

Abstract
In the CNS, microglia are activated in response to injury or infection and in neurodegenerative diseases. The endocytic and cell signaling receptor, LDL receptor-related protein-1 (LRP1), is reported to suppress innate immunity in macrophages and oppose microglial activation. The goal of this study was to identify novel mechanisms by which LRP1 may regulate microglial activation. Using primary cultures of microglia isolated from mouse brains, we demonstrated that LRP1 gene silencing increases expression of proinflammatory mediators; however, the observed response was modest. By contrast, the LRP1 ligand, receptor-associated protein (RAP), robustly activated microglia, and its activity was attenuated in LRP1-deficient cells. An important element of the mechanism by which RAP activated microglia was its ability to cause LRP1 shedding from the plasma membrane. This process eliminated cellular LRP1, which is anti-inflammatory, and generated a soluble product, shed LRP1 (sLRP1), which is potently proinflammatory. Purified sLRP1 induced expression of multiple proinflammatory cytokines and the mRNA encoding inducible nitric-oxide synthase in both LRP1-expressing and -deficient microglia. LPS also stimulated LRP1 shedding, as did the heat-shock protein and LRP1 ligand, calreticulin. Other LRP1 ligands, including α2-macroglobulin and tissue-type plasminogen activator, failed to cause LRP1 shedding. Treatment of microglia with a metalloproteinase inhibitor inhibited LRP1 shedding and significantly attenuated RAP-induced cytokine expression. RAP and sLRP1 both caused neuroinflammation in vivo when administered by stereotaxic injection into mouse spinal cords. Collectively, these results suggest that LRP1 shedding from microglia may amplify and sustain neuroinflammation in response to proinflammatory stimuli.
Keywords: lipoprotein receptor-related protein (LRP), metalloprotease, microglia, neuroinflammation, shedding, receptor-associated protein
Introduction
Microglia constitute 8–12% of the cells in the brain (1, 2). These cells are regulators of innate immunity and are related in function to cells of the monocyte–macrophage lineage (1, 2). A principal function of microglia is surveillance. In response to injury or infection, microglia become activated and express cytokines and other mediators, which help orchestrate the inflammatory response. However, in various forms of neurodegeneration, including Alzheimer's disease, chronically activated microglia may accelerate disease progression (1–6). Injury to peripheral nerves activates microglia in the spinal cord, which promotes central sensitization and neuropathic pain (7–9). Understanding pathways that control microglial activation is an important problem.
LDL receptor-related protein-1 (LRP1)2 is an endocytic and cell signaling receptor with over 100 ligands, including proteins released from injured and dying cells (10–14). The structure of membrane-anchored LRP1 includes the 515-kDa α-chain, which is entirely extracellular and coupled to the cell surface by non-covalent interactions with the 85-kDa β-chain. The α-chain is responsible for most of the ligand-binding activity of LRP1. The β-chain includes an ectodomain, a transmembrane domain, and the intracellular tail that becomes phosphorylated when LRP1 functions in cell signaling.
In peripheral macrophages, LRP1 gene deletion is proinflammatory in vitro and in vivo in mice (15–20). In macrophages that express LRP1, ligands control its activity in a ligand-specific manner (20, 21). Tissue-type plasminogen activator (tPA) and activated α2-macroglobulin (α2M*) amplify the anti-inflammatory activity of LRP1, whereas receptor-associated protein (RAP) and lactoferrin inhibit it (20). The proinflammatory response induced by RAP in macrophages is similar in magnitude to that caused by LRP1 gene deletion.
LRP1 is expressed by microglia, particularly at sites of inflammation (22–25), and may be involved in regulating microglial activation. Silencing LRP1 gene expression in microglia or treating microglia with RAP activates NFκB and c-Jun N-terminal kinase (JNK) and increases expression of inflammatory cytokines (25). The LRP1 ligand, apolipoprotein E, suppresses microglial activation by a mechanism that requires LRP1 (26, 27). LRP1 gene deletion in microglia in vivo exacerbates experimental autoimmune encephalomyelitis (24).
LRP1 is shed from cell surfaces by the metalloproteinases ADAM10, ADAM17, and MMP-14 (28–31). In shed LRP1 (sLRP1), the β-chain is truncated; however, the entire α-chain is intact and detectable in plasma, brain, cerebrospinal fluid, and the peripheral nervous system (29, 32, 33). The concentration of sLRP1 is increased in the plasma of mice treated with lipopolysaccharide (LPS) and in humans with rheumatoid arthritis or systemic lupus erythematosus (30). sLRP1 also is increased in osteoarthritic cartilage and in broncho-alveolar lavage fluid from patients with adult respiratory distress syndrome (31, 34). Although the biological activity of sLRP1 remains incompletely understood, sLRP1 promotes expression of inflammatory mediators by macrophages (30). sLRP1 also may bind biologically active proteins such as ADAMTS-5, MMP-13, and TIMP-3, preventing their endocytosis (31, 35). Once sLRP1 is released from the cell surface, the residual LRP1 fragment may be further processed by γ-secretase to generate an intracellular product that opposes inflammation (17). Phylogenetically, LRP1 shedding is conserved throughout mammalian, avian, and reptilian species (36), supporting the hypothesis that shedding may be physiologically significant.
Herein, we demonstrate that LRP1 is shed from microglia exposed to the proinflammatory mediators LPS and RAP. We also identify calreticulin (CRT), a known LRP1 ligand, as an activator of microglia, which induces LRP1 shedding. Purified sLRP1 was potently proinflammatory when added to primary cultures of microglia and when injected into spinal cords in mice. When LRP1 shedding was inhibited with GM6001, expression of proinflammatory mediators in response to RAP was largely attenuated. These results suggest a model in which LRP1 shedding converts an anti-inflammatory receptor into a proinflammatory product. sLRP1 may amplify and sustain neuroinflammation.
Results
LRP1 gene silencing modestly increases cytokine expression by microglia
Microglia were isolated from 8-week-old mice that were homozygous for the floxed LRP1 gene (LRP1fl/fl) and LysM–Cre-positive (20). LysM–Cre drives expression of Cre recombinase in monocytes, macrophages, neutrophils, and microglia, although the level of Cre recombinase expressed in microglia may depend on whether the cells are activated or in culture (37, 38). As a control, microglia were isolated from LRP1fl/fl–LysM–Cre-negative mice. RNA and protein were harvested from cells without culturing. Fig. 1A shows that LRP1 mRNA was decreased 72 ± 8% in cells from LRP1fl/fl–LysM–Cre-positive mice. LRP1 protein was decreased 85 ± 8%, as determined by immunoblot analysis and densitometry (Fig. 1, B and C).
Figure 1.

Effects of LRP1 deficiency on cytokine expression by microglia. A–F, microglia were isolated from LRP1fl/fl–LysM–Cre-positive (black bar) and LysM–Cre-negative (open bar) adult mice. A, RT-qPCR was performed to quantify expression of LRP1 mRNA. B and C, cell extracts were immunoblotted to detect LRP1 β-chain. GAPDH was used as loading control. Densitometry analysis was performed to determine the relative level of LRP1 protein, standardized against the loading control, in extracts from Cre-negative (N) and Cre-positive (P) cells (mean ± S.E.; n = 4; **, p < 0.01, paired t test). D–F, RT-qPCR was performed to compare relative quantities (RQ) of mRNA for TNF-α, IL-6, and IL-1β in microglia from Cre-positive (P) and Cre-negative (N) mice (mean ± S.E.; n = 4; paired t test). G–K, microglia were isolated from C57BL/6J mouse pups and transfected with LRP1-specific or NTC siRNA. RT-qPCR was performed to determine mRNA levels for LRP1 (G) and TNF-α (H) (mean ± S.E.; n = 4; ***, p < 0.001; paired t test). I, cells transfected with LRP1-specific siRNA, and NTC siRNA were allowed to condition medium for 48 h. CM was recovered, and ELISAs were performed to quantify TNF-α protein (mean ± S.E. n = 3). NC shows medium that was “not conditioned.” J and K, mRNA levels were determined for IL-6 and IL-1β (mean ± S.E.; n = 4; *, p < 0.05; paired t test).
LRP1-deficient microglia from LRP1fl/fl–LysM–Cre-positive mice demonstrated slight increases in TNF-α mRNA (Fig. 1D), IL-6 mRNA (Fig. 1E), and IL-1β mRNA (Fig. 1F); however, none of these increases achieved statistical significance at the p < 0.05 level (n = 4). We therefore applied a second approach to induce LRP1 deficiency. Microglia were harvested from wild-type C57BL/6 mouse pups and established in primary culture. LRP1 gene expression was silenced with siRNA. Control cells were transfected with non-targeting control (NTC) siRNA. LRP1 mRNA was decreased by 69 ± 5% in cells transfected with LRP1-specific siRNA (Fig. 1G). A modest but non-significant increase in TNF-α mRNA was observed in these cells (p = 0.2, Fig. 1H). TNF-α protein was unchanged, as determined by analyzing conditioned medium (CM) by ELISA (Fig. 1I). LRP1 gene silencing significantly increased expression of IL-6 mRNA (p < 0.05) (Fig. 1J) and IL-1β mRNA (p < 0.01) (Fig. 1K).
RAP robustly increases expression of proinflammatory mediators by microglia
Next, we studied the effects of RAP on expression of proinflammatory mediators by microglia. For these studies, microglia were isolated from wild-type mouse pups and established in primary culture. Cells were treated with 150 nm RAP, LPS (1 μg/ml), or vehicle (phosphate-buffered saline/PBS) for 24 h. Because RAP is expressed as a glutathione S-transferase (GST)-fusion protein, as an additional control, cells were treated with purified GST (150 nm).
RAP robustly increased expression of TNF-α mRNA (p < 0.001), as did LPS but not GST (Fig. 2A). RAP and LPS also substantially increased the level of TNF-α protein in CM (Fig. 2B). Pre-boiling LPS at 100 °C for 5 min had no effect on its ability to increase TNF-α protein secretion, as anticipated (21). By contrast, pre-boiling RAP eliminated its ability to stimulate TNF-α protein expression, arguing against LPS contamination as contributing to the activity of RAP. Pre-boiling RAP also completely blocked its ability to induce expression of TNF-α mRNA (Fig. 2C).
Figure 2.

RAP induces expression of inflammatory mediators by microglia. A–H, microglia were isolated from C57BL/6J mouse pups and cultured in the presence of LPS (1 μg/ml), boiled LPS (bLPS, 1 μg/ml), RAP (150 nm), or boiled RAP (bRAP, 150 nm). Control cultures were treated with vehicle (PBS; 10 μl/ml) or purified GST (150 nm). After 24 h, RT-qPCR was performed to quantify mRNA levels for TNF-α (A and C); IL-6 (D); IL-1β (F); and iNOS (G). CM was recovered from microglia after treatment for 24 h and tested to determine the following: B, TNF-α protein; E, IL-6 protein; and H, nitrite (mean ± S.E.; n = 4–6; ns, not significant, *, p < 0.05; **, p < 0.01; ***, p < 0.001, one-way ANOVA followed by Tukey's post hoc analysis). I–L, microglia cultured in 0.5% FBS-supplemented medium were pre-treated with CHX (10 or 50 μm) or 2 μl/ml DMSO (listed as “0” μm CHX) for 30 min and then treated with 150 nm RAP (+RAP) or vehicle (−RAP) for 6 h. RT-qPCR was performed to determine mRNA for TNF-α (I) and IL-6 (J). CM was recovered and protein levels of TNF-α (K) and IL-6 (L) were determined (mean ± S.E.; n = 3; *, p < 0.05; **, p < 0.01; ***, p < 0.001, one-way ANOVA with Tukey's post hoc analysis).
Treating microglia with RAP increased expression of IL-6 mRNA (Fig. 2D), IL-6 protein (Fig. 2E), and IL-1β mRNA (Fig. 2F). In each case, the response elicited by RAP was either equivalent in magnitude to that elicited by LPS or only slightly decreased. No response was observed with GST. Pre-boiling RAP completely blocked its ability to induce expression of IL-6 and IL-1β. Pre-boiling LPS did not significantly decrease its activity. The increases in expression of IL-6 and IL-1β, observed in microglia treated with RAP for 24 h, were 100–1,000-fold greater than the increases observed in LRP1 gene-silenced cells.
Inducible nitric-oxide synthase (iNOS) is a proinflammatory enzyme expressed by activated microglia (39). Fig. 2G shows that RAP robustly increased iNOS mRNA expression in microglia. The response was similar in magnitude to that caused by LPS. To assess iNOS activity, we measured nitrite in CM. RAP significantly increased nitrite levels in CM (p < 0.01), mimicking the response observed with LPS (Fig. 2H). Boiling RAP blocked its ability to increase nitrite production, but boiling LPS had no effect. Purified GST did not increase nitrite levels.
In RAP-treated macrophages, TNF-α functions as an early mediator that increases expression of other proinflammatory cytokines, such as IL-6, in a secondary wave (20). Fig. 2I shows that the protein synthesis inhibitor, cycloheximide (CHX), failed to inhibit the increase in TNF-α mRNA observed 6 h after adding RAP. By contrast, CHX significantly attenuated the increase in IL-6 mRNA caused by RAP (Fig. 2J). CHX decreased TNF-α protein (Fig. 2K) and IL-6 protein (Fig. 2L) in CM from RAP-treated cells, as anticipated. The effects of CHX on IL-6 mRNA expression suggest that a protein intermediate or intermediates are involved in the pathway by which RAP increases expression of this cytokine.
RAP regulates microglial cell morphology, proliferation, and migration
Non-activated microglia demonstrate a ramified morphology, which upon activation transforms into a more rounded, amoeboid shape (40). We compared the morphology of microglia after incubation with RAP, LPS, or GST by phalloidin staining. Fig. 3A shows that LPS and RAP induced similar changes in microglial morphology. In response to both agents, the cells adopted a bloated amoeboid shape. Image analysis was conducted by an investigator who was blinded to group identity. Cells were classified as amoeboid or ramified. LPS and RAP both induced significant increases (p < 0.001) in the percentage of cells with amoeboid morphology (Fig. 3B). GST had no effect on cell morphology.
Figure 3.
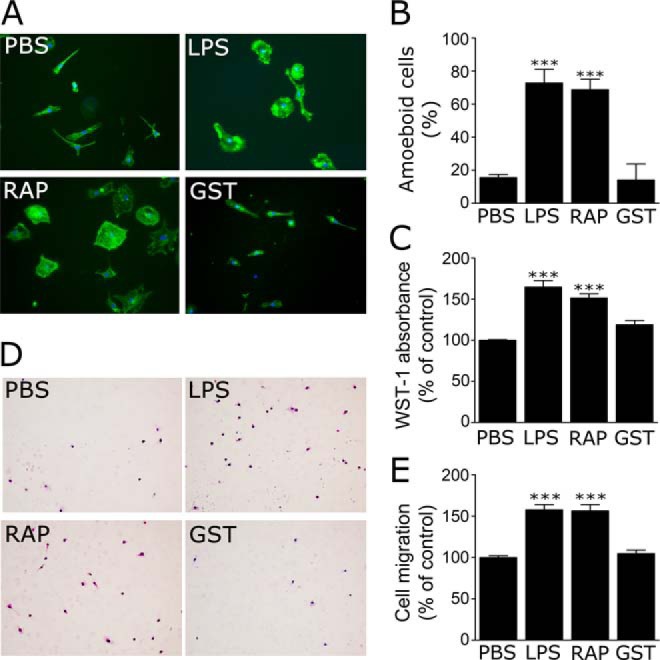
RAP regulates microglial morphology, proliferation, and migration. A and B, microglia isolated from wild-type mouse pups were treated with LPS (1 μg/ml), RAP (150 nm), GST (150 nm), or vehicle for 24 h. A, images of phalloidin-stained microglia are shown. Nuclei are stained with DAPI (blue) (scale bar, 10 μm). B, percentage of cells with an amoeboid phenotype was determined by an investigator who was blinded to groups. C, microglial cells were cultured in the presence or absence of LPS or RAP for 48 h, and proliferation was determined by WST-1 assay. D, migration of microglia was determined using transwells. Representative photomicrographs of cells that migrated through the membranes are shown (scale bar, 50 μm). E, quantification of cell migration results. Migration is presented as a percentage of that observed with cells treated with vehicle. All results presented are mean ± S.E.; n = 4; ***, p < 0.001, one-way ANOVA with Dunnett's post hoc analysis.
Next, we compared the effects of LPS and RAP on microglial proliferation and migration. Increased proliferation and migration are characteristic of microglial activation (1, 2, 40). Fig. 3C shows that treating cells with RAP or LPS for 48 h significantly increased microglial cell proliferation (p < 0.001). RAP and LPS also apparently promoted microglial cell migration. Representative images of Transwell membranes, showing cells that migrated through membrane pores, are shown in Fig. 3D. The results of four separate experiments are summarized in Fig. 3E. Because RAP and LPS promoted microglial cell proliferation, we cannot exclude the possibility that the measured effects of these reagents on cell migration were artifactually increased due to an increase in the number of cells present during the course of the 16-h assay.
LRP1 deficiency protects microglia from the proinflammatory effects of RAP
Members of the LDL receptor gene family in addition to LRP1 bind RAP (41). We therefore conducted RAP ligand-blotting studies, as described previously (42), to identify RAP-binding proteins in microglia. Microglia from LRP1fl/fl–LysM–Cre-positive and Cre-negative mice were compared. In LRP1-expressing microglia from LysM–Cre-negative mice, a single band with a mass of ∼500 kDa was detected, consistent with the known mass of the LRP1 α-chain (Fig. 4A). The absence of bands with lower molecular masses indicated that RAP-binding receptors, such as the VLDL receptor and ApoER2, were not present in substantial quantity. In LRP1-deficient microglia, isolated from LRP1fl/fl–LysM–Cre-positive mice, the 500-kDa band was nearly absent, confirming the identity of that band as the LRP1 α-chain. When equivalent blots were probed with purified GST instead of GST-RAP, no bands were detected. These results demonstrate that LRP1 is the principal RAP-binding protein in microglia and the most likely target for RAP in cultured microglia, as reported previously by Pocivavsek et al. (27).
Figure 4.

Activation of microglia by RAP requires LRP1. Microglia were isolated from 8-week-old LRP1fl/fl–LysM–Cre-positive and LysM–Cre-negative mice. A, cell extracts were subjected to RAP-ligand blotting to detect RAP-binding proteins (blot labeled RAP). Control membranes were probed with purified GST (100 nm) followed by GST antibody (blot labeled GST). β-Actin immunoblots were performed as a control for load on extracts from LRP1fl/fl–LysM–Cre-negative (N) and LysM–Cre-positive (P) microglia. Images are representative of four independent experiments. B–F, microglia isolated from adult mice were cultured for 5 days in complete medium and then treated with RAP (150 nm) or vehicle (PBS) for 24 h in low-serum medium. RT-qPCR was performed to determine mRNA for TNF-α (B), IL-6 (D), and IL-1β (F). TNF-α protein (C) and IL-6 protein (E) in CM were determined by ELISA. Cells from LRP1fl/fl–LysM–Cre-negative mice are shown with open bars; cells from LRP1fl/fl–LysM–Cre-positive mice are shown with black bars (mean ± S.E.; n = 4; ns, not significant, **, p < 0.01; ***, p < 0.001, one-way ANOVA followed by Tukey's post hoc analysis). IL-1β protein levels were not determined.
RAP is generally considered an LRP1 antagonist, which blocks binding of other ligands to LRP1, including ligands added exogenously or produced endogenously by cells in culture (11, 41). We therefore conducted experiments to test why the effects of RAP on inflammatory mediator expression by microglia appeared so much greater than the effects of LRP1 gene silencing or deletion. At first, we hypothesized that the modest effects of LRP1 gene silencing and deletion reflected residual LRP1. To test this hypothesis, microglia from LRP1fl/fl–LysM–Cre-positive and LysM–Cre-negative mice were treated with RAP. The goal of this experiment was to neutralize residual LRP1 in cells from LysM–Cre-positive mice. Fig. 4, B and C, shows that RAP increased TNF-α mRNA and TNF-α protein in LRP1-deficient and -expressing cells. Following RAP treatment, the levels of TNF-α mRNA and protein were not significantly different in the two cell types.
RAP increased IL-6 mRNA expression in microglia from LysM–Cre-positive and -negative mice; however, unexpectedly, the quantity of IL-6 mRNA detected in RAP-treated LRP1-deficient cells remained significantly decreased, compared with that detected in RAP-treated LRP1-expressing cells (Fig. 4D). RAP also was substantially less effective at inducing expression of IL-6 protein (Fig. 4E) and IL-1β mRNA (Fig. 4F) in LRP1-deficient cells from LysM–Cre-positive mice, compared with LRP1-expressing cells from LysM–Cre-negative mice. Taken together, these results demonstrate that the modest effects of LRP1 gene silencing and deletion on cytokine expression are not entirely due to incomplete LRP1 neutralization. Furthermore, LRP1 gene deletion is at least partially protective against the proinflammatory effects of RAP.
RAP and CRT promote LRP1 shedding
It is reported that the proinflammatory mediators, LPS and interferon-γ, decrease LRP1 mRNA expression in microglia (22, 25). We treated microglia with 150 nm RAP or 1.0 μg/ml LPS for 24 h and demonstrated that LRP1 mRNA levels are significantly decreased (Fig. 5A). Both treatments also decreased the abundance of LRP1 protein, as determined by immunoblot analysis and densitometry (Fig. 5, B and C).
Figure 5.

RAP and LPS induce LRP1 shedding from microglia. Microglia isolated from mouse pups were treated with LPS (1 μg/ml), RAP (150 nm), purified GST (150 nm), or vehicle (PBS) for 24 h. A, RT-qPCR was performed to determine LRP1 mRNA (mean ± S.E.; n = 4; ***, p < 0.001, one-way ANOVA followed by Dunnett's post hoc test). Cell extracts were subjected to immunoblot to detect LRP1 and β-actin (B) and densitometry analysis was performed (C) (mean ± S.E.; n = 4; **, p < 0.01, one-way ANOVA followed by Dunnett's post hoc test). Microglia were treated with LPS (1 μg/ml), RAP (150 nm), or vehicle (PBS) for up to 24 h (D). At the indicated times, CM was recovered. sLRP1 was detected in CM by RAP ligand blotting, as described in Fig. 4A (upper panels). CM also was subjected to immunoblot analysis to detect LRP1 α-chain (middle panels) and LRP1 β-chain (lower panels). Molecular mass standards are shown to the left of each blot. Results are representative of three experiments. E and F, densitometry analysis was performed on RAP ligand blots to quantify the increase of sLRP1 in the CM after treatment with LPS (E) and RAP (F) (mean ± S.E.; n = 3; *, p < 0.05; **, p < 0.01, one-way ANOVA followed by Dunnett's post hoc test). G, microglia were treated with increasing concentrations of RAP. After 24 h, CM was recovered and subjected to RAP ligand blotting to detect sLRP1. Results are representative of two independent experiments.
Next, we tested whether LPS and RAP induce LRP1 shedding by subjecting CM to RAP ligand blotting. Fig. 5D shows that both LPS and RAP induced time-dependent shedding of a high molecular mass RAP-binding protein consistent with the known mass of the LRP1 α-chain (∼515-kDa). The abundance of the high molecular mass protein in CM from LPS- and RAP-treated cells was similar. No other RAP-binding species were detected.
To confirm that the RAP-binding protein was sLRP1, CM samples were subjected to immunoblot analysis. LRP1 α-chain was detected; the mobility of the α-chain was equivalent to that of the protein detected by RAP ligand blotting. The 85-kDa LRP1 β-chain was not detected using an antibody that recognizes an intracellular epitope, arguing against contamination of CM with cells or cell fragments that have full-length cellular LRP1. RAP-binding proteins and LRP1 α-chain were absent in CM from microglia treated with PBS instead of RAP or LPS. Densitometry analysis summarizing the results of three separate LRP1 shedding experiments are presented in Fig. 5, E and F. Fig. 5G shows that RAP induced detectable LRP1 shedding at concentrations down to 15 nm.
tPA and α2M* are LRP1 ligands that induce anti-inflammatory responses in peripheral macrophages (20, 21). Fig. 6A shows that enzymatically-inactive tPA (EI-tPA) (12 nm) and α2M* (10 nm) both failed to induce IL-6 mRNA expression in microglia. Fig. 6B shows that EI-tPA and α2M* also failed to induce expression of IL-1β mRNA. The concentrations of EI-tPA and α2M* studied in these experiments were selected to match those that generate maximum anti-inflammatory responses in macrophages (20, 21). When microglia were treated with 12 nm EI-tPA or 10 nm α2M* for 24 h, LRP1 shedding was not observed (Fig. 6C). In separate experiments, we studied EI-tPA at concentrations up to 100 nm; again LRP1 shedding was not observed (results not shown). These results suggest that LRP1 ligands do not, in general, induce LRP1 shedding.
Figure 6.

Select LRP1 ligands induce LRP1 shedding. A–C, microglia were cultured in low serum medium in the presence of LPS (1 μg/ml), EI-tPA (12 nm), or α2M* (10 nm). After 24 h, mRNA levels for IL-6 (A) and IL-1β (B) were determined by RT-qPCR (mean ± S.E.; n = 4; ***, p < 0.001, one-way ANOVA followed by Dunnett's post hoc test). C, CM was recovered and subjected to RAP ligand blotting to detect sLRP1. D–F, microglia were cultured in the presence of increasing concentrations of recombinant human CRT. After 24 h, CM was recovered. TNF-α protein (D) and IL-6 protein (E) in CM were determined by ELISA. F, sLRP1 released into CM was determined by RAP ligand blotting (mean ± S.E.; n = 3; **, p < 0.01; ***, p < 0.001, one-way ANOVA followed by Dunnett's post hoc test).
Although lactoferrin generates proinflammatory responses in macrophages (20, 21), in microglia, lactoferrin, at concentrations up to 100 nm, did not significantly regulate TNF-α mRNA expression or induce LRP1 shedding (results not shown). We did not explore why microglia do not respond to lactoferrin; however, we did study another LRP1 ligand with known proinflammatory activity. CRT is a heat-shock protein known to bind directly to LRP1, induce LRP1-dependent inflammatory responses in antigen presenting cells, and function together with LRP1 in efferocytosis (43–46). Fig. 6, D and E, shows that CRT increased expression of TNF-α protein and IL-6 protein in CM from microglia. CRT also induced LRP1 shedding (Fig. 6F). These results suggest that the ability of a reagent to stimulate LRP1 shedding from microglia correlates with its ability to stimulate a proinflammatory response. Stimulation of shedding is a property of some but not all LRP1 ligands.
Shed LRP1 activates microglia in vitro
To test whether sLRP1 regulates cell signaling and gene expression in microglia, sLRP1 was purified from human plasma. A single major band with a mobility consistent with that of the 515-kDa LRP1 α-chain was detected by SDS-PAGE (Fig. 7A). RAP ligand blotting confirmed that the high molecular mass band was the LRP1 α-chain.
Figure 7.

Purified sLRP1 induces inflammatory mediator expression in microglia in vitro. A, Coomassie-stained gel (left panel) and RAP-ligand blot (right panel) showing purified human sLRP1. B–F, microglia from mouse pups were cultured in the presence of increasing concentrations purified sLRP1 for 6 h. Control cells were treated with vehicle (0 ng/ml sLRP1) or boiled sLRP1. B, RT-qPCR was performed to determine TNF-α mRNA (*, p < 0.05; one-way ANOVA with Tukey's post hoc test). C, TNF-α protein in CM was determined by ELISA (mean ± S.E.; n = 4; *, p < 0.05; ***, p < 0.001; one-way ANOVA with Tukey's post hoc test). D–F, RT-qPCR was performed to determine mRNA levels for IL-6, IL-1β, and iNOS. G–I, microglia were isolated from adult LRP1fl/fl–LysM–Cre-positive (black bar) and LysM–Cre-negative (open bar) mice and treated with increasing concentrations of sLRP1 for 6 h. mRNA levels of TNF-α, IL-6, and IL-1β were determined by RT-qPCR (mean ± S.E.; n = 3; one-way ANOVA followed by Tukey's post hoc analysis). iNOS mRNA expression was not studied in this experiment.
Wild-type microglia were treated with increasing concentrations of purified sLRP1 in 0.5% serum-supplemented medium for 6 h. sLRP1 robustly increased expression of TNF-α mRNA (Fig. 7B) and stimulated TNF-α protein accumulation in CM (Fig. 7C). sLRP1 also increased expression of the mRNAs for IL-6 (Fig. 7D), IL-1β (Fig. 7E), and iNOS (Fig. 7F). In all four mRNA expression studies, the responses were sLRP1 concentration-dependent and statistically significant with 60 ng/ml sLRP1 (0.15 nm). A significant increase in TNF-α protein accumulation in CM was observed with 30 ng/ml sLRP1. Boiling sLRP1 at 100 °C for 5 min neutralized its ability to induce cytokine expression at the mRNA and protein levels, excluding LPS contamination as an explanation for the activity of sLRP1.
Next, we compared the ability of sLRP1 to increase cytokine expression in LRP1-expressing and -deficient microglia, isolated from LRP1fl/fl–LysM–Cre-positive and LysM–Cre-negative mice. sLRP1 increased expression of TNF-α (Fig. 7G), IL-6 (Fig. 7H), and IL-1β (Fig. 7I) similarly in LRP1-expressing and -deficient cells. These results suggest that membrane-anchored LRP1 is not essential in the pathway by which sLRP1 induces expression of proinflammatory mediators in microglia.
To rule out the possibility that species differences between the cells (mouse) and sLRP1 (human) contributed to the results observed, we repeated the cytokine expression studies using full-length LRP1, purified from mouse liver (mLRP1). In bone marrow-derived macrophages, sLRP1 and mLRP1 are equally effective at inducing inflammatory responses (30). Purified mLRP1 robustly increased expression of the mRNAs for TNF-α (supplemental Fig. 1A), IL-6 (supplemental Fig. 1B), IL-1β (supplemental Fig. 1C), and iNOS (supplemental Fig. 1D). Boiling purified mLRP1 blocked its activity or significantly inhibited it.
sLRP1 and mLRP1 were purified by affinity chromatography using GST-RAP covalently coupled to Sepharose (30). Because of the covalent coupling method, it is unlikely that RAP co-eluted with and contaminated purified LRP1 preparations. The low concentrations of sLRP1 and mLRP1 necessary to induce cytokine expression (100 pm or less) further argue against RAP contamination as an explanation for the activity of these proteins. When purified sLRP1 and mLRP1 (up to 1.0 μg of each protein) were subjected to immunoblot analysis to detect GST-RAP, no signal was observed (supplemental Fig. 2).
sLRP1 amplifies the microglial response to RAP
We hypothesized that LRP1 shedding contributes to the proinflammatory response observed in RAP-treated microglia. To test this hypothesis, first we examined cell signaling in microglia treated with RAP or sLRP1. Yang et al. (25) demonstrated that RAP activates NFκB and JNK in microglia.
To examine RAP-initiated cell signaling in an unbiased manner, we treated microglia with RAP or vehicle for 1 h and identified protein phosphorylation events using the phosphoprotein proteome-profiler from R&D Systems, which profiles 43 distinct protein phosphorylation events. IκBα is not represented in the array; however, we did observe increased phosphorylation of c-Jun N-terminal kinase (JNK), together with its downstream substrate c-Jun (Fig. 8, A and B). ERK1/2 and p38 MAPK were phosphorylated, as was Akt at Ser-473 and its downstream target GSK3β. Phosphorylation of GSK3β by Akt results in GSK3β inactivation (47). The transcription factor, cAMP-response element-binding protein (CREB), is a target for multiple kinases (48).
Figure 8.

RAP and sLRP1 activate cell signaling in microglia. A–D, microglia isolated from mouse pups were treated with RAP (150 nm) or vehicle (PBS) for 1 h. A, protein phosphorylation was determined using the Phospho-kinase Array Proteome ProfilerTM. The membranes were processed simultaneously and have internal controls to ensure equal loading of cell extracts and equal exposure. The second blot is annotated with proteins that demonstrated altered phosphorylation. B, arrays were subjected to densitometry. Changes in phosphorylation are shown (fold-increase, n = 1). C, to confirm the results of the array, immunoblot analysis was performed to detect the indicated phosphorylated proteins, including phospho-p38 MAPK, phospho-ERK1/2, phospho-c-Jun, and phospho-Ser-473 in Akt. Blots were probed to detect β-actin as a loading control. D, immunoblots like those shown in C were subjected to densitometry. The results of three separate experiments were averaged (mean ± S.E.; *, p < 0.05; ***, p < 0.001, unpaired t test). E, microglia were cultured in low serum medium for 30 min and then treated with sLRP1 (60 ng/ml) for the indicated times. Immunoblot analysis was performed to detect the phosphorylated forms of p38 MAPK, ERK1/2, and Akt Ser-473. Blots were re-probed for β-actin as a loading control. Results are representative of two independent experiments.
The results of the phosphoprotein array experiment were confirmed in separate immunoblotting studies. Fig. 8C shows that RAP caused phosphorylation of p38 MAPK, ERK1/2, c-Jun, and Akt Ser-473. The results of three separate immunoblotting experiments are summarized in Fig. 8D.
Next, microglia were treated with sLRP1 (60 ng/ml) for up to 8 h. Fig. 8E shows that p38 MAPK was phosphorylated, and this response was sustained. ERK1/2 activation also was observed; this response was apparent throughout the time course but appeared to maximize at 2 h. Ser-473 in Akt was phosphorylated transiently within the 8-h incubation. These results demonstrate overlap in the phosphorylation events caused by RAP and sLRP1.
To test whether inhibiting LRP1 shedding may attenuate the response of microglia to RAP, microglia were pre-treated with the general metalloproteinase inhibitor GM6001 (50 μm) and then with RAP. LRP1 shedding was largely blocked when assessed 12 h after adding RAP (Fig. 9A) and remained substantially decreased 24 h after adding RAP (Fig. 9B). Cell viability was not compromised (results not shown). Next, we examined expression of the cytokines IL-6 and IL-1β. GM6001 markedly inhibited expression of IL-6 mRNA (Fig. 9C) and IL-1β mRNA (Fig. 9D) in cells treated with RAP for 12 or 24 h.
Figure 9.

Preventing LRP1 shedding inhibits RAP-induced cytokine expression. A–D, microglia were treated with the metalloproteinase inhibitor, GM6001 (50 μm), or DMSO for 3 h in low-serum medium and then with RAP (150 nm) or vehicle (PBS) for 12 or 24 h, as indicated. CM was recovered and subjected to RAP ligand blotting to detect sLRP1 at 12 h (A) and 24 h (B). RT-qPCR was performed to determine mRNA for IL-6 (C) and IL-1β (D) (mean ± S.E.; n = 4; **, p < 0.01; ***, p < 0.001, one-way ANOVA followed by Tukey's post hoc test). E–G, microglia were pre-treated with DAPT (10 μm) or DMSO for 2 h and then with RAP (150 nm) or PBS for 24 h. RT-qPCR was performed to determine expression of the following: E, TNF-α; F, IL-6; and G, IL-1β (mean ± S.E.; n = 4; ns, not significant, one-way ANOVA with Tukey's post hoc analysis).
When sLRP1 is released from cells, the residual membrane-anchored LRP1 fragment may be further processed by γ-secretase to release a cytoplasmic fragment, which has been reported to attenuate inflammation (17). We therefore tested whether inhibiting γ-secretase further increases the proinflammatory response to RAP in microglia. Cells were pre-treated with 10 μm N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) for 2 h and then with RAP or vehicle. The concentration of DAPT selected for this experiment was previously shown to block processing of LRP1 by γ-secretase in mouse macrophages (17). Although DAPT slightly increased expression of TNF-α (Fig. 9E), IL-6 (Fig. 9F), and IL-1β (Fig. 9G) in RAP-treated cells, the increases were not statistically significant.
RAP and sLRP1 induce neuroinflammation when injected into the spinal cord
To test whether RAP induces neuroinflammation in vivo, 120 pmol of RAP (2 μl of 60 μm stock solution) or vehicle (PBS) was injected directly into the right dorsal horn of the spinal cord (T10–T11) of wild-type adult mice using a stereotaxic instrument. Tissue was harvested 24 h later and immunostained for Iba-1 to assess microgliosis (Fig. 10A). RAP induced a significant increase in the density of Iba1-immunopositive cells, as determined by image analysis (Fig. 10B).
Figure 10.
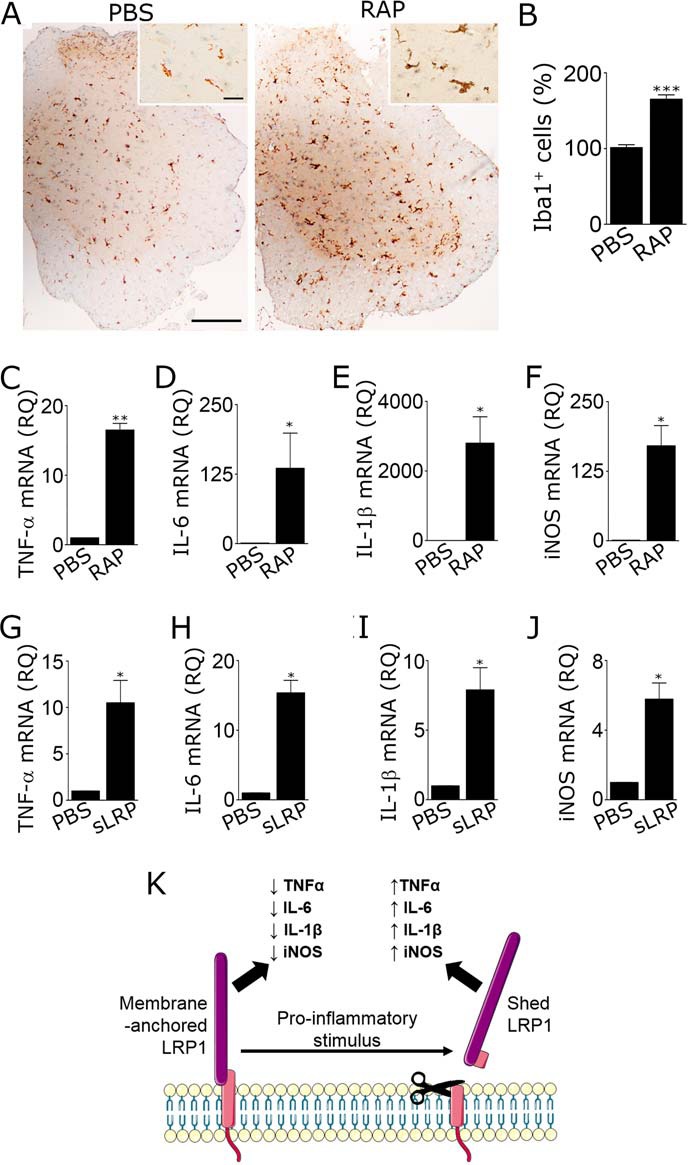
RAP and sLRP1 induce neuroinflammation in spinal cords. A–F, adult C57BL6/J mice were injected with 2 μl of RAP (stock solution 60 μm) or PBS into the right dorsal horn of the spinal cord (T10–T11). Tissue was recovered 24 h later. A, IHC was performed to detect Iba1 immunoreactivity (scale bar, 250 μm for main figure and 25 μm for insets). B, number of Iba1-positive cells in the ipsilateral dorsal horn was determined by a blinded investigator (mean ± S.E.; n = 3; ***, p < 0.001, unpaired t test). C–F, mRNA encoding TNF-α, IL-6, IL-1β, and iNOS was determined by RT-qPCR (mean ± S.E.; n = 4; *, p < 0.05; **, p < 0.01, paired t test). G–J, sLRP1 (2 μl of 0.1 μm stock solution) was injected into spinal cords following the same protocol, and RT-qPCR was performed to determine mRNA for TNF-α, IL-6, IL-1β, and iNOS (mean ± S.E.; n = 4; *, p < 0.05, paired t test). K, model showing how membrane-anchored and shed LRP1 oppose each other in controlling activation of microglia and neuroinflammation. The 515-kDa LRP1 α-chain is shown in purple. The 85-kDa β-chain is shown in pink. Shedding truncates the β-chain so that a small fragment of its ectodomain is associated with sLRP1.
Next, 120 pmol of RAP, 0.2 pmol of sLRP1 (2 μl of 0.1 μm stock solution), or vehicle was injected into spinal cords, using the equivalent procedure. RNA was isolated from the ipsilateral side, 24 h later. Expression of proinflammatory mediators was determined by RT-qPCR. RAP significantly increased expression of TNF-α, IL-6, IL-1β, and iNOS (Fig. 10, C–F). sLRP1 also increased expression of TNF-α, IL-6, IL-1β, and iNOS (Fig. 10, G–J). sLRP1 is thus capable of inducing neuroinflammation in vivo. Overall, our results support a model in which LRP1 shedding from microglia converts an anti-inflammatory receptor into a proinflammatory product in the CNS (Fig. 10K).
Discussion
LRP1 gene deletion is embryonic lethal in mice, implying a critical function for LRP1 in development (49). In adult mammals, the function of LRP1 remains incompletely understood; however, there is increasing evidence that LRP1 regulates the activity of cells that respond to tissue injury (11, 14). Previous studies suggest that in microglia, LRP1 may function to oppose activation (24–27). Our results suggest a more complicated model in which the effects of LRP1 on microglial activation reflect a balance between the opposing activities of membrane-anchored and shed LRP1. The balance may be controlled by signals in the microglial microenvironment that either promote or attenuate LRP1 shedding.
Our work that led to the identification of LRP1 shedding as a proinflammatory pathway were initiated in an attempt to explain why RAP treatment was so much more robust at inducing cytokine expression in microglia, compared with LRP1 gene silencing or deletion. A key observation was the ability of RAP to cause LRP1 shedding, like LPS. We then showed that sLRP1 is potently proinflammatory against cultured microglia. When LRP1 shedding was inhibited with GM6001, expression of IL-6 and IL-1β in response to RAP was attenuated. Similarly, expression of IL-6 and IL-1β in response to RAP was substantially decreased in LRP1-deficient microglia isolated from LRP1fl/fl–LysM–Cre-positive mice. We interpret this result to reflect a decreased capacity for LRP1-deficient microglia to generate sLRP1.
In addition to RAP and LPS, we showed that CRT also induces LRP1 shedding from microglia. CRT was selected for study because it is known to bind to LRP1 and also to trigger proinflammatory cell signaling in antigen-presenting cells (43, 45). We did not confirm that CRT-induced LRP1 shedding resulted from an interaction with LRP1. CRT may interact with other cell surface-associated proteins such as C1q as well, either independently of LRP1 or as part on an LRP1-containing multiprotein complex (50, 51). Results obtained with RAP, LPS, and CRT suggest that LRP1 shedding may represent a common pathway by which diverse proinflammatory mediators promote microglial activation.
Purified sLRP1 was potently proinflammatory at concentrations under 1.0 nm in experiments with cultured microglia. The mechanism by which sLRP1 activates cell signaling in microglia and induces cytokine expression remains to be determined. Because sLRP1 binds ligands similarly to membrane-anchored LRP1, sLRP1 may serve as a “receptor decoy” competing for endogenously-produced ligands (31, 35, 52), including proteins that stimulate anti-inflammatory responses if they bind to cells. Alternatively, sLRP1 may interact directly with microglia. This interaction, if essential, does not appear to require membrane-anchored LRP1. The response to sLRP1 is similar in microglia and peripheral macrophages (30); however, in Schwann cells, sLRP1 has an apparently opposite effect, attenuating cellular activation, which induces cytokine expression and recruits macrophages to injured peripheral nerves (33). Direct binding of sLRP1 to Schwann cell surfaces was demonstrated (33). Furthermore, Schwann cells may be pre-conditioned by sLRP1 and resist subsequent challenges with inflammatory agents in the absence of sLRP1. It is therefore likely that the mechanism by which sLRP1 regulates Schwann cell physiology is different from that which is operational in microglia and macrophages.
To test whether RAP and sLRP1 induce neuroinflammation in vivo, these proteins were injected directly into spinal cords in mice. Induction of cytokine expression was observed. In mice injected with RAP, we observed microglial activation; however, microglia may not be the only cells responsible for the changes in gene expression in the spinal cord. Diverse cells in the CNS express LRP1, including neurons and astrocytes (53–55), and thus may respond to RAP. Furthermore, cells in the CNS in addition to microglia may be targets for exogenously-administered sLRP1 and contribute to the proinflammatory responses observed.
Chuang et al. (24) demonstrated that LRP1 expression is increased in microglia in association with multiple sclerosis lesions and that microglial LRP1 is protective in experimental autoimmune encephalomyelitis. Because multiple sclerosis lesions are considered the focus of inflammation, the study by Chuang et al. (24) emphasizes the importance of identifying mediators that regulate microglial LRP1 expression in vivo. If LRP1 expression is increased in vivo, the quantity of substrate available for shedding also may be increased, and the resulting sLRP1 may contribute to the chronicity of inflammation. In macrophages, LRP1 expression is up-regulated by colony-stimulating factor-1 and then decreased by factors such as interferon-γ and LPS (56–58). This example of dynamic regulation of gene expression suggests that the abundance of LRP1 may be fine-tuned to the degree of differentiation and activation in cells of monocytic lineage.
Although our data indicate that sLRP1 contributes to the potency of RAP in activating microglia, the mechanism by which RAP initially triggers a cellular response remains to be determined. In this study, we added a fairly high concentration of RAP (150 nm), assuming that RAP functions by blocking autocrine LRP1 signaling initiated by endogenously-produced ligands. However, RAP at concentrations as low as 15 nm was effective at inducing LRP1 shedding. The ability of RAP to activate cell signaling in microglia, as demonstrated here and elsewhere (25), raises the possibility that RAP regulates microglial cell physiology autonomously and directly through LRP1, as opposed to functioning as an antagonist of other ligands. This model is supported by results obtained with macrophages (20). In these cells, IκBα phosphorylation was observed within 5 min of adding RAP. For all LRP1 ligands that trigger cell-signaling responses, there is now evidence that diverse essential co-receptors may be involved (21, 59, 60). The presence or absence of an essential co-receptor may explain why the response to a specific ligand, such as lactoferrin, may be cell type-specific.
Overall, we view two pathways by which the cell-surface abundance of LRP1 may be regulated in microglia in response to proinflammatory mediators. First, LRP1 gene expression may be down-regulated (22, 25). Second, LRP1 is subject to shedding. In both cases, an anti-inflammatory receptor was removed from the cell surface. With LRP1 shedding, a proinflammatory agent was added to the microglial microenvironment. The same proinflammatory mediators may stimulate both processes simultaneously.
In conclusion, we have identified sLRP1 as a biologically active product, capable of activating microglia and promoting neuroinflammation. The opposing activities of membrane-anchored and shed LRP1 suggest that proteinases, which release LRP1 from the cell surface, also may be potent regulators of microglial activation. Understanding the function of this novel biochemical system is an important goal for future work.
Experimental procedures
Proteins and reagents
RAP was expressed as a GST fusion protein in bacteria and purified as described (61). As a control, free GST was expressed in bacteria transformed with the empty vector, pGEX-2T. GST fusion proteins were subjected to chromatography on Detoxi-Gel endotoxin-removing columns (Pierce). Recombinant human CRT was purchased from Sino Biological. LPS, serotype 055.B5, was from Sigma. DAPT and GM6001 were from EMD Millipore. Primers and probes for RT-qPCR experiments were purchased from Thermo Fisher Scientific.
sLRP1 was purified from human plasma by the method of Gorovoy et al. (30). In brief, fresh-frozen human plasma was supplemented with proteinase inhibitors, dialyzed against 50 mm Tris-HCl, 150 mm NaCl, pH 7.5, with 1 mm CaCl2 (TBS-Ca) for 12 h at 4 °C, and then subjected to affinity chromatography on GST-RAP covalently coupled to Sepharose 4 Fast Flow (GE Healthcare). RAP-associated proteins were eluted in 0.1 m sodium acetate, 0.5 m NaCl, pH 4, and neutralized by rapid mixing with 50 mm Tris-HCl, pH 8.0. Each sLRP1 preparation was examined for integrity and purity by SDS-PAGE with Coomassie staining and by RAP ligand blotting.
mLRP1 was purified from mouse livers, as described previously (30). EI-tPA was from Molecular Innovations. α2M was purified from human plasma and activated for binding to LRP1 as described previously (56).
Mice
Wild-type C57BL/6J mice were from The Jackson Laboratory. Mice in which the promoter and first two exons of the LRP1 gene are flanked by loxP sites were originally generated by Rohlmann et al. (62). Mice that are homozygous for the floxed LRP1 gene (LRP1fl/fl) were bred with mice that express Cre recombinase under the control of the lysozyme-M promoter (LysM–Cre) in the C57BL/6J background to generate LRP1fl/fl–LysM–Cre-positive mice. Littermate controls were LRP1fl/fl and LysM–Cre-negative. All animal experiments were approved by the Institutional Animal Care and Use Committee at University of California San Diego.
Isolation of microglia
Microglia were isolated from C57BL/6J mouse pups, as described previously (63). In brief, brains were harvested from postnatal day 1–6 mice. The cortices were dissected from the forebrain, and the surrounding meninges were removed. Intact cortices were mechanically and enzymatically dissociated using the neural tissue dissociation kit (Miltenyi Biotec). Mixed glial cultures were established in Dulbecco's modified Eagle's medium/F-12 (DMEM/F-12) supplemented with GlutaMAXTM (Life Technologies, Inc.), 10% fetal bovine serum (FBS, Hyclone), and 100 units/ml Fungizon® (Life Technologies, Inc.). After culturing for 10–14 days, microglia were harvested by shaking the mixed cultures at 200 rpm for 30 min at 37 °C. The floating cells were collected by centrifugation (5 min, 1500 rpm) and re-plated at 3 × 105 cells/well. Culture purity was >96% as determined by immunofluorescence microscopy for Iba1 (positive), glial fibrillary acidic protein (negative), β-III tubulin (negative), and OLIG1 (negative). Experiments were performed within 24 h of completing cell isolations.
Microglia also were isolated from 8-week-old male LRP1fl/fl–LysM–Cre-positive and LysM–Cre-negative mice. Brains were harvested and dissociated using the neural tissue dissociation kit. Myelin debris was removed using Myelin Removal Beads II (Miltenyi Biotec). Microglia were then isolated by magnetic cell sorting using CD11b microbeads (Miltenyi Biotec). Cells were plated in medium supplemented with 10% FBS and 5 ng/ml GM-CSF (R&D Systems) and studied within 5 days.
LRP1 gene silencing
Mouse LRP1-specific siRNA pool (ON-TARGETplus mouse LRP1 siRNA; L-040764-00-0005) and NTC siRNA (ON-TARGETplus non-targeting pool; D-001810-10-20) were from GE Dharmacon. siRNA (20 pmol) was transfected into primary microglia (4 × 105) using Lipofectamine 2000 (Invitrogen). The cells were studied 24–48 h after transfection.
RT-qPCR
Total RNA was isolated using the NucleoSpin® RNA kit (Macherey-Nagel) and converted into cDNA using the iScript cDNA synthesis kit (Bio-Rad). RT-qPCR was performed with TaqMan® gene expression products and an AB Step One Plus Real-Time PCR System (Applied Biosystems). The relative change in gene expression was calculated using the 2ΔΔCt method and GAPDH mRNA as a normalizer. The following primer-probe sets were used: GAPDH (Mm99999915_g1); TNF-α (Mm00443258_m1); IL-6 (Mm00–446190_m1); IL-1β (Mm00434228_m1); iNOS (Mm00440502_m1); and LRP1 (Mm00464608_m1).
Immunoblot analysis and RAP ligand blotting
Microglia were extracted in RIPA buffer (PBS with 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with Complete Protease Inhibitor Mixture (Roche Diagnostics) and sodium orthovanadate. The protein concentration in cell extracts was determined by bicinchoninic acid assay. An equivalent amount of cellular protein was subjected to SDS-PAGE and electrotransferred to PVDF membranes (Bio-Rad). The membranes were blocked with 5% nonfat dry milk in TBS with 0.1% Tween 20 (TBS-T buffer) and incubated with primary antibodies targeting the following: LRP1 β-chain (L2170; Sigma); LRP1 α-chain (L2295; Sigma); phospho-p38 MAPK (9211; Cell Signaling Technologies); phospho-ERK1/2 (4695; Cell Signaling Technologies); phospho-c-Jun (9165; Cell Signaling Technologies); phospho-Ser-473 in Akt (9271; Cell Signaling Technologies); GAPDH (GTX627408; GeneTex); and β-actin (A5316; Sigma). Primary antibody was detected with horseradish peroxidase-conjugated species-specific secondary antibody (Cell Signaling Technologies).
In RAP ligand-blotting studies, blocked PVDF membranes were incubated with 100 nm RAP-GST in 5% nonfat milk for 1 h at 22 °C. As a control, equivalent membranes were incubated with 100 nm free GST. The membranes were washed three times and then incubated with GST-specific antibody coupled to horseradish peroxidase (84–814; Genesee Scientific). Conjugated antibody was detected with ECL PlusTM (GE Healthcare) and HyBlot CL autoradiography film (Denville Scientific). Blots were scanned (Canoscan), and densitometry was performed using ImageJ software.
Analysis of conditioned medium
Microglia were allowed to condition medium that contained 0.5% FBS. TNF-α and IL-6 in CM were determined with mouse quantikine ELISA kits (R&D Systems). sLRP1 was determined by immunoblot analysis without concentrating samples. Nitric oxide was determined by measuring nitrite in CM with the Griess reagent system (Promega). In these studies, CM (50 μl) was incubated with 100 μl of Griess reagent in 96-well plates for 30 min at 22 °C. The absorbance was determined at 540 nm using a SpectraMax M2e microplate reader (Molecular Biodevices).
Phosphoprotein array studies
Phosphorylated proteins were detected in an unbiased manner using the human Phospho-kinase Array Proteome ProfilerTM (R&D System). Although this system was originally developed to examine human proteins, we have demonstrated its effectiveness using rodent cell extracts (21, 64). Microglia in 0.5% serum-supplemented medium were treated with RAP (150 nm) or vehicle (PBS) for 1 h. Protein extracts were prepared and applied to the membranes. The membranes were developed using ECL, as described by the manufacturer. Blots were scanned (Canoscan), and densitometry was performed using ImageJ software.
Cell proliferation assay
Microglia were plated in 96-well plates at a density of 105 cells per well and cultured in low-serum medium for 48 h in the presence of LPS (1 μg/ml), RAP (150 nm), GST (150 nm), or vehicle (PBS). Cell proliferation was determined using the Cayman's WST-1 assay according to the manufacturer's instructions. Briefly after 48 h, cells were incubated at 37 °C for 2 h with WST-1 mixture. Absorbance at 450 nm was measured using a SpectraMax M2e microplate reader (Molecular Biodevices).
Transwell cell migration assays
Microglia (1 × 104) were treated with LPS (1 μg/ml), RAP (150 nm), GST (150 nm), or vehicle (PBS) for 10 min at 37 °C and then added with the same reagents in 0.5% FBS-supplemented medium to the top chamber of 24-well Transwell units with 8.0-μm pores (Corning Glass). The underside of each membrane was coated with 10 μg/ml fibronectin (Millipore). The bottom chamber contained 10% FBS. Cells were allowed to migrate at 37 °C in 5% CO2 for 16 h. Non-migrating cells were removed from the upper surface using a cotton swab. The lower surfaces were stained with Hema 3 (Thermo Fisher Scientific). Stained membranes were mounted on microscope slides and imaged using a Leica DMIRE2 microscope. The number of migrated cells was determined in four representative fields, selected by a blinded investigator, using ImageJ software. Three separate membranes from four independent experiments were analyzed for each condition.
Stereotaxic injection of spinal cords
Mice were anesthetized and placed in a stereotaxic frame. Under sterile conditions, an incision was made from thoracic vertebrae T8 to T12, and the spinal cord was exposed by laminectomy between T10 and T11. A total volume of 2 μl of each experimental solution (RAP, sLRP1, vehicle) was slowly injected into the right dorsal horn of the spinal cord using a Hamilton neuro-syringe. The needle was withdrawn after 5 min to avoid efflux of the injected solution. The wound was closed with 6-0 nylon suture.
Immunohistochemistry
Spinal cords were harvested 24 h after stereotaxic injections. Mice were deeply anesthetized and subjected to intracardiac perfusion with fresh PBS followed by 4% paraformaldehyde. Tissues were paraffin-embedded, and 4-μm sections were prepared (at least 3/tissue). Tissue sections were incubated with 10% nonfat milk and then with primary antibody against Iba-1 (019-19741; Wako) for 1 h. Next, sections were incubated with anti-rabbit antibody conjugated to HRP and developed with 3′,3-diaminobenzidine. Control sections were treated with secondary antibody only. Light microscopy was performed using a Leica DFC420 microscope with Leica Imaging Software 2.8.1 (Leica Microsystems). IHC studies were subjected to image analysis using ImageJ software (National Institutes of Health). The total number of Iba1-positive cells on the ipsilateral side of the spinal cord was determined by a blinded investigator.
Fluorescence microscopy
Microglia were cultured on NuncTM Lab-TekTM II CC2TM chamber slides (Thermo Fisher Scientific). The cells were fixed in 4% paraformaldehyde, permeabilized in 0.3% Triton X-100 (Sigma), and blocked with 10% normal donkey serum (Sigma). Cells were stained with Oregon Green-Phalloidin (Molecular Probes). Slides were mounted using Prolong Gold Antifade reagent with DAPI (Molecular Probes) and viewed under an inverted fluorescence microscope.
Statistics
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software Inc.). All results are expressed as the mean ± S.E. Comparisons between two groups were performed using paired or unpaired t test. Results from more than two groups were analyzed by one-way ANOVA followed by Tukey's or Dunnett's post hoc analysis as stated. p < 0.05 was considered as statistically significant.
Author contributions
C. B. and S. L. G. conceived of and designed the overall study. All authors contributed to experimental design. A. S. G. purified required proteins, including sLRP1 and mLRP1. C. B., E. L., and M. B. performed the experiments. C. B., E. L., and S. L. G. analyzed the data. C. B. and S. L. G. wrote the paper. All authors read and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Dr. Don Pizzo for assistance with the immunohistochemistry studies.
This work was supported by Grants R01 HL136395 and R01 NS097590 from the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1 and S2.
- LRP1
- LDL receptor-related protein-1
- tPA
- tissue-type plasminogen activator
- EI-tPA
- enzymatically-inactive tPA
- α2M*
- activated α2-macroglobulin
- RAP
- receptor-associated protein
- CRT
- calreticulin
- NTC
- non-targeting control
- CM
- conditioned medium
- iNOS
- inducible nitric-oxide synthase
- CHX
- cycloheximide
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- qPCR
- quantitative PCR
- ANOVA
- analysis of variance
- IHC
- immunohistochemistry.
References
- 1. Dheen S. T., Kaur C., and Ling E.-A. (2007) Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14, 1189–1197 [DOI] [PubMed] [Google Scholar]
- 2. Kreutzberg G. W. (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318 [DOI] [PubMed] [Google Scholar]
- 3. Guillot-Sestier M.-V., Doty K. R., and Town T. (2015) Innate immunity fights Alzheimer's disease. Trends Neurosci. 38, 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G. M., Cooper N. R., Eikelenboom P., Emmerling M., Fiebich B. L., Finch C. E., Frautschy S., Griffin W. S., Hampel H., Hull M., Landreth G., et al. (2000) Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Glass C. K., Saijo K., Winner B., Marchetto M. C., and Gage F. H. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heppner F. L., Ransohoff R. M., and Becher B. (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 [DOI] [PubMed] [Google Scholar]
- 7. Scholz J., and Woolf C. J. (2007) The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci. 10, 1361–1368 [DOI] [PubMed] [Google Scholar]
- 8. Austin P. J., and Moalem-Taylor G. (2010) The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J. Neuroimmunol. 229, 26–50 [DOI] [PubMed] [Google Scholar]
- 9. Schomberg D., and Olson J. K. (2012) Immune responses of microglia in the spinal cord: contribution to pain states. Exp. Neurol. 234, 262–270 [DOI] [PubMed] [Google Scholar]
- 10. Herz J., and Strickland D. K. (2001) LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gonias S. L., and Campana W. M. (2014) LDL receptor-related protein-1: a regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. Am. J. Pathol. 184, 18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernandez-Castaneda A., Arandjelovic S., Stiles T. L., Schlobach R. K., Mowen K. A., Gonias S. L., and Gaultier A. (2013) Identification of the low density lipoprotein (LDL) receptor-related protein-1 interactome in central nervous system myelin suggests a role in the clearance of necrotic cell debris. J. Biol. Chem. 288, 4538–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Gool B., Dedieu S., Emonard H., and Roebroek A. J. (2015) The matricellular receptor LRP1 forms an interface for signaling and endocytosis in modulation of the extracellular tumor environment. Front. Pharmacol. 6, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Flütsch A., Henry K., Mantuano E., Lam M. S., Shibayama M., Takahashi K., Gonias S. L., and Campana W. M. (2016) Evidence that LDL receptor-related protein 1 acts as an early injury detection receptor and activates c-Jun in Schwann cells. Neuroreport 27, 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Overton C. D., Yancey P. G., Major A. S., Linton M. F., and Fazio S. (2007) Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ. Res. 100, 670–677 [DOI] [PubMed] [Google Scholar]
- 16. Gaultier A., Arandjelovic S., Niessen S., Overton C. D., Linton M. F., Fazio S., Campana W. M., Cravatt B. F. 3rd, and Gonias S. L. (2008) Regulation of tumor necrosis factor receptor-1 and the IKK-NF-κB pathway by LDL receptor-related protein explains the antiinflammatory activity of this receptor. Blood 111, 5316–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zurhove K., Nakajima C., Herz J., Bock H. H., and May P. (2008) γ-Secretase limits the inflammatory response through the processing of LRP1. Sci. Signal. 1, ra15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Staudt N. D., Jo M., Hu J., Bristow J. M., Pizzo D. P., Gaultier A., VandenBerg S. R., and Gonias S. L. (2013) Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res. 73, 3902–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. May P., Bock H. H., and Nofer J.-R. (2013) Low density receptor-related protein 1 (LRP1) promotes anti-inflammatory phenotype in murine macrophages. Cell Tissue Res. 354, 887–889 [DOI] [PubMed] [Google Scholar]
- 20. Mantuano E., Brifault C., Lam M. S., Azmoon P., Gilder A. S., and Gonias S. L. (2016) LDL receptor-related protein-1 regulates NFκB and microRNA-155 in macrophages to control the inflammatory response. Proc. Natl. Acad. Sci. U.S.A. 113, 1369–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mantuano E., Azmoon P., Brifault C., Banki M. A., Gilder A. S., Campana W. M., and Gonias S. L. (2017) Tissue-type plasminogen activator regulates macrophage activation and innate immunity. Blood 130, 1364–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marzolo M. P., von Bernhardi R., Bu G., and Inestrosa N. C. (2000) Expression of α(2)-macroglobulin receptor/low density lipoprotein receptor-related protein (LRP) in rat microglial cells. J. Neurosci. Res. 60, 401–411 [DOI] [PubMed] [Google Scholar]
- 23. Hendrickx D. A., Koning N., Schuurman K. G., van Strien M. E., van Eden C. G., Hamann J., and Huitinga I. (2013) Selective upregulation of scavenger receptors in and around demyelinating areas in multiple sclerosis. J. Neuropathol. Exp. Neurol. 72, 106–118 [DOI] [PubMed] [Google Scholar]
- 24. Chuang T.-Y., Guo Y., Seki S. M., Rosen A. M., Johanson D. M., Mandell J. W., Lucchinetti C. F., and Gaultier A. (2016) LRP1 expression in microglia is protective during CNS autoimmunity. Acta Neuropathol. Commun. 4, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L., Liu C.-C., Zheng H., Kanekiyo T., Atagi Y., Jia L., Wang D., N'songo A., Can D., Xu H., Chen X.-F., and Bu G. (2016) LRP1 modulates the microglial immune response via regulation of JNK and NF-κB signaling pathways. J. Neuroinflammation. 13, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pocivavsek A., Burns M. P., and Rebeck G. W. (2009) Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia 57, 444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pocivavsek A., Mikhailenko I., Strickland D. K., and Rebeck G. W. (2009) Microglial low-density lipoprotein receptor-related protein 1 modulates c-Jun N-terminal kinase activation. J. Neuroimmunol. 214, 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. von Arnim C. A., Kinoshita A., Peltan I. D., Tangredi M. M., Herl L., Lee B. M., Spoelgen R., Hshieh T. T., Ranganathan S., Battey F. D., Liu C.-X., Bacskai B. J., Sever S., Irizarry M. C., Strickland D. K., and Hyman B. T. (2005) The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J. Biol. Chem. 280, 17777–17785 [DOI] [PubMed] [Google Scholar]
- 29. Liu Q., Zhang J., Tran H., Verbeek M. M., Reiss K., Estus S., and Bu G. (2009) LRP1 shedding in human brain: roles of ADAM10 and ADAM17. Mol. Neurodegener. 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gorovoy M., Gaultier A., Campana W. M., Firestein G. S., and Gonias S. L. (2010) Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. J. Leukocyte Biol. 88, 769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamamoto K., Santamaria S., Botkjaer K. A., Dudhia J., Troeberg L., Itoh Y., Murphy G., and Nagase H. (2017) Inhibition of shedding of low-density lipoprotein receptor-related protein 1 reverses cartilage matrix degradation in osteoarthritis. Arthritis Rheumatol. 69, 1246–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quinn K. A., Pye V. J., Dai Y. P., Chesterman C. N., and Owensby D. A. (1999) Characterization of the soluble form of the low density lipoprotein receptor-related protein (LRP). Exp. Cell Res. 251, 433–441 [DOI] [PubMed] [Google Scholar]
- 33. Gaultier A., Arandjelovic S., Li X., Janes J., Dragojlovic N., Zhou G. P., Dolkas J., Myers R. R., Gonias S. L., and Campana W. M. (2008) A shed form of LDL receptor-related protein-1 regulates peripheral nerve injury and neuropathic pain in rodents. J. Clin. Invest. 118, 161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wygrecka M., Wilhelm J., Jablonska E., Zakrzewicz D., Preissner K. T., Seeger W., Guenther A., and Markart P. (2011) Shedding of low-density lipoprotein receptor-related protein-1 in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 184, 438–448 [DOI] [PubMed] [Google Scholar]
- 35. Scilabra S. D., Troeberg L., Yamamoto K., Emonard H., Thøgersen I., Enghild J. J., Strickland D. K., and Nagase H. (2013) Differential regulation of extracellular tissue inhibitor of metalloproteinases-3 levels by cell membrane-bound and shed low density lipoprotein receptor-related protein 1. J. Biol. Chem. 288, 332–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grimsley P. G., Quinn K. A., Chesterman C. N., and Owensby D. A. (1999) Evolutionary conservation of circulating soluble low density lipoprotein receptor-related protein-like (“LRP-like”) molecules. Thromb. Res. 94, 153–164 [DOI] [PubMed] [Google Scholar]
- 37. Clausen B. E., Burkhardt C., Reith W., Renkawitz R., and Förster I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 [DOI] [PubMed] [Google Scholar]
- 38. Kavanagh E., Burguillos M. A., Carrillo-Jimenez A., Oliva-Martin M. J., Santiago M., Rodhe J., Joseph B., and Venero J. L. (2015) Deletion of caspase-8 in mouse myeloid cells blocks microglia proinflammatory activation and confers protection in MPTP neurodegeneration model. Aging 7, 673–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murphy S. (2000) Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia 29, 1–13 [DOI] [PubMed] [Google Scholar]
- 40. Kettenmann H., Hanisch U.-K., Noda M., and Verkhratsky A. (2011) Physiology of microglia. Physiol. Rev. 91, 461–553 [DOI] [PubMed] [Google Scholar]
- 41. Strickland D. K., Gonias S. L., and Argraves W. S. (2002) Diverse roles for the LDL receptor family. Trends Endocrinol. Metab. 13, 66–74 [DOI] [PubMed] [Google Scholar]
- 42. Webb D. J., Nguyen D. H., Sankovic M., and Gonias S. L. (1999) The very low density Lipoprotein receptor regulates urokinase receptor catabolism and breast cancer cell motility in vitro. J. Biol. Chem. 274, 7412–7420 [DOI] [PubMed] [Google Scholar]
- 43. Basu S., Binder R. J., Ramalingam T., and Srivastava P. K. (2001) CD91 is a common receptor for heat-shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14, 303–313 [DOI] [PubMed] [Google Scholar]
- 44. Gardai S. J., McPhillips K. A., Frasch S. C., Janssen W. J., Starefeldt A., Murphy-Ullrich J. E., Bratton D. L., Oldenborg P.-A., Michalak M., and Henson P. M. (2005) Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 [DOI] [PubMed] [Google Scholar]
- 45. Pawaria S., and Binder R. J. (2011) CD91-dependent programming of T-helper cell responses following heat-shock protein immunization. Nat. Commun 2, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fricker M., Oliva-Martín M. J., and Brown G. C. (2012) Primary phagocytosis of viable neurons by microglia activated with LPS or Aβ is dependent on calreticulin/LRP phagocytic signalling. J. Neuroinflammation 9, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., and Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 48. Johannessen M., Delghandi M. P., and Moens U. (2004) What turns CREB on? Cell. Signal. 16, 1211–1227 [DOI] [PubMed] [Google Scholar]
- 49. Herz J., Clouthier D. E., and Hammer R. E. (1992) LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 71, 411–421 [DOI] [PubMed] [Google Scholar]
- 50. Lillis A. P., Greenlee M. C., Mikhailenko I., Pizzo S. V., Tenner A. J., Strickland D. K., and Bohlson S. S. (2008) Murine low-density lipoprotein receptor-related protein 1 (LRP1) is required for phagocytosis of targets bearing LRP ligands but is not required for C1q-triggered enhancement of phagocytosis. J. Immunol. 181, 364–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duus K., Hansen E. W., Tacnet P., Frachet P., Arlaud G. J., Thielens N. M., and Houen G. (2010) Direct interactions between CD91 and C1q. FEBS J. 277, 3526–3537 [DOI] [PubMed] [Google Scholar]
- 52. Quinn K. A., Grimsley P. G., Dai Y. P., Tapner M., Chesterman C. N., and Owensby D. A. (1997) Soluble low density lipoprotein receptor-repeated protein (LRP) circulates in human plasma. J. Biol. Chem. 272, 23946–23951 [DOI] [PubMed] [Google Scholar]
- 53. Wolf B. B., Lopes M. B., VandenBerg S. R., and Gonias S. L. (1992) Characterization and immunohistochemical localization of α2-macroglobulin receptor (low-density lipoprotein receptor-related protein) in human brain. Am. J. Pathol. 141, 37–42 [PMC free article] [PubMed] [Google Scholar]
- 54. Moestrup S. K., Gliemann J., and Pallesen G. (1992) Distribution of the α2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 269, 375–382 [DOI] [PubMed] [Google Scholar]
- 55. Lopes M. B., Bogaev C. A., Gonias S. L., and VandenBerg S. R. (1994) Expression of α2-macroglobulin receptor/low density lipoprotein receptor-related protein is increased in reactive and neoplastic glial cells. FEBS Lett. 338, 301–305 [DOI] [PubMed] [Google Scholar]
- 56. Hussaini I. M., Srikumar K., Quesenberry P. J., and Gonias S. L. (1990) Colony-stimulating factor-1 modulates alpha 2-macroglobulin receptor expression in murine bone marrow macrophages. J. Biol. Chem. 265, 19441–19446 [PubMed] [Google Scholar]
- 57. LaMarre J., Wolf B. B., Kittler E. L., Quesenberry P. J., and Gonias S. L. (1993) Regulation of macrophage α2-macroglobulin receptor/low density lipoprotein receptor-related protein by lipopolysaccharide and interferon-γ. J. Clin. Invest. 91, 1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Costales P., Castellano J., Revuelta-López E., Cal R., Aledo R., Llampayas O., Nasarre L., Juarez C., Badimon L., and Llorente-Cortés V. (2013) Lipopolysaccharide downregulates CD91/low-density lipoprotein receptor-related protein 1 expression through SREBP-1 overexpression in human macrophages. Atherosclerosis 227, 79–88 [DOI] [PubMed] [Google Scholar]
- 59. Shi Y., Mantuano E., Inoue G., Campana W. M., and Gonias S. L. (2009) Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci. Signal. 2, ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mantuano E., Lam M. S., and Gonias S. L. (2013) LRP1 assembles unique co-receptor systems to initiate cell signaling in response to tissue-type plasminogen activator and myelin-associated glycoprotein. J. Biol. Chem. 288, 34009–34018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Herz J., Goldstein J. L., Strickland D. K., Ho Y. K., and Brown M. S. (1991) 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J. Biol. Chem. 266, 21232–21238 [PubMed] [Google Scholar]
- 62. Rohlmann A., Gotthardt M., Hammer R. E., and Herz J. (1998) Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J. Clin. Invest. 101, 689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ni M., and Aschner M. (2010) Neonatal rat primary microglia: isolation, culturing, and selected applications. Curr. Protoc. Toxicol. Chapter 12, Unit 12.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Campana W. M., Mantuano E., Azmoon P., Henry K., Banki M. A., Kim J. H., Pizzo D. P., and Gonias S. L. (2017) Ionotropic glutamate receptors activate cell signaling in response to glutamate in Schwann cells. FASEB J. 31, 1744–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.