

Abstract
Purpose
Idiopathic calcium oxalate kidney stones form while attached to Randall plaques, the subepithelial deposits on renal papillary surfaces. Plaque formation and growth mechanisms are poorly understood. Plaque formation elsewhere in the body is triggered by reactive oxygen species and oxidative stress. This review explores possible reactive oxygen species involvement in plaque formation and calcium oxalate nephrolithiasis.
Materials and Methods
A search of various databases for the last 8 years identified literature on reactive oxygen species involvement in calcium oxalate nephrolithiasis. The literature was reviewed and results are discussed.
Results
Under normal conditions reactive oxygen species production is controlled, increasing as needed and regulating crystallization modulator production. Reactive oxygen species overproduction or decreased antioxidants lead to oxidative stress, inflammation and injury, and are involved in stone comorbidity. All major chronic inflammation markers are detectable in stone patient urine. Patients also have increased urinary excretion of the IαI and the thrombin protein families. Results of a recent study of 17,695 participants in NHANES III (National Health and Nutrition Examination Survey) showed significantly lower antioxidants, carotene and β-cryptoxanthin in those with a kidney stone history. Animal model and tissue culture studies revealed that high oxalate, calcium oxalate and calcium phosphate crystals provoked renal cell reactive oxygen species mediated inflammatory responses. Calcium oxalate crystals induce renin up-regulation and angiotensin II generation. Nonphagocytic NADPH oxidase leads to reactive oxygen species production mediated by protein kinase C. The P-38 MAPK/JNK transduction pathway is turned on. Transcriptional and growth factors, and generated secondary mediators become involved. Chemoattractant and osteopontin production is increased and macrophages infiltrate the renal interstitium around the crystal. Phagocytic NADPH oxidase is probably activated, producing additional reactive oxygen species. Localized inflammation, extracellular matrix and fibrosis develop. Crystallization modulators have a significant role in inflammation and tissue repair.
Conclusions
Based on available data, Randall plaque formation is similar to extracellular matrix mineralization at many body sites. Renal interstitial collagen becomes mineralized, assisting plaque growth through the interstitium until the mineralizing front reaches papillary surface epithelium. Plaque exposure to pelvic urine may also be a result of reactive oxygen species triggered epithelial sloughing.
Keywords: kidney, nephrolithiasis, calcium oxalate, reactive oxygen species, calcification, physiologic
It is estimated that symptomatic stone disease develops in 5% to 15% of the American population by age 70 years. The worldwide prevalence of urolithiasis in men by age 70 years varies from approximately 4% in England to 20% in Saudi Arabia. Data indicate that between 1976 to 1980 and 1988 to 1994 the incidence of kidney stones increased by 37% in each gender in the United States. Geographically, kidney stones are more common in places with high humidity and temperature. With the expected elevated temperature worldwide an increase of 1.6 to 2.2 million lifetime cases of kidney stones by 2050 has been predicted, particularly in the southeastern regions of the United States.1
Urolithiasis is a chronic disease with a 60% chance of recurrence within 10 years. Reports indicate that the incidence in children has increased. Unfortunately, they have more difficulty passing calculi and often require invasive surgery. In addition, the increased CaP, particularly brushite in recurrent stones, leads to increased stone hardness, making them more difficult to comminute by shock wave lithotripsy. A cycle of tissue damage and recurrent stone formation is thought to be the result of lithotripsy.2
Despite increased understanding of crystallization and stone formation,3 and effective current therapies advances in medical management have been modest because of the slow progress in determining stone formation pathophysiology. CaOx stone formation is associated with a number of metabolic disorders that cause urine to become sufficiently supersaturated for CaOx/CaP crystals to form. Renal epithelial cells become exposed to slightly higher levels of Ox, CaP and/or CaOx crystals during stone formation. Based on experimental investigations, we hypothesized that stone formation depends on the type of renal cellular response to the changing urinary environment with significant ROS involvement.4 This article provides a brief overview of experimental and clinical data supporting the role of ROS and inflammation in CaOx stone formation.
REACTIVE OXYGEN SPECIES
Free radicals, atoms or molecules with unpaired electrons and their metabolites are collectively called ROS. Under normal conditions ROS are generated by tightly controlled enzymes. They serve as mediators in various regulatory processes and signaling pathways, including the proliferation, activation or inactivation of regulatory biomolecules and regulation of transcriptional activity. Major cellular ROS include superoxide anion (O2−●), nitric oxide radical (NO●), hydroxyl radical (OH●) and hydrogen peroxide (H2O2), which are generated by several pathways. O2−● anions are produced by NADPH oxidase, xanthine oxidase, lipooxygenase, cyclooxygenase and heme oxygenase, and they are a byproduct of the mitochondrial respiratory chain.4 Lipid radicals can also produce O2−●. NO● radicals are produced by endothelial nitric oxide synthase mediated oxidation of L-arginine. Endothelial nitric oxide synthase can also produce O2−●. The reaction between superoxide and nitric oxide can produce the highly reactive peroxynitrite (ONOO−). Signaling molecules regulated by ROS include but are not limited to protein tyrosine kinases and phosphatases, serine/theorine kinases and phosphatases, RAS, various phospholipases and many calcium signals. ROS also regulate such genes as c-fos, c-myc and c-jun, and transcription factor AP-1 and NF-κB. In addition, ROS also participate in the initiation and implementation of apoptosis.
Since ROS have many significant regulatory roles, they normally occur at steady state levels, are generated as needed, and are then cleared by the activity of various antioxidants and scavengers. They are short lived with tightly controlled actions and localized to specific sites where needed. They do not travel too far because of their potential to do harm.
ROS can also produce chemical modifications of and damage to proteins, lipids, carbohydrates and nucleotides, and they modulate renal and cardiovascular systems through redox dependent signaling pathways. Uncontrolled generation of reactive oxygen and/or a decrease in endogenous antioxidant capacity creates OS, which may lead to inflammation and injury.5 Most cells respond to OS by boosting levels of intracellular antioxidants, such as glutathione. Pathological changes may result from the damaging effects of ROS and from ROS mediated changes in gene expression and signal transduction. Because most ROS are short lived and do not travel a long distance, the presence of OS is generally recognized by the abundance of byproducts of ROS interaction with cell constituents. Malondialdehyde, isoprostanes and oxidized lipids are among the most common byproducts of ROS induced OS.
CLINICAL DATA
Analysis of stone patient urine suggests that ROS induced renal cellular injury and inflammation are likely involved in the pathogenesis of idiopathic stone disease. Higher than normal levels of renal enzymes, γ-glutamyl transpeptidase, angiotensin 1 converting enzyme, β-galactosidase and NAG, indicative of tubular injury, were found in the urine of idiopathic CaOx stone formers.6 Results of recent studies indicate that urine from CaOx stone patients had significantly increased thiobarbituric acid-reactive substances, in addition to renal enzymes, indicating that renal injury was most likely caused by ROS production. Urinary 8-hydroxydeoxyguanosine, a marker of DNA oxidative damage, was increased in stone patients and correlated positively with tubular damage, as assessed by urinary NAG excretion.7 Recently, a group reported urinary excretion of the anti-inflammatory proteins calgranulin, α-defensin and myeloperoxidase8 by stone patients and the presence of these proteins in the inner core of CaOx stones, indicating a putative role in nephrolithiasis.
Other studies of recurrent idiopathic calcium stone formers suggest an association between stone formation and decreased antioxidant levels.9 Investigation revealed that antioxidant deficit is common and unrelated to the presence or absence of stones. The investigators concluded that stone formation started with oxidatively damaged cells. In another recent study serum levels of antioxidants were examined in adult participants of 1988 to 1994 NHANES III.10 Mean levels of antioxidants, α-carotene, β-carotene and β-cryptoxanthin were significantly lower in participants with a self-reported history of kidney stones, while those with higher serum β-carotene and β-cryptoxanthin showed a tendency toward a decreasing prevalence of kidney stones.
Stones, such as cystine, brushite and CaOx calculi in primary hyperoxaluria, calculi that develop after bariatric surgery and CaP stones in those with primary hyperparathyroidism, are attached to tubular crystal deposits in the ducts of Bellini.3 Crystal deposition is associated with renal cell injury, cell loss, inflammation and fibrosis.3,11 Most idiopathic stones form while connected to RPs, which start as deposits of poorly crystalline biological apatite in the basement membrane of the loops of Henle,3,12 collecting ducts13 or vasa recta.14 Although Randall considered plaque a product of a natural repair process for some form of tubule damage,15 recent reports indicate absent tissue injury and inflammation in association with RP in idiopathic stone formers.3,12 Collagen is a part of interstitial CaP deposits and it is mineralized during plaque formation (fig. 1).16
Figure 1.
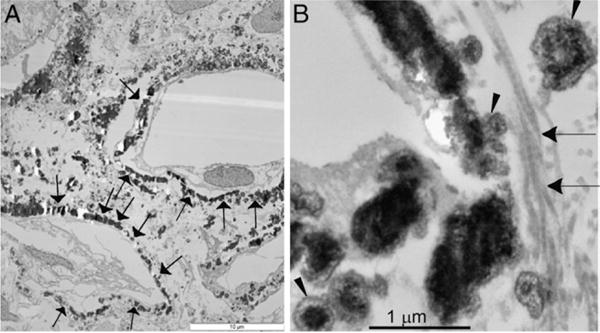
Transmission electron microscopy. A, spherulitic CaP deposits in kidneys of idiopathic stone former in interstitium and around necrosing tubules (arrows). Scale bar indicates 10 μm. B, higher magnification reveals CaP (arrowhead) and collagen fibers (arrows). Bar indicates 1 μm.
OS is also a common feature of many stone comorbidities, such as hypertension, diabetes mellitus, atherosclerosis and myocardial infarction.17 Interestingly, adherence to a DASH (Dietary Approaches to Stop Hypertension) diet, which reduces the risk of stroke and cardiovascular disease, also reduces the risk of stone formation up to 45%.18 The DASH diet decreases blood pressure by increasing antioxidant capacity and lowering OS. Another study of 57,326 hyperlipidemic active duty and retired members of the United States Armed Forces and their dependents showed a 50% decreased risk of stone formation in patients who used statins compared to those who did not. Statins not only lower cholesterol but also decrease the expression and activity of NADPH oxidase and inhibit ROS production.
TISSUE CULTURE
Tissue culture studies, in which renal epithelial or other cells are exposed to Ox and/or CaOx or CaP crystals, have provided new insights into the pathogenesis of nephrolithiasis. The cell response is time and concentration dependent, and cell specific. Crystals bind rapidly to the surface of epithelial cells and are internalized. Finger-like cell surface projections or microvilli appear to probe the crystals, followed by crystal endocytosis into the cells (fig. 2).
Figure 2.
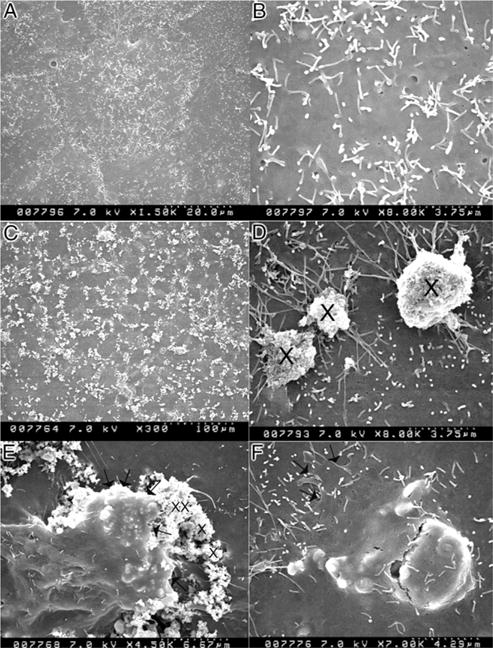
Scanning electron microscopy shows NRK52E cells on exposure to 133 μg/cm2 hydroxyapatite. A, normal cells. B, higher magnification shows surface covered with short microvilli. C, crystals are evenly dispersed on cell surface. D, at 3 hours of treatment crystals (x) appear to be in contact with thin projections from cell surface. E, cell appears to take in crystals. Arrows indicate cell leading edge. F, epithelial cell appears to have phagocytosed crystals. Arrows indicate cell leading edge.
The response of renal epithelial cells to CaOx monohydrate crystals is characterized by increased expression of specific encoding genes, transcriptional activators, extracellular matrix regulators, growth factors, and production of proinflammatory and anti-inflammatory molecules, such as OPN, MCP-1, prostaglandin E2, bikunin and components of IαI, α-1 microglobulin, CD-44, calgranulin, heparan sulfate, osteonectin, fibronectin and matrix gla protein.11,19,20 Although many of these molecules are integral to inflammation and fibrosis, they also modulate biomineralization (see Appendix).21
Renal cells exposed to CaOx crystals secrete superoxide in real time, as measured by an electrochemical superoxide biosensor.22 Mitochondria4,23 and NADPH oxidase11,19,24 are involved in ROS production. Ox or crystal induced cellular responses can be inhibited by antioxidants as well as by NADPH oxidase inhibitors. Cellular injury could be ameliorated by antioxidants and free radical scavengers.4,19,25 Catalase decreased LDH release, and hydrogen peroxide and 8-isoprostane production by LLC-PK1 and MDCK cells exposed to brushite crystals. Similarly, free radical scavengers, catalase and superoxide dismutase provided protection from Ox induced injury to the 2 cell types and reduced MCP-1 mRNA as well as protein in Ox, CaOx, CaP and uric acid treated NRK52E renal epithelial cells. The NADPH oxidase inhibitor diphenyleneiodium also decreased Ox, CaOx, brushite and uric acid induced up-regulation of OPN and MCP-1 in renal epithelial cells.19 MCP-1 mRNA expression and protein production were increased after exposure of the human renal tubular cell line HK-2 to Ox or CaOx crystals.26 Pretreatment with free radical scavengers, catalase and superoxide dismutase significantly decreased the expression and production of MCP-1.
NADPH oxidase is a major source of ROS in the kidneys, particularly in the presence of angiotensin II. Exposure of the human renal epithelial derived cell line HK-2 to Ox and CaOx crystals resulted in increased LDH release, 8-isoprotane production, increased NADPH oxidase activity and superoxide production.24 Exposure to CaOx crystals resulted in significantly higher activity than Ox exposure alone. Exposure to Ox and CaOx crystals affected the expression of various NADPH oxidase subunits with consistent increases in the expression of membrane bound p22phox and cytosolic p47phox. Strong, significant correlations were seen between NADPH oxidase activity, and p22phox and p47phox expression, superoxide production and LDH release when cells were exposed to CaOx crystals. Expression of neither p22phox nor p47phox correlated significantly with increased NADPH oxidase activity after Ox exposure. Ox induced injury is mediated by Rac-1, and PKC-α and δ dependent activation of NADPH oxidase.27,28
Mitochondria are generally the most common source of superoxide and H2O2 in most cells and tissues. Selective probes, substrates and inhibitors show that mitochondria are a major site of CaOx crystal induced superoxide production and glutathione depletion in LLC-PK1 and MDCK cells. Exposure of LLC-PK1 cells to Ox significantly increased cellular ceramides, which could be blocked by N-acetylcysteine pretreatment. Furthermore, Ox exposure can decrease mitochondrial membrane potential in MDCK cells. Isolated mitochondria responded to Ox exposure by the accumulation of ROS, lipid peroxides and oxidized thiol proteins. Administration of exogenous citrate to LLC-PK1 and MDCK cells bolstered antioxidant defenses and decreased cellular injury caused by exposure to increased Ox and CaOx crystals.29 Citrate in culture medium was associated with a significant increase in glutathione, and decreased production of H2O2 and 8-isoprostane, an end product of lipid breakdown. Cell viability significantly improved, as demonstrated by decreased LDH release and increased trypan blue exclusion.
ANIMAL MODEL
A number of animal models have been developed to investigate the pathogenesis of different types of kidney stones.30 CaOx stone disease is established by inducing hyperoxaluria by administering Ox or precursors such as glyoxylate, ethylene glycol and hydroxyl-L-proline, which lead to intratubular CaOx deposition (fig. 3). Experimental induction of hypercalciuria by dietary or genetic manipulation leads to CaP crystal deposition in the tubular lumina and/or pelvic space. Thus, neither hyperoxaluria nor hypercalciuria produces Ca deposits similar to idiopathic stones. However, animal model studies provide information on the renal cellular response to hypercalciuria, hyperoxaluria, and CaOx and CaP crystals. Hyperoxaluria and CaOx crystal deposition triggers various morphological and pathophysiological changes in the kidneys and alters urinary composition.30,31 These changes indicate an inflammatory response by renal cells to high Ox and CaOx crystals. Renal expression of OPN,20 IαI, α-1 microglobulin,32 calgranulin, heparan sulfate and matrix gla protein33 is increased (see Appendix and fig. 4). The expression of NF-κB, kidney injury molecule, proliferating cell nuclear antigen, CD44 and e-cadherin was also increased in the renal tubules of hyperoxaluric rats (fig. 4), particularly in association with CaOx crystal deposits.31 Urinary excretion of many of these molecules is increased.11 As indicated by ED4 positive cells, monocytes and macrophages migrate to crystal deposition sites (fig. 4).
Figure 3.
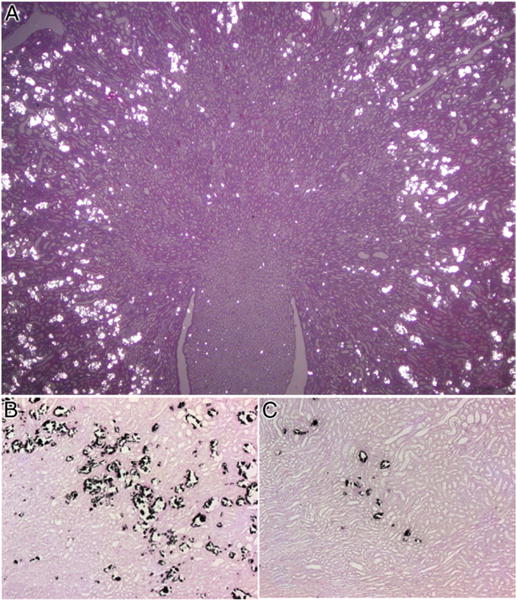
Light microscopy. A, rat kidney with significant CaOx crystal deposition as seen under polarized light. H & E, reduced from ×1.2. B, in kidney section crystals appear as dark deposits. Pizzalato stain, reduced from ×10. C, kidney of hyperoxaluric rat treated with apocynin demonstrates highly significant decrease in CaOx crystal deposition. Reduced from ×10.
Figure 4.
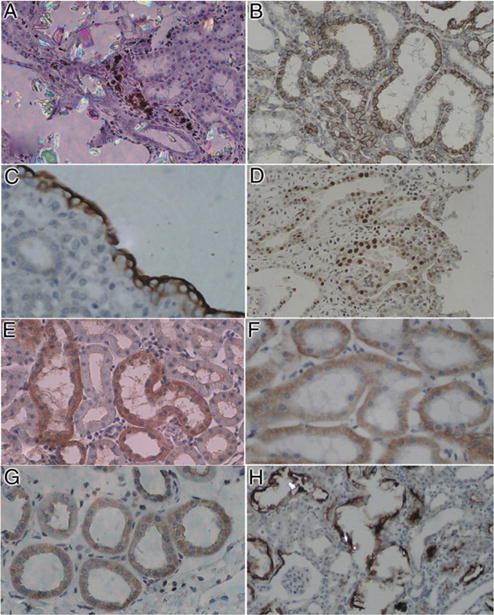
Immunohistochemical staining of kidneys of hyperoxaluric rats. A, ED-1 positive cells were present in renal interstitium next to birefringent CaOx crystal containing tubules, indicating infiltration of monocytes and macrophages (inflammatory response). Reduced from ×20. B, tubular epithelial cells stained positive for CD-44, which is receptor for hyaluronic acid and other ligands, such as OPN and collagens. Reduced from ×20. C, renal papillary surface epithelium in renal fornix, particularly outer surface, shows heavy staining for OPN, modulator of biomineralization and inflammation. Reduced from ×40. D, tubular epithelial cells demonstrate positive staining for proliferating cell nuclear antigen, indicating cellular regeneration and proliferation. Reduced from ×20. E, increased staining for OPN in renal cortical tubules. Reduced from ×20. F, epithelial cells lining cortical renal tubules stained positive for E-cadherin, which has important role in cell adhesion. Reduced from ×20. G, tubular epithelial cells showed increased staining for NF-κB, mediator of inflammation and injury. H, crystal deposition was associated with increased expression of kidney injury molecule in tubular cells and their luminal surfaces. Reduced from ×20.
Animal model studies also provide evidence that renal responses to Ox and crystals are mediated by ROS. Lipid peroxides increased in renal tissue and urine in rats with hyperoxaluria and CaOx nephrolithiasis.34 Vitamin E treatment improved tissue levels of antioxidant enzymes, decreased peroxidative tissue injury and totally eliminated CaOx crystal deposition in the kidneys. CaOx crystal deposition in the kidneys was associated with a reduction in total renal cellular glutathione and an increase in lipid peroxides. Rats that received the angiotensin converting enzyme inhibitor losartan, which is known to decrease OS, showed a significant increase in glutathione and a decrease in thiobarbituric acid-reactive substances in the kidneys. Catalase and Mn superoxide dismutase activities increased in the kidneys, while α and μ-glutathione-S-transferase increased in the urine of hyperoxaluric rats.35
ROS are at least in part produced with the involvement of NADPH oxidase through activation of the renin-angiotensin system. Decreasing angiotensin production by inhibiting angiotensin converting enzyme as well as blocking angiotensin receptor reduces crystal deposition and ameliorates the associated inflammatory response. CaOx crystal deposition in rat kidneys activated the renin-angiotensin system, increased renin expression in the kidneys and serum, and blocked angiotensin receptor decreased OPN expression and CaOx crystal deposition in hyperoxaluric rats.20
NADPH oxidase inhibition by apocynin treatment reduced ROS production and CaOx crystal renal deposition in hyperoxaluric rats.31 Atorvastatin, which decreases the expression of Nox-1 and p22phox subunits of NADPH oxidase, also inhibited crystal deposition in rats with experimentally induced hyperoxaluria.36
DISCUSSION
Experimental studies suggest that exposure to high Ox, and/or CaOx or CaP crystals results in increased gene expression and production of molecules involved in tissue remodeling, inflammation and biomineralization (fig. 5). Stone formation is influenced by a number of crystallization modulators/inhibitors, which affect crystal nucleation, growth, aggregation and retention, and can be modified by ROS. The loss of inhibitory activity of THP in patients with hyperoxaluria was suggested to be mediated primarily by oxidative damage.37 Almost all macromolecules regarded as crystallization inhibitors/modulators are known participants in inflammatory processes and are generated through redox dependent signaling pathways (see Appendix).
Figure 5.
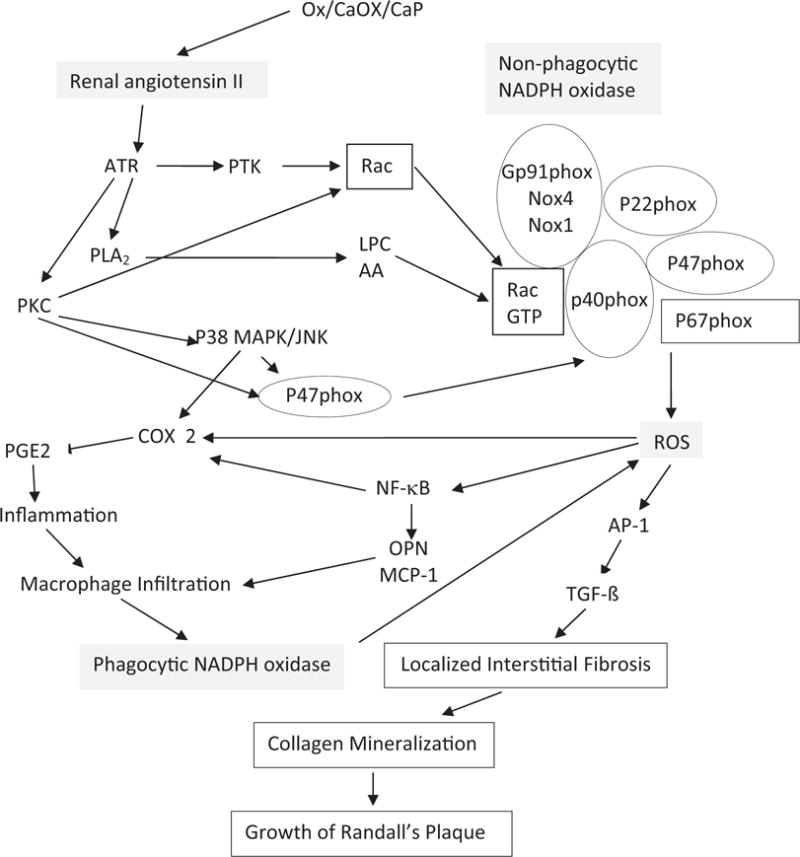
Pathways involved in NADPH oxidase regulation and ROS production. Renal cell exposure to high Ox, CaOx/CaP crystals and mechanical stress associated with crystal deposition in kidneys leads to renin up-regulation and angiotensin II generation. Nonphagocytic NADPH oxidase is activated and ROS are produced, mediated by PKC. Activation involves phosphorylation of p47phox, and translocation of Rac1 and p47phox to membrane. P-38 mitogen activated protein kinase/jun N-terminal kinase (MAPK/JNK) transduction pathway is turned on. Various transcriptional and growth factors, including NF-κB, AP-1 and transforming growth factor-β (TGF- β), become involved. Secondary mediators are generated, such as isoprostanes, cytoplasmic phospholipase A2 and prostaglandin. Production of chemoattractants, such as MCP-1 and crystallization modulator OPN, is increased. Macrophages infiltrate renal interstitium around crystal deposits. Activation of phagocytic NADPH oxidase results in additional ROS production. Inflammation, fibrosis, collagen deposition and mineralization develop, leading to growth of interstitial CaP deposits or RP. AA, arachidonic acid. ATR, angiotensin II type 1 receptor. COX 2, cyclooxygenase 2. LPC, lysophosphatidylcholine. PGE2, prostaglandin E2. PLA2, phospholipase A2. PTK, protein tyrosine kinase. Rac GTP, ρ-related guanosine triphosphate.
The up-regulated macromolecules have significant roles in the inflammatory process. Heparan sulfate regulates extracellular matrix production. Bikunin, a constituent of IαI, is a proteinase inhibitor associated with inflammation that stabilizes the extracellular matrix. Acute inflammatory conditions are known to up-regulate or down-regulate transcription of IαI genes. THP, seen in the renal interstitium in several forms of tubulointerstitial disease, produces interstitial inflammation and scarring. Interestingly, inactivating the THP gene in mouse embryonic stem cells resulted in spontaneous formation of CaOx crystals in adult kidneys.38 THP can activate alternate pathways, interact with neutrophils and bind to certain cytokines. Prothrombin is the precursor of thrombin, and fragments 1 and 2. Thrombin is involved in platelet aggregation and blood coagulation, and it has a major role in the recruitment and activation of infiltrating immune cells.
Inflammation is above all a protective response. In the case of pathological mineralization (stone formation), one would expect the body to produce mineralization inhibitors/modulators. For example, OPN, which is involved in inflammation and fibrosis,39 is a known mineralization inhibitor as well as a chemoattractant.11,21 Its presence would attract monocytes/macrophages to the mineral formation, first to inhibit crystallization and in the case of mineral formation attract inflammatory cells to then eliminate the crystals.11 Thus, being part of the inflammatory cascade and being a mineralization inhibitor are not mutually exclusive.
Most idiopathic stones form while connected to RP, which starts as deposits of poorly crystalline biological apatite in the basement membrane of the loops of Henle, collecting ducts13 or vasa recta.14 It was proposed that the deposits, consisting of aggregated CaP spherules, then move through the interstitium toward the renal papillary epithelium, where they eventually ulcerate to the surface. A number of questions remain unanswered. Does urinary supersaturation affect CaP crystallization in the renal interstitium without intraluminal crystallization? How and why do the crystals deposit in the basement membrane? How do the crystals move through the interstitium and then ulcerate to the papillary surface? Crystals generally stay where they are formed except when they deposit unattached in the tubular lumina. We noted that collagen is a part of interstitial CaP deposits (fig. 1).16 It is mineralized during plaque formation and may have a significant role in plaque growth and development. Collagen is deposited during fibrosis.40 It is an excellent nucleator of CaP and has a critical role in the biomineralization processes of the body.41
Interestingly, all RPs are not associated with stones and the kidneys of nonstone formers also show RP.42 It was suggested that RP is formed without causing renal injury and inflammation. Since RPs are CaP deposits at abnormal sites, RP should be recognized as a foreign body and an inflammatory response should be expected unless it is actively suppressed. Alternatively, the inflammatory response may be localized, short lived and quickly resolved, leaving only telltale signs, or perhaps plaque is a relic of an earlier injury, as suggested by Randall.15
Our examinations of RP have revealed injured tubules. The presence of molecules such as OPN,12 heavy chain of IαI,12 collagen42–44 and zinc45 in interstitial plaques strongly suggests that inflammation may be an early local participant.11 Biopsies are taken at stone removal many months after its formation and only a small amount of tissue from a limited number of patients has been investigated to date. Inflammation is a complex biological response to irritants, in this case high Ox, and/or CaOx and CaP crystals. OPN and IαI may be protective mediators that are most likely produced to inhibit crystallization. In normal human kidneys OPN is localized primarily to the distal nephron and strongly expressed in the thick ascending limbs of the loops of Henle and papillary surface epithelium. OPN expression is increased during inflammation and interstitial fibrosis.46
A number of pathways of RP formation have been proposed14,47,48 but to our knowledge none have been experimentally tested to date. As mentioned, idiopathic stones start as deposits of biological apatite or RP in the renal papillary interstitium, at the junction of the loops of Henle, and the collecting ducts and vasa recta. Changes in local availability of Ca, phosphate and citrate as well as pH may have a critical role in crystal formation. After CaP crystals are deposited, nearby cells respond to the foreign body by generating ROS, producing chemoattractants such as MCP-1 and OPN. Monocytes/macrophages infiltrate the interstitium. Crystals are phagocytized and eliminated or fenced by a wall of macromolecules adsorbed to crystal surfaces, rendering them harmless.49 This is likely the case with interstitial plaques, which are common in the kidneys of stone formers and nonstone formers. However, if the conditions that promoted crystal formation persist and crystals continue to form, localized injury and inflammation develop, followed by fibrosis and collagen deposition around the CaP crystal deposits, initiating the second phase of stone pathogenesis. Collagen is mineralized, leading to plaque growth, which eventually reaches the papillary epithelium, ulcerates to the surface and develops into a stone nidus. After it is exposed to pelvic urine, the plaque is overgrown by CaOx crystals, leading to the formation of an idiopathic CaOx kidney stone attached to the subepithelial RP.11
CONCLUSIONS
Experimental and clinical studies provide evidence for ROS production and OS development in the kidneys of stone forming patients. Initially ROS are involved in the production of crystallization inhibitors, preventing stone formation. However, decreased antioxidant capacity or persistently supersaturated urine, leading to increased crystallization, may result in OS and stone disease. Pretreatment with antioxidants and inhibitors of ROS generating enzymes substantially decreases renal CaOx crystal deposition, which is a surrogate marker of nephrolithiasis in animal models. OS is also suggested to be a mediator of other chronic renal diseases that simultaneously occur with kidney stones. OS generation in one renal disease may predispose the kidneys to another disease. Surgical treatment, particularly shock wave lithotripsy, also generates ROS, which may in part be responsible for stone recurrence. A combination of antioxidants and free radical scavengers may provide renal protection against shock wave induced renal injury50 as well as stone recurrence.
We hypothesize that localized OS induced inflammation resulting from interstitial CaP deposition in the renal papillae promotes fibrosis and collagen formation. Collagen mineralization promotes CaP propagation and RP growth. Antioxidants and ant-iinflammatory therapy may impede RP growth and inhibit stone recurrence.
Acknowledgments
Supported by National Institutes of Health Grant RO1-DK078602 and the University of Florida Center for the Study of Lithiasis.
Abbreviation and Acronyms
- AP-1
activator protein 1
- CaP
biological calcium phosphate
- IαI
inter-α-inhibitor
- LDH
lactate dehydrogenase
- MCP-1
monocyte chemoattractant protein-1
- NADPH
nicotinamide adenine dinucleotide phosphate
- NAG
N-acetyl-β-glucosaminidase
- NF-κB
nuclear factor κ-light-chain-enhancer of activated B cells
- OPN
osteopontin
- OS
oxidative stress
- Ox
oxalate
- phox
phagocytic oxidase
- PKC
protein kinase
- ROS
reactive oxygen species
- RP
Randall plaque
- THP
Tamm-Horsfall protein
APPENDIX
Crystallization Modulating Macromolecules in Renal Epithelial Cells in response to Ox or CaOx crystal exposure
| Nephrolithiasis Role | Inflammation + Repair Role | Ox + CaOx Crystal Response | |
|---|---|---|---|
| THP | Crystal aggregation inhibitor | Renoprotective, elicits immune response | Up-regulation, down-regulation, no change |
| OPN | Free OPN inhibits crystal nucleation, growth, aggregation + attachment, + immobilized OPN promotes crystal attachment | Ca binding, renoprotective, tissue repair + inflammation, monocyte/macrophage chemoattractant | Up-regulation |
| Prothrombin fragment-1 | Crystal growth + aggregation inhibitor | Ca binding, coagulation | Up-regulation, no change |
| Bikunin + IαI family | Crystal nucleation, growth, aggregation + attachment inhibitor | Metastasis, tissue repair + remodeling | Up-regulation |
| α-1 Microglobulin | Crystallization inhibitor | Immunosuppressive, mitogenic | Up-regulation |
| Hyaluronic acid | Major stone matrix constituent | Important extracellular matrix constituent | |
| CD-44 | Crystal attachment promoter | Tissue repair + remodeling | Up-regulation |
| Calgranulin | Crystal growth + aggregation inhibitor | Ca binding, tissue remodeling + inflammation | Up-regulation |
| Heparan sulfate | Crystal aggregation + attachment inhibitor | Tissue remodeling | Up-regulation |
| Osteonectin | Ca binding, tissue remodeling | Up-regulation | |
| Fibronectin | Crystal aggregation, attachment + endocytosis inhibitor | Morphogenesis, wound healing + metastasis | Up-regulation |
| Matrix gla protein | Crystal deposition inhibitor | Biomineralization inhibitor | Up-regulation |
References
- 1.Brikowski TH, Lotan Y, Pearle MS. Climate-related increase in the prevalence of urolithiasis in the United States. Proc Natl Acad Sci U S A. 2008;105:9841. doi: 10.1073/pnas.0709652105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McAteer JA, Evan AP. The acute and longterm adverse effects of shock wave lithotripsy. Semin Nephrol. 2008;28:200. doi: 10.1016/j.semnephrol.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coe FL, Evan AP, Lingeman JE, et al. Plaque and deposits in nine human stone diseases. Urol Res. 2010;38:239. doi: 10.1007/s00240-010-0296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan SR. Hyperoxaluria-induced oxidative stress and antioxidants for renal protection. Urol Res. 2005;33:349. doi: 10.1007/s00240-005-0492-4. [DOI] [PubMed] [Google Scholar]
- 5.Manea A. NADPH oxidase-derived reactive oxygen species: involvement in vascular physiology and pathology. Cell Tissue Res. 2010;342:325. doi: 10.1007/s00441-010-1060-y. [DOI] [PubMed] [Google Scholar]
- 6.Baggio B, Gambaro G, Ossi E, et al. Increased urinary excretion of renal enzymes in idiopathic calcium oxalate nephrolithiasis. J Urol. 1983;129:1161. doi: 10.1016/s0022-5347(17)52619-1. [DOI] [PubMed] [Google Scholar]
- 7.Boonla C, Wunsuwan R, Tungsanga K, et al. Urinary 8-hydroxydeoxyguanosine is elevated in patients with nephrolithiasis. Urol Res. 2007(35):185. doi: 10.1007/s00240-007-0098-0. [DOI] [PubMed] [Google Scholar]
- 8.Mushtaq S, Siddiqui AA, Naqvi ZA, et al. Identification of myeloperoxidase, alpha-defensin and calgranulin in calcium oxalate renal stones. Clin Chim Acta. 2007;384:41. doi: 10.1016/j.cca.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Schwille PO, Manoharan M, Schmiedl A. Is idiopathic recurrent calcium urolithiasis in males a cellular disease? Laboratory findings in plasma, urine and erythrocytes, emphasizing the absence and presence of stones, oxidative and mineral metabolism: an observational study. Clin Chem Lab Med. 2005;43:590. doi: 10.1515/CCLM.2005.103. [DOI] [PubMed] [Google Scholar]
- 10.Holoch PA, Tracy CR. Antioxidants and self-reported history of kidney stones: the National Health and Nutrition Examination Survey. J Endourol. 2011;25:1903. doi: 10.1089/end.2011.0130. [DOI] [PubMed] [Google Scholar]
- 11.Khan SR. Crystal-induced inflammation of the kidneys: results from human studies, animal models, and tissue-culture studies. Clin Exp Nephrol. 2004;8:75. doi: 10.1007/s10157-004-0292-0. [DOI] [PubMed] [Google Scholar]
- 12.Evan AP. Physiopathology and etiology of stone formation in the kidney and the urinary tract. Pediatr Nephrol. 2010;25:831. doi: 10.1007/s00467-009-1116-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weller RO, Nester B, Cooke SAR. Calcification in the human renal papilla: an electron-microscope study. J Pathol. 1972;107:211. doi: 10.1002/path.1711070308. [DOI] [PubMed] [Google Scholar]
- 14.Stoller ML, Meng MV, Abrahams HM, et al. The primary stone event: a new hypothesis involving a vascular etiology. J Urol. 2004;171:1920. doi: 10.1097/01.ju.0000120291.90839.49. [DOI] [PubMed] [Google Scholar]
- 15.Randall A. The origin and growth of renal calculi. Ann Surg. 1937;105:1009. doi: 10.1097/00000658-193706000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan SR, Rodriguez DE, Gower LB, et al. Association of Randall plaque with collagen fibers and membrane vesicles. J Urol. 2012;187:1094. doi: 10.1016/j.juro.2011.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan SR. Is oxidative stress, a link between nephrolithiasis and obesity, hypertension, diabetes, chronic kidney disease, metabolic syndrome? Urol Res. 2012;40:95. doi: 10.1007/s00240-011-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol. 2009;20:2253. doi: 10.1681/ASN.2009030276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Umekawa T, Byer K, Uemura H, et al. Diphenyleneiodium (DPI) reduces oxalate ion- and calcium oxalate monohydrate and brushite crystal-induced upregulation of MCP-1 in NRK 52E cells. Nephrol Dial Transplant. 2005;20:870. doi: 10.1093/ndt/gfh750. [DOI] [PubMed] [Google Scholar]
- 20.Umekawa T, Hatanaka Y, Kurita T, et al. Effect of angiotensin II receptor blockage on osteopontin expression and calcium oxalate crystal deposition in rat kidneys. J Am Soc Nephrol. 2004;15:635. doi: 10.1097/01.asn.0000113321.49771.2d. [DOI] [PubMed] [Google Scholar]
- 21.Khan SR, Kok DJ. Modulators of urinary stone formation. Front Biosci. 2004;9:1450. doi: 10.2741/1347. [DOI] [PubMed] [Google Scholar]
- 22.Gáspár S, Niculite C, Cucu D, et al. Effect of calcium oxalate on renal cells as revealed by real-time measurement of extracellular oxidative burst. Biosens Bioelectron. 2010;25:1729. doi: 10.1016/j.bios.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 23.Meimaridou E, Jacobson J, Seddon AM, et al. Crystal and microparticle effects on MDCK cell superoxide production: oxalate-specific mitochondrial membrane potential changes. Free Radic Biol Med. 2005;38:1553. doi: 10.1016/j.freeradbiomed.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 24.Khan SR, Khan A, Byer KJ. Temporal changes in the expression of mRNA of NADPH oxidase subunits in renal epithelial cells exposed to oxalate or calcium oxalate crystals. Nephrol Dial Transplant. 2011;26:1778. doi: 10.1093/ndt/gfq692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang HS, Chen J, Chen CF, et al. Vitamin E attenuates crystal formation in rat kidneys: roles of renal tubular cell death and crystallization inhibitors. Kidney Int. 2006;70:699. doi: 10.1038/sj.ki.5001651. [DOI] [PubMed] [Google Scholar]
- 26.Habibzadegah-Tari P, Byer KG, Khan SR. Reactive oxygen species mediated calcium oxalate crystal-induced expression of MCP-1 in HK-2 cells. Urol Res. 2006;34:26. doi: 10.1007/s00240-005-0007-3. [DOI] [PubMed] [Google Scholar]
- 27.Thamilselvan V, Menon M, Thamilselvan S. Selective Rac1 inhibition protects renal tubular epithelial cells from oxalate-induced NADPH oxidase-mediated oxidative cell injury. Urol Res. 2012;40:415. doi: 10.1007/s00240-011-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thamilselvan V, Menon M, Thamilselvan S. Oxalate-induced activation of PKC-alpha and -delta regulates NADPH oxidase-mediated oxidative injury in renal tubular epithelial cells. Am J Physiol Renal Physiol. 2009;297:F1399. doi: 10.1152/ajprenal.00051.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Byer K, Khan SR. Citrate provides protection against oxalate and calcium oxalate crystal induced oxidative damage to renal epithelium. J Urol. 2005;173:640. doi: 10.1097/01.ju.0000143190.49888.c7. [DOI] [PubMed] [Google Scholar]
- 30.Khan SR, Glenton PA, Byer KJ. Modeling of hyperoxaluric calcium oxalate nephrolithiasis: experimental induction of hyperoxaluria by hydroxy-L-proline. Kidney Int. 2006;70:914. doi: 10.1038/sj.ki.5001699. [DOI] [PubMed] [Google Scholar]
- 31.Zuo J, Khan A, Glenton PA, et al. Effect of NADPH oxidase inhibition on the expression of kidney injury molecule and calcium oxalate crystal deposition in hydroxy-L-proline-induced hyperoxaluria in the male Sprague-Dawley rats. Nephrol Dial Transplant. 2011;26:1785. doi: 10.1093/ndt/gfr035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grewal JS, Tsai JY, Khan SR. Oxalate-inducible AMBP gene and its regulatory mechanism in renal tubular epithelial cells. Biochem J. 2005;387:609. doi: 10.1042/BJ20041465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okada A, Yasui T, Fujii Y, et al. Renal macrophage migration and crystal phagocytosis via inflammatory-related gene expression during kidney stone formation and elimination in mice: detection by association analysis of stone-related gene expression and microstructural observation. J Bone Miner Res. 2010;25:2701. doi: 10.1002/jbmr.158. [DOI] [PubMed] [Google Scholar]
- 34.Thamilselvan S, Menon M. Vitamin E therapy prevents hyperoxaluria-induced calcium oxalate crystal deposition in the kidney by improving renal tissue antioxidant status. BJU Int. 2005;96:117. doi: 10.1111/j.1464-410X.2005.05579.x. [DOI] [PubMed] [Google Scholar]
- 35.Huang HS, Ma MC, Chen J, et al. Changes in the oxidant-antioxidant balance in the kidney of rats with nephrolithiasis induced by ethylene glycol. J Urol. 2002;167:2584. [PubMed] [Google Scholar]
- 36.Tsujihata M, Yoshioka I, Tsujimura A, et al. Why does atorvastatin inhibit renal crystal retention? Urol Res. 2011;39:379. doi: 10.1007/s00240-011-0370-1. [DOI] [PubMed] [Google Scholar]
- 37.Sumitra K, Pragasam V, Sakthivel R, et al. Beneficial effect of vitamin E supplementation on the biochemical and kinetic properties of Tamm-Horsfall glycoprotein in hypertensive and hyperoxaluric patients. Nephrol Dial Transplant. 2005;20:1407. doi: 10.1093/ndt/gfh794. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Mo L, Goldfarb DS, et al. Progressive renal papillary calcification and ureteral stone formation in mice deficient for Tamm-Horsfall protein. Am J Physiol Renal Physiol. 2010;299:F469. doi: 10.1152/ajprenal.00243.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urtasun R, Lopategi A, George J, et al. Osteopontin, an oxidant stress-sensitive cytokine, up-regulates collagen-I via integrin α(V)β(3) engagement and PI3K/pAkt/NFκB signaling. Hepatology. 2012;55:594. doi: 10.1002/hep.24701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeisberg M, Strutz F, Muller GA. Renal fibrosis: an update. Curr Opin Nephrol Hypertens. 2001;10:315. doi: 10.1097/00041552-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Murshed M, McKee MD. Molecular determinants of extracellular matrix mineralization in bone and blood vessels. Curr Opin Nephrol Hypertens. 2010;19:359. doi: 10.1097/MNH.0b013e3283393a2b. [DOI] [PubMed] [Google Scholar]
- 42.Haggitt RC, Pitcock JA. Renal medullary calcifications: a light and electron microscopic study. J Urol. 1971;106:342. doi: 10.1016/s0022-5347(17)61284-9. [DOI] [PubMed] [Google Scholar]
- 43.Cooke SAR. The site of calcification in the human renal papilla. Br J Surg. 1970;57:890. doi: 10.1002/bjs.1800571205. [DOI] [PubMed] [Google Scholar]
- 44.Evan AP, Lingeman JE, Coe FL, et al. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest. 2003;111:607. doi: 10.1172/JCI17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carpentier X, Bazin D, Combes C, et al. High Zn content of Randall’s plaque: A μ-X-ray fluorescence investigation. J Trace Elem Med Biol. 2011;25:160. doi: 10.1016/j.jtemb.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Xie Y, Sakatsume M, Nishi S, et al. Expression, roles, receptors, and regulation of osteopontin in the kidney. Kidney Int. 2001;60:1645. doi: 10.1046/j.1523-1755.2001.00032.x. [DOI] [PubMed] [Google Scholar]
- 47.Gambaro G, D’Angelo A, Fabris A, et al. Crystals, Randall’s plaques and renal stones: do bone and atherosclerosis teach us something? J Nephrol. 2004;17:774. [PubMed] [Google Scholar]
- 48.Coe FL, Evan AP, Worcester EM, et al. Three pathways for human kidney stone formation. Urol Res. 2010;38:147. doi: 10.1007/s00240-010-0271-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Escobar C, Byer KJ, Khan SR. Naturally produced crystals obtained from kidney stones are less injurious to renal tubular epithelial cells than synthetic crystals. BJU Int. 2007;100:891. doi: 10.1111/j.1464-410X.2007.07002.x. [DOI] [PubMed] [Google Scholar]
- 50.Sarica K, Yencilek F. Prevention of shockwave induced functional and morphological alterations: an overview. Arch Ital Urol Androl. 2008;80:27. [PubMed] [Google Scholar]