

Abstract
Background
Acute respiratory distress syndrome (ARDS) results in vascular leakage, inflammation and respiratory failure. There are currently no approved pharmacological treatments for ARDS and standard of care involves treatment of the underlying cause, and supportive care. The vascular leakage may be related to reduced concentrations of local adenosine, which is involved in maintaining endothelial barrier function. Interferon (IFN) beta-1a up-regulates the cell surface ecto-5′-nucleotidase cluster of differentiation 73 (CD73), which increases adenosine levels, and IFN beta-1 may, therefore, be a potential treatment for ARDS. In a phase I/II, open-label study in 37 patients with acute lung injury (ALI)/ARDS, recombinant human IFN beta-1a was well tolerated and mortality rates were significantly lower in treated than in control patients.
Methods/design
In this phase III, double-blind, randomized, parallel-group trial, the efficacy and safety of recombinant human IFN beta-1a (FP-1201-lyo) will be compared with placebo in adult patients with ARDS. Patients will be randomly assigned to receive 10 μg FP-1201-lyo or placebo administered intravenously once daily for 6 days and will be monitored for 28 days or until discharged from the intensive care unit. Follow-up visits will then take place at days 90, 180 and 360. The primary endpoint is a composite endpoint including any cause of death at 28 days and days free of mechanical ventilation within 28 days among survivors. Secondary endpoints include: all-cause mortality at 28, 90, 180 and 360 days; organ failure-free days; length of hospital stay; pharmacodynamic assessment including measurement of myxovirus resistance protein A concentrations; and measures of quality of life, respiratory and neurological function at 180 and 360 days. The estimated sample size to demonstrate a reduction in the primary outcome between groups from 30% to 15% is 300 patients, and the study will be conducted in 70–80 centers in nine countries across Europe.
Discussion
There are no effective specific treatments for patients with ARDS and mortality rates remain high. The results from this study will provide evidence regarding the efficacy of a potential new therapeutic agent, FP-1201-lyo, in improving the clinical course and outcome for patients with moderate/severe ARDS.
Trial registration
European Union Clinical Trials Register, no: 2014-005260-15. Registered on 15 July 2017.
Electronic supplementary material
The online version of this article (doi:10.1186/s13063-017-2234-7) contains supplementary material, which is available to authorized users.
Keywords: Interferon, ARDS, Vascular leakage, CD73
Background
Acute respiratory distress syndrome (ARDS)
ARDS is a serious clinical disorder which follows a variety of severe lung insults including, among others, pneumonia, aspiration of gastric contents, non-pulmonary sepsis and major trauma. ARDS is a type of acute diffuse lung injury, characterized by acute lung inflammation with injury to the endothelial barriers and alveolar epithelium of the lung, increased pulmonary vascular permeability, and protein-rich pulmonary edema leading to acute respiratory failure. In the Berlin definition of ARDS (Table 1) [1], severity is graded from mild, through moderate, to severe ARDS.
Table 1.
The Berlin ARDS definition [1]
| Characteristic | Mild ARDS | Moderate ARDS | Severe ARDS |
|---|---|---|---|
| Timing | Acute onset within 1 week of a known clinical insult or new or worsening respiratory symptoms | ||
| Hypoxemia | PaO2/FiO2
> 200– ≤ 300 mmHg with PEEP or CPAP ≥ 5 cmH2O |
PaO2/FiO2
> 100– ≤ 200 mmHg with PEEP ≥ 5 cmH2O |
PaO2/FiO2
≤ 100 mmHg with PEEP ≥ 5 cmH2O |
| Origin of edema | Respiratory failure associated with known ARDS risk factors and not fully explained by cardiac failure or fluid overload. An objective assessment of cardiac failure or fluid overload is needed if no ARDS risk factors are present | ||
| Radiological abnormalities (chest X-ray or CT scan) | Bilateral opacities not fully explained by effusions, nodules, masses or lobar/lung collapse | ||
ARDS acute respiratory distress syndrome, CPAP continuous positive airway pressure, CT computed tomography, PaO 2 /FiO 2 partial pressure of oxygen/fraction of inspired oxygen, PEEP positive end-expiratory pressure
Although mortality from ARDS has decreased in the last decade due to improvements in supportive care and in the treatment of underlying conditions [2, 3], it remains high at levels of 20 to 40% across all severities and even higher when associated with dysfunction in other organs [1, 2, 4, 5].
ARDS is also costly in health economics terms. Patients with ARDS consume significantly more resources than other groups of critically ill patients [6] because they have longer intensive care unit (ICU) and hospital stays. The quality of life (QoL) of these patients may also be significantly impacted [7], with 35% of patients with moderate or severe ARDS unable to return to work 24 months after hospital discharge [8, 9].
There are currently no approved pharmacological therapies for ARDS and treatment relies on management of the underlying cause and supportive care.
Interferon beta-1a
A key pathophysiological feature of ARDS is increased vascular leakage, which has been suggested to be related to a lack of local adenosine which acts to enhance endothelial barrier function [10]. Therefore, any biological substances that can increase local adenosine levels may reduce vascular leakage and thus be of benefit in ARDS. Such a substance is cluster of differentiation 73 (CD73) – a cell surface ecto-5′-nucleotidase enzyme that converts soluble AMP into locally active adenosine [11]. Interferons, such as interferon (IFN) beta-1a, have been shown to up-regulate CD73 and could, therefore, represent a potential treatment for ARDS. Preclinical studies have shown that CD73 expression on endothelial cells is up-regulated by IFN beta-1a treatment in a time- and dose-dependent fashion [12–14]. Furthermore, IFN beta-1a treatment has been shown to prevent leakage in animal models of acute lung injury (ALI) [12]. Enhanced adenosine production also controls leukocyte infiltration [15], thus reducing the escalation of inflammation in the lungs.
Recombinant human IFN beta-1a (FP-1201-lyo) was assessed for the treatment of ALI and ARDS, as defined using the AECC definitions [16], in a phase I/II study [13]. This open-label study, conducted in eight ICUs in the UK, included 37 patients with ALI/ARDS and the optimum tolerated dose of FP-1201-lyo was shown to be 10 μg daily. The primary efficacy endpoint, 28-day mortality, was 8% in the treated patients compared to 32% in the control patients; treatment with FP-1201-lyo was associated with an 81% reduction in the odds of death at 28 days (odds ratio 0.19 (95% CI 0.03–0.72); p = 0.01) [13]. The beneficial effects on outcomes were still present at 6-month follow-up. Pyrexia was the most common drug-related treatment-emergent adverse event in the study. All pyrexia events resolved rapidly without sequelae. There were no other safety concerns during the study period. Subcutaneous recombinant human IFN beta-1a is already an approved treatment for patients with relapsing-remitting multiple sclerosis and its safety profile in such patients is well characterized.
Following the promising results of the phase I/II study [13], the current phase III study was designed to confirm the beneficial effects of FP-1201-lyo in a larger population of patients with ARDS. Herein, we describe the final protocol (version 6.0; 29 Aug 2017) for this study, written in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines. The SPIRIT Checklist is provided as Additional file 1.
Methods/design
This is a multicenter, phase III, double-blind, randomized, parallel-group comparison study of the efficacy and safety of FP-1201-lyo compared with placebo in adult patients with moderate or severe ARDS. The study will be conducted in 70–80 centers in nine European countries. The study is registered on the European Union Clinical Trials Register, no: 2014-005260-15. We will conduct the study in accordance with the principles of the Declaration of Helsinki [17] and the International Conference on Harmonization (ICH) guidelines on Good Clinical Practice. The Local Ethics Committee for each center will approve the study (approvals already in place shown in Additional file 2).
Study population
Study site investigators will screen patients in the ICU for eligibility and obtain written informed consent. Study sites will review all ICU patients daily in order to identify potential patients.
Inclusion criteria include:
Patient is aged ≥ 18 years
Patient's trachea is intubated and they are receiving mechanical ventilation
- Patient has a diagnosis of moderate or severe ARDS according to the Berlin definition of ARDS [1]:
- Acute onset of respiratory failure within 1 week of a known clinical insult or new or worsening respiratory symptoms
- Respiratory failure associated with known ARDS risk factors and not fully explained by either cardiac failure or fluid overload (an objective assessment of cardiac failure or fluid overload is needed if no risk factors for ARDS are present)
- Radiological abnormalities on chest X-ray or on computed tomography (CT) scan, i.e., bilateral opacities that are not fully explained by effusions, nodules, masses or lobar/lung collapse
- Hypoxemia:
- Moderate ARDS: PaO2/FiO2 > 100 mmHg (>13.3 kPa) to ≤ 200 mmHg (≤ 26.6 kPa) with positive end-expiratory pressure (PEEP) ≥ 5 cmH2O
- Severe ARDS: PaO2/FiO2 ≤ 100 mmHg (≤ 13.3 kPa) with PEEP ≥ 5 cmH2O
- The radiological and hypoxemia criteria (3 (c) and (d)) must occur within the same 24-h period. The time of onset of ARDS is defined as the time when the last of these two ARDS criteria is met
- Administration of the first dose of study drug must be planned to take place within 48 h of moderate or severe ARDS diagnosis
- A signed written informed consent form from the patient or the patient’s personal legal representative or a professional legal representative must be available
Exclusion criteria include:
Woman known to be pregnant, lactating or with a positive (urine or serum test) or indeterminate (serum test) pregnancy test
Patient simultaneously taking part in another pharmacotherapy protocol
Patient not expected to survive for 24 h
Patient has an underlying clinical condition where, in the opinion of the investigator, it would be extremely unlikely that the patient would be able to come off ventilation, e.g., motor neurone disease, Duchenne muscular dystrophy, or rapidly progressive interstitial pulmonary fibrosis
Patient has severe chronic obstructive pulmonary disease (COPD) requiring long-term home oxygen therapy or mechanical ventilation (non-invasive ventilation or via tracheotomy) except for continuous positive airway pressure (CPAP) or bi-level positive airway pressure (BIPAP) used solely for sleep-disordered breathing
Patient has congestive heart failure, defined as New York Heart Association class IV
Patient has acute left ventricular failure
Patient has liver failure (Child-Pugh grade C)
Patient has received any prior IFN
Patient has known hypersensitivity to natural or recombinant IFN beta or to any of the excipients
Patient is receiving renal dialysis therapy for chronic renal failure
Patient is receiving extracorporeal membrane oxygenation, high-frequency oscillatory ventilation (HFOV) or any form of extracorporeal lung support
Patient has had any form of mechanical ventilation (invasive or non-invasive, excluding CPAP alone) for longer than 48 h prior to the diagnosis of ARDS. Non-invasive ventilation has to be continuously applied for at least 12 h per day in these 48 h
Patient has burns to ≥ 15% of their total body surface area
Study drug
All study drugs will be supplied in identical vials and will be similar in color and appearance, enabling double-blind conditions. The powdered FP-1201-lyo or placebo (powdered lyophilisate) will be diluted in water for injection near the patient/in the ICU using a MixJect® transfer device (West Pharmaceutical Services GmbH, Germany/Medimop Medical Projects, Ra’anana, Israel). Once prepared, the dose must be administered to the patient immediately. The diluted FP-1201-lyo or placebo will be administered as an intravenous bolus injection via a central or peripheral line. The injection will be followed with a 5-mL flush of sterile saline. FP-1201-lyo and placebo injections will be given once daily for 6 days. The injection will be given at the same time each day ± 1 h providing the patient’s condition allows this. If for any reason this is not possible, the treatment window may be extended by up to 4 h. Subsequent doses should not be delayed and should revert to the original time schedule. No dose modifications or temporary cessations of study drug administration are allowed. If a delay beyond the 4-h window described above is required, the patient must be withdrawn from study drug but all data must continue to be collected per protocol.
Study protocol
Local study investigators will use an interactive, web-response system to randomize the patients on a 1:1 basis using country and ARDS severity as stratification parameters. To ensure that conclusions are not dominated by data from a small number of centers, and also to obtain a broad spread of patients and centers within the constraints of the inclusion/exclusion criteria, each center will be allowed to include up to, but no more than, 30 patients. Study investigators at each center will enter all study-related data using electronic Case Report Forms (e-CRFs).
Following randomization, patients will receive FP-1201-lyo 10 μg or placebo administered intravenously as a bolus each day for 6 days. The first dose of drug must be administered within 48 h after diagnosis of moderate or severe ARDS. The APACHE II score [18] will be calculated within 24 h of ICU admission. Patients will undergo daily assessments while in the ICU for a maximum of 28 days to include blood sampling for hematology and biochemical variables, CD73 and myxovirus resistance protein A (MxA) concentrations; dipstick urinalysis; PaO2/FiO2; fluid balance; vital signs (heart rate, respiratory rate, arterial blood pressure, body temperature); and Sequential Organ Failure Assessment (SOFA) score variables [19]. The SOFA score will be calculated pre-dose on day 1, then daily up to day 14, at day 21 and at day 28 while the patient is in the ICU, based on worst daily values. Concomitant medications will be recorded. Blood samples for IFN beta-1a neutralizing antibodies will be taken 1 h prior to the first dose and on the day that the patient leaves the ICU or on day 28 if the patient is still on the ICU. Long-term follow-up will occur at day 90 (visit or telephone contact) and day 180. On day 180 (± 14 days), the EuroQol 5-Dimensions 3-Levels questionnaire (EQ-5D-3 L), the forced expiratory volume in 1 s (FEV1) and the 6-minute Walk Test (6MWT) will be assessed. The Clinical Study Report (CSR) will be completed once all the unblinded data up to day 90 have been collected and verified and analyzed according to the statistical plan. Long-term and extended follow-ups will take place at day 180 and day 360 and will include assessments of 6- and 12-month mortality, EQ-5D-3 L, 6MWT and FEV1. The extended follow-up data will be reported as addendums to the CSR. During the long-term and extended follow-up periods, and after the end of the extended follow-up visit, patient care will follow normal hospital procedures. The study will be completed when the final patient completes their day 360 study assessment. No interim analyses are planned between days 1–180. The study design is summarized in Fig. 1 and timings of assessments and procedures detailed in Fig. 2.
Fig. 1.
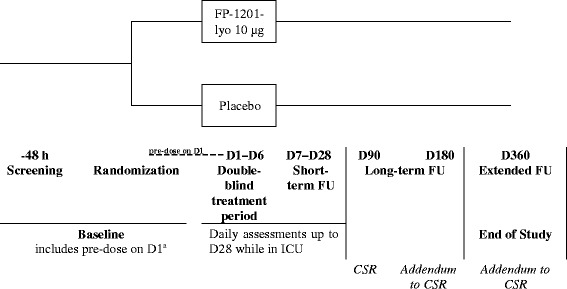
Study design. aNot more than 48 h may elapse between confirmation of moderate or severe acute respiratory distress syndrome (ARDS) during screening and administration of the first dose of study drug on day 1. Once eligibility has been met, randomization can occur during screening or pre-dose on day 1
Fig. 2.
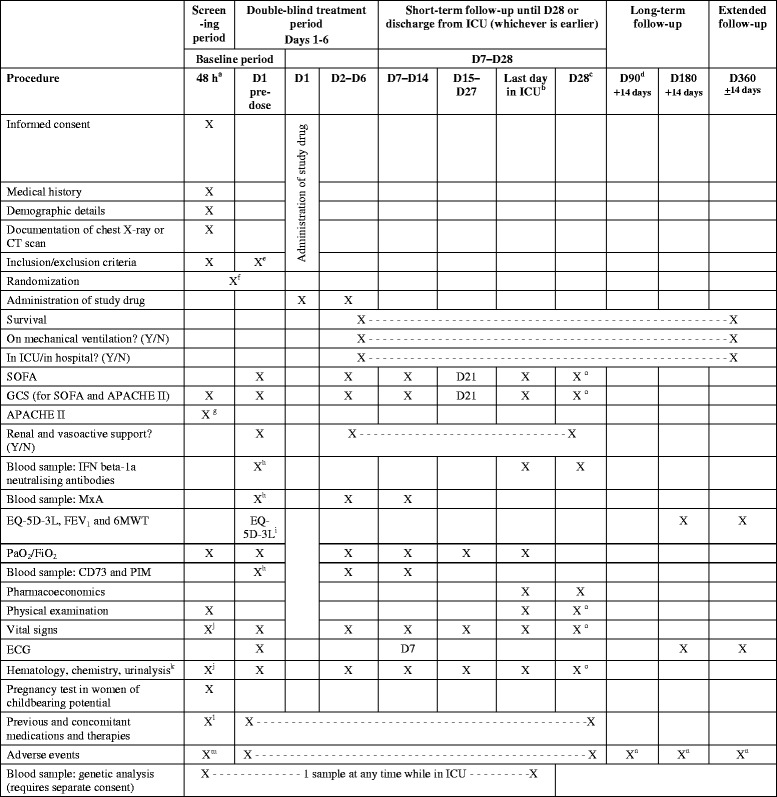
Schedule of procedures. aNo more than 48 hours may elapse between confirmation of moderate or severe ARDS and administration of the first dose of study drug. bThese assessments will be done on the day the patient leaves the ICU, which will either be on D28 or earlier, according to the clinical progress of the patient. If the patient is still in the ICU on D28, the next visit or telephone contact will be at D90. If a patient leaves the ICU before D28, the survival status and other endpoints must be assessed on D28. cD28 procedures apply for patients leaving the ICU before D28 and for patients withdrawing from the study before D28. For patients withdrawing from the study before D28 a sample should be taken for neutralizing antibodies on the day they leave the ICU. dD90 can either be a visit or telephone contact. eReconfirm inclusion/exclusion criteria before dosing, including that patient requires mechanical ventilation and is in the ICU. fRandomize after consent obtained and once eligibility criteria confirmed. gWithin 24 hours of ICU admission. h1 hour pre-dose. iBaseline EQ-5D-3L to be obtained from relatives and checked later with patient. jFor APACHE II scoring. kSamples should be taken in the morning between 04:00 and 10:00. lMedicines and therapies in previous month. mAdverse events will be recorded after informed consent is obtained. nDeaths are reported as SAE. 0if it is possible to be performed by the investigator. APACHE II Acute Physiology and Chronic Health Evaluation, CD Cluster of differentiation, CT Computerized tomography, D Study day, ECG Electrocardiogram, EQ-5D-3L EuroQol 5-Dimensions 3-Levels questionnaire, FEV1 Forced expiratory volume in 1 second, GCS Glasgow Coma Scale, ICU Intensive care unit, IFN Interferon, MxA Myxovirus resistance protein A, PaO2/FiO2 Partial pressure of oxygen/fraction of inspired oxygen, PIM Potential inflammatory marker, SOFA Sequential Organ Failure Assessment, 6MWT 6-minute walk test
General patient management
Apart from administration of the study drug, patients will be managed according to best practice as detailed in Additional file 3. In particular, mechanical ventilation should be provided using a lung-protective ventilation approach incorporating a low-tidal-volume strategy [20]. Any mode of ventilation capable of delivering the prescribed tidal volume (6 mL/kg predicted body weight (PBW) ± 2 mL/kg) within the pressure limitation (plateau pressure limitation ≤ 30 cmH2O) may be used. The use of extracorporeal membrane oxygenation (ECMO) is allowed as rescue therapy. Weaning should follow local ICU protocols if available or the guidelines given in Additional file 3. Fluid management should be unrestricted during episodes of shock, but in patients not in shock, a conservative fluid approach should be adopted (see the table in Additional file 3) [21].
Adverse events
All serious adverse events (SAEs) that occur between the signing of informed consent and day 28 will be recorded. Events occurring after day 28 will be reported only if they are considered to be causally related to the investigational drug; however, all deaths up to day 180 will be reported as SAEs. An SAE is defined as any untoward medical occurrence that at any dose:
Results in death
Is life-threatening
Requires inpatient hospitalization or prolongation of existing hospitalization
Results in persistent or significant disability or incapacity
Causes a congenital anomaly or birth defect
Is an important medical event that may not be immediately life-threatening or result in death or hospitalization but that may jeopardize the patient or require intervention to prevent one of the above outcomes
The study design incorporates an Independent Data Monitoring Committee (IDMC) comprising four members: one independent biostatistician and three senior clinicians with significant experience in ARDS who are not involved in the study. The IDMC will review ongoing safety data in an unblinded manner with meetings scheduled to take place after the data has been received from the last patient of approximately 30, 60, 120, 200 and 300 patients who either have completed 14 days in the study following their first dose of study medication or have been withdrawn for any reason (including death). At each meeting, the IDMC will make a blinded recommendation to the sponsor regarding the study to continue without change, modify study or enrollment to be placed on hold, or study termination.
Outcome measurements
The primary endpoint is a composite of death and days free of mechanical ventilation within 28 days among survivors. A patient will be considered as ventilator free after two consecutive calendar days of unassisted breathing, defined as breathing spontaneously with a face mask, nasal prong oxygen or room air; T-piece breathing; tracheostomy mask breathing; CPAP ≤ 5 cmH2O without pressure support or intermittent mandatory ventilation assistance; or use of CPAP or BIPAP solely for sleep apnea management.
Secondary endpoints can be divided into efficacy, safety and exploratory categories.
Efficacy
All-cause mortality assessed at 28, 90, 180 and 360 days
Days free of organ failure (assessed using the SOFA score), days free of renal support, days free of vasoactive support, days free of mechanical ventilation, number of intensive care unit (ICU)-free days, assessed at day 28 (or on the last day in the ICU if the patient leaves the ICU before day 28)
Length of hospital stay
Immunogenicity of FP-1201-lyo assessed by change in level of neutralizing antibodies to IFN beta-1a between baseline and day 28 (or on the last day in the ICU if the patient leaves the ICU before day 28)
FEV1, neurological functioning (6MWT) and QoL (EQ-5D-3 L) assessed at 180 and 360 days
Safety
Adverse events (assessed by seriousness, intensity and causality) up to day 28, and up to day 360 if the investigator considers that there is a causal relationship with the study drug
Physical examination, vital signs and laboratory results up to day 28 (or last day in ICU if patient leaves the ICU earlier)
Exploratory
Gas exchange (partial pressure of oxygen/fraction of inspired oxygen (PaO2/FiO2) ratio) during mechanical ventilation as an indicator of improving lung function on treatment. This categorical endpoint is defined as improvement, no change, or worsening in terms of gas exchange (PaO2/FiO2 ratio) from baseline to day 28
Change in the concentration of CD73 and MxA and potential inflammatory markers, including interleukin-6 and interleukin-8, from baseline to day 14
Genetic testing to identify factors that may be involved in the response or nonresponse of diseases to FP-1201-lyo. In patients who have provided separate consent for this procedure, a 10-mL blood sample for deoxyribonucleic acid (DNA) extraction will be collected at any time during the patient’s ICU stay. All such samples will be destroyed 15 years after completion of the study. If the patient withdraws the consent, the sample will be destroyed immediately
Data collection and management
The sponsor or sponsor’s designee will conduct a site visit to each study center to verify the qualifications of each investigator, inspect the site facilities and inform the investigator of their responsibilities and the procedures for ensuring adequate and correct documentation.
The investigators will be given access to an online, web-based, electronic data-capture system that is compliant with US Food and Drug Administration Title 21 Code of Federal Regulations Part 11. Access rights to the electronic data-capture system will be carefully controlled and configured according to each individual’s role throughout the study. Computerized data-check programs and manual checks will identify any data discrepancies for resolution. All discrepancies must be resolved online directly by the investigator. Only the investigator will be able to enter and correct data in the e-CRF.
All study findings and documents will be regarded as confidential. Patients will be identified on the e-CRF by their patient number and/or birth date, not by name. Documents that identify the patient must be maintained in confidence by the investigator so that the anonymity of participating patients is ensured.
During the study, the Contract Research Organization (CRO) will make regular site visits to review protocol compliance, conduct source data verification, assess drug accountability and management, assess laboratory procedures and ensure that the study is being conducted according to pertinent regulatory and protocol requirements.
The study blind should only be broken in a medical emergency (where knowledge of the study drug received would affect the treatment of the emergency) or as a regulatory requirement (e.g., for serious adverse events or death).
Withdrawal of consent
Patients may withdraw from the study at any time and for any reason and such a decision will not affect the ongoing care given to the patient. Data recorded up to the point of withdrawal will be included in the study analyses, unless consent for use of the data has also been withdrawn. If a patient requests termination of the administration of the study drug during the treatment period, then the administration of the study drug will be stopped but the patient will continue in the study and all follow-up assessments will be performed. If a patient withdraws consent during or after the treatment period then no further active study assessments will be performed from that time point. However, permission will be sought to access the patient’s medical records to obtain data relevant to the study (e.g., outcome status).
Statistical analysis
For 90% power and a two-sided Mann-Whitney U test at the significance level of 0.05, a total of 272 patients are required based on the following assumptions:
Mortality rate of 30% in the control group and 15% in the FP-1201-lyo group at day 28
20% of patients survive but with zero ventilator-free days in the control group
A mean difference (FP-1201-lyo minus control) of 3.0 days in mean ventilator-free days where patients who die are assigned a score of 0
However, assuming that 5% of patients will drop out and a further 4% of the remaining patients will not be evaluable for the efficacy analysis, we plan to randomize 300 patients.
The full analysis set (FAS) will consist of all randomized and treated patients. The per-protocol set (PPS) will consist of patients in the FAS excluding those with major protocol violations. We will perform statistical analyses for the primary and secondary endpoints on both the FAS and PPS.
The safety set will consist of all patients who receive at least one dose of the study drug. The safety and tolerability analyses will be based on this analysis set. A patient who receives the wrong treatment according to the randomization will be analyzed for safety and tolerability in the treatment group corresponding to the treatment received.
The non-parametric analysis of the primary composite endpoint, any cause of death at day 28 and days free of mechanical ventilation within 28 days among survivors (VFDsurv), will use a scoring scheme with patients who do better getting a higher score. We will assign a VFDsurv score of − 1 to all patients who die before 28 days. For those patients who survive to day 28 the VFDsurv score will be equal to the number of VFDs calculated according to the above definition. The statistical method for group comparison of this endpoint will be based on the van Elteren test adjusting for the country, ARDS severity and key baseline characteristics. The statistical methodology for the scoring scheme is as set down in Finkelstein and Schoenfeld [22].
In general, data will not be imputed for the primary efficacy analysis or the safety analysis. For other efficacy analyses, where relevant, imputations will use the last-observation-carried-forward method. All statistical tests will be two-sided and will be performed at the significance level of 0.05.
Discussion
At present, there is no approved pharmacological treatment for ARDS so there is no possible comparator for studies of potential ARDS therapies. The only currently available treatment for ARDS patients is supportive care. Hence, the current approach to ARDS study design should be to show superiority of the investigated study drug over current standard of care. The study drug will be used in addition to supportive care and, therefore, it is most appropriate to use placebo as a comparator. The pathogenesis of ARDS can be divided into three distinct phases – acute (days 1–6), sub-acute (days 7–14) and chronic (day 15 +). Almost all patients who fail to improve or deteriorate after 1 week of ventilation have evidence of lung fibrosis, so that administering treatment beyond 6 days would add little value to patients included in this study. A 6-day dosing regimen was, therefore, selected as the optimal treatment regimen.
The composite endpoint was chosen as likely to be more sensitive than just 28-day mortality to detect an effect signal. In addition, this study has a double-blind design, so the decision to wean the patient from mechanical ventilation should not be influenced by the treatment group. Owing to the randomization procedure and the double-blind nature of this study, the potential bias of the study results is minimized.
If this study demonstrates improved outcomes in patients receiving FP-1201-lyo, this agent will represent the first specific pharmacological treatment for ARDS and a major advance in the management of these patients.
Trial status
The first patient was enrolled on 28 December 2015 and the study is ongoing.
Additional files
SPIRIT Checklist. (PDF 172 kb)
List of Ethical Committee approvals already in place. (PDF 403 kb)
Guidelines for general support. (PDF 286 kb)
Sample Consent Form. (PDF 133 kb)
Acknowledgements
The authors want to thank those who provided feedback from participating sites of the INTEREST trial. Also, the financial contribution from the European Union in the form of a research grant is much appreciated.
Funding
The study is funded by Faron Pharmaceuticals Ltd. (Joukahaisenkatu 6, 20520 Turku, Finland) and is also supported, in part, by the European Union Seventh Framework Program (FP7/2007-2013) under grant agreement number [305853].
The study was designed jointly by the sponsor and the investigators. The funder has insurance to cover any study-related injuries according to national legislation and local laws.
Availability of data and materials
Only the designated trial investigators will have access to the personal data of participants and to the final data set. The original e-CRF pages generated during the study will become the property of the sponsor.
Abbreviations
- ALI
Acute lung injury
- ARDS
Acute respiratory distress syndrome
- CD73
Cell surface ecto-5′-nucleotidase
- COPD
Chronic obstructive pulmonary disease
- CPAP
Continuous positive airway pressure
- CT
Computed tomography
- FAS
Full analysis set
- HFOV
High-frequency oscillatory ventilation
- ICU
Intensive care unit
- IDMC
Independent Data Monitoring Committee
- IFN
Interferon
- MxA
Myxovirus resistance protein A
- PAS
Per-protocol set
- PEEP
Positive end-expiratory pressure
- QoL
Quality of life
- SOFA
Sequential Organ Failure Assessment
- APACHE II
Acute Physiology and Chronic Health Evaluation
- CD
Cluster of differentiation
- CT
Computerized tomography
- D
Study day
- ECG
Electrocardiogram
- EQ-5D-3L
EuroQol
- 5
Dimensions
- 3
Levels questionnaire
- FEV1
Forced expiratory volume in 1 second
- GCS
Glasgow Coma Scale
- A
PaO2/FiO2=partial pressure of oxygen/fraction of inspired oxygen
- PIM
Potential inflammatory marker
Authors’ contributions
JLV and VMR drafted the current manuscript. GB, DB, JM, AM, NP, VP, MQ, MM, MJ and IP critically reviewed and revised the draft report. All authors have read and approved the final version which was also approved by the sponsor.
Ethics approval and consent to participate
The Local Ethics Committee at each site will approve the study protocol (approvals already in place shown in Additional file 2). Any modifications to the protocol will be immediately communicated to all responsible authorities. All patients, or their legal representative, must give written informed consent before study participation (model form, Additional file 4). The patient or their representative will also be asked to give separate consent for a genetic sample to be taken. Consent for genetic sampling is not a prerequisite for study participation.
Consent for publication
Results obtained in this trial will be published in an international journal and may be presented at international scientific meetings. This will be included in the patient consent form. There are no plans for data sharing. Results suggested for presentation or publication will be circulated to the Steering Committee members representing each participating country and the sponsor, Faron Pharmaceuticals Ltd. Authorship of any publications will be granted based on standard authorship criteria and will be decided based on the contributions to the design, conduct, interpretation, and reporting of the INTEREST trial. Disputes regarding authorship will be settled by the sponsor. Professional medical writers may be contracted to improve clarity and structure in trial-related reports.
Competing interests
Mikael Maksimow, Markku Jalkanen and Ilse Piippo are employed by Faron Pharmaceuticals and hold Faron shares and/or options for shares. The other authors are members of the INTEREST trial Steering Committee and have received expenses only for participation in required study meetings.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13063-017-2234-7) contains supplementary material, which is available to authorized users.
Contributor Information
Geoff Bellingan, Email: geoff.bellingan@uclh.nhs.uk.
David Brealey, Email: david.brealey@uclh.nhs.uk.
Jordi Mancebo, Email: JMancebo@santpau.cat.
Alain Mercat, Email: AlMercat@chu-angers.fr.
Nicolò Patroniti, Email: nicolo.patroniti@unimib.it.
Ville Pettilä, Email: ville.pettila@hus.fi.
Michael Quintel, Email: mquintel@gwdg.de.
Jean-Louis Vincent, Email: jlvincent@intensive.org.
Mikael Maksimow, Email: mikael.maksimow@faronpharmaceuticals.com.
Markku Jalkanen, Email: markku.jalkanen@faronpharmaceuticals.com.
Ilse Piippo, Email: ilse.piippo@faronpharmaceuticals.com.
V. Marco Ranieri, Email: marco.ranieri@uniroma1.it.
References
- 1.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–33. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 2.Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. 2008;133:1120–7. doi: 10.1378/chest.07-2134. [DOI] [PubMed] [Google Scholar]
- 3.Cochi SE, Kempker JA, Annangi S, Kramer MR, Martin GS. Mortality trends of acute respiratory distress syndrome in the United States from 1999–2013. Ann Am Thorac Soc. 2016;13:1742–51. doi: 10.1513/AnnalsATS.201512-841OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, et al. Has mortality from acute respiratory distress syndrome decreased over time?: A systematic review. Am J Respir Crit Care Med. 2009;179:220–7. doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- 5.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315:788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 6.Rossi C, Simini B, Brazzi L, Rossi G, Radrizzani D, Iapichino G, et al. Variable costs of ICU patients: a multicenter prospective study. Intensive Care Med. 2006;32:545–52. doi: 10.1007/s00134-006-0080-2. [DOI] [PubMed] [Google Scholar]
- 7.Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364:1293–304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 8.Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF., Jr Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2005;171:340–7. doi: 10.1164/rccm.200406-763OC. [DOI] [PubMed] [Google Scholar]
- 9.Cheung AM, Tansey CM, Tomlinson G, Diaz-Granados N, Matte A, Barr A, et al. Two-year outcomes, health care use, and costs of survivors of acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006;174:538–44. doi: 10.1164/rccm.200505-693OC. [DOI] [PubMed] [Google Scholar]
- 10.Umapathy NS, Fan Z, Zemskov EA, Alieva IB, Black SM, Verin AD. Molecular mechanisms involved in adenosine-induced endothelial cell barrier enhancement. Vascul Pharmacol. 2010;52:199–206. doi: 10.1016/j.vph.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiss J, Yegutkin GG, Koskinen K, Savunen T, Jalkanen S, Salmi M. IFN-beta protects from vascular leakage via up-regulation of CD73. Eur J Immunol. 2007;37:3334–8. doi: 10.1002/eji.200737793. [DOI] [PubMed] [Google Scholar]
- 13.Bellingan G, Maksimow M, Howell DC, Stotz M, Beale R, Beatty M, et al. The effect of intravenous interferon-beta-1a (FP-1201) on lung CD73 expression and on acute respiratory distress syndrome mortality: an open-label study. Lancet Respir Med. 2014;2:98–107. doi: 10.1016/S2213-2600(13)70259-5. [DOI] [PubMed] [Google Scholar]
- 14.Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–37. doi: 10.4049/jimmunol.178.12.8127. [DOI] [PubMed] [Google Scholar]
- 15.Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol. 2014;88:10214–27. doi: 10.1128/JVI.01068-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–24. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 17.World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–4. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 18.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–29. doi: 10.1097/00003246-198510000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Vincent JL, Moreno R, Takala J, Willatts S, De Mendonca A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22:707–10. doi: 10.1007/BF01709751. [DOI] [PubMed] [Google Scholar]
- 20.The Acute Respiratory Distress Syndrome Network Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–8. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 21.Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, de Boisblanc B, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354:2564–75. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 22.Finkelstein DM, Schoenfeld DA. Combining mortality and longitudinal measures in clinical trials. Stat Med. 1999;18:1341–54. doi: 10.1002/(SICI)1097-0258(19990615)18:11<1341::AID-SIM129>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SPIRIT Checklist. (PDF 172 kb)
List of Ethical Committee approvals already in place. (PDF 403 kb)
Guidelines for general support. (PDF 286 kb)
Sample Consent Form. (PDF 133 kb)
Data Availability Statement
Only the designated trial investigators will have access to the personal data of participants and to the final data set. The original e-CRF pages generated during the study will become the property of the sponsor.