

Abstract
For most individuals, the respiratory control system produces a remarkably stable and coordinated motor output—recognizable as a breath—from birth until death. Very little is understood regarding the processes by which the respiratory control system maintains network stability in the presence of changing physiological demands and network properties that occur throughout life. An emerging principle of neuroscience is that neural activity is sensed and adjusted locally to assure that neurons continue to operate in an optimal range, yet to date, it is unknown whether such homeostatic plasticity is a feature of the neurons controlling breathing. Here, we review the evidence that local mechanisms sense and respond to perturbations in respiratory neural activity, with a focus on plasticity in respiratory motor neurons. We discuss whether these forms of plasticity represent homeostatic plasticity in respiratory control. We present new analyses demonstrating that reductions in synaptic inputs to phrenic motor neurons elicit a compensatory enhancement of phrenic inspiratory motor output, a form of plasticity termed inactivity-induced phrenic motor facilitation (iPMF), that is proportional to the magnitude of activity deprivation. Although the physiological role of iPMF is not understood, we hypothesize that it may have an important role in protecting the drive to breathe during conditions of prolonged or intermittent reductions in respiratory neural activity, such as following spinal cord injury or during central sleep apnea.
INTRODUCTION
Throughout life, the brain exhibits a phenomenal capacity to produce a stable, rhythmic motor output that achieves adequate gas exchange, yet maintains sufficient dynamic range to respond to acute respiratory challenges. This feat is particularly astonishing given the myriad of changes an organism may experience that perturb respiratory control, such as development, aging, coordination of breathing with other motor behaviors, pregnancy, gain/loss of weight, ascent to altitude, etc. While plasticity in the respiratory control system likely confers flexibility in response to physiological or environmental challenges (Feldman et al., 2003, Johnson and Mitchell, 2013), unrestrained changes in neural activity can lead to network instability (Turrigiano, 2008, Turrigiano, 2012). Thus, we hypothesize that to maintain the dynamic, yet reliable properties of the respiratory system in the presence of the many modulatory and plastic mechanisms that confer system flexibility, there likely are also mechanisms that ensure stability of respiratory motor output. One way stability may be maintained is through surveillance and maintenance of respiratory neural activity within specified limits.
The concept of promoting stability in complex physiological systems by homeostatically regulating a physiological parameter around an ideal value has long been appreciated (Cannon, 1932). For example, it is well known that body temperature, blood pressure or blood glucose levels must necessarily be maintained within a narrow range (i.e., a “set-point”) to be compatible for life. As such, environmental or physiological changes that push the system away from its set-point trigger compensatory responses that reestablish that set-point. Recently it has become clear that neuronal activity itself is subject to such homeostatic regulation (Turrigiano et al., 1998, Turrigiano, 2008, Turrigiano, 2012). For example, when cortical or hippocampal neurons are forced to fire outside their normal range (either more or less) for an extended period of time, mechanisms of plasticity are induced that alter cellular properties in the right direction to restore normal firing rates (Turrigiano et al., 1998, Turrigiano, 2012, Tatavarty et al., 2013). Recently, we discovered that activity in respiratory motor neurons might also be subject to such homeostatic regulation: when synaptic inputs to respiratory motor neurons are reduced (in the absence of changing blood gases), local mechanisms of plasticity are induced that counteract this reduction and increase respiratory motor output (Streeter and Baker-Herman, 2014a).
In this review, we discuss evidence that local mechanisms sense and rapidly respond to changes in respiratory neural activity, with a focus on respiratory motor neurons. Although homeostatic mechanisms monitor and adjust neural activity bidirectionally, we highlight mechanisms underlying plasticity induced by hypoactivity. Finally, we put forth the hypothesis that plasticity induced by reductions in respiratory neural activity may be involved in compensatory adaptations during disease and that a failure of such mechanisms may contribute to ventilatory control disorders.
Neural stability is achieved through homeostatic plasticity
Synapse-specific, correlation based changes in synaptic strength are widely thought to be an important mechanism whereby neural circuits learn and store information (e.g., Hebbian plasticity). These experience-based adaptations enable flexibility, but also generate a powerful destabilizing force on neural circuitry, making neurons and networks vulnerable to synaptic saturation or synaptic silencing through positive feedback mechanisms (Miller, 1996, Abbott and Nelson, 2000, Turrigiano, 2008, Turrigiano, 2012). However, in most cases, neural transmission does not unravel as a result of plasticity. Instead, dependable yet dynamic neurotransmission accompany long-term synaptic changes and memory formation in healthy neural networks. Reliability of neurotransmission in a system that readily and continuously adapts its synaptic and network properties is thought to be forged by mechanisms that bidirectionally maintain neural activity within an optimal range for information processing (Abbott and Nelson, 2000, Davis, 2006, Turrigiano, 2008, Turrigiano, 2012, Davis, 2013). Indeed, in organisms ranging from flies to humans, and in a variety of mammalian circuits including cortex (Turrigiano et al., 1998, Watt et al., 2000, Hengen et al., 2013), hippocampus (Lissin et al., 1998, Burrone et al., 2002, Thiagarajan et al., 2005) spinal cord (O’Brien et al., 1998, Gonzalez-Islas and Wenner, 2006) and neuromuscular junction (Davis and Bezprozvanny, 2001), neurons respond to long-term perturbations in neuronal activity by rebalancing cellular properties to restore set-point basal activity levels. Importantly, while the overall output of the neuron/network is homeostatically maintained, the relative strength of individual synapses is preserved, thereby maintaining information storage. This so-called “homeostatic plasticity” is emerging as an important mechanism whereby neuronal function is stabilized in the face of repeated or prolonged perturbations that act to push neuronal activity away from its set-point (Turrigiano, 2008, Turrigiano, 2012).
Since a neuron’s firing rate is a complex interplay between synaptic inputs and intrinsic firing properties, it is not surprising that distinct forms of homeostatic plasticity have been identified that regulate synaptic information flow and intrinsic excitability. For example, deviations in neuronal activity can drive changes in synaptic strength at excitatory and/or inhibitory synapses that are in the right direction to restore baseline firing rates (Turrigiano et al., 1998, Davis, 2006, Turrigiano, 2008, Pozo and Goda, 2010, Turrigiano, 2012, Davis, 2013). Homeostatic synaptic plasticity can manifest as a change in synapse number, presynaptic neurotransmitter release and/or neurotransmitter receptor expression (Davis and Bezprozvanny, 2001; Thiagarajan et al. 2007; Turrigiano, 2011; Yu and Goda 2009). Deviations in neuronal activity can also result in homeostatic adjustments that re-establish baseline firing rates by altering intrinsic excitability (Turrigiano et al., 1994, Desai et al., 1999, Marder and Prinz, 2003). Homeostatic intrinsic plasticity alters the balance of inward and outward voltage-dependent conductances, thereby modulating a neuron’s response to a given synaptic input. Although it is useful to categorize these distinct mechanisms as separate entities for discussion, these mechanisms do not necessarily operate independently, but rather, may be simultaneously expressed within a single cell (Lambo and Turrigiano, 2013). Further complicating matters, mechanisms of homeostatic plasticity can operate locally at a single synapse (Hou et al., 2008, Hou and Man, 2012), or globally throughout a single cell, a group of neurons, or on a network level (Turrigiano, 2008), and can develop after several minutes (Frank et al., 2006), hours (Sutton et al., 2006, Ibata et al., 2008) or days (Stellwagen and Malenka, 2006) of perturbed neuronal activity. Rules that govern when and where these different forms of plasticity are employed are not at all understood.
One key feature of homeostatic plasticity is that it is a quantitatively accurate form of modulation. In this sense, the homeostatic rebalancing of cellular properties that offset a perturbation achieves firing properties that are often indistinguishable from pre-perturbation levels. This requires that a cell has an endogenous set-point that defines the baseline output of the system, a sensor that detects deviations from that set-point to generate an error signal, and mechanisms that drive compensatory changes in response to that error signal to reestablish the set-point (Davis, 2006). It is clear that this defined set-point must be cell type specific since different neurons can have different “jobs” in the nervous system and as such, have vastly different firing properties. Firing properties can be as diagnostic of cell identity as any other attribute, including cell size, dendrite shape, or neurotransmitter composition (Davis, 2013). Mechanisms that determine the set-point may involve cell specific expression of “terminal selector” transcription factors, which control the expression of genes that define cellular identity (Hobert, 2011, Davis, 2013). A further prediction is that the time course for homeostatic adjustments will differ based on cell type. For example, inspiratory motor neurons operate at a high level of activity compared to the sporadic activity of other motor pools (Sieck et al., 2012); as such, perturbations in activity that push critical inspiratory motor neurons outside of their set-point and ideal dynamic range are expected to be sensed and responded to more rapidly than less active neurons.
Mechanisms giving rise to homeostatic plasticity are diverse, which is fitting given the diverse forms of homeostatic plasticity that have been identified. Many cellular/molecular players have been implicated in participating in homeostatic plasticity—far too many to discuss here; thus, the reader is directed to several excellent reviews (Turrigiano, 2011, Fernandes and Carvalho, 2016). Since evidence suggests that many forms of homeostatic plasticity are cell-autonomous, in which neurons sense changes in their own activity through changes in calcium influx, it is not surprising that many calcium sensors have been shown to play a key role, such as calcium/calmodulin dependent kinases (CaMKII, CaMKIV and CaMKK; Thiagarajan et al., 2002, Ibata et al., 2008 Goold and Nicoll, 2010) and calcineurin (Arendt et al., 2015). As neuronal information flow tends to converge on AMPA receptor activation (Turrigiano, 2012), many of the molecules identified in homeostatic plasticity are known to regulate AMPA receptor trafficking, such as stargazin (Louros et al., 2014), B3 integrins (Cingolani et al., 2008), PICK1 (Anggono et al., 2011), PSD-95 and PSD-93 (Sun and Turrigiano, 2011), GRIP1 (Gainey et al., 2015, Tan et al., 2015), the immediate early gene product Arc/Arg3.1 (Shepherd et al., 2006) and Homer1a (Hu et al., 2010). Additional mediators like BDNF (Rutherford et al., 1998), NMDA receptors (Lee and Chung, 2014), retinoic acid (Aoto et al., 2008), Plk2-CDK5 signaling (Seeburg et al., 2008), Eph4A (Fu et al., 2011), MeCP2 (Qiu et al., 2012), immune molecule MHC1 (Goddard et al., 2007) and TNFα (Beattie et al., 2002, Stellwagen and Malenka, 2006) have also been identified. While a laundry list of potential “key” molecules has been generated, precise pathways and how these molecules all fit together to establish a neuron’s set-point, sense deviations from that set-point and then re-establish and maintain set-point are unknown. Understanding the unique roles for the modulators identified in homeostatic plasticity and the interactions between distinct homeostatic signaling pathways may reveal redundancy and/or convergence of mechanisms to ensure appropriate activity rebalancing across neural circuits.
Is homeostatic plasticity a feature of the control of breathing?
As mechanisms of homeostatic plasticity are unraveled across species and a variety of neural circuits, it is becoming clear that homeostatic plasticity is not a unique adaptation, but rather a conserved feature of neural networks. Within the respiratory control system, which is constantly adapting to perturbations, preservation of life is dependent on its ability to produce stable, yet dynamic output. This leads to the hypothesis that the respiratory control system has a robust set of homeostatic mechanisms to maintain stable breathing in the face of prolonged or repeated changes in respiratory neural activity that push network output outside an ideal range (Koch et al., 2011, Strey et al., 2013). However, to date, very little is known regarding the mechanisms of homeostatic plasticity in the control of breathing, or the role it may play in maintaining stable breathing throughout life.
Plasticity in respiratory motor neurons in response to reduced neural activity
To our knowledge, the first indication that a reduction in synaptic inputs to respiratory motor neurons elicits mechanisms that oppose that reduction and enhance respiratory motor output was a serendipitous observation by Castro-Moure and Goshgarian (1996). These investigators focally cooled the ventral cervical spinal cord for 4 hours to block cervical spinal axon conduction unilaterally, silencing descending inputs to ipsilateral phrenic motor neurons. As expected, blocking phrenic synaptic inputs silenced ipsilateral diaphragm EMG activity; however, to the investigators’ surprise, upon restoration of axon conduction, EMG activity in the hemidiaphragm ipsilateral (but not contralateral) to cold block was significantly increased relative to pre-block levels (Castro-Moure and Goshgarian, 1996). Although this study could not differentiate whether observed effects were due to central neural versus diaphragm neuromuscular junction plasticity, morphological changes in the ipsilateral phrenic motor pool suggested local mechanisms of plasticity were initiated within the phrenic motor pool in response to activity deprivation (Castro-Moure and Goshgarian, 1997). To note, these observations were confounded by the possibility of fluctuating blood gases during the cold block since the rats were breathing spontaneously.
Later studies conclusively demonstrated that reduced respiratory neural activity in the absence of changing blood gases elicited central neural plasticity. In anesthetized, mechanically ventilated, vagotmized rats, brainstem respiratory activity was reversibly reduced (i.e., neural apnea) for ~30 min using one of three different treatments: pharmacological respiratory depression with isoflurane, hyperventilation to reduce arterial PCO2 below the threshold for inspiratory neural activity (i.e., apneic threshold), and mechanoreflex inhibition with high frequency ventilation (Mahamed et al., 2011). Although these conditions created a neural apnea by different means, all three treatments resulted in a similar, long-lasting increase in phrenic motor output upon resumption of respiratory neural activity; thus, we suggested that reduced respiratory neural activity induces a compensatory form of plasticity in the respiratory control system that enhances phrenic motor output. Importantly, since arterial PO2 was constant throughout the protocol in all treatments, and in two out of three treatments, arterial PCO2 was constant, a role for fluctuations in blood gases in eliciting this form of plasticity could be ruled out. We termed this form of plasticity inactivity-induced phrenic motor facilitation (iPMF; Mahamed et al., 2011), although this term is a bit of a misnomer since complete suppression of respiratory neural activity is not necessary to induce iPMF (Streeter and Baker-Herman, 2014a; Figure 1). Later studies revealed that plasticity following reduced respiratory neural activity is also expressed in hypoglossal motor output (inactivity-induced hypoglossal motor facilitation, iHMF; Baker-Herman and Strey, 2011) and inspiratory intercostal EMG activity (inactivity-induced intercostal motor facilitation, iIMF; Strey et al., 2013). Since hypoglossal motor neurons control the diameter of the upper airway and intercostals are accessory muscles important in inspiration and expiration, these findings suggest that a compensatory enhancement of inspiratory motor output in response to reductions in respiratory neural activity might be a general feature of respiratory motor control. However, in these studies, respiratory neural activity was reduced throughout the brain and spinal cord by manipulating (and silencing) brainstem respiratory rhythm generation. As such, it was unknown where in the respiratory neural control system activity was being sensed.
Figure 1. Reduced respiratory neural activity elicits rebound increases in phrenic burst amplitude.
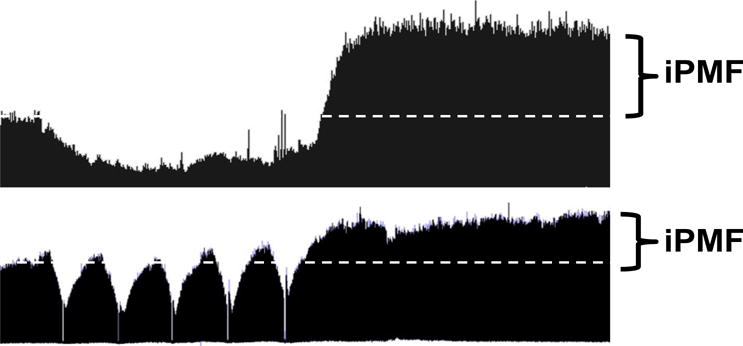
Compressed phrenic neurograms (~60 min recording) depicting baseline, a 30 min neural hypopnea (top) or 5, brief (~1 min) hypopneas separated by 5 min (bottom). Enhanced phrenic amplitude after neural hypopnea indicates iPMF.
To better understand the role for local mechanisms in sensing and responding to reductions in respiratory neural activity, we developed a novel model whereby synaptic inputs to one phrenic motor pool were reduced while respiratory neural activity was maintained at normal levels in the brainstem and contralateral phrenic nerve (Streeter and Baker-Herman, 2014a). In anesthetized, mechanically ventilated rats (in which blood gases were held constant), we microinjected procaine into the C2 ventrolateral funiculus on one side of the spinal cord to reversibly block axon conduction in descending bulbospinal tracts providing respiratory-related synaptic inputs to ipsilateral phrenic motor neurons. In these studies, ipsilateral phrenic motor output was “clamped” at a level that was <40% of baseline for ~30 min. Following activity deprivation, ipsilateral phrenic motor output was significantly increased relative to pre-activity deprivation levels, indicating that mechanisms local to the phrenic motor pool sensed and responded to a reduction in synaptic inputs to elicit iPMF. Since ipsilateral phrenic motor output was only reduced, not eliminated, during the period of activity deprivation, we sought to understand the relationship between the amount of activity deprivation and the magnitude of the resulting iPMF; here, we present additional analysis of data presented in Streeter and Baker-Herman, 2014a (Figure 2). Linear regression analysis was used to determine the relationship between the average reduction in phrenic burst amplitude during axon conduction block and the resulting iPMF. A significant positive relationship (R2 = 0.52, p<0.01) was found, indicating that plasticity in response to a withdrawal of respiratory related synaptic inputs is proportional to the degree of activity deprivation; i.e., the more phrenic synaptic inputs are reduced, the larger the resulting compensatory response. If these results can be extended to other respiratory motor pools, these data suggest that local mechanisms sense and respond to a reduction in synaptic inputs to respiratory motor neurons to elicit a compensatory increase in inspiratory motor output that is proportional to the degree of activity deprivation. The “sensors” and “responders” for this form of plasticity are currently unknown but could include any combination of motor neurons, glia, or interneurons.
Figure 2. iPMF magnitude is proportional to the magnitude of phrenic activity deprivation.
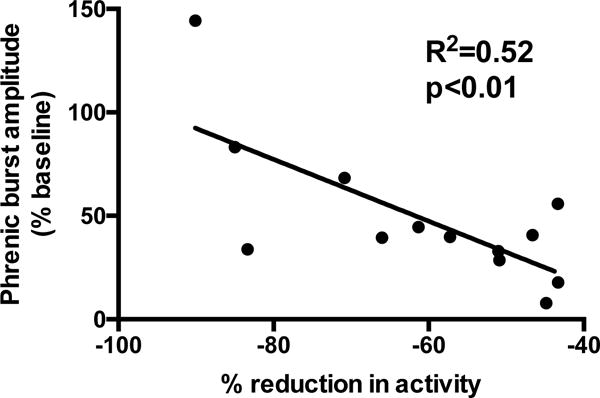
Descending synaptic inputs to phrenic motor neurons on one side of the spinal cord were reversibly impaired with intraspinal procaine injections into the C2 ventrolateral funiculus in anesthetized, ventilated rats (Streeter and Baker-Herman, 2014a). Linear regression analysis indicates a significant relationship between the average decrease in phrenic motor output during axon conduction block and the resulting increase in phrenic burst amplitude (i.e., iPMF) observed 15 min following reversal of conduction block. These data indicate that iPMF magnitude is proportional to the degree of phrenic activity deprivation.
While these studies demonstrated a fundamental, and previously unknown, property of respiratory motor neurons, it was difficult to imagine a scenario in which these mechanisms might actually come into play physiologically since a 30 min reduction in phrenic motor output generally is not compatible with life, except under the most extreme of circumstances (i.e., mechanical ventilation). Although we do not know the exact duration of reduced respiratory neural activity needed to initiate these mechanisms, it is clear that <7 min is not enough (Baertsch and Baker-Herman, 2013)–as such, most mammals would long be dead before these mechanisms might kick in. Thus, we sought to experimentally replicate situations in which reduced respiratory neural activity might be commonly experienced. To model physiologically relevant occurrences of reduced inspiratory neural activity, we induced brief, intermittent neural apneas (Baertsch and Baker-Herman, 2013, 2015), which are a feature of sleep in both healthy individuals and individuals with pathological sleep disordered breathing (Dempsey et al., 2010). In anesthetized, mechanically ventilated rats, five brief (~1 min) neural apneas were induced (separated by 5 min), while recording phrenic inspiratory activity (Baertsch and Baker-Herman, 2013). Generally, after the first neural apnea, phrenic inspiratory activity resumed baseline levels; however, with each subsequent neural apnea, phrenic inspiratory activity progressively increased, such that after the fifth neural apnea, phrenic inspiratory activity was well above baseline levels in a phenotypically similar way as that following a prolonged (~30 min) neural apnea. Since combining the total duration of reduced respiratory neural activity (~7 min) experienced in each intermittent bout into one sustained episode did not enhance phrenic inspiratory motor output, we concluded that intermittent reductions in inspiratory neural activity more efficiently induce iPMF than prolonged neural apnea (Baertsch and Baker-Herman, 2013), which is not surprising since intermittent exposure to other stimuli, (e.g. hypoxia and interference of vagal transmission; Bach and Mitchell, 1996, Tadjalli et al., 2010) more efficiently elicit respiratory plasticity than sustained exposures (Baker et al., 2001, Tadjalli et al., 2010, Devinney et al., 2013).
We have begun to understand key components of the mechanisms giving rise to plasticity following reduced respiratory neural activity. Surprisingly, mechanisms that induce iPMF following intermittent versus prolonged reduced respiratory neural activity are different (Strey et al., 2012, Broytman et al., 2013, Baertsch and Baker-Herman, 2015). iPMF elicited by prolonged reductions in respiratory neural activity requires spinal tumor necrosis factor alpha (TNFα; Strey et al., 2012, Broytman et al., 2013), whereas iPMF elicited by intermittent reductions does not (Baertsch and Baker-Herman, 2015). Mechanisms that induce iPMF following intermittent reductions in respiratory neural activity are currently unknown, but preliminary results indicate a key role for retinoic acid (Baertsch and Baker-Herman, unpublished observations). Of note, both TNFα and retinoic acid are known mediators of homeostatic synaptic plasticity in the hippocampus and cortex (Beattie et al., 2002, Stellwagen and Malenka, 2006, Aoto et al., 2008, Maghsoodi et al., 2008, Steinmetz and Turrigiano, 2010, Chen et al., 2014). Our evidence suggests that both TNFα and retinoic acid ultimately converge on the atypical protein kinase C isozyme, protein kinase C zeta (PKCζ; Strey et al., 2012, Baertsch and Baker-Herman, 2015), which stabilizes early, transient increases in phrenic burst amplitude post-activity deprivation into a long-lasting iPMF (Strey et al., 2012, Streeter and Baker-Herman, 2014a, Baertsch and Baker-Herman, 2015). Collectively, these data suggest that while different mechanisms “sense” prolonged versus intermittent reductions in respiratory neural activity, they ultimately result in the activation of PKCζ, likely within phrenic motor neurons (Guenther et al., 2010, Strey et al., 2012), to induce long-lasting increases in phrenic inspiratory output by an unknown mechanism.
It is important to note that in the models that have been developed thus far, activity deprivation has been rapidly induced, artificially clamped at a low level for a defined period of time, and then rapidly reversed. As a result, the compensatory plasticity (i.e., iPMF) that resulted from reductions in respiratory neural activity resulted in an “overshoot” upon reversal of activity deprivation. If iPMF represents a homeostatic response to activity deprivation, then one prediction is that a counter-mechanism would be activated in response to the relative hyperactivity observed when activity deprivation is relieved. Indeed, the definition of homeostatic plasticity implies that a set-point (i.e., baseline) level of activity is sought and maintained, and clearly, in the artificial conditions we used to elicit iPMF, baseline was not maintained–at least not following reversal of the activity deprivation “clamp.” Nevertheless, consistent with the hypothesis that baseline activity is the regulated variable for such forms of plasticity, inactivity-induced respiratory motor facilitation is reversible; however, the time course for reversibility is variable, depending on motor pool and rat substrain. For example, in the hypoglossal and inspiratory intercostal motor pools, iHMF and iIMF last ~15–30 min before inspiratory motor output declines back to baseline levels (i.e., pre-activity deprivation set-point; Baker-Herman and Strey, 2011, Strey et al., 2013). Similarly, in some rat substrains, iPMF is also transient, and baseline levels of phrenic burst amplitude are restored within 60 min following resumption of respiratory neural activity (Streeter and Baker-Herman, 2014b). It is not currently known if the transiency of inactivity-induced plasticity in these examples reflects activation of a hyperactivity-induced counter mechanism or a lack of stabilization of iPMF, nor do we understand why some rat substrains express a longer duration of inactivity-induced plasticity than others. However, similar to homeostatic downscaling following elevated levels of neural activity (Hou et al., 2011, Lee and Chung, 2014, Siddoway et al., 2014), NMDAR activation during early iPMF expression appears to be key for dis-facilitating phrenic motor output and restoring baseline phrenic burst amplitude (Streeter and Baker-Herman, 2014b), suggesting that the iPMF-induced “overshoot” of phrenic burst amplitude activates an NMDA receptor-dependent counter-mechanism that restores baseline phrenic activity.
Three features of iPMF are consistent with the hypothesis that iPMF homeostatically adjusts and stabilizes phrenic burst amplitude. First, as already discussed, the magnitude of iPMF is proportional to the amount of activity deprivation (Figure 2). Secondly, mechanisms underlying iPMF also proportionally increase the response of phrenic burst amplitude to a respiratory challenge (hypercapnia; Baertsch and Baker-Herman, 2013, Streeter and Baker-Herman, 2014a, Baertsch and Baker-Herman, 2015). Respiratory motor output operates within a limited range beginning at zero (neural apnea) and spanning to a maximum value that can be elicited during a potent respiratory challenge, such as high CO2, or during a non-ventilatory behavior (e.g., coughing). During resting/eupneic breathing, only a fraction of this range is utilized (Mantilla and Sieck, 2011). As such, preservation of “normal” dynamic range following iPMF induction may be the result of a compensatory shift in overall phrenic excitability that restores inspiratory motor output when respiratory neural activity is reduced, while at the same time preserving the capacity to generate maximal respiratory responses. Thirdly, iPMF is associated with a shift in the CO2 threshold for inspiratory activity that makes it harder for the system to experience reduced respiratory neural activity again (Baertsch and Baker-Herman, this issue; and see below for further discussion). In other words, activity deprivation induces a reconfiguration of the neurons generating breathing that makes the system more likely to remain active to maintain stable breathing. It is currently unknown whether such shifts in the CO2 apneic threshold are due to the same local mechanisms as iPMF, or whether they are due to distinct mechanisms of plasticity operating elsewhere in the respiratory control network.
Plasticity in respiratory motor neurons in response to increased neural activity
While not the focus of this review, we will briefly discuss available evidence that mechanisms sense and respond to elevations in respiratory neural activity. One of the first studies to demonstrate that respiratory neurons respond to chronic elevations in activity by downregulating cellular properties is derived from work investigating the response of neurons in the nucleus tractus solitarius (NTS) to chronic intermittent hypoxia (CIH; Kline et al., 2007). Hypoxia is sensed by peripheral chemoreceptors in the carotid body, which send afferent information to the central nervous system via the carotid sinus nerve. The NTS is the first relay for carotid chemoafferent feedback, where it is integrated with other visceral afferent information, then transmitted to other regions of the brain important in respiratory control. During hypoxia, carotid body chemoafferent activity increases, resulting in a reflexive increase in breathing; when normoxia is restored, chemoafferent activity and breathing return to baseline levels. However, if animals are exposed to repeated episodes of hypoxia (i.e., CIH), a form of plasticity is induced in this chemoreflex loop that results in augmented chemoafferent discharge in both hypoxia and normoxia (Peng and Prabhakar, 2003, 2004, Kline et al., 2007), thereby increasing the traffic of information to the NTS. Further, CIH also induces an increase in NTS neuronal activity, driven by an increase in spontaneous neurotransmitter release (Kline, 2010). However, these CIH-induced increases in afferent activity are counterbalanced by a reduction in evoked synaptic transmission between sensory afferents and NTS second-order cells (Kline et al., 2007, Almado et al., 2012), which may serve to somewhat “normalize” activity in NTS neurons. Mechanisms that underlie this activity-dependent synaptic downscaling may be due to Ca2+ dependent reductions in evoked neurotransmitter release (Kline et al., 2007) and/or a loss in the number of active synapses (Almado et al., 2012). Regulation of activity in the NTS may be bidirectional; indeed, chronic ozone exposure results in a decrease in chemoafferent glutamate release, which is counterbalanced by a concomitant increase in NTS postsynaptic excitability (Chen et al., 2003).
Relatively less is understood regarding compensatory responses in respiratory motor neurons to increased respiratory neural activity. Artificially increasing phrenic burst amplitude with high frequency electrical stimulation of descending bulbospinal tracts onto phrenic motor neurons does not appear to elicit a counterbalancing depression in phrenic motor output (Hayashi et al., 2003). This lack of effect may be due to the limited duration of electrical stimulation; in fact, it would be undesirable for inspiratory descending pathways to “downscale” following a brief augmented respiratory maneuver, such as a sigh, cough, or sneeze. However, compensatory changes in respiratory motor neurons may occur following more long-lasting increases in respiratory neural activity. Of note, prolonged or intermittent exposures to elevated levels of CO2 elicit a rebound depression in phrenic burst amplitude (Baker et al., 2001, Valic et al., 2016), although the mechanisms underlying this depression are incompletely understood.
Perhaps the clearest evidence for homeostatic plasticity following chronic elevations in respiratory motor neuron activity is work investigating hypoglossal motor neuron properties following developmental nicotine exposure. Nicotine is a powerful neuroteratogen, resulting in an array of developmental abnormalities, including in neurons underlying breathing (Pilarski et al., 2011; Jaiswal et al., 2013, 2016). Developmental nicotine exposure disrupts normal growth of hypoglossal motor neurons (Powell et al., 2016) and increases their excitability (Pilarski et al., 2011, Jaiswal et al., 2013, Jaiswal et al., 2016). However, these changes are counterbalanced by decreased glutamatergic (Jaiswal et al., 2013) and enhanced GABAergic synaptic transmission (Jaiswal et al., 2016) between brainstem neurons generating respiratory rhythm and pattern and hypoglossal motor neurons. Since hypoglossal motor output (i.e, the population response) remains largely unchanged following developmental nicotine exposure (Pilarski et al., 2011, Jaiswal et al., 2016), these adaptations may serve to homeostatically mitigate chronic nicotine-induced increases in neuronal excitability (Pilarski and Fregosi, 2009, Jaiswal et al., 2016). While these adaptations may preserve hypoglossal motor output in the face of developmental abnormalities in network circuitry, they are imperfect and do not come without cost: impaired ventilatory responses to critical stressors, such as hypoxia and hypercapnia (Hafstrom et al., 2005, Huang et al., 2010, Taylor et al., 2013).
Does plasticity induced by reductions in respiratory neural activity play a role in clinical disorders?
While the models discussed above were developed to investigate where, how and under what conditions the respiratory control system senses and adapts to changes in neural activity, it is important to note that these models are often studied in well-controlled preparations designed to limit contributions from other respiratory modulators (e.g., hypoxia, sensory feedback). Whether homeostatic mechanisms influence respiratory motor output in physiological or pathophysiological conditions during which respiratory neural activity is either not properly generated or not successfully transmitted to the motor pools required for breathing remains uninvestigated. Consistent with the hypothesis that the central respiratory network can exhibit some degree of compensation, persistent deficits in individual respiratory neurons or components of the respiratory network have been identified in animal models of several clinical disorders prior to the onset of overt breathing abnormalities (Viemari et al., 2005, Johnson and Mitchell, 2013). We have selected representative clinical conditions characterized by either a prolonged reduction (spinal cord injury) or brief, intermittent reductions (central sleep apnea) in respiratory neural activity to use as frameworks to postulate whether homeostatic mechanisms contribute to the restoration of appropriate respiratory motor output and/or whether a failure of homeostatic plasticity contributes to breathing instability.
Spinal injury: a role for iPMF in spontaneous recovery of phrenic burst amplitude?
The resilience of the respiratory neural network is exemplified through spontaneous recovery of respiratory motor output in motor neuron pools caudal to a spinal cord injury (SCI). SCI disrupts descending inputs from brainstem respiratory rhythm and pattern generating neurons to spinal motor neurons that innervate muscles required for breathing. Since respiratory motor neurons are found in cervical, thoracic and lumbar spinal segments, injury at any level of the spinal cord results in some type of respiratory impairment (Hoh et al., 2013). The most devastating respiratory impairments occur with upper cervical SCI since this injury disrupts inputs to phrenic motor neurons innervating the diaphragm, in addition to motor neurons innervating accessory inspiratory (e.g., intercostal) and expiratory (e.g., abdominal and intercostal) muscles found in the thoracic and lumbar segments. Remarkably, many investigators have reported that an initially silent phrenic nerve/hemidiaphragm ipsilateral to a high cervical lesion will spontaneously recover rhythmic inspiratory activity in the minutes-to-hours following SCI (Ghali and Marchenko, 2015), with improvements continuing in the days, weeks, and months following injury in both experimental models and in humans (Mansel and Norman, 1990, El-Bohy and Goshgarian, 1999, Nantwi et al., 1999, Raineteau and Schwab, 2001, Kaegi et al., 2002, Goshgarian, 2003, Golder et al., 2011, Lane, 2011, Lane et al., 2012, Gransee et al., 2013, Hoh et al., 2013, Gransee et al., 2015). A suite of mechanisms have been proposed to contribute to this spontaneous recovery of ipsilateral phrenic/hemidiaphragm activity, including remodeling of spinal neural circuits (Lane, 2011, Lane et al., 2012, Hoh et al., 2013), recruitment of latent pathways (Crossed Phrenic Phenomenon; Golder et al., 2003, Goshgarian, 2003, 2009, Lane et al., 2009), enhancement of spared ipsilateral synaptic inputs (Vinit et al., 2008, Vinit and Kastner, 2009) and neuromuscular junction/diaphragm muscle plasticity (Mantilla and Sieck, 2009). Precise mechanisms that promote recovery of function following SCI are very poorly understood, but a prominent role for the neurotrophin, brain derived neurotrophic factor (BDNF), and its receptor, TrkB, have been implicated (Gransee et al., 2013, Mantilla et al., 2013, Mantilla et al., 2014, Gill et al., 2015).
We hypothesize that mechanisms of plasticity that sense and respond to reduced phrenic synaptic inputs and/or phrenic neural activity are a previously unrecognized component of spontaneous recovery. Consistent with this hypothesis, atypical PKC expression and activity are increased in the phrenic motor pool within the time frame of phrenic motor recovery post-SCI (Guenther et al., 2012). Indeed, upregulation of the atypical PKC isozyme, PKCζ, has been shown to promote plasticity in spinal dorsal horn neurons post-SCI and contribute to neuropathic pain (Narita et al., 2004, Marchand et al., 2011). Similarly, immediately following SCI, TNFα levels increase in the spinal cord (Wang et al., 1996), as does the expression of TNF receptors, TNFR1 and TNFR2, on spinal motor neurons (Yan et al., 2003). While some of this elevation in TNFα and its receptors is due to the microglial response to tissue trauma (Hausmann, 2003, Loane and Byrnes, 2010), reduced neural activity resulting from disruption in synaptic inputs may also play a key role (Stellwagen and Malenka, 2006).
TNFα has the requisite characteristics to underlie spontaneous, compensatory plasticity in the phrenic motor system following SCI by strengthening spared ipsilateral pathways and/or recruiting latent pathways. TNFα increases BDNF synthesis and TrkB expression (Saha et al., 2006, Balkowiec-Iskra et al., 2011), which is hypothesized to play a key role in augmenting neuronal excitability following SCI (Lin et al., 2011; Mantilla et al., 2014). Secondly, TNFα rapidly (within minutes) increases surface expression of Ca2+ permeable AMPA receptors (Ferguson et al., 2008, Beattie et al., 2010, Huie et al., 2015, Jiang et al., 2015) and NMDA receptors (Han and Whelan, 2010) on spinal motor neurons. Additionally, TNFα increases glutamate release from glia (Bezzi et al., 2001, Takeuchi, 2006) and decreases glutamate transporter expression on glia (Sitcheran et al., 2005, Zou and Crews, 2005, Tilleux and Hermans, 2007). The impact of TNFα on GABA receptor expression appears more complex. While in vitro studies suggest that TNFα decreases GABA receptor expression in the hippocampus (Stellwagen et al., 2005, Pribiag and Stellwagen, 2013), in vivo studies suggest that TNFα increases synaptic GABA receptors in motor neurons (Stuck et al., 2012). Since SCI also results in a shift in the balance of chloride co-transporters on spinal motor neurons that diminish GABAergic inhibitory neurotransmission and may even render GABA excitatory (Cramer et al., 2008, Boulenguez et al., 2010), the electrophysiological consequences of TNFα-induced increases in GABA receptor expression post-SCI are unclear. Collectively, these data point to an important role for TNFα in inducing homeostatic increases in excitatory synaptic transmission, in combination with down-regulation of inhibition, to enhance spared synaptic inputs and improve spinal motor function; however, in the injured spinal cord in which excessive TNFα is present, this may come at a cost–spasticity, excitotoxicity and cell death (Olmos and Llado, 2014). Consistent with this hypothesis, while reducing elevated levels of TNFα post-injury may be beneficial in preventing excitotoxic damage and cell death (Genovese et al., 2006, Genovese et al., 2007, Genovese et al., 2008, Dinomais et al., 2009, Chen et al., 2011), a complete lack of TNFα signaling may actually prevent post-SCI circuit remodeling and worsen motor recovery (Scherbel et al., 1999, Kim et al., 2001, Oshima et al., 2009; Yang et al., 2010).
Central sleep apnea: a role for iPMF in preventing recurrent apnea/hypopnea?
Recurrent episodes of absent (apnea) or markedly reduced (hypopnea) respiratory motor output is a hallmark feature of central sleep apnea (CSA); by contrast, obstructive sleep apnea (OSA) is characterized by continued (futile) central motor output in the presence of a closed or reduced airway. Although less common in the general population than OSA, CSA is relatively common in patients with heart failure (Levy et al., 2013), neurological disease (Deak and Kirsch, 2014), chronic spinal cord injuries (Berlowitz et al., 2005), and long-term opioid use (Wang and Teichtahl, 2007). It is becoming increasingly recognized that both CSA and OSA often co-exist in the same patient (“mixed” apnea; Xie et al., 2011, Javaheri and Dempsey, 2013) or CSA may develop during treatment of OSA for reasons that are not clearly understood (“complex” sleep apnea; Morgenthaler et al., 2006, Dernaika et al., 2007, Lehman et al., 2007). Repetitive central neural apnea is also commonly observed in infants (Eichenwald et al., 1997, Ng and Chan, 2013) and in otherwise normal individuals upon ascent to altitude (Pack, 2011). Although the root cause of central apneas in these diverse populations may be subtly different, recurrent lapses in inspiratory motor output is a common feature.
We hypothesize that iPMF-like mechanisms may be one of several endogenous homeostatic mechanisms that prevent the occurance of central apnea/hypopnea by actively monitoring and responding to repetitive reductions in respiratory neural activity. Indeed, even in otherwise healthy individuals, recurrent reductions in respiratory neural activity are common during sleep (Javaheri, 2010; Dempsey et al., 2010), although the frequency and duration of these apneas/hypopneas are typically minimal and not considered to be clinically relevant. One possibility is that these minimal disruptions in respiratory neural activity “train” the respiratory control system, resetting respiratory neuron properties throughout the neuraxis to defend an optimal activity level and prevent cessations in breathing. Mechanisms by which iPMF might stabilize inspiratory motor output to correct reductions in respiratory neural activity are not entirely clear. It is possible that enhanced phrenic burst amplitude following iPMF induction augments phrenic motor neuron responsiveness to otherwise sub-threshold descending respiratory drive (Batsel, 1967; Ezure et al., 2003; Garcia et al., 2016; Kam et al., 2013; St. John, 1998), thereby preventing apneas and hypopneas due to “transmission” failure. However, enhanced phrenic motor output (and subsequent increases in breathing) is also expected to result in a lowering of arterial PaCO2. A critical determinant of ventilatory stability and the occurrence of CSA is the difference between the prevailing arterial PCO2 during spontaneous breathing versus the arterial PCO2 at which breathing efforts cease (the CO2 apneic threshold; Dempsey et al., 2010). As such, a reasonable prediction would be that iPMF might actually destabilize breathing and promote central neural apneas by bringing eupneic PaCO2 closer to the apneic threshold. However, we recently discovered that iPMF induction with either intermittent or prolonged neural apnea elicited a long-lasting decrease in the apneic threshold for phrenic inspiratory activity (Baertsch and Baker-Herman, this issue). Thus, reductions in respiratory neural activity transform the respiratory control network in a way that lowers the level of PaCO2 necessary to trigger the next apnea, thereby minimizing the likelihood of recurrent apneas. While the location of apneic threshold plasticity remains unknown, we suggest that plasticity in respiratory motor neurons induced by reductions in respiratory neural activity attempt to homeostatically maintain stable breathing by strengthening inspiratory motor neuron responsiveness to descending respiratory drive and lowering the threshold for further apneas.
Why, then, does this built-in security system not prevent the recurring apneas characteristic of patients with CSA? One possibility is that the expression of iPMF and other forms of plasticity induced by reduced respiratory neural activity might be constrained in these patients. In addition to the frequency of apneas, one differentiating feature between individuals with clinically relevant CSA/OSA and otherwise heatlhy individuals with normal respiratory pauses during sleep is the duration of such events— in other words, the presence or absence of concurrent hypoxia (Dempsey et al., 2010). Literature reports support the existence of an endogenous form of compensatory plasticity that “self-corrects” motor output instability and suggest the intruiguing possibility that it may be constained by concurrent hypoxia. First, many patients with mixed/complex sleep apnea receiving CPAP or tracheostomy will continue to have central apnea/hypoponea initially upon treatment; however, in many of these patients, central events will gradually, and spontaneously, self-resolve through mechanisms that are not understood (Arzt et al., 2009, Salloum et al., 2010, Deacon and Catcheside, 2015). Secondly, many patients with CSA experience a reduction in central events following treatment with inspired oxygen; this effect is not apparent immediately, but occurs gradually over time (Chowdhuri et al., 2012). Collectively, these data are consistent with the hypothesis that an endogenous form of compensatory plasticity corrects respiratory motor output instability, but it is only operative when underlying hypoxia is resolved. Further studies are necessary to address whether iPMF-like mechanisms are one component of this endogenous self-correction, and whether these mechanisms are constrained by hypoxia.
CONCLUSION
The brain is charged with a remarkable task–command several respiratory muscles to contract in a carefully coordinated sequence to generate a breath that maintains arterial blood gases at tightly controlled levels without fail, with only the briefest of pauses, and without error throughout the life of the organism. But it does not stop there. The system must also be precise enough to produce a motor output that does not result in fluctuations in blood gases, dynamic enough to respond quickly and effectively to acute respiratory challenges, and it must faithfully adapt during a myriad of life changes that perturb respiratory control, such as development, aging, pregnancy, weight gain/loss, ascent to altitude, etc. And finally, this respiratory motor output must be coordinated with other non-respiratory motor behaviors, such as vocalization, swallowing and airway clearance. Yet, throughout all of these tasks, producing and maintaining a stable motor output is paramount. A major gap in the field is understanding the mechanisms that enable the respiratory control system to be flexible and adaptive, yet persistently tenacious.
It is increasingly clear that individual neurons and neural networks sense and homeostatically respond to perturbations in their activity levels to maintain stable function (Turrigiano, 2008). We hypothesize that the absolute requirement for stable breathing necessitates homeostatic mechanisms that sense and rapidly compensate for changes in respiratory neural activity within networks controlling breathing, and suggest that iPMF represents only one component in a continuum of homeostatic plasticity mechanisms that assure appropriate respiratory motor output throughout life. Indeed, similar mechanisms are also likely to be operative in other respiratory neuron pools essential for breathing, such as brainstem neurons that generate respiratory rhythm and pattern. We suggest that such plasticity subtlly adjusts respiratory motor output on a continual basis throughout life, and may help to preserve respiratory motor output following the onset of pathophysiological disorders of ventilatory control (Strey et al., 2013). In the phrenic motor system (and by extension, other respiratory motor pools) we propose that plasticity induced by reductions in respiratory-related synaptic inputs ensures reliable and dynamic respiratory activity via two mechanisms: 1) strengthening phrenic motor output, which in turn leads to a 2) lowering of the threshold for phrenic inspiratory activity. While it is still unknown whether such plasticity represents homeostatic plasticity in the control of breathing per se, enhancement of phrenic motor output that is proportional to its activity deprivation (Figure 2) and the increase in phrenic dynamic range observed with iPMF are suggestive.
Several open questions remain. First, respiratory neural activity is not static throughout life; the level and pattern of appropriate respiratory neural activity is vastly different under different vigilance states (i.e., sleep versus wakefulness), during exercise, development/aging or under conditions of hypoxia/hypercapnia. A key feature of homeostatic plasticity is that it balances neural or network activity around an ideal set-point, but how homeostatic mechanisms are refined and regulated to permit a range of state-dependent “set-points” is completely unknown. One possibility is that state-dependent differences in the expression profile of a medley of different neuromodulators known to impact breathing (Saper et al., 2001, Jones, 2005, Saper et al., 2005, Horner, 2008) determine the appropriate set-point for respiratory neural activity and influence the capacity for homeostatic regulation of respiratory neural activity. However, this hypothesis remains to be investigated. A second major unanswered question is: How does plasticity induced by changes in respiratory neural activity interact with other forms of plasticity to shape necessary long-term adaptations in the respiratory control system? Rarely do changes in respiratory neural activity occur without concomitant changes in blood gases or mechanosensory feedback, both of which have been shown to elicit plasticity in isolation (Baker et al., 2001; Tadjalli et al., 2010). However, how distinct forms of plasticity interact to promote or constrain plasticity to sculpt the final output of the system—a breath—has not been explored. Consideration of the interplay of diverse forms of respiratory plasticity and the impact of neuromodulators will be important to advance our understanding of how breathing is stabilized in response to perturbations that act to push respiratory neural activity outside its limits.
Acknowledgments
This work was supported by a grant from the National Heart, Lung and Blood Institute (NHLBI), grant number HL105511. We thank Joel Weltman for his insightful comments on this manuscript.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nature neuroscience. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Almado CE, Machado BH, Leao RM. Chronic intermittent hypoxia depresses afferent neurotransmission in NTS neurons by a reduction in the number of active synapses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:16736–16746. doi: 10.1523/JNEUROSCI.2654-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Clem RL, Huganir RL. PICK1 loss of function occludes homeostatic synaptic scaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:2188–2196. doi: 10.1523/JNEUROSCI.5633-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60:308–320. doi: 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Zhang Z, Ganesan S, Hintze M, Shin MM, Tang Y, Cho A, Graef IA, Chen L. Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2015 doi: 10.1073/pnas.1510239112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzt M, Schulz M, Schroll S, Budweiser S, Bradley TD, Riegger GA, Pfeifer M. Time course of continuous positive airway pressure effects on central sleep apnoea in patients with chronic heart failure. Journal of sleep research. 2009;18:20–25. doi: 10.1111/j.1365-2869.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respiration physiology. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baertsch NA, Baker-Herman TL. Inactivity-induced phrenic and hypoglossal motor facilitation are differentially expressed following intermittent vs. sustained neural apnea. Journal of applied physiology. 2013;114:1388–1395. doi: 10.1152/japplphysiol.00018.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertsch NA, Baker-Herman TL. Intermittent reductions in respiratory neural activity elicit spinal TNFalpha-independent, atypical PKC-dependent inactivity-induced phrenic motor facilitation. American journal of physiology Regulatory, integrative and comparative physiology ajpregu 00359 02014. 2015 doi: 10.1152/ajpregu.00359.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respiration physiology. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Strey KA. Similarities and differences in mechanisms of phrenic and hypoglossal motor facilitation. Respiratory physiology & neurobiology. 2011;179:48–56. doi: 10.1016/j.resp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec-Iskra E, Vermehren-Schmaedick A, Balkowiec A. Tumor necrosis factor-alpha increases brain-derived neurotrophic factor expression in trigeminal ganglion neurons in an activity-dependent manner. Neuroscience. 2011;180:322–333. doi: 10.1016/j.neuroscience.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Ferguson AR, Bresnahan JC. AMPA-receptor trafficking and injury-induced cell death. The European journal of neuroscience. 2010;32:290–297. doi: 10.1111/j.1460-9568.2010.07343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlowitz DJ, Brown DJ, Campbell DA, Pierce RJ. A longitudinal evaluation of sleep and breathing in the first year after cervical spinal cord injury. Archives of physical medicine and rehabilitation. 2005;86:1193–1199. doi: 10.1016/j.apmr.2004.11.033. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nature neuroscience. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nature medicine. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Broytman O, Baertsch NA, Baker-Herman TL. Spinal TNF is necessary for inactivity-induced phrenic motor facilitation. The Journal of physiology. 2013;591:5585–5598. doi: 10.1113/jphysiol.2013.256644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O’Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Cannon WB. The Wisdom of the body. New York: W.W. Norton CO., Inc; 1932. [Google Scholar]
- Castro-Moure F, Goshgarian HG. Reversible cervical hemispinalization of the rat spinal cord by a cooling device. Experimental neurology. 1996;141:102–112. doi: 10.1006/exnr.1996.0143. [DOI] [PubMed] [Google Scholar]
- Castro-Moure F, Goshgarian HG. Morphological plasticity induced in the phrenic nucleus following cervical cold block of descending respiratory drive. Experimental neurology. 1997;147:299–310. doi: 10.1006/exnr.1997.6615. [DOI] [PubMed] [Google Scholar]
- Chen KB, Uchida K, Nakajima H, Yayama T, Hirai T, Watanabe S, Guerrero AR, Kobayashi S, Ma WY, Liu SY, Baba H. Tumor necrosis factor-alpha antagonist reduces apoptosis of neurons and oligodendroglia in rat spinal cord injury. Spine. 2011;36:1350–1358. doi: 10.1097/BRS.0b013e3181f014ec. [DOI] [PubMed] [Google Scholar]
- Chen L, Lau AG, Sarti F. Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:3–12. doi: 10.1016/j.neuropharm.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhuri S, Ghabsha A, Sinha P, Kadri M, Narula S, Badr MS. Treatment of central sleep apnea in U.S. veterans. Journal of clinical sleep medicine: JCSM: official publication of the American Academy of Sleep Medicine. 2012;8:555–563. doi: 10.5664/jcsm.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, Goda Y. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58:749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SW, Baggott C, Cain J, Tilghman J, Allcock B, Miranpuri G, Rajpal S, Sun D, Resnick D. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Molecular pain. 2008;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annual review of neuroscience. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80:718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annual review of physiology. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- Deacon NL, Catcheside PG. The role of high loop gain induced by intermittent hypoxia in the pathophysiology of obstructive sleep apnoea. Sleep medicine reviews. 2015;22:3–14. doi: 10.1016/j.smrv.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Deak MC, Kirsch DB. Sleep-disordered breathing in neurologic conditions. Clinics in chest medicine. 2014;35:547–556. doi: 10.1016/j.ccm.2014.06.009. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiological reviews. 2010;90:47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dernaika T, Tawk M, Nazir S, Younis W, Kinasewitz GT. The significance and outcome of continuous positive airway pressure-related central sleep apnea during split-night sleep studies. Chest. 2007;132:81–87. doi: 10.1378/chest.06-2562. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nature neuroscience. 1999;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Devinney MJ, Huxtable AG, Nichols NL, Mitchell GS. Hypoxia-induced phrenic long-term facilitation: emergent properties. Annals of the New York Academy of Sciences. 2013;1279:143–153. doi: 10.1111/nyas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinomais M, Stana L, Egon G, Richard I, Menei P. Significant recovery of motor function in a patient with complete T7 paraplegia receiving etanercept. Journal of rehabilitation medicine. 2009;41:286–288. doi: 10.2340/16501977-0329. [DOI] [PubMed] [Google Scholar]
- Eichenwald EC, Aina A, Stark AR. Apnea frequently persists beyond term gestation in infants delivered at 24 to 28 weeks. Pediatrics. 1997;100:354–359. doi: 10.1542/peds.100.3.354. [DOI] [PubMed] [Google Scholar]
- El-Bohy AA, Goshgarian HG. The use of single phrenic axon recordings to assess diaphragm recovery after cervical spinal cord injury. Experimental neurology. 1999;156:172–179. doi: 10.1006/exnr.1999.7013. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annual review of neuroscience. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes D, Carvalho AL. Mechanisms of homeostatic plasticity in the excitatory synapse. Journal of neurochemistry. 2016 doi: 10.1111/jnc.13687. [DOI] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu AK, Hung KW, Fu WY, Shen C, Chen Y, Xia J, Lai KO, Ip NY. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nature neuroscience. 2011;14:181–189. doi: 10.1038/nn.2715. [DOI] [PubMed] [Google Scholar]
- Gainey MA, Tatavarty V, Nahmani M, Lin H, Turrigiano GG. Activity-dependent synaptic GRIP1 accumulation drives synaptic scaling up in response to action potential blockade. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E3590–3599. doi: 10.1073/pnas.1510754112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muia C, Bramanti P, Cuzzocrea S. Immunomodulatory effects of etanercept in an experimental model of spinal cord injury. The Journal of pharmacology and experimental therapeutics. 2006;316:1006–1016. doi: 10.1124/jpet.105.097188. [DOI] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muia C, Esposito E, Bramanti P, Cuzzocrea S. TNF-alpha blockage in a mouse model of SCI: evidence for improved outcome. Shock. 2008;29:32–41. doi: 10.1097/shk.0b013e318059053a. [DOI] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Crisafulli C, Esposito E, Di Paola R, Muia C, Di Bella P, Meli R, Bramanti P, Cuzzocrea S. Combination of dexamethasone and etanercept reduces secondary damage in experimental spinal cord trauma. Neuroscience. 2007;150:168–181. doi: 10.1016/j.neuroscience.2007.06.059. [DOI] [PubMed] [Google Scholar]
- Ghali MG, Marchenko V. Dynamic changes in phrenic motor output following high cervical hemisection in the decerebrate rat. Experimental neurology. 2015;271:379–389. doi: 10.1016/j.expneurol.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Gill LC, Gransee HM, Sieck GC, Mantilla CB. Functional recovery after cervical spinal cord injury: Role of neurotrophin and glutamatergic signaling in phrenic motoneurons. Respiratory physiology & neurobiology. 2015 doi: 10.1016/j.resp.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Davenport PW, Johnson RD, Reier PJ, Bolser DC. Respiratory motor recovery after unilateral spinal cord injury: eliminating crossed phrenic activity decreases tidal volume and increases contralateral respiratory motor output. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:2494–2501. doi: 10.1523/JNEUROSCI.23-06-02494.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Lovett-Barr MR, Vinit S, Resnick DK, Mitchell GS. Breathing patterns after mid-cervical spinal contusion in rats. Experimental neurology. 2011;231:97–103. doi: 10.1016/j.expneurol.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Wenner P. Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron. 2006;49:563–575. doi: 10.1016/j.neuron.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon: a model for plasticity in the respiratory pathways following spinal cord injury. Journal of applied physiology. 2003;94:795–810. doi: 10.1152/japplphysiol.00847.2002. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon and recovery of function following spinal cord injury. Respiratory physiology & neurobiology. 2009;169:85–93. doi: 10.1016/j.resp.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gransee HM, Zhan WZ, Sieck GC, Mantilla CB. Targeted delivery of TrkB receptor to phrenic motoneurons enhances functional recovery of rhythmic phrenic activity after cervical spinal hemisection. PloS one. 2013;8:e64755. doi: 10.1371/journal.pone.0064755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gransee HM, Zhan WZ, Sieck GC, Mantilla CB. Localized delivery of brain-derived neurotrophic factor-expressing mesenchymal stem cells enhances functional recovery following cervical spinal cord injury. Journal of neurotrauma. 2015;32:185–193. doi: 10.1089/neu.2014.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CH, Vinit S, Windelborn JA, Behan M, Mitchell GS. Atypical protein kinase C expression in phrenic motor neurons of the rat. Neuroscience. 2010;169:787–793. doi: 10.1016/j.neuroscience.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CH, Windelborn JA, Tubon TC, Jr, Yin JC, Mitchell GS. Increased atypical PKC expression and activity in the phrenic motor nucleus following cervical spinal injury. Experimental neurology. 2012;234:513–520. doi: 10.1016/j.expneurol.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafstrom O, Milerad J, Sandberg KL, Sundell HW. Cardiorespiratory effects of nicotine exposure during development. Respiratory physiology & neurobiology. 2005;149:325–341. doi: 10.1016/j.resp.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Han P, Whelan PJ. Tumor necrosis factor alpha enhances glutamatergic transmission onto spinal motoneurons. Journal of neurotrauma. 2010;27:287–292. doi: 10.1089/neu.2009.1016. [DOI] [PubMed] [Google Scholar]
- Hausmann O. Post-traumatic inflammation following spinal cord injury Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Hinrichsen CF, McCrimmon DR. Short-term plasticity of descending synaptic input to phrenic motoneurons in rats. Journal of applied physiology. 2003;94:1421–1430. doi: 10.1152/japplphysiol.00599.2002. [DOI] [PubMed] [Google Scholar]
- Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG. Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron. 2013;80:335–342. doi: 10.1016/j.neuron.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. Regulation of terminal differentiation programs in the nervous system. Annual review of cell and developmental biology. 2011;27:681–696. doi: 10.1146/annurev-cellbio-092910-154226. [DOI] [PubMed] [Google Scholar]
- Hoh DJ, Mercier LM, Hussey SP, Lane MA. Respiration following spinal cord injury: evidence for human neuroplasticity. Respiratory physiology & neurobiology. 2013;189:450–464. doi: 10.1016/j.resp.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner RL. Neuromodulation of hypoglossal motoneurons during sleep. Respiratory physiology & neurobiology. 2008;164:179–196. doi: 10.1016/j.resp.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Hou Q, Gilbert J, Man HY. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron. 2011;72:806–818. doi: 10.1016/j.neuron.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q, Man HY. Input-specific homeostatic regulation of AMPA receptor accumulation at central synapses. Communicative & integrative biology. 2012;5:553–556. doi: 10.4161/cib.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Park JM, Park S, Xiao B, Dehoff MH, Kim S, Hayashi T, Schwarz MK, Huganir RL, Seeburg PH, Linden DJ, Worley PF. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron. 2010;68:1128–1142. doi: 10.1016/j.neuron.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Brown AR, Cross SJ, Cruz J, Rice A, Jaiswal S, Fregosi RF. Influence of prenatal nicotine exposure on development of the ventilatory response to hypoxia and hypercapnia in neonatal rats. Journal of applied physiology. 2010;109:149–158. doi: 10.1152/japplphysiol.01036.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie JR, Stuck ED, Lee KH, Irvine KA, Beattie MS, Bresnahan JC, Grau JW, Ferguson AR. AMPA Receptor Phosphorylation and Synaptic Colocalization on Motor Neurons Drive Maladaptive Plasticity below Complete Spinal Cord Injury(1,2,3) eNeuro. 2015;2 doi: 10.1523/ENEURO.0091-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Jaiswal SJ, Buls Wollman L, Harrison CM, Pilarski JQ, Fregosi RF. Developmental nicotine exposure enhances inhibitory synaptic transmission in motor neurons and interneurons critical for normal breathing. Developmental neurobiology. 2016;76:337–354. doi: 10.1002/dneu.22318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal SJ, Pilarski JQ, Harrison CM, Fregosi RF. Developmental nicotine exposure alters AMPA neurotransmission in the hypoglossal motor nucleus and pre-Botzinger complex of neonatal rats. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:2616–2625. doi: 10.1523/JNEUROSCI.3711-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaheri S. Central sleep apnea. Clin Chest Med. 2010;31(2):235–48. doi: 10.1016/j.ccm.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Javaheri S, Dempsey JA. Central sleep apnea. Comprehensive Physiology. 2013;3:141–163. doi: 10.1002/cphy.c110057. [DOI] [PubMed] [Google Scholar]
- Jiang L, Voulalas P, Ji Y, Masri R. Post-translational modification of cortical GluA receptors in rodents following spinal cord lesion. Neuroscience. 2015 doi: 10.1016/j.neuroscience.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Mitchell GS. Common mechanisms of compensatory respiratory plasticity in spinal neurological disorders. Respiratory physiology & neurobiology. 2013;189:419–428. doi: 10.1016/j.resp.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE. From waking to sleeping: neuronal and chemical substrates. Trends in pharmacological sciences. 2005;26:578–586. doi: 10.1016/j.tips.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Kaegi S, Schwab ME, Dietz V, Fouad K. Electromyographic activity associated with spontaneous functional recovery after spinal cord injury in rats. The European journal of neuroscience. 2002;16:249–258. doi: 10.1046/j.1460-9568.2002.02076.x. [DOI] [PubMed] [Google Scholar]
- Kim GM, Xu J, Xu J, Song SK, Yan P, Ku G, Xu XM, Hsu CY. Tumor necrosis factor receptor deletion reduces nuclear factor-kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:6617–6625. doi: 10.1523/JNEUROSCI.21-17-06617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:4663–4673. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD. Chronic intermittent hypoxia affects integration of sensory input by neurons in the nucleus tractus solitarii. Respiratory Physiology and Neurobiology. 2010;174(1–2):29–36. doi: 10.1016/j.resp.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch H, Garcia AJ, 3rd, Ramirez JM. Network reconfiguration and neuronal plasticity in rhythm-generating networks. Integrative and comparative biology. 2011;51:856–868. doi: 10.1093/icb/icr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambo ME, Turrigiano GG. Synaptic and intrinsic homeostatic mechanisms cooperate to increase L2/3 pyramidal neuron excitability during a late phase of critical period plasticity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:8810–8819. doi: 10.1523/JNEUROSCI.4502-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA. Spinal respiratory motoneurons and interneurons. Respiratory physiology & neurobiology. 2011;179:3–13. doi: 10.1016/j.resp.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Lane MA, Lee KZ, Fuller DD, Reier PJ. Spinal circuitry and respiratory recovery following spinal cord injury. Respiratory physiology & neurobiology. 2009;169:123–132. doi: 10.1016/j.resp.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Lee KZ, Salazar K, O’Steen BE, Bloom DC, Fuller DD, Reier PJ. Respiratory function following bilateral mid-cervical contusion injury in the adult rat. Experimental neurology. 2012;235:197–210. doi: 10.1016/j.expneurol.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Chung HJ. NMDA receptors and L-type voltage-gated Ca(2)(+) channels mediate the expression of bidirectional homeostatic intrinsic plasticity in cultured hippocampal neurons. Neuroscience. 2014;277:610–623. doi: 10.1016/j.neuroscience.2014.07.038. [DOI] [PubMed] [Google Scholar]
- Lehman S, Antic NA, Thompson C, Catcheside PG, Mercer J, McEvoy RD. Central sleep apnea on commencement of continuous positive airway pressure in patients with a primary diagnosis of obstructive sleep apnea-hypopnea. Journal of clinical sleep medicine: JCSM: official publication of the American Academy of Sleep Medicine. 2007;3:462–466. [PMC free article] [PubMed] [Google Scholar]
- Levy P, Ryan S, Oldenburg O, Parati G. Sleep apnoea and the heart. European respiratory review: an official journal of the European Respiratory Society. 2013;22:333–352. doi: 10.1183/09059180.00004513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Ro LS, Wang HL, Chen JC. Up-regulation of dorsal root ganglia BDNF and trkB receptor in inflammatory pain: an in vivo and in vitro study. Journal of neuroinflammation. 2011;8:126. doi: 10.1186/1742-2094-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M, Hardy S, Nicoll RA, Malenka RC, von Zastrow M. Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7097–7102. doi: 10.1073/pnas.95.12.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louros SR, Hooks BM, Litvina L, Carvalho AL, Chen C. A role for stargazin in experience-dependent plasticity. Cell reports. 2014;7:1614–1625. doi: 10.1016/j.celrep.2014.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, Chen L. Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16015–16020. doi: 10.1073/pnas.0804801105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Strey KA, Mitchell GS, Baker-Herman TL. Reduced respiratory neural activity elicits phrenic motor facilitation. Respiratory physiology & neurobiology. 2011;175:303–309. doi: 10.1016/j.resp.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansel JK, Norman JR. Respiratory complications and management of spinal cord injuries. Chest. 1990;97:1446–1452. doi: 10.1378/chest.97.6.1446. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Gransee HM, Zhan WZ, Sieck GC. Motoneuron BDNF/TrkB signaling enhances functional recovery after cervical spinal cord injury. Experimental neurology. 2013;247:101–109. doi: 10.1016/j.expneurol.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Greising SM, Stowe JM, Zhan WZ, Sieck GC. TrkB kinase activity is critical for recovery of respiratory function after cervical spinal cord hemisection. Experimental neurology. 2014;261:190–195. doi: 10.1016/j.expneurol.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Sieck GC. Neuromuscular adaptations to respiratory muscle inactivity. Respiratory physiology & neurobiology. 2009;169:133–140. doi: 10.1016/j.resp.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Sieck GC. Phrenic motor unit recruitment during ventilatory and non-ventilatory behaviors. Respiratory physiology & neurobiology. 2011;179:57–63. doi: 10.1016/j.resp.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand F, D’Mello R, Yip PK, Calvo M, Muller E, Pezet S, Dickenson AH, McMahon SB. Specific involvement of atypical PKCzeta/PKMzeta in spinal persistent nociceptive processing following peripheral inflammation in rat. Molecular pain. 2011;7:86. doi: 10.1186/1744-8069-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Prinz AA. Current compensation in neuronal homeostasis. Neuron. 2003;37:2–4. doi: 10.1016/s0896-6273(02)01173-x. [DOI] [PubMed] [Google Scholar]
- Miller KD. Synaptic economics: competition and cooperation in synaptic plasticity. Neuron. 1996;17:371–374. doi: 10.1016/s0896-6273(00)80169-5. [DOI] [PubMed] [Google Scholar]
- Morgenthaler TI, Kagramanov V, Hanak V, Decker PA. Complex sleep apnea syndrome: is it a unique clinical syndrome? Sleep. 2006;29:1203–1209. doi: 10.1093/sleep/29.9.1203. [DOI] [PubMed] [Google Scholar]
- Nantwi KD, El-Bohy AA, Schrimsher GW, Reier PJ, Goshgarian HG. Spontaneous functional recovery in a paralyzed hemidiaphragm following upper cervical spinal cord injury in adult rats. Neurorehabilitation and neural repair. 1999;13:225–234. [Google Scholar]
- Narita M, Oe K, Kato H, Shibasaki M, Narita M, Yajima Y, Yamazaki M, Suzuki T. Implication of spinal protein kinase C in the suppression of morphine-induced rewarding effect under a neuropathic pain-like state in mice. Neuroscience. 2004;125:545–551. doi: 10.1016/j.neuroscience.2004.02.022. [DOI] [PubMed] [Google Scholar]
- Ng DK, Chan CH. A review of normal values of infant sleep polysomnography. Pediatrics and neonatology. 2013;54:82–87. doi: 10.1016/j.pedneo.2012.11.011. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Olmos G, Llado J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators of inflammation. 2014;2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima T, Lee S, Sato A, Oda S, Hirasawa H, Yamashita T. TNF-alpha contributes to axonal sprouting and functional recovery following traumatic brain injury. Brain research. 2009;1290:102–110. doi: 10.1016/j.brainres.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Pack AI. Central sleep apnea. Handbook of clinical neurology. 2011;98:411–419. doi: 10.1016/B978-0-444-52006-7.00027-7. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. Journal of applied physiology. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. Journal of applied physiology. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. discussion 1196. [DOI] [PubMed] [Google Scholar]
- Pilarski JQ, Fregosi RF. Prenatal nicotine exposure alters medullary nicotinic and AMPA-mediated control of respiratory frequency in vitro. Respiratory physiology & neurobiology. 2009;169:1–10. doi: 10.1016/j.resp.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarski JQ, Wakefield HE, Fuglevand AJ, Levine RB, Fregosi RF. Developmental nicotine exposure alters neurotransmission and excitability in hypoglossal motoneurons. Journal of neurophysiology. 2011;105:423–433. doi: 10.1152/jn.00876.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell GL, Gaddy J, Xu F, Fregosi RF, Levine RB. Developmental Nicotine Exposure disrupts dendritic arborization patterns of hypoglossal motoneurons in the neonatal rat. Developmental Neurobiology. 2016 doi: 10.1002/dneu.22379. Jan 28 epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H, Stellwagen D. TNF-alpha downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:15879–15893. doi: 10.1523/JNEUROSCI.0530-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A. The Rett syndrome protein MeCP2 regulates synaptic scaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:989–994. doi: 10.1523/JNEUROSCI.0175-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineteau O, Schwab ME. Plasticity of motor systems after incomplete spinal cord injury. Nature reviews Neuroscience. 2001;2:263–273. doi: 10.1038/35067570. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. Journal of neuroimmune pharmacology: the official journal of the Society on NeuroImmune Pharmacology. 2006;1:212–222. doi: 10.1007/s11481-006-9020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloum A, Rowley JA, Mateika JH, Chowdhuri S, Omran Q, Badr MS. Increased propensity for central apnea in patients with obstructive sleep apnea: effect of nasal continuous positive airway pressure. American journal of respiratory and critical care medicine. 2010;181:189–193. doi: 10.1164/rccm.200810-1658OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Cano G, Scammell TE. Homeostatic, circadian, and emotional regulation of sleep. The Journal of comparative neurology. 2005;493:92–98. doi: 10.1002/cne.20770. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends in neurosciences. 2001;24:726–731. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DT, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway B, Hou H, Xia H. Molecular mechanisms of homeostatic synaptic downscaling. Neuropharmacology. 2014;78:38–44. doi: 10.1016/j.neuropharm.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieck DC, Zhan WZ, Fang YH, Ermilov LG, Sieck GC, Mantilla CB. Structure-activity relationships in rodent diaphragm muscle fibers vs. neuromuscular junctions. Respiratory physiology & neurobiology. 2012;180:88–96. doi: 10.1016/j.resp.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitcheran R, Gupta P, Fisher PB, Baldwin AS. Positive and negative regulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlled repression. The EMBO journal. 2005;24:510–520. doi: 10.1038/sj.emboj.7600555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-John WM. Alterations in respiratory neuronal activities in the medullary ‘pre-Botzinger’ region in hypocapnia. Respiration physiology. 1998;114:119–131. doi: 10.1016/s0034-5687(98)00088-7. [DOI] [PubMed] [Google Scholar]
- Steinmetz CC, Turrigiano GG. Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Streeter KA, Baker-Herman TL. Decreased spinal synaptic inputs to phrenic motor neurons elicit localized inactivity-induced phrenic motor facilitation. Experimental neurology. 2014a;256:46–56. doi: 10.1016/j.expneurol.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeter KA, Baker-Herman TL. Spinal NMDA receptor activation constrains inactivity-induced phrenic motor facilitation in Charles River Sprague Dawley rats. Journal of applied physiology. 2014b doi: 10.1152/japplphysiol.00342.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strey KA, Baertsch NA, Baker-Herman TL. Inactivity-induced respiratory plasticity: protecting the drive to breathe in disorders that reduce respiratory neural activity. Respiratory physiology & neurobiology. 2013;189:384–394. doi: 10.1016/j.resp.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strey KA, Nichols NL, Baertsch NA, Broytman O, Baker-Herman TL. Spinal atypical protein kinase C activity is necessary to stabilize inactivity-induced phrenic motor facilitation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:16510–16520. doi: 10.1523/JNEUROSCI.2631-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuck ED, Christensen RN, Huie JR, Tovar CA, Miller BA, Nout YS, Bresnahan JC, Beattie MS, Ferguson AR. Tumor necrosis factor alpha mediates GABA(A) receptor trafficking to the plasma membrane of spinal cord neurons in vivo. Neural plasticity. 2012;2012:261345. doi: 10.1155/2012/261345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Turrigiano GG. PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:6800–6808. doi: 10.1523/JNEUROSCI.5616-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Tadjalli A, Duffin J, Peever J. Repeated obstructive apneas induce long-term facilitation of genioglossus muscle tone. Advances in experimental medicine and biology. 2010;669:297–301. doi: 10.1007/978-1-4419-5692-7_61. [DOI] [PubMed] [Google Scholar]
- Takeuchi T. TNF targetting therapy for rheumatoid arthritis. Nihon Naika Gakkai zasshi, The Journal of the Japanese Society of Internal Medicine. 2006;95:1787–1794. doi: 10.2169/naika.95.1787. [DOI] [PubMed] [Google Scholar]
- Tan HL, Queenan BN, Huganir RL. GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:10026–10031. doi: 10.1073/pnas.1512786112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavarty V, Sun Q, Turrigiano GG. How to scale down postsynaptic strength. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:13179–13189. doi: 10.1523/JNEUROSCI.1676-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BE, Brundage CM, McLane LH. Chronic nicotine and ethanol exposure both disrupt central ventilatory responses to hypoxia in bullfrog tadpoles. Respiratory physiology & neurobiology. 2013;187:234–243. doi: 10.1016/j.resp.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Malgaroli A, Tsien RW. LTP and adaptation to inactivity: overlapping mechanisms and implications for metaplasticity. Neuropharmacology. 2007;52(1):156–75. doi: 10.1016/j.neuropharm.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. Journal of neuroscience research. 2007;85:2059–2070. doi: 10.1002/jnr.21325. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annual review of neuroscience. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harbor perspectives in biology. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Valic M, Pecotic R, Pavlinac Dodig I, Valic Z, Stipica I, Dogas Z. Intermittent hypercapnia-induced phrenic long-term depression is revealed after serotonin receptor blockade with methysergide in anaesthetized rats. Experimental physiology. 2016;101:319–331. doi: 10.1113/EP085161. [DOI] [PubMed] [Google Scholar]
- Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Pena F, Zanella S, Bevengut M, Barthelemy-Requin M, Herzing LB, Moncla A, Mancini J, Ramirez JM, Villard L, Hilaire G. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinit S, Darlot F, Stamegna JC, Sanchez P, Gauthier P, Kastner A. Long-term reorganization of respiratory pathways after partial cervical spinal cord injury. The European journal of neuroscience. 2008;27:897–908. doi: 10.1111/j.1460-9568.2008.06072.x. [DOI] [PubMed] [Google Scholar]
- Vinit S, Kastner A. Descending bulbospinal pathways and recovery of respiratory motor function following spinal cord injury. Respiratory physiology & neurobiology. 2009;169:115–122. doi: 10.1016/j.resp.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Wang CX, Nuttin B, Heremans H, Dom R, Gybels J. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. Journal of neuroimmunology. 1996;69:151–156. doi: 10.1016/0165-5728(96)00080-x. [DOI] [PubMed] [Google Scholar]
- Wang D, Teichtahl H. Opioids, sleep architecture and sleep-disordered breathing. Sleep medicine reviews. 2007;11:35–46. doi: 10.1016/j.smrv.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron. 2000;26:659–670. doi: 10.1016/s0896-6273(00)81202-7. [DOI] [PubMed] [Google Scholar]
- Xie A, Bedekar A, Skatrud JB, Teodorescu M, Gong Y, Dempsey JA. The heterogeneity of obstructive sleep apnea (predominant obstructive vs pure obstructive apnea) Sleep. 2011;34:745–750. doi: 10.5665/SLEEP.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Liu N, Kim GM, Xu J, Xu J, Li Q, Hsu CY, Xu XM. Expression of the type 1 and type 2 receptors for tumor necrosis factor after traumatic spinal cord injury in adult rats. Experimental neurology. 2003;183:286–297. doi: 10.1016/s0014-4886(03)00135-3. [DOI] [PubMed] [Google Scholar]
- Yang J, You Z, Kim HH, Hwang SK, Khuman J, Guo S, Lo EH, Whalen MJ. Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma. 2010;27(6):1037–46. doi: 10.1089/neu.2009.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu LM, Goda Y. Dendritic signalling and homeostatic adaptation. Current Opinion in Neurobiolology. 2009;19(3):327–35. doi: 10.1016/j.conb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain research. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]