IRF5 is activated downstream of IRAK4 kinase.
Figure 3.

IRAK4 kinase controls IRF5 but not NFKβ activation and translocation to the nucleus. ImageStream files of human PBMC were gated for single, focused cells and CD14+ monocytes, then analyzed for nuclear translocation using similarity score (SS). A, Brightfield, CD14, DAPI, IRF5, and NFκB images and overlays from samples with and without R848 stimulation, with and without IRAK4 inhibitor treatment. B, single parameter histogram overlays depicting the distribution of monocyte nuclear similarity scores with and without R848 stimulation, with and without IRAK4 inhibitor treatment. C, quantification of IRF5 and NFκB nuclear translocation by similarity score percent positive events. n = 4 human PBMC samples (mean ± S.E.; *, p < 0.0001). D, analysis of IRAK4 kinase inhibitor–mediated inhibition of IκB degradation by SCNP flow cytometry analysis in PBMC from n = 59 healthy human volunteer blood donors. L2DiffAF = log2(MFI-MFI AF), where MFI = mean fluorescence intensity and AF = autofluorescence (**, p < 2.2 × 10(−16)).
IRAK4 kinase acts through IKKβ to activate IRF5
The mechanism by which TLRs activate IRF5 is currently unclear. Recently, it has been shown that IKKβ can phosphorylate IRF5 in vitro and induce dimerization and nuclear translocation in human monocytic and dendritic cell lines (15,16). Additionally, Bergstrom et al. (17) have shown that TLR activation of IFNβ and IL12 in primary human monocytes involves activation of IRF5 through TAK1 and IKKβ. Therefore, we investigated whether IRAK4 kinase acts through IKKβ and TAK1 to control IRF5 activation. We treated human monocytes with IRAK4 kinase inhibitor and then stimulated for 15 and 30 min with R848. Fig. 4A (densitometry in supplemental Fig. 2) shows that the IRAK4 inhibitor blocks IRAK4 kinase autophosphorylation under the same conditions that block IRF5 nuclear translocation (Fig. 3C). Similar to IRAK4 kinase inhibition, we found that pharmacologically inhibiting IKKβ (27) or TAK1 (28) in human monocytes blocks inflammatory cytokine production (Fig. 4B) and IRF5 translocation to the nucleus (Fig. 4C), but does not impact NFκB p65 nuclear translocation (Fig. 4C). The IRAK4 inhibitor also blocks the phosphorylation of IKKβ at Ser-177 but has no effect on the phosphorylation and degradation of IκB (Fig. 4A). Inhibition of IKKβ blocks phosphorylation and degradation of IκB but appears to enhance the phosphorylation of Ser-177 on IKKβ. This increase in phosphorylated IKK upon its inhibition has been observed previously but the mechanism is not known (29). A TAK1 inhibitor blocks IKKβ and IκB phosphorylation but not IRAK4 phosphorylation following R848 stimulation (Fig. 4A), demonstrating that TAK1 lies between IRAK4 and IKKβ in the signaling cascade. Taken together these data delineate a pathway by which IRAK4 kinase activates TAK1 to phosphorylate and activate IKKβ which in turn phosphorylates IRF5 to induce the translocation of IRF5 to the nucleus and the transcription of pro-inflammatory cytokines (Fig. 5). Therefore, inhibition of IRAK4 kinase activity is sufficient to block this signaling cascade and cytokine production without impacting NFκB activation.
Figure 4.

IRAK4 kinase acts through IKKβ to activate IRF5. A and B, 10 million monocytes from healthy human blood donors were reconstituted in 2 ml of RPMI with PenStepGln/HEPES/0.5% heat-inactivated fetal bovine serum (Thermo Fisher) and incubated with 1 μm IRAK4i (IRAK4 inhibitor), 1 μm TAK1i (TAK1 inhibitor 5Z-7 oxozeaenol, Tocris), 1 μm IKKβi (IKKβ inhibitor TPCA, Sigma), or 0.1% DMSO for 30 min prior to stimulation with 0.5 μg/ml R848 for 15 or 30 min prior to analysis of cell lysates by Western blot (A) or 20 h prior to analysis of supernatant cytokines by ELISA (B). A, Western blot image representative of one of three donors. Quantitation by LI-COR image analysis software shown in supplemental Fig. S2. B, cytokines represent mean ± S.E. for n = 3 donors, comparison by one-way analysis of variance (ANOVA). C, 2-ml cultures of freshly isolated human PBMC were stimulated 30 min with 1 μg/ml R848 after incubation with or without inhibitors for 30 min and then subjected to Amnis ImageStream analysis of IRF5 and NFκB nuclear localization (mean ± S.E.; n = 4 donors).
Figure 5.
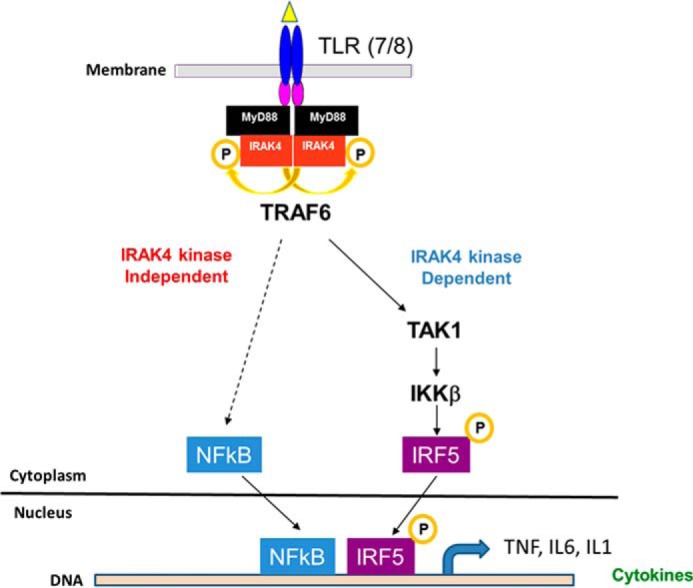
A proposed model of IRAK4 regulation of TLR7/8 signaling pathway in human monocytes is shown. Ligation of endosomal TLR7 and/or TLR8 by R848 leads to cytoplasmic assembly of the myddosome and autophosphorylation of IRAK4. Inflammatory cytokine production is induced by a signaling cascade including TAK1 and IKKβ that results in phosphorylation, nuclear translocation, and transcription mediated by IRF5 (right). At the same time, an as yet unidentified signaling cascade (left, dashed line) that requires IRAK4 scaffolding activity but not IRAK4 kinase activity but is independent of TAK1 and IKKβ, induces translocation of NFκB to the nucleus. Binding of both NFκB and IRF5 to the promoters of inflammatory cytokines is required for transcription.
Discussion
We show that IRAK4 kinase inhibition in R848-treated human monocytes blocks inflammatory cytokine mRNA and protein production, including IL1, IL6, and TNF. Transcriptional pathway analysis suggests that this block in mRNA and cytokine production is mediated by IRF5, and this is confirmed by demonstrating the reduction of IRF5 nuclear translocation and IRF5 binding to the promoters of target genes following IRAK4 inhibition. Interestingly, and in agreement with previous reports from our lab, the effect of an IRAK4 kinase inhibitor on the NFκB nuclear translocation and binding to promoters of target genes is minimal. Finally, we find that IRAK4 kinase acts through IKKβ and TAK1 to activate IRF5.
Activation of IRF5
Exactly which kinases phosphorylate and activate IRF5 is not known, and there are likely different mechanisms depending on cell type and stimulation. In vitro studies have shown that TBK1 and RIP2 can phosphorylate IRF5 (30). Overexpression studies also identified IKKE and TBK1 as activators of IRF5 (31). However Tbk1 deficiency in mouse macrophages had no effect on the TLR7/8/9-induced cytokine production, in contrast to Irak4- and Irf5-deficient cells (32). These data suggest that TBK1 is not involved in IRAK4 regulation of the IRF5 pathway. Most recently, IKKβ was identified as the kinase that phosphorylates IRF5 on Ser-462 and causes its dimerization and activation in myeloid cells (16, 33). Here we show that inhibition of IRAK4 blocks IKKβ phosphorylation and IRF5 nuclear translocation, similar to the inhibition of TAK1 or IKKβ. Our data suggest IRAK4 kinase activity regulates inflammatory cytokine production in human monocytes via an IRAK4-TAK1-IKK-IRF5 axis (17).
IKKβ activates IRF5 but not NFκB in TLR signaling
Our data show that IRAK4 controls the activation of IKKβ to activate IRF5 but not NFκB. Of note, and entirely consistent with our results, Dudhgaonkar et al. (34) recently demonstrated inhibition of IKKβ phosphorylation and cytokine release from TLR ligand stimulated human PBMC using an undisclosed highly potent and selective IRAK4 inhibitor, but did not present data regarding NFκB. IKKβ is thought to be the canonical activator of NFκB, yet our data demonstrate that NFκB is minimally affected with IRAK4 kinase inhibition in R848-treated human monocytes, as is phosphorylation and degradation of IκB. This phenomenon has been observed previously as TLR/IL1-induced NFκB activation was shown to occur through a TAK1/IKKβ-independent pathway (35, 36). These studies found that IRAK4 kinase activity led to TAK1-dependent IKKα/β phosphorylation and IKKβ activation as TAK1 and IKKβ were neither phosphorylated nor activated in cells from IRAK4 kinase–inactive mice. Interestingly, our data show that NFκB was activated by R848 in the absence of IRAK4 kinase activity in human monocytes treated with an IRAK4 kinase inhibitor. In contrast, NFκB was not activated if IRAK4 expression was ablated (1), indicating that a TAK1/IKKβ-independent pathway of NFκB activation is controlled by IRAK4 scaffolding activity. This IRAK4 scaffold–dependent activation of NFκB was ablated when Mekk3 expression was eliminated, suggesting that MEKK3 is the kinase mediating activation of NFκB through the IRAK4 scaffold (37). Furthermore the authors found that the MEKK3-dependent pathway utilized IKKγ phosphorylation and IKKα activation. Yamazaki et al. (38) further confirmed and added to this knowledge, showing that TAK1-dependent signaling required a complex for signaling that included TRAF6, MEKK3, and TAK1. After TAK1 initially activates NFκB, MEKK3 subsequently activated NFκB independently of TAK1 for continuous NFκB activation and cytokine production. Thus it is tempting to speculate that inhibiting IRAK4 kinase blocks the TAK1-dependent pathway through IKKβ with a resultant inhibition of IRF5 activation, but the MEKK3-dependent pathway is untouched, allowing near normal NFκB activity. Further studies need to be carried out to prove this hypothesis and further clarify the mechanisms involved. Indeed, multiple kinases have been shown to have the ability to phosphorylate and activate NFκB (39). In addition, post-transcriptional modifications other than phosphorylation, such as ubiquitination, and their role in this signaling cascade need further exploration.
Proposed model for IRAK4 regulation of IRF5
Based on our data reported here and published previously, we propose the following model for the regulation of TLR responses by IRAK4 in human monocytes. IRAK4 regulates the TLR response through both IRAK4 kinase activity–dependent and –independent (scaffold function–dependent) pathways. In response to TLR7/8 stimulation by ligands such as R848, IRAK4 is activated by recruitment into the receptor complex via interaction with MyD88. The resulting myddosome formation promotes autophosphorylation of IRAK4 and increases the kinase activity of IRAK4. IRAK4 kinase activity is responsible for the activation of TAK1 and IKKβ, likely mediated through IRAK1 and TRAF6. The activated IKKβ can then phosphorylate IRF5 and induce nuclear translocation of IRF5, leading to the expression of inflammatory cytokines. In addition to the IRAK4 kinase activity–dependent pathway, the scaffold function of IRAK4, independent of kinase activity, is essential for the activation of the MAP kinase and NFκB pathways. Future studies are required to understand the signaling components in both pathways. Our data indicate that an IRAK4 inhibitor will only affect the IRAK4 kinase–dependent pathways in human myeloid cells while preserving the scaffold function in both myeloid and structural cells (5). The effect on the kinase-dependent pathway appears sufficient to provide a protective effect in mouse models of autoimmune diseases such as RA or multiple sclerosis based on mouse studies with kinase-inactive knock-in mice (42, 43) or IRAK4 inhibitor studies.8 Whether inhibition of the IRAK4 kinase–dependent pathway is sufficient to provide efficacy will await data from ongoing clinical trials.
Experimental procedures
Monocyte enrichment and treatment
Leukopaks from healthy donors (discarded byproducts of blood component donation, Massachusetts General Hospital, Boston) were incubated with RosetteSep Monocyte Enrichment Cocktail (STEMCELL Technologies) and purified according to the manufacturer. The isolated monocytes were then resuspended for culture in RPMI plus 0.5% FBS (Thermo Fisher), pretreated with 1 μm inhibitor compounds or DMSO (Sigma) for 30 min and then stimulated with 0.5 μg/ml R848 (InvivoGen) for 0, 15, 30 min (phosphoprotein) or 1 and 4 h (RNA and cytokines).
ELISA
Culture supernatants were transferred to 4-plex pro-inflammatory II plates (Mesoscale Discovery) and developed according to the manufacturer's instructions. Statistical significance was determined by Student's t-tests.
RNA isolation and qRT-PCR
RNA was isolated utilizing the RNeasy Mini Kit (Qiagen) and quality and concentration assessed utilizing an Agilent 2100 bioanalyzer (Santa Clara, CA) and NanoDrop ND-1000 (Wilmington, DE), respectively. Total RNA (10 ng) was then processed using TaqMan Gene Expression Assays (Applied Biosystems) and qScript 1-Step qRT-PCR Kit, with ROX (Quanta Biosciences) according to the manufacturer's protocol. qRT-PCR and analysis were performed on the Viia7 Real-Time PCR system (Applied Biosystems). Statistical significance was determined by Student's t-tests.
Affymetrix array
Total RNA was processed following the NuGen (San Carlos, CA) RNA amplification protocol to produce double-stranded cDNA. 3.75 μg of the cDNA was fragmented to between 35 and 200 bases in length and used as a template to generate biotin-targeted cDNA following the manufacturer's recommendations. Biotin-labeled cDNA was hybridized onto the Affymetrix (Santa Clara, CA) GeneChipTM genome array HG-U133 Plus overnight in the GeneChip Hybridization Oven 640. The following day the arrays were washed and then stained in a GeneChip Fluidics Station 450. Scanning was carried out with the GeneChip Scanner 3000 and image analysis was performed using the GeneChip Operating Software.
Bioinformatics analysis
Signal values were determined using GeneChip Operating System 1.0 (GCOS) (Affymetrix). For each array, all probe sets were normalized to a mean signal intensity value of 100. The default GeneChip Operating System statistical values were used for all analysis. Signal values and absolute detection calls were imported into Expressionist (GeneData, Lexington, MA) for analysis. A gene was considered detectable if at least 80% of the samples within a group had an expression greater than 20 signal units. Two independent Welch's t test analyses on the normalized signal values were performed between the R848-stimulated samples and the control samples and between the samples pretreated with 1 μm inhibitor compounds with R848 stimulation and the R848-stimulated samples. The 860 probes with a difference of at least 4.0-fold (p < 0.01) in the R848 stimulated versus control comparison were used for further analysis. For some computations, the expression values for each qualifier were normalized to a mean of zero and a standard deviation of 1 (z-score normalization). This allowed a direct comparison of patterns within the data without respect to absolute expression levels.
ChIP-qPCR
50 million cells per sample of primary human monocytes were pretreated with IRAK4 kinase inhibitor or DMSO for 30 min and stimulated with R848 or media control for 30 min. The cells were then fixed according to the cell fixation protocol recommended by Active Motif. Briefly, the cells were fixed in formaldehyde for cross-linking of proteins to DNA, and fixation was stopped with glycine. Cells were then washed in PBS (Life Technologies), IGEPAL (Sigma), and PMSF (Sigma) at 1 mm. After a final centrifugation, cell pellets were snap frozen on dry ice and shipped to Active Motif for ChIP-qPCR processing and analysis. Antibodies used for immunoprecipitation were IRF5 (Abcam, ab2932) and p65 (Santa Cruz Biotechnology, sc-109). Binding was tested for negative control regions (Untr12) and positive control regions (IRF2BP2 and IFNGR2), as well as binding sites for IRF5 and NFκB in CXCL10 andTNF. The primers used for IRF5 binding CXCL10 were TGTGGGAGGAGAGGAAGAAG and TTGCACCATCATCCAATCAG; for IRF5 binding TNF, TCATACAACCTCCGAAACTGAG and GAAGGGTAAGACTGGGCTGAG; for p65 binding CXCL10, GAGTCTGCAACATGGGACTTC and TTTCCCTCCCTAATTCTGATTG; and for p65 binding TNF, TGTGAGGGGTATCCTTGATG and GCACCTTCTGTCTCGGTTTC. Quantitative PCR analyses were done in triplicate and represented as percent input after normalization for primer efficiency and starting material per manufacturer's instructions.
Transcription factor nuclear translocation
Peripheral blood mononuclear cells were prepared from fresh heparinized peripheral whole blood (Pfizer volunteer donation program, Institutional Review Board–approved protocol) using Ficoll-Paque Plus (GE Lifesciences), suspended in complete RPMI (RPMI 1640 supplemented with l-glutamine, sodium pyruvate, nonessential amino acids, penicillin-streptomycin, and 10% heat-inactivated fetal bovine serum (Thermo Fisher)). Aliquots of 2 × 106 cells were rested in a 96-well polypropylene deep-well plate (Axygen) at 37 °C in a 5% CO2 incubator for 1 h, incubated with 1 μm compounds for 1 h, and stimulated 30 min with 1 μg/ml R848 (InvivoGen). After centrifugation, cells were fixed 15 min with Cytofix (BD Biosciences), washed, and permeabilized with phosphate-buffered saline containing 2% FBS and 0.1% Triton X-100 (Sigma). Cells were incubated with phycoerythrin-labeled CD14 antibody (Beckman Coulter) and unlabeled rabbit antibodies to IRF5 (Abcam) or NFκB p65 (Cell Signaling Technology) for 30 min. After washing, secondary Alexa 647–labeled goat anti-rabbit IgG and the DNA dye DAPI were incubated with cells for 20 min, and fixation with Cytofix was repeated for 10 min after washing. At least 100,000 cellular events were collected on an Amnis ImageStream Mark II imaging cytometer. Single fluorophore controls were used to construct a compensation matrix for each experiment, monocytes were gated by CD14 staining, and nuclear localization was quantified using pixel similarity scoring with a nuclear mask generated from the DAPI image. A gate was set on the 10% highest similarity scores in the unstimulated sample and applied to the remaining samples to quantify changes in nuclear translocation of the target molecule. The identical experiment was performed on blood from four volunteers on three different days, and compared with a Student's t test.
Analysis of intracellular signaling by SCNP technology
Single cell network profiling (SCNP) was carried out as described previously (40). Briefly, 59 healthy donor PBMC were plated and incubated at 100,000 cells/well in 96-well plates. The IRAK4 inhibitor PF-06426779 at 500 nm was applied for 1 h prior to TLR7/8 modulation (R848, InvivoGen, catalog no. tlrl-r848, 2 μg/ml) for 15 min after which all wells were fixed, permeabilized, and incubated with a mixture of fluorochrome-conjugated antibodies that recognize extracellular lineage markers (CD20 (BD Biosciences, catalog no. 558021, 1:4 from neat), CD14 (Beckman Coulter, catalog no. B01175, 1:16 from neat)) and intracellular epitopes (cPARP (BD Biosciences, catalog no. 560640, 1:8 from neat), IκBα (Cell Signaling Technology, catalog no. 7523BC, 1:4 from neat)). Cleaved PARP was examined in every well to enable assessment of cell health and to allow gating on cPARP negative (i.e. nonapoptotic) monocytes (41). Data were acquired using FACS Diva software (BD Biosciences) on a Canto II (BD) flow cytometer. Flow Cytometry Standard Data (FCS) files were gated using WinList (Verity House Software, Topsham, ME) and all data were stored in a MySQL database for access and querying.
Western blotting
Cells were lysed in 200 μl of 1× RIPA lysis buffer (Cell Signaling Technology) according to the manufacturer's instructions. They were then heated to 95 °C for 5 min, separated by SDS-PAGE, and transferred to nitrocellulose membranes. The antibodies phospho-IRAK4 (Thr-345/Ser-346) rabbit monoclonal (5), IRAK4 (mouse, Abcam, catalog no. 119942), phospho-IκBα (mouse mAb, Cell Signaling Technology, catalog no. 9246), IκBα (rabbit, Cell Signaling Technology, catalog no. 4812), phospho-IKKβ Ser-177 (rabbit, Cell Signaling Technology, catalog no. 2078), and actin (mouse, Cell Signaling Technology, catalog no. 3700) were incubated with membranes blocking in Odyssey Blocking Buffer (LI-COR) for 1 h at room temperature and then incubated overnight in primary antibody at 4 °C. Membranes were washed four times for 5 min each in 1× PBS-T, incubated in secondary antibody for 1 h at room temperature, washed again four times for 5 min each in PBS-T, rinsed in PBS, and imaged on the Odyssey CLx.
Author contributions
L. C., A. W., V. R. R., M. F., and L.-L. L. contributed to experimental design. L. C. and A. W. wrote the manuscript. L. C. performed monocyte stimulations and analyzed protein and mRNA expression as well as ChIP-Seq. A. W. performed nuclear translocation assays. S. A. J. performed bioinformatics analyses. K. L. designed IRAK4 inhibitors. W. K. and R. H. designed and supervised SCNP experiments. V. R. R. performed cell signaling phosphoprotein analysis. L.-L. L. and V. R. R. edited the manuscript. All authors reviewed the manuscript.
Supplementary Material
Acknowledgments
We are thankful for the following contributions: Erik Evensen and Andrew Conroy at Nodality performed and analyzed SCNP assays and John Cheng at Pfizer assisted in establishing IRF5 nuclear translocation assay. We thank Shizuo Akira of the Japan Science and Technology Agency, Osaka, Japan, for the generous gift of Irak4 knock-in mice.
W. K. and R. H. were employees of Nodality, contracted by Pfizer to perform SCNP studies with the IRAK4 inhibitor. All other authors were Pfizer employees.
This article contains supplemental Figs. 1 and 2 and supplemental Table 1.
Data are available in the Gene Expression Omnibus (GEO) database under accession no. GSE86234.
L. Cushing, A. Winkler, S. A. Jelinsky, K. Lee, V. R. Rao, M. Fleming, and L.-L. Lin, unpublished data.
- IRAK4
- interleukin-1 receptor–associated kinase 4
- IRF
- interferon regulatory factor
- TLR
- Toll-like receptor
- IL1R
- interleukin-1 receptor
- RA
- rheumatoid arthritis
- hPBMC
- human peripheral blood mononuclear cells
- SCNP
- single cell network profiling.
References
- 1. Picard C., Puel A., Bonnet M., Ku C. L., Bustamante J., Yang K., Soudais C., Dupuis S., Feinberg J., Fieschi C., Elbim C., Hitchcock R., Lammas D., Davies G., Al-Ghonaium A., et al. (2003) Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 299, 2076–2079 [DOI] [PubMed] [Google Scholar]
- 2. Suzuki N., Suzuki S., Duncan G. S., Millar D. G., Wada T., Mirtsos C., Takada H., Wakeham A., Itie A., Li S., Penninger J. M., Wesche H., Ohashi P. S., Mak T. W., and Yeh W.-C. (2002) Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416, 750–756 [DOI] [PubMed] [Google Scholar]
- 3. Seganish W. M. (2016) Inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4): A patent review. (2012–2015). Expert Opin. Ther. Pat. 26, 917–932 [DOI] [PubMed] [Google Scholar]
- 4. Flannery S., and Bowie A. G. (2010) The interleukin-1 receptor-associated kinases: Critical regulators of innate immune signalling. Biochem. Pharmacol. 80, 1981–1991 [DOI] [PubMed] [Google Scholar]
- 5. Cushing L., Stochaj W., Siegel M., Czerwinski R., Dower K., Wright Q., Hirschfield M., Casanova J. L., Picard C., Puel A., Lin L. L., and Rao V. R. (2014) Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J. Biol. Chem. 289, 10865–10875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim T. W., Staschke K., Bulek K., Yao J., Peters K., Oh K. H., Vandenburg Y., Xiao H., Qian W., Hamilton T., Min B., Sen G., Gilmour R., and Li X. (2007) A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J. Exp. Med. 204, 1025–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kawagoe T., Sato S., Jung A., Yamamoto M., Matsui K., Kato H., Uematsu S., Takeuchi O., and Akira S. (2007) Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor–mediated immune responses but not in TCR signaling. J. Exp. Med. 204, 1013–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baechler E. C., Batliwalla F. M., Karypis G., Gaffney P. M., Ortmann W. A., Espe K. J., Shark K. B., Grande W. J., Hughes K. M., Kapur V., Gregersen P. K., and Behrens T. W. (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U.S.A. 100, 2610–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yao Y., Richman L., Morehouse C., de los Reyes M., Higgs B. W., Boutrin A., White B., Coyle A., Krueger J., Kiener P. A., and Jallal B. (2008) Type I interferon: Potential therapeutic target for psoriasis? PLoS One 3, e2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baechler E. C., Bilgic H., and Reed A. M. (2011) Type I interferon pathway in adult and juvenile dermatomyositis. Arthritis Res. Ther. 13, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wright H. L., Thomas H. B., Moots R. J., and Edwards S. W. (2015) Interferon gene expression signature in rheumatoid arthritis neutrophils correlates with a good response to TNFi therapy. Rheumatology (Oxf.) 54, 188–193 [DOI] [PubMed] [Google Scholar]
- 12. Barnes B. J., Moore P. A., and Pitha P. M. (2001) Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon α genes. J. Biol. Chem. 276, 23382–23390 [DOI] [PubMed] [Google Scholar]
- 13. Schoenemeyer A., Barnes B. J., Mancl M. E., Latz E., Goutagny N., Pitha P. M., Fitzgerald K. A., and Golenbock D. T. (2005) The interferon regulatory factor, IRF5, is a central mediator of Toll-like receptor 7 signaling. J. Biol. Chem. 280, 17005–17012 [DOI] [PubMed] [Google Scholar]
- 14. Saliba D. G., Heger A., Eames H. L., Oikonomopoulos S., Teixeira A., Blazek K., Androulidaki A., Wong D., Goh F. G., Weiss M., Byrne A., Pasparakis M., Ragoussis J., and Udalova I. A. (2014) IRF5:RelA interaction targets inflammatory genes in macrophages. Cell Rep. 8, 1308–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hayden M. S., and Ghosh S. (2014) Innate sense of purpose for IKKβ. Proc. Natl. Acad. Sci. U.S.A. 111, 17348–17349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopez-Pelaez M., Lamont D. J., Peggie M., Shpiro N., Gray N. S., and Cohen P. (2014) Protein kinase IKKβ-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc. Natl. Acad. Sci. U.S.A. 111, 17432–17437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bergstrøm B., Aune M. H., Awuh J. A., Kojen J. F., Blix K. J., Ryan L., Flo T. H., Mollnes T. E., Espevik T., and Stenvik J. (2015) TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-β production via a TAK1-IKKβ-IRF5 signaling pathway. J. Immunol. 195, 1100–1111 [DOI] [PubMed] [Google Scholar]
- 18. Dideberg V., Kristjansdottir G., Milani L., Libioulle C., Sigurdsson S., Louis E., Wiman A. C., Vermeire S., Rutgeerts P., Belaiche J., Franchimont D., Van Gossum A., Bours V., and Syvänen A. C. (2007) An insertion-deletion polymorphism in the interferon regulatory factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Hum. Mol. Genet. 16, 3008–3016 [DOI] [PubMed] [Google Scholar]
- 19. Dieguez-Gonzalez R., Calaza M., Perez-Pampin E., de la Serna A. R., Fernandez-Gutierrez B., Castañeda S., Largo R., Joven B., Narvaez J., Navarro F., Marenco J. L., Vicario J. L., Blanco F. J., Fernandez-Lopez J. C., Caliz R., et al. (2008) Association of interferon regulatory factor 5 haplotypes, similar to that found in systemic lupus erythematosus, in a large subgroup of patients with rheumatoid arthritis. Arthritis Rheum. 58, 1264–1274 [DOI] [PubMed] [Google Scholar]
- 20. Sigurdsson S., Nordmark G., Göring H. H., Lindroos K., Wiman A. C., Sturfelt G., Jönsen A., Rantapää-Dahlqvist S., Möller B., Kere J., Koskenmies S., Widén E., Eloranta M. L., Julkunen H., Kristjansdottir H., Steinsson K., Alm G., Rönnblom L., and Syvänen A. C. (2005) Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am. J. Hum. Genet. 76, 528–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Richez C., Barnetche T., Miceli-Richard C., Blanco P., Moreau J. F., Rifkin I., and Schaeverbeke T. (2010) Role for interferon regulatory factors in autoimmunity. Joint Bone Spine 77, 525–531 [DOI] [PubMed] [Google Scholar]
- 22. Feng D., Yang L., Bi X., Stone R. C., Patel P., and Barnes B. J. (2012) Irf5-deficient mice are protected from pristane-induced lupus via increased Th2 cytokines and altered IgG class switching. Eur. J. Immunol. 42, 1477–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., Kano S., Honda K., Ohba Y., Mak T. W., and Taniguchi T. (2005) Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249 [DOI] [PubMed] [Google Scholar]
- 24. Lee K. L., Ambler C. M., Anderson D. R., Boscoe B. P., Bree A. G., Brodfuehrer J. I., Chang J. S., Choi C., Chung S., Curran K. J., Day J. E., Dehnhardt C. M., Dower K., Drozda S. E., Frisbie R. K., et al. (2017) Discovery of clinical candidate 1-[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy-7-methoxyisoquinoline-6-carboxamide (PF-06650833), a potent, selective inhibitor of interleukin-1 receptor associated kinase 4 (IRAK4), by fragment-based drug design. J. Med. Chem. 60, 5521–5542 [DOI] [PubMed] [Google Scholar]
- 25. Honda K., and Taniguchi T. (2006) IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658 [DOI] [PubMed] [Google Scholar]
- 26. Rieckmann J. C., Geiger R., Hornburg D., Wolf T., Kveler K., Jarrossay D., Sallusto F., Shen-Orr S. S., Lanzavecchia A., Mann M., and Meissner F. (2017) Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol. 18, 583–593 [DOI] [PubMed] [Google Scholar]
- 27. Podolin P. L., Callahan J. F., Bolognese B. J., Li Y. H., Carlson K., Davis T. G., Mellor G. W., Evans C., and Roshak A. K. (2005) Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IκB kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced t cell proliferation. J. Pharmacol. Exper. Ther. 312, 373–381 [DOI] [PubMed] [Google Scholar]
- 28. Ninomiya-Tsuji J., Kajino T., Ono K., Ohtomo T., Matsumoto M., Shiina M., Mihara M., Tsuchiya M., and Matsumoto K. (2003) A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J. Biol. Chem. 278, 18485–18490 [DOI] [PubMed] [Google Scholar]
- 29. Zhang J., Clark K., Lawrence T., Peggie M. W., and Cohen P. (2014) An unexpected twist to the activation of IKKβ: TAK1 primes IKKβ for activation by autophosphorylation. Biochem. J. 461, 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang Foreman H. C., Van Scoy S., Cheng T. F., and Reich N. C. (2012) Activation of interferon regulatory factor 5 by site specific phosphorylation. PLoS One 7, e33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng T. F., Brzostek S., Ando O., Van Scoy S., Kumar K. P., and Reich N. C. (2006) Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 transcription factors during viral infection. J. Immunol. 176, 7462–7470 [DOI] [PubMed] [Google Scholar]
- 32. Marchlik E., Thakker P., Carlson T., Jiang Z., Ryan M., Marusic S., Goutagny N., Kuang W., Askew G. R., Roberts V., Benoit S., Zhou T., Ling V., Pfeifer R., Stedman N., Fitzgerald K. A., Lin L. L., and Hall J. P. (2010) Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J. Leukocyte Biol. 88, 1171–1180 [DOI] [PubMed] [Google Scholar]
- 33. Ren J., Chen X., and Chen Z. J. (2014) IKKβ is an IRF5 kinase that instigates inflammation. Proc. Natl. Acad. Sci. U.S.A. 111, 17438–17443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dudhgaonkar S., Ranade S., Nagar J., Subramani S., Prasad D. S., Karunanithi P., Srivastava R., Venkatesh K., Selvam S., Krishnamurthy P., Mariappan T. T., Saxena A., Fan L., Stetsko D. K., Holloway D. A., Li X., et al. (2017) Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. J. Immunol. 198, 1308–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fraczek J., Kim T. W., Xiao H., Yao J., Wen Q., Li Y., Casanova J. L., Pryjma J., and Li X. (2008) The kinase activity of IL-1 receptor-associated kinase 4 is required for interleukin-1 receptor/Toll-like receptor-induced TAK1-dependent NFκB activation. J. Biol. Chem. 283, 31697–31705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qin J., Yao J., Cui G., Xiao H., Kim T. W., Fraczek J., Wightman P., Sato S., Akira S., Puel A., Casanova J. L., Su B., and Li X. (2006) TLR8-mediated NF-κB and JNK activation are TAK1-independent and MEKK3-dependent. J. Biol. Chem. 281, 21013–21021 [DOI] [PubMed] [Google Scholar]
- 37. Yao J., Kim T. W., Qin J., Jiang Z., Qian Y., Xiao H., Lu Y., Qian W., Gulen M. F., Sizemore N., DiDonato J., Sato S., Akira S., Su B., and Li X. (2007) Interleukin-1 (IL-1)-induced TAK1-dependent versus MEKK3-dependent NFκB activation pathways bifurcate at IL-1 receptor-associated kinase modification. J. Biol. Chem. 282, 6075–6089 [DOI] [PubMed] [Google Scholar]
- 38. Yamazaki K., Gohda J., Kanayama A., Miyamoto Y., Sakurai H., Yamamoto M., Akira S., Hayashi H., Su B., and Inoue J. (2009) Two mechanistically and temporally distinct NF-κB activation pathways in IL-1 signaling. Sci. Signal. 93, ra66. [DOI] [PubMed] [Google Scholar]
- 39. Buss H., Dörrie A., Schmitz M. L., Hoffmann E., Resch K., and Kracht M. (2004) Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKϵ, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 279, 55633–55643 [DOI] [PubMed] [Google Scholar]
- 40. Cesano A., Rosen D. B., O'Meara P., Putta S., Gayko U., Spellmeyer D. C., Cripe L. D., Sun Z., Uno H., Litzow M. R., Tallman M. S., and Paietta E. (2012) Functional pathway analysis in acute myeloid leukemia using single cell network profiling assay: Effect of specimen source (bone marrow or peripheral blood) on assay readouts. Cytometry B Clin. Cytom. 82, 158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cesano A., Putta S., Rosen D. B., Cohen A. C., Gayko U., Mathi K., Woronicz J., Hawtin R. E., Cripe L., Sun Z., Tallman M. S., and Paietta E. (2013) Functional pathway analysis using SCNP of FLT3 receptor pathway deregulation in AML provides prognostic information independent from mutational status. PLoS One 8, e56714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koziczak-Holbro M., Littlewood-Evans A., Pöllinger B., Kovarik J., Dawson J., Zenke G., Burkhart C., Müller M., and Gram H. (2009) The critical role of kinase activity of interleukin-1 receptor-associated kinase 4 in animal models of joint inflammation. Arthritis Rheum. 60, 1661–1671 [DOI] [PubMed] [Google Scholar]
- 43. Staschke K. A., Dong S., Saha J., Zhao J., Brooks N. A., Hepburn D. L., Xia J., Gulen M. F., Kang Z., Altuntas C. Z., Tuohy V. K., Gilmour R., Li X., and Na S. (2009) IRAK4 kinase activity is required for Th17 differentiation and Th17-mediated disease. J. Immunol. 183, 568–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}