

The periplasmic form of dipeptidyl aminopeptidase IV from P. mexicana WO24 was crystallized in complex with the tripeptide Lys-Pro-Tyr. X-ray diffraction data were collected to 1.90 Å resolution.
Keywords: DAP, dipeptidyl aminopeptidase, DPP4, DPP IV, Pseudoxanthomonas mexicana
Abstract
Dipeptidyl aminopeptidase IV (DAP IV or DPP IV) from Pseudoxanthomonas mexicana WO24 (PmDAP IV) preferentially cleaves substrate peptides with Pro or Ala at the P1 position [NH2-P2-P1(Pro/Ala)-P1′-P2′…]. For crystallographic studies, the periplasmic form of PmDAP IV was overproduced in Escherichia coli, purified and crystallized in complex with the tripeptide Lys-Pro-Tyr using the hanging-drop vapour-diffusion method. Kinetic parameters of the purified enzyme against a synthetic substrate were also determined. X-ray diffraction data to 1.90 Å resolution were collected from a triclinic crystal form belonging to space group P1, with unit-cell parameters a = 88.66, b = 104.49, c = 112.84 Å, α = 67.42, β = 68.83, γ = 65.46°. Initial phases were determined by the molecular-replacement method using Stenotrophomonas maltophilia DPP IV (PDB entry 2ecf) as a template and refinement of the structure is in progress.
1. Introduction
Dipeptidyl aminopeptidase (DAP or DPP; EC 3.4.14) catalyses the removal of dipeptides from the amino-termini of peptides and proteins. In microorganisms, we have reported the identification, purification and characterization of DAP BI (bacterial dipeptidyl aminopeptidase; Ogasawara, Ochiai et al., 1996 ▸; Ogasawara et al., 1998 ▸); DAP BII (Ogasawara, Kobayashi et al., 1996 ▸; Suzuki et al., 2014 ▸; Sakamoto et al., 2014 ▸); DAP BIII (Ogasawara, Kobayashi et al., 1996 ▸); DAP IV (in this paper, we designate bacterial DPP IV as DAP IV; Ogasawara, Ogawa et al., 1996 ▸; Ogasawara et al., 2005 ▸) and prolyloligopeptidase (POP; Ogasawara et al., unpublished work) from Pseudoxanthomonas mexicana WO24, which is a Gram-negative aerobic bacteria isolated from the wastewater of a bean-curd (tofu) factory. We have demonstrated that DAP BI, DAP BIII, DAP IV and POP belong to the POP family (Kanatani et al., 1991 ▸; Rawlings et al., 1991 ▸) and that they are classified into clan SC, family S9 in the MEROPS database (Rawlings et al., 2014 ▸), whereas DAP BII is classified into the clan PA, family S46 (Suzuki et al., 2014 ▸; Sakamoto et al., 2014 ▸). On the basis of the enzymological data that we have obtained, we proposed that bacterial DAPs should be classified in a different manner to mammalian DPPs (Ogasawara, Kobayashi et al., 1996 ▸), except for DAP IV. Bacterial DAP IVs and mammalian DPP IVs liberate dipeptides from the free amino-terminus and have a specificity for proline and alanine residues at the penultimate position of peptides, and are classified under the same EC number, EC 3.4.14.5. DPP IV has been isolated from bacteria (Yoshimoto & Tsuru, 1982 ▸; Mayo et al., 1991 ▸; Ogasawara, Ogawa et al., 1996 ▸) and fungi (Tachi et al., 1992 ▸; Beauvais et al., 1997 ▸), as well as mammals (Hopsu-Havu & Glenner, 1966 ▸; Barth et al., 1974 ▸; Yoshimoto & Walter, 1977 ▸), and both the bacterial and mammalian enzymes form a dimer in solution. In mammals, soluble and membrane-bound forms of DPP IV have been characterized. Because DPP IV is responsible for the degradation of incretins such as GLP-1 and plays a major role in glucose metabolism, DPP IV is the well known target of a new class of oral hypoglycaemics (Barnett, 2006 ▸). Several reviews of the structural and functional studies of mammalian DPP IV and related enzymes are available (Gorrell, 2005 ▸; Wagner et al., 2016 ▸; Klemann et al., 2016 ▸). On the other hand, DAP IV plays an important nutritional function in asaccharolytic bacteria, which utilize proteins or peptides as an energy source, in cooperation with other peptidases. Because the DAP IV gene is found in many pathogenic bacteria, such as Porphyromonas gingivalis (causative of periodontitis) and Stenotrophomonas maltophilia (causative of opportunistic infectious diseases), high-resolution crystal structure analyses of bacterial DAP IV in complex with an inhibitor (or substrate analogue) would assist in the development of selective inhibitors of the pathogenic DAP IVs. To date, several crystal structures of mammalian DPP IVs have been reported and substrate-recognition mechanisms of the mammalian DPP IVs have been discussed in detail (Engel et al., 2003 ▸; Hiramatsu et al., 2003 ▸; Rasmussen et al., 2003 ▸). However, crystal structure analysis of a bacterial DAP IV has been only reported at medium resolution (2.8 Å) for DAP IV from S. maltophilia (SmDAP IV; Nakajima et al., 2008 ▸) in a peptide-free form. The structural analysis of SmDAP IV revealed that the overall structure of SmDAP IV is similar to those of mammalian DPP IVs; however, an active-site arginine residue (Arg125 in human DPP IV) which is responsible for the recognition of the carbonyl group of the P2 residue of a substrate peptide is not conserved in bacterial DAP IVs. Amino-acid sequence identities between bacterial DAP IVs and mammalian DPP IVs are below 25%. In the absence of a high-resolution crystal structure of a bacterial DAP IV complexed with an inhibitor (or substrate analogue), a complete structural understanding of the DAP IV–substrate interactions has been impossible. Here, we report the purification, kinetic characterization, crystallization and X-ray crystallographic analysis of the periplasmic 82 kDa form of DAP IV from P. mexicana WO24.
2. Materials and methods
2.1. Overproduction and purification
A gene coding for PmDAP IV (residues 1–745) was cloned from a P. mexicana WO24 genomic DNA library using a cultivation-plate assay based on the activity of DPP IV in the hydrolysis of Gly-Pro-β-naphthylamide (Ogasawara, Ogawa et al., 1996 ▸). The N-terminal region (residues 1–22) has a signal sequence typical of Gram-negative bacteria. In an Escherichia coli expression system, PmDAP IV was expressed as 82 and 84 kDa isoforms (corresponding to residues 23–745 and 12–745, respectively), with two translation-initiation codons, Met1 and Met12 (Ogasawara et al., 2005 ▸). The PmDAP IV M12I mutant produced only the 82 kDa molecule owing to translation from Met1 and removal of the signal sequence. The periplasmic form of PmDAP IV, residues 23–745, is composed of 723 amino acids with a theoretical molecular weight of 79 981.58 and a theoretical isoelectric point of 5.80 (Gasteiger et al., 2005 ▸). In this study, an E. coli JM109 (Takara Bio) transformant harbouring the full-length PmDAP IV M12I mutant sequence inserted into the pUC19 (Takara Bio) expression plasmid was used for production of the periplasmic form of PmDAP IV (Table 1 ▸). The cells were grown in 2× YT medium at 310 K. Overproduction of PmDAP IV was performed by IPTG induction (final concentration 0.1 mM) at an OD600 of approximately 0.6. 16 h after induction, the cells were harvested by centrifugation at 8000g. The cells were disrupted using BugBuster Protein Extraction Reagent (Novagen). The cell extract was obtained by centrifugation at 27 000g for 30 min. The PmDAP IV in the cell extract was precipitated by 35–70% saturated ammonium sulfate and the precipitate was then dissolved in 30% saturated ammonium sulfate solution in buffer A (50 mM Tris–HCl pH 8.0). The enzyme solution was filtrated with a 0.22 µm pore filter and applied onto a 20 ml HiPrep 16/10 Butyl column (GE Healthcare) equilibrated with wash buffer (30% saturated ammonium sulfate in buffer A). After sample injection, the column was washed with two column volumes of wash buffer. After washing, PmDAP IV was eluted with five column volumes of a gradient from 30 to 0% saturated ammonium sulfate in buffer A. The fractions containing PmDAP IV were pooled and applied onto a 50 ml HiPrep 26/10 Desalting column (GE Healthcare) equilibrated with buffer B (20 mM Tris–HCl pH 8.0). The PmDAP IV was then applied onto a 1 ml Mono Q 5/50 GL anion-exchange chromatography column (GE Healthcare) equilibrated with buffer B. The column was washed with five column volumes of buffer B. After washing, PmDAP IV was eluted with 30 column volumes of a gradient from 0 to 0.3 M sodium chloride in buffer B. The fractions containing PmDAP IV were pooled and the buffer was exchanged to 80 mM Tris–HCl pH 8.5 and concentrated to 10 mg ml−1 using a Vivaspin 20 concentrator (GE Healthcare). The protein concentration was determined by the Bradford assay using bovine serum albumin as a standard (Bio-Rad). The above-mentioned column chromatography and other purification steps were performed at 298 and 277 K, respectively.
Table 1. Expression system for PmDAP IV.
| Source organism | P. mexicana WO24 |
| DNA source | Genomic DNA library of P. mexicana WO24 |
| Cloning method | Screening by the DPP IV activity in an agar plate containing the DPP IV substrate Gly-Pro-β-naphthylamide (Ogasawara, Ogawa et al., 1996 ▸) |
| Cloning vector | pBluescript II KS(+) |
| Expression vector | pUC19 |
| Expression host | E. coli JM109 |
| Complete amino-acid sequence of the construct produced† | MRLALFALFALITVATALPAHAEKLTLEAITGSAPLSGPTLTKPQIAPDGSRVTFLRGKDRDRNRLDLWEYDIASGQTRLLVDSSVVLPGEEVLSDEEKARRERQRIAALSGIVDYQWSPDGKALLFPLGGELYFYDLTKSGRDAVRKLTNGGGFATDPKISPKGGFVSFIRDRNLWAIDLASGKEVQLTRDGSDTIGNGVAEFVADEEMDRHTGYWWAPDDAAIAFARIDETPVPVQKRYEVYPDRTEVVEQRYPAAGDHNVRVQLGVIAPKTGARPRWIDLGKDPDIYLARVDWRDPQRLTFQRQSRDQKKIELIETTLTNGTQRTLVTETSTTWVPLHNDLRFLKDGRFLWSSERSGFEHLYVASEDGSTLTALTQGEWVVDSLLAIDEAAGLAYVSGTRDGATEAHVYAVPLSGGEPRRLTQAPGMHAATFARNASVFVDSWSSDTTLPQIELFKADGTKLATLLVNDVSDATHPYAKYRAAHQPTAYGTLTAADGTTPLHYSLIKPAGFDPKKQYPVVVFVYGGPAAQTVTRAWPGRSDSFFNQYLAQQGYVVFTLDNRGTPRRGAAFGGALYGKQGTVEVDDQLRGIEWLKSQAFVDPARIGVYGWSNGGYMTLMLLAKHDEAYACGVAGAPVTDWALYDTHYTERYMDLPKANEAGYREASVFTHVDGIGAGKLLLIHGMADDNVLFTNSTKLMSELQKRGTPFELMTYPGAKHGLRGSDLLHRYRLTEDFFARCLKP |
The signal sequence (residues 1–22; Met12 was mutated to Ile) is underlined. Ile12 is shown in bold.
2.2. Determination of kinetic parameters
Kinetic parameters were determined by fitting the experimental data to the Michaelis–Menten equation using Excel Solver (Microsoft) by nonlinear least-squares fitting with different concentrations (0.78, 1.56, 3.13, 6.25, 12.5, 25, 50 and 100 µM) of glycyl-l-proline 4-methylcoumaryl-7-amide (Gly-Pro-MCA; Peptide Institute) as a substrate. The enzyme reaction was performed in a reaction buffer consisting of 50 mM sodium phosphate buffer pH 8.0, 5 mM EDTA, 0.005% Tween 20 at 298 K for 20 min and standard deviations were calculated from three independent experiments. The fluorescence intensity of the released MCA was measured with excitation at 355 nm and emission at 460 nm using an Infinite 200 PRO microplate reader (Tecan).
2.3. Crystallization
Initial sparse-matrix crystal screening (Jancarik & Kim, 1991 ▸) was conducted using Crystal Screen, Crystal Screen 2, Crystal Screen Cryo, PEG/Ion, PEG/Ion 2 and Index (Hampton Research), Wizard I, II and III, Ozma 1K, 4K, 8K and 10K, Cryo I and II (Rigaku Reagents) and The PACT and JCSG+ Suites (Qiagen). Crystallization was carried out using the hanging-drop method, in which 1 µl protein solution (10 mg ml−1) was mixed with the same volume of crystallization buffer and incubated at 293 K. The drops were suspended over 200 µl reservoir solution in 48-well plates.
2.4. X-ray data collection and processing
Since the optimized crystallization condition of PmDAP IV described below contained 20%(v/v) glycerol in the reservoir solution, X-ray data collection could be performed under cryogenic conditions without further addition of cryoprotectant. Crystals from the hanging drop were directly mounted in nylon loops and flash-cooled in a cold nitrogen-gas stream at 95 K just prior to data collection. Data collection was performed by a helical scan method at 95 K with an ADSC Q270 CCD detector using synchrotron radiation (λ = 0.9800) on beamline BL17A of the Photon Factory, Tsukuba, Japan. The helical scan adds a small translation to the crystal in the rotation method, so that the crystal is shifted to expose a fresh part for each image. The total translation of the crystal was 0.3 mm during 200° of rotation. The Laue group and unit-cell parameters were determined using xia2 (Winter, 2010 ▸). Self-rotation and native Patterson functions were calculated using POLARRFN and FFT from the CCP4 suite (Winn et al., 2011 ▸).
3. Results and discussion
3.1. Overproduction and purification
The periplasmic form of PmDAP IV was successfully overproduced and purified to homogeneity with an approximate yield of 12 mg of protein per litre of bacterial culture at the end of the purification process. SDS–PAGE of the purified enzyme revealed a single 82 kDa protein band by Coomassie Brilliant Blue staining (Fig. 1 ▸).
Figure 1.
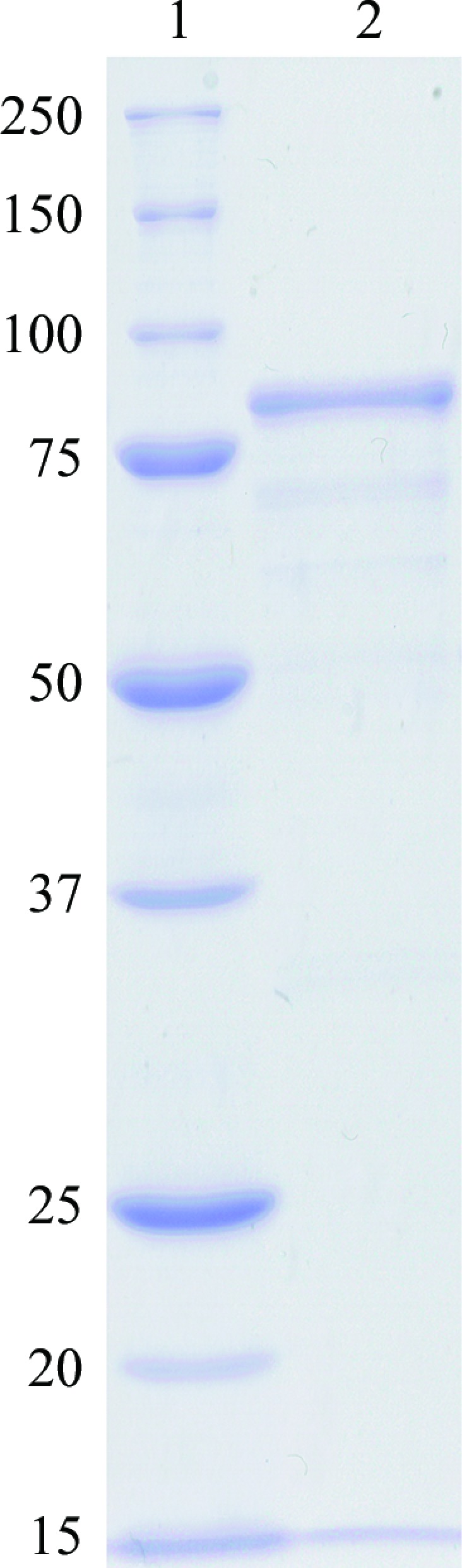
Coomassie-stained 10% SDS–PAGE analysis of PmDAP IV. Lane 1, molecular-weight markers; lane 2, purified PmDAP IV after the final buffer exchange. Molecular-weight markers are labelled in kDa.
3.2. Kinetic parameters
Supplementary Fig. S1 shows a substrate concentration versus reaction rate curve. The Michaelis constant (K m), the catalytic rate (k cat) and the catalytic efficiency (k cat/K m) of PmDAP IV for the synthetic substrate Gly-Pro-MCA were determined to be 19.4 ± 0.8 µM, 19.2 ± 0.4 s−1 and 0.989 ± 0.035 µM −1 s−1, respectively. The K m values of a mammalian DPP IV for various peptide substrates range from 4 to 66 µM (Rahfeld et al., 1991 ▸).
3.3. Crystallization
Initial crystal screening produced microcrystals within two months. Several crystals grew from condition No. 22 of Crystal Screen 2 from Hampton Research [0.1 M MES pH 6.5, 12%(w/v) PEG 20 000] and condition No. 46 of The PACT Suite from Qiagen [0.1 M Tris–HCl pH 8.0, 0.2 M magnesium chloride, 20%(w/v) PEG 8000]. To obtain crystals suitable for X-ray analysis, a droplet was prepared by mixing equal volumes (1 µl + 1 µl) of protein solution containing the tripeptide Lys-Pro-Tyr (9 mg ml−1 protein and 4 mM Lys-Pro-Tyr in 80 mM Tris–HCl pH 8.5) and reservoir solution [20% glycerol, 12%(w/v) PEG 20 000, 80 mM MES pH 6.5] and was suspended over 200 µl reservoir solution in 48-well plates (Table 2 ▸). Plate-shaped crystals with typical dimensions of approximately 0.7 × 0.2 × 0.01 mm grew within 5 d (Fig. 2 ▸). Addition of the Lys-Pro-Tyr tripeptide was required to obtain well diffracting crystals. Although the cytosolic 84 kDa form of PmDAP IV was also successfully overproduced and purified to homogeneity, with an approximate yield of 7 mg of protein per litre of bacterial culture (Ogasawara et al., 2005 ▸), crystallization of the 84 kDa PmDAP IV resulted in the formation of poorly diffracting crystals both in the presence and absence of additional peptides.
Table 2. Crystallization of PmDAP IV.
| Method | Hanging-drop vapour diffusion |
| Plate type | Hampton Research 48-well plates |
| Temperature (K) | 293 |
| Protein concentration (mg ml−1) | 9 |
| Peptide (Lys-Pro-Tyr) concentration (mM) | 4 |
| Buffer composition of protein solution | 80 mM Tris–HCl pH 8.5 |
| Composition of reservoir solution | 80 mM MES pH 6.5, 12%(w/v) PEG 20 000, 20%(v/v) glycerol |
| Volume of drop | 1 µl protein + 1 µl reservoir |
| Volume of reservoir (µl) | 200 |
Figure 2.

A triclinic crystal of PmDAP IV.
3.4. X-ray analysis
The Laue group of the PmDAP IV crystals was found to be −1 and the unit-cell parameters were a = 88.66, b = 104.49, c = 112.84 Å, α = 67.42, β = 68.83, γ = 65.46°. The current best diffraction data from a PmDAP IV crystal were collected to 1.90 Å resolution (Fig. 3 ▸). Data-collection statistics are summarized in Table 3 ▸. The presence of four subunits (two dimers) per asymmetric unit (see below) led to a V M value of 2.67 Å3 Da−1, corresponding to a solvent content of 53.9% (Matthews, 1968 ▸).
Figure 3.

X-ray diffraction image from a PmDAP IV crystal. The circles indicate resolutions of 7.00, 3.00 and 1.90 Å.
Table 3. Data-collection statistics for PmDAP IV.
Values in parentheses are for the outer shell.
| Diffraction source | BL17A, Photon Factory |
| Wavelength (Å) | 0.9800 |
| Temperature (K) | 95 |
| Detector | ADSC Q270 |
| Crystal-to-detector distance (mm) | 250.1 |
| Rotation range per image (°) | 0.15 |
| Total rotation range (°) | 200 |
| Exposure time per image (s) | 1 |
| Space group | P1 |
| a, b, c (Å) | 88.66, 104.49, 112.84 |
| α, β, γ (°) | 67.42, 68.83, 65.46 |
| Mosaicity (°) | 0.11 |
| Resolution range (Å) | 50.0–1.90 (2.00–1.90) |
| Total No. of reflections | 540678 (59278) |
| No. of unique reflections | 245086 (27199) |
| Completeness (%) | 94.2 (71.5) |
| Multiplicity | 2.2 (2.2) |
| 〈I/σ(I)〉 | 6.5 (2.0) |
| R merge † | 0.082 (0.316) |
| Overall B factor from Wilson plot (Å2) | 12.5 |
R
merge = 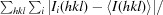
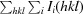 , where Ii(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the average of symmetry-related (or Friedel-related) observations of a unique reflection.
, where Ii(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the average of symmetry-related (or Friedel-related) observations of a unique reflection.
3.5. Initial phase determination
Initial phase determination for the PmDAP IV crystal was performed by the molecular-replacement technique using the coordinates of one protomer of SmDAP IV (PDB entry 2ecf; Nakajima et al., 2008 ▸), which has approximately 74% amino-acid sequence identity to PmDAP IV, as a search model. Bound water molecules were removed from the search model. Cross-rotation and translation functions were calculated using MOLREP (Vagin & Teplyakov, 2010 ▸) from the CCP4 suite (Winn et al., 2011 ▸). The results showed a clear solution [correlation coefficient of 0.318 (0.270 for the first noise) and R factor of 0.610 (0.627 for the first noise) in the resolution range 50.0–1.90 Å] with two dimers of PmDAP IV in the asymmetric unit for space group P1. After the first round of restrained refinement, the R and R free factors decreased to 0.409 and 0.449, respectively, at 1.90 Å resolution. Automatic model building and refinement using the programs ARP/wARP (Langer et al., 2008 ▸) and REFMAC5 (Murshudov et al., 2011 ▸) and further iterative manual model building and refinement with Coot (Emsley et al., 2010 ▸) and REFMAC5 are currently in progress. In parallel to the refinement, we are preparing crystals of PmDAP IV complexed with various peptidase inhibitors and oligopeptides in order to study their mode of interaction with the enzyme.
Supplementary Material
Supplementary Figure S1.. DOI: 10.1107/S2053230X17014911/nw5063sup1.pdf
Acknowledgments
We thank Drs Y. Yamada and N. Matsugaki of the Photon Factory and Drs K. Hasegawa, A. Higashiura and E. Yamashita of SPring-8 for their help with data collection at the synchrotron facilities. The synchrotron-radiation experiments were performed on BL17A at Photon Factory (Proposal Nos. 2011G090, 2013G138 and 2017G162) and BL44XU and BL41XU at SPring-8 (Proposal Nos. 2013A6822, 2013B6822, 2014A6924, 2014B6924, 2015A6521, 2015B6521, 2016B6620, 2017A6721 and 2017B6721). This work was supported in part by the Platform for Drug Discovery, Informatics and Structural Life Science (to NT and Y. Sakamoto) and the Collaborative Researcher Program of The Institute for Protein Research, Osaka University (Proposal Nos. CR1405, CR1505, CR1605 and CR1705; to NT and Y. Sakamoto).
Funding Statement
This work was funded by Japan Society for the Promotion of Science grants 25462872, 16K08322, and 16H04902. Takeda Science Foundation grant to Yasumitsu Sakamoto. Nagai Foundation Tokyo grant to Saori Roppongi.
References
- Barnett, A. H. (2006). Int. J. Clin. Pract. 60, 1454–1470. [DOI] [PubMed]
- Barth, A., Schulz, H. & Neubert, K. (1974). Acta Biol. Med. Ger. 32, 157–174. [PubMed]
- Beauvais, A., Monod, M., Wyniger, J., Debeaupuis, J. P., Grouzmann, E., Brakch, N., Svab, J., Hovanessian, A. G. & Latgé, J. P. (1997). Infect. Immun. 65, 3042–3047. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engel, M., Hoffmann, T., Wagner, L., Wermann, M., Heiser, U., Kiefersauer, R., Huber, R., Bode, W., Demuth, H.-U. & Brandstetter, H. (2003). Proc. Natl Acad. Sci. USA, 100, 5063–5068. [DOI] [PMC free article] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Gorrell, M. D. (2005). Clin. Sci. 108, 277–292. [DOI] [PubMed]
- Hiramatsu, H., Kyono, K., Higashiyama, Y., Fukushima, C., Shima, H., Sugiyama, S., Inaka, K., Yamamoto, A. & Shimizu, R. (2003). Biochem. Biophys. Res. Commun. 302, 849–854. [DOI] [PubMed]
- Hopsu-Havu, V. K. & Glenner, G. G. (1966). Histochemie, 7, 197–201. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Kanatani, A., Masuda, T., Shimoda, T., Misoka, F., Lin, X. S., Yoshimoto, T. & Tsuru, D. (1991). J. Biochem. 110, 315–320. [DOI] [PubMed]
- Klemann, C., Wagner, L., Stephan, M. & von Hörsten, S. (2016). Clin. Exp. Immunol. 185, 1–21. [DOI] [PMC free article] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mayo, B., Kok, J., Venema, K., Bockelmann, W., Teuber, M., Reinke, H. & Venema, G. (1991). Appl. Environ. Microbiol. 57, 38–44. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nakajima, Y., Ito, K., Toshima, T., Egawa, T., Zheng, H., Oyama, H., Wu, Y.-F., Takahashi, E., Kyono, K. & Yoshimoto, T. (2008). J. Bacteriol. 190, 7819–7829. [DOI] [PMC free article] [PubMed]
- Ogasawara, W., Kobayashi, G., Ishimaru, S., Okada, H. & Morikawa, Y. (1998). Gene, 206, 229–236. [DOI] [PubMed]
- Ogasawara, W., Kobayashi, G., Okada, H. & Morikawa, Y. (1996). J. Bacteriol. 178, 6288–6295. [DOI] [PMC free article] [PubMed]
- Ogasawara, W., Ochiai, K., Ando, K., Yano, K., Yamasaki, M., Okada, H. & Morikawa, Y. (1996). J. Bacteriol. 178, 1283–1288. [DOI] [PMC free article] [PubMed]
- Ogasawara, W., Ogawa, Y., Yano, K., Okada, H. & Morikawa, Y. (1996). Biosci. Biotechnol. Biochem. 60, 2032–2037. [DOI] [PubMed]
- Ogasawara, W., Tanaka, C., Suzuki, M., Kobayashi, G., Ogawa, Y., Okada, H. & Morikawa, Y. (2005). Protein Expr. Purif. 41, 241–251. [DOI] [PubMed]
- Rahfeld, J., Schierhorn, M., Hartrodt, B., Neubert, K. & Heins, J. (1991). Biochim. Biophys. Acta, 1076, 314–316. [DOI] [PubMed]
- Rasmussen, H. B., Branner, S., Wiberg, F. C. & Wagtmann, N. (2003). Nature Struct. Biol. 10, 19–25. [DOI] [PubMed]
- Rawlings, N. D., Polgar, L. & Barrett, A. J. (1991). Biochem. J. 279, 907–909. [DOI] [PMC free article] [PubMed]
- Rawlings, N. D., Waller, M., Barrett, A. J. & Bateman, A. (2014). Nucleic Acids Res. 42, D503–D509. [DOI] [PMC free article] [PubMed]
- Sakamoto, Y. et al. (2014). Sci. Rep. 4, 4977. [DOI] [PMC free article] [PubMed]
- Suzuki, Y., Sakamoto, Y., Tanaka, N., Okada, H., Morikawa, Y. & Ogasawara, W. (2014). Sci. Rep. 4, 4292. [DOI] [PMC free article] [PubMed]
- Tachi, H., Ito, H. & Ichishima, E. (1992). Phytochemistry, 31, 3707–3709.
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Wagner, L., Klemann, C., Stephan, M. & von Hörsten, S. (2016). Clin. Exp. Immunol. 184, 265–283. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Winter, G. (2010). J. Appl. Cryst. 43, 186–190.
- Yoshimoto, T. & Tsuru, D. (1982). J. Biochem. 91, 1899–1906. [DOI] [PubMed]
- Yoshimoto, T. & Walter, R. (1977). Biochim. Biophys. Acta, 485, 391–401. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1.. DOI: 10.1107/S2053230X17014911/nw5063sup1.pdf