

Abstract
Background
Ehrlichia ruminantium is the causal agent of heartwater, a fatal tropical disease affecting ruminants with important economic impacts. This bacterium is transmitted by Amblyomma ticks and is present in sub-Saharan Africa, islands in the Indian Ocean and the Caribbean, where it represents a threat to the American mainland.
Methods
An automated DNA extraction method was adapted for Amblyomma ticks and a new qPCR targeting the pCS20 region was developed to improve E. ruminantium screening capacity and diagnosis. The first step in the preparation of tick samples, before extraction, was not automated but was considerably improved by using a Tissue Lyser. The new pCS20 Sol1 qPCR and a previously published pCS20 Cow qPCR were evaluated with the OIE standard pCS20 nested PCR.
Results
pCS20 Sol1 qPCR was found to be more specific than the nested PCR, with a 5-fold increase in sensitivity (3 copies/reaction vs 15 copies/reaction), was less prone to contamination and less time-consuming. As pCS20 Sol1 qPCR did not detect Rickettsia, Anasplasma and Babesia species or closely related species such as Panola Mountain Ehrlichia, E. chaffeensis and E. canis, its specificity was also better than Cow qPCR. In parallel, a tick 16S qPCR was developed for the quality control of DNA extraction that confirmed the good reproducibility of the automated extraction. The whole method, including the automated DNA extraction and pCS20 Sol1 qPCR, was shown to be sensitive, specific and highly reproducible with the same limit of detection as the combined manual DNA extraction and nested PCR, i.e. 6 copies/reaction. Finally, 96 samples can be tested in one day compared to the four days required for manual DNA extraction and nested PCR.
Conclusions
The adaptation of an automated DNA extraction using a DNA/RNA viral extraction kit for tick samples and the development of a new qPCR increased the accuracy of E. ruminantium epidemiological studies, as well as the diagnostic capabilities and turn-over time for surveillance of heartwater. This new method paves the way for large-scale screening of other bacteria and viruses in ticks as well as genetic characterization of ticks and tick-pathogen coevolution studies.
Electronic supplementary material
The online version of this article (10.1186/s13071-017-2490-0) contains supplementary material, which is available to authorized users.
Keywords: Ehrlichia ruminantium, Ticks, pCS20, Automated DNA extraction, qPCR
Background
Ehrlichia ruminantium is an obligate intracellular bacterium that causes heartwater, an infectious, virulent, transmissible but non-contagious disease of ruminants [1]. Its main vectors are Amblyomma hebraeum ticks in southern Africa and A. variegatum ticks that transmit the disease to the rest of sub-Saharan Africa, and to islands in the Indian Ocean and the Caribbean [2, 3]. Heartwater is one of the major obstacles to the introduction of high-producing animals to upgrade and replace local stock in Africa [4]. Its economic impact is high, estimated at US $44.7 million per year for the SADC region (Southern Africa Development Community) [5]. The U.S. Homeland Security Department for the American mainland lists heartwater as one of the 12 most important animal transboundary diseases [6]. Effective vaccines to control the disease are needed [1], but to date, experimental vaccines including recombinant, attenuated and inactivated vaccines have not been particularly successful, presumably owing to the antigenic variability of the pathogen [7, 8]. In this context, characterization of field strains is indispensable to design appropriate vaccines including regional strains.
Ideally, a large number of ticks need to be collected and tested to accurately evaluate the prevalence of E. ruminantium in ticks from endemic areas, and further characterize the genetic diversity and population structure of E. ruminantium from several geographical areas [9, 10]. In addition, given that certain areas (e.g. the American mainland) are at high risk of the introduction of heartwater, it is advantageous to have rapid high-throughput molecular tools in preparation for the possible introduction of the pathogen in a previously pathogen-free area. Various methods for manual extraction of tick DNA are currently available and result in high DNA yields [11, 12]. However, all these methods have a low sample processing capacity and are time consuming. A few automated DNA extraction methods have been tested for arthropods including spiders and flies [13–15]. Moriarity et al. [16] developed a high-throughput DNA extraction method specifically for ticks, including Ixodes scapularis and optimized a qPCR for the detection of Rickettsia rickettsii, R. sibirica, R. africae and R. prowazekii using the Promega Wizard SV96 genomic DNA purification system [16]. Crowder et al. [17] automated a Qiagen (Courtaboeuf, France) MiniElute Virus extraction kit and detected the presence of Borrelia burgdorferi and Powassan virus in I. scapularis ticks [17].
Several molecular methods targeting different specific E. ruminantium genes or regions have been developed to diagnose heartwater in ruminants and to screen E. ruminantium in ticks. Some of these methods target the pCS20 region, a highly conserved and specific gene region of E. ruminantium. The pCS20 nested PCR, recommended by OIE, the World Organization for Animal Health, has been tested on a wide range of E. ruminantium strains from cattle and tick samples [18–20]. However, it has two main disadvantages: it is time consuming and the risk of contamination is high. To solve these problems and to address the need for quantitative results, qPCRs have been developed for the detection and quantification of E. ruminantium. A SYBR Green and TaqMan qPCRs targeting two different regions of the map-1 gene were used to quantify E. ruminantium during vaccine production and growth in endothelial cell culture [21, 22]. Likewise, E. ruminantium map1–1 transcripts were quantified in tick midguts and salivary glands as well as in E. ruminantium infected endothelial cell cultures by SYBR Green RT-qPCR targeting the map-1-1 gene [23]. However, due to the polymorphism of the map1 multigenic family, assays targeting these genes are not suitable for diagnostic tests. A pCS20 quantitative real-time PCR based on a TaqMan probe, CowTqM, was developed in 2008 by Steyn et al. [24] to detect E. ruminantium in livestock blood and ticks from the fin l [24]. However, CowTqM qPCR cross-reacts with E. chaffeensis and E. canis. Since E. chaffeensis is widespread in the USA and E. canis or related species, have recently been observed in the Caribbean [25], CowTqM qPCR cannot be used as a diagnostic tool in mainland USA and in the Caribbean. Nakao et al. [26] developed another method for rapid low cost detection of E. ruminantium using loop-mediated isothermal amplification (LAMP) targeting the pCS20 and sodB gene regions, and tested it on blood samples and ticks [26]. However, this technique, which was tested on 16 E. ruminantium strains, was less sensitive than CowTqM qPCR due to the inhibitory effects of A. variegatum ticks. These results prevented the use of this LAMP method for the detection of E. ruminantium in ticks and no additional data have become available since its publication in 2010.
In addition, multi-pathogen qPCRs including E. ruminantium detection have been developed. Sayler et al. [27] developed and validated a dual-plex TaqMan qPCR assay targeting the groEL gene of Panola Mountain Ehrlichia (PME) and E. ruminantium in field samples from ruminants or from ticks, which enabled the differentiation of the two species in the USA [27]. A generic Ehrlichia FRET-qPCR targeting the 16S rRNA gene has also been developed [25]. Based on melting point analysis, this method made it possible to distinguish eight Ehrlichia species from four groups: E. ruminantium (Group 1); E. chaffeensis and E. ewingii (Group 2); E. canis, E. muris, E. ovina and Ehrlichia sp. BOV2010 (Group 3) and PME (Group 4).
Since all these techniques failed to address the question of the scalability of tick sample processing and screening, we decided to adapt a commercial automated DNA extraction kit to ticks and to develop a new qPCR assay with improved specificity and sensitivity compared to the OIE standard nested PCR and the published CowTqM qPCR. To this end, a high-throughput DNA extraction method and a new pCS20 Sol1 qPCR assay were optimized.
The new pCS20 Sol1 qPCR (both, SYBR Green, Sol1SG qPCR and TaqMan, Sol1TqM qPCR), the previously published CowTqM qPCR and the OIE standard pCS20 nested PCR were evaluated. In parallel, a tick 16S rDNA qPCR was developed to check the quality of tick DNA extraction and the absence of PCR inhibitors. The whole method, including automated DNA extraction and Sol1TqM qPCR, was then compared to manual DNA extraction and nested PCR reference methods. The advantages of this new qPCR over those already published are discussed.
Methods
Development of pCS20 Sol1TqM and Sol1SG qPCRs
Design of pCS20 Sol1 primers and probes
For the design of Sol1 primers and probes, we identified the most conserved gene region of E. ruminantium, pCS20, through multiple alignments of nucleotide sequences from 13 strains available in GenBank (detailed in Additional file 1).
We developed the new pCS20 Sol1 qPCR using two types of chemistry, one based on SYBR Green (SG) and the second using the TaqMan (TqM) technology. The cycling conditions are described in detail in Additional file 1: Table S1.
Efficiency, limit of detection and reproducibility
Ehrlichia ruminantium from the strain Gardel passage 48, was grown in bovine aorta endothelial cells as previously described [28]. DNA was then extracted using the QiaAmp DNA minikit (Qiagen, Courtaboeuf, France) according to the manufacturer’s instructions and following the protocol of Frutos et al. [29]. The extracted DNA was quantified by a map-1 TqM qPCR [22]. We tested 10-fold serial dilutions of E. ruminantium ranging from 3 × 106 to 30 copies/reaction in triplicate with Sol1TqM and Sol1SG qPCRs at annealing temperatures ranging from 48 °C to 56 °C, in order to optimize the qPCR efficiency as previously described [30].
At optimal temperatures, we determined the limit of detection of both Sol1 qPCRs in testing the same serial dilutions of E. ruminantium Gardel DNA passage 48 and an additional sample with 3 copies/reaction. To comparatively assess the performance of the new pCS20 Sol1 qPCRs, the limit of detection was also determined for the conventional pCS20 nested PCR and pCS20 CowTqM qPCR as described by Steyn et al. [24]. For the nested PCR, only 1 μl of DNA was run instead of 2 μl for the two qPCRs, with final concentrations ranging from 1.5 × 106 to 1.5 copies/reaction. For pCS20 CowTqM, the running temperature of 48 °C recommended by the authors was used and 56 °C, which is close to the theoretical annealing temperature of its probe, was also tested [24]. The detailed cycling conditions are described in Additional file 1: Table S1.
Analytical sensitivity and specificity
We evaluated the analytical sensitivity of the pCS20 Sol1TqM qPCR with DNA extracted from 16 E. ruminantium strains isolated in different geographical areas (Sudan, Burkina Faso, Senegal, South Africa, Zambia, Ghana, Cameroon, Mozambique and Guadeloupe) (Table 1) [31, 32]. In addition, we tested 10 isolates from South Africa (Kruger National Park) and Mozambique [three sites from Maputo Province: Matutuine (MAT), Chobela (CHOB) and Changalane (CHA)], collected in a previous study [33].
Table 1.
pCS20 Sol1TqM qPCR sensitivity and specificity test on Ehrlichia ruminantium strains and other tick-borne pathogens
| Name | Origin | Geographical origin | Nested pCS20 PCR | Sol1TqM QPCR | Reference |
|---|---|---|---|---|---|
| E. ruminantium strains | |||||
| Blonde | CC p8 | Guadeloupe | + | + | [31] |
| Gardel | CC p48 | + | + | [29] | |
| Sara 455 | CC p10 | Burkina Faso | + | + | [8] |
| Bekuy 255 | CC p9 | + | + | [31] | |
| Bankouma 421 | CC p15 | + | + | [8] | |
| Banankeledaga | CC p1 | + | w+ | [8] | |
| Lamba 479 | CC p16 | + | + | [31] | |
| Cameroun | CC p9 | Cameroon | + | + | [31] |
| Pokoase 412 | CC p10 | Ghana | + | + | [31] |
| Senegal | CC p60 | Senegal | + | + | [31] |
| Umbaneim | B | Sudan | + | + | [31] |
| Mara | CC p1 | South Africa | + | + | [31] |
| Welgevonden | CC p12 | + | + | [29] | |
| Lutale | CC p6 | Zambia | + | + | [31] |
| Sankat 430 | CC p16 | Ghana | + | + | [32] |
| Umpala | CC p6 | Mozambique | + | + | [31] |
| MAT2-17MH1 | T | + | w+ | [33] | |
| MAT2-26MH1 | + | + | [33] | ||
| MAT2-32MH2 | + | w+ | [33] | ||
| CHOB4MH2 | + | w + | [33] | ||
| CHOB25MH1 | + | w+ | [33] | ||
| CHA2-32MH1 | + | w+ | [33] | ||
| KNP51MH1 | T | South Africa | + | w+ | [33] |
| KNPC15MH1 | w+ | w+ | [33] | ||
| KNPC2MH1 | + | + | [33] | ||
| KNPC2MH2 | + | + | [33] | ||
| Other tick-borne pathogens | |||||
| A. marginale | Argentina | – | – | ||
| A. phagocytophilum | CC | USA | – | – | |
| A. platys (E. platys) | CC | – | – | ||
| B. bovis | Argentina | – | – | ||
| B. bigemina | – | – | |||
| E. canis | CC | USA | – | – | [34] |
| E. muris | CC | – | – | [35] | |
| PME 160055 | T | + | – | ||
| PME 160178 | – | – | |||
| PME 160277 | + | – | |||
| PME 160359 | – | – | |||
| PME 160366 | + | – | |||
| PME 160491 | + | – | |||
| R. felis | CC | – | – | [36] | |
| R. parkeri | CC | – | – | [37] | |
| 9 uninfected A. variegatum | T | Guadeloupe | – | – | |
E. ruminantium was detected from A. variegatum and A. hebraeum ticks while PME was detected from A. americanum ticks
Abbreviations: T ticks, B blood, CC p cell culture passage, + positive, − negative, w + weak positive
The analytical specificity of Sol1TqM qPCR was evaluated using 10 tick-borne pathogens, species of Anaplasma, Babesia, Ehrlichia and Rickettsia (Table 1) [34–37]. In addition, nine DNA samples extracted from non-infected A. variegatum adult ticks were obtained from the tick rearing stock in the CIRAD laboratory and used as negative controls. Using BLAST, sequences of Sol1 primers and probe were checked against available E. chaffeensis sequences to evaluate the amplification capacity of Sol1TqM qPCR in silico.
Development of tick 16S rDNA qPCR for DNA extraction and PCR control
Design of 16S rDNA primers
To develop a SYBR Green qPCR, targeting the mitochondrial 16S ribosomal DNA (rDNA) gene, hereafter referred to as 16SSG rDNA qPCR, the forward primer 16SF was selected from a previous paper [38] and a new reverse primer 16SR2 was designed based on eight available sequences using Primer3 [39] to obtain an optimal product size of 181 bp for the qPCR (Additional file 1: Table S1). The Rhipicephalus, Amblyomma and Ixodes nucleic sequences used to design the primers are described in Additional file 1.
Efficiency, limit of detection and reproducibility
To evaluate the analytical performance of the new 16SSG rDNA qPCR, we tested 10-fold serial dilutions of DNA extracted from a single field tick (A. variegatum), with three replicates to define the optimal temperature and then with five replicates to obtain accurate measurements of efficiency at this temperature.
We set up a new protocol for sample preparation before DNA extraction, based on tick grinding in the TissueLyser II (Qiagen, Courtaboeuf, France). The detailed protocol for grinding and manual DNA extraction is given in Additional file 2. The amplification efficiency (E) and percentage of efficiency were calculated as described in Bustin et al. [30]. Average Ct and standard deviation (± SD) were calculated for the replicates to assess the reproducibility of the 16SSG rDNA qPCR.
Quality control criteria
In order to set the threshold of the new 16SSG rDNA qPCR, a panel of 37 field ticks (A. hebraeum and A. variegatum), collected in Mozambique and South Africa, were individually extracted on the automated platform as described below and tested. We calculated the mean Ct value for these 37 tests and set the upper limit to validate both the automated extraction of nucleic acids and the absence of inhibitors in the real-time qPCR, using the following formula:
Ctsample < mean Ct value 37 ticks + 2 SD.
Validation of the whole method: Performance of the automated tick DNA extraction and pCS20 Sol1TqM qPCR
Limit of detection
To assess the limit of detection, pools of unfed naïve tick lysates were spiked with serial dilutions of E. ruminantium passage 43 from infected cell cultures [28] at a concentration ranging from 6 × 103 to 6 copies/reaction (details on the preparation of the samples are given in Additional file 2). The limit of detection of the new method [automated extraction on Biomek 4000 (Beckman Coulter, Villepinte, France) with the kit viral RNA and DNA (Macherey-Nagel, Hoerdt, France) in a 96-well plate format and Sol1TqM qPCR) was compared in two independent assays on the same samples with the standard method based on manual DNA extraction (QiaAmp DNA minikit, Qiagen, Courtaboeuf, France) and nested PCR.
Performance of the manual and automated DNA extraction
The performance of the automated and manual extractions was compared using pCS20 nested PCR and Sol1TqM qPCR on 17 tick lysates spiked with E. ruminantium cell cultures (as described above) and lysates of 30 adult ticks moulted from nymphs experimentally engorged on infected goats. The production of these adult ticks hereafter named “experimentally infected ticks” is detailed in Additional file 3. The 47 samples were subjected to automated and manual DNA extractions in parallel and then to either Sol1TqM qPCR or nested PCR (94 final results).
The quantitative and qualitative results of the nested PCR and of the Sol1TqM qPCR after manual or automated extraction were converted into positive or negative, excluding any doubtful results by either nested PCR (multiple band PCR product) or by qPCR (Ct > limit of positivity). The relative specificity sensitivity and accuracy of automated extraction as compared to manual extraction were calculated as described in Additional file 4. The degree of agreement between the two extraction methods was calculated using kappa statistics [40]. Kappa values were interpreted as follows: very good agreement: ≥ 0.81; good agreement: 0.61–0.80; moderate agreement: 0.41–0.6; fair agreement: 0.21–0.4; and poor agreement: ≤ 0.20 [40].
The distributions of the Ct values generated by pCS20 Sol1TqM qPCR (n = 17) on experimentally infected ticks or serial dilutions of E. ruminantium in tick lysates both extracted automatically and manually, were represented onto a 2-D dot plot. One dot corresponds to the Cts obtained for one sample with the pCS20 Sol1 qPCRTqM on nucleic acids extracted manually (y-axis) and automatically (x-axis) [41].
Relative sensitivity and specificity of pCS20 Sol1TqM qPCR
We determined the relative sensitivity, specificity and accuracy of the pCS20 Sol1TqM qPCR on 60 field A. hebraeum and A. variegatum adult ticks from Mozambique and South Africa [42], extracted either manually or automatically. The true status (positive or negative) of these ticks was established by the combined results of two tests, hereafter referred to as the reference method. The first test included in the reference method was the OIE gold standard pCS20 nested PCR [11]. The second test was based on multilocus sequence typing (MLST), performed according to Cangi et al. [33].
A sample was considered as negative when both tests were scored as negative. When multiple bands were detected with the nested pCS20 PCR but MLST was negative, the sample was also considered negative. In all other cases, the sample was scored as positive.
Relative sensitivity (Se) and specificity (Sp) were defined as the percentages of samples scored by the pCS20 Sol1TqM qPCR as positive and negative, respectively, out of all the samples scored by the reference method (pCS20 nested PCR and MLST) as positive and negative (Additional file 4) [43]. Relative accuracy (Ac) was defined as the degree of agreement between the results obtained by the pCS20 Sol1TqM qPCR and by the reference method. The formulas used to calculate Se, Sp and Ac are detailed in Additional file 4.
Results of the Sol1TqM qPCR and the pCS20 nested PCR in combination with MLST were cross-tabulated (2 × 2 table). In addition, the kappa agreement between the two methods was determined as described above.
Relative sensitivity and specificity of the whole method
Finally, we compared the whole method (automated extraction + pCS20 Sol1TqM qPCR) and the standard method (manual extraction + nested pCS20 PCR) using 47 samples: 17 samples spiked with serial 10-fold E. ruminantium dilutions from infected cell cultures and 30 experimentally infected ticks as described above. No doubtful samples with multiple band PCR products or Ct greater than the limit of positivity were included in the panel. Sensitivity, specificity, accuracy and kappa agreement between the two methods were determined based on positive and negative results obtained by both methods (Additional file 4).
Reproducibility and processing time
To estimate the reproducibility of automated DNA extraction coupled with the pCS20 Sol1TqM qPCR, E. ruminantium strain Gardel passage 43 was appropriately diluted and added to a supernatant of tick lysates to reach final concentrations ranging between 6 and 60 copies/reaction. Spiked tick supernatants were extracted in triplicate in separate procedures and further tested by pCS20 Sol1TqM qPCR. Last, sample processing time was estimated for automated and manual DNA extraction as well as for nested PCR and Sol1TqM qPCR.
Results
Development of pCS20 Sol1TqM and Sol1SG qPCRs
Optimization and efficiency of pCS20 Sol1 TqM and Sol1SG qPCR
The pCS20 Sol1TqM and Sol1SG qPCR efficiencies (%) were measured at different temperatures (from 50 °C to 56 °C) using 10-fold serial dilutions of E. ruminantium Gardel DNA in three separate experiments. With pCS20 Sol1SG qPCR, the optimal annealing temperature was 51 °C with 98.1 ± 1.9% efficiency and the mean expected temperature of dissociation was 74.2 ± 0.5 °C. The maximum PCR efficiency of Sol1TqM was obtained at 55 °C with 94.4 ± 3.6%. At 54 °C and 56 °C, efficiencies were 89.1 ± 6.1% and 93.8 ± 5.8% with more variation between the tests. Consequently, the optimal PCR conditions were defined for the new pCS20 Sol1TqM and Sol1SG qPCR as annealing temperatures of 55 °C and 51 °C, respectively.
With CowTqM qPCR, the use of the optimal temperature of 48 °C recommended by Steyn et al. [24], only allowed the detection of 3 × 104 copies/reaction in one out of three independent assays. We then tested CowTqM qPCR at 56 °C, a temperature closer to the melting temperature of the probe, which resulted in a low PCR efficiency of 69.2 ± 3.1%.
Limit of detection and reproducibility of pCS20 Sol1 TqM and Sol1SG qPCR
At the optimal annealing temperatures, pCS20 Sol1TqM and Sol1SG qPCRs were performed on E. ruminantium Gardel DNA at 3 × 106 to 3 copies/reaction in parallel with pCS20 nested PCR and CowTqM qPCR. The mean Ct values of three independent runs and standard deviations are listed in Table 2. The results of the CowTqM qPCR are not shown because the detection limit achieved with this test in optimal conditions was very poor (3 × 103 copies of bacteria per reaction). The limit of detection of the pCS20 Sol1 qPCRs was better than that of pCS20 nested PCR, with detection limits of 3 and 15 copies/reaction, respectively (Table 2). The detection threshold for pCS20 Sol1TqM was then set at a Ct of 37. With Sol1SG qPCR, the Ct was 30.5 ± 1.3 and 34 ± 0.7 for 30 and 3 copies, respectively. However, a biphasic dissociation curve was found for 3 copies, highlighting the presence of both primer dimers and pCS20 amplicons. Sol1SG qPCR detected a signal for non-template control (NTC) with a Ct of 35 ± 1.1 due to primer dimers, as evidenced by a lower dissociation temperature than that of the target (data not shown). The positive threshold for Sol1SG qPCR was established at 35 Ct.
Table 2.
Limit of detection of pCS20 Sol1TqM qPCR, Sol1SG qPCR and pCS20 nested PCR
| DNA E. ruminantium strain Gardel (copies/reaction)a |
Sol1TqM qPCR (Th = 55 °C) Ct ± SDb |
Sol1SG qPCR (Th = 51 °C) Ct ± SDb |
pCS20 nested PCR signalc |
|---|---|---|---|
| 3.106 | 17.0 ± 0.3 | 13.6 ± 0.6 | + |
| 3.105 | 20.3 ± 0.1 | 16.8 ± 0.4 | + |
| 3.104 | 23.6 ± 0.0 | 20.0 ± 0.5 | + |
| 3.103 | 27.1 ± 0.6 | 23.5 ± 0.4 | + |
| 3.102 | 31.0 ± 0.5 | 26.9 ± 1.0 | + |
| 30 | 34.2 ± 0.3 | 30.5 ± 1.3 | w+ |
| 3 | 36.7d | 34.0 ± 0.7 | – |
| NTC | Undet | 35.0 ± 1.1 | – |
aBacterial load used for qPCR: the nested PCR samples were amplified from 1 μl of DNA, containing half the quantity from 1.5 ×106 to 1.5 copies/reaction
bThe average Ct value is indicated for each dilution (bacteria copy number) and standard deviation was derived from 3 replicates
cConventional PCR done in duplicate
dTested once
Abbreviations: Th temperature of hybridization, w + weak positive, NTC non-template control, Undet undetermined, SD standard deviation
With both pCS20 qPCRs, the Ct standard deviations were extremely low, ranging from 0 to 1.3, demonstrating the good reproducibility of both assays (Table 2).
Sensitivity and specificity of pCS20 Sol1TqM
Twenty-six E. ruminantium strains from different geographic origins (Table 1) were successfully amplified by both pCS20 Sol1TqM qPCR and the gold standard test pCS20 nested PCR. Concerning the specificity of the assay, A. marginale, A. phagocytophilum, A. platys, B. bovis, B. bigemina, E. canis and E. muris, R. felis or R. parkeri were not detected by either pCS20 Sol1TqM qPCR or pCS20 nested PCR. Moreover, PME was not detected by pCS20 Sol1TqM qPCR whereas 4 out of 6 samples were scored positive by pCS20 nested PCR (Table 1). Nine uninfected A. variegatum DNA samples from the CIRAD rearing facilities were scored negative by pCS20 Sol1TqM qPCR and pCS20 nested PCR. Concerning E. chaffeensis, using BLAST sequence analysis, the Sol1 TaqMan probe did not align with the E. chaffeensis pCS20 gene region and only 13 out of 20 nucleotides of the Sol1R primer, in the middle of the primer, matched this gene. Based on these in silico analyses, no positive results can be expected using Sol1 qPCRTqM on E. chaffeensis.
Development of tick 16SSG rDNA qPCR as internal control for DNA extraction and PCR
16SSG rDNA qPCR efficiency and limit of detection
Serial dilutions (from 10−1 to 10−5) of A. variegatum DNA were amplified in triplicate with 16SSG rDNA qPCR at different temperatures ranging from 58 °C to 61 °C. Efficiency levels ranged from 80 to 84% and did not differ significantly with the hybridization temperature. However, Ct values were higher at 60 °C and 61 °C than at 58 °C and 59 °C, with an increment of two to seven Cts depending on dilution. Furthermore, the dilution 10−5 of the 16S rDNA was not detected at 61 °C. The optimal hybridization temperature for the 16SSG rDNA qPCR was defined as 59 °C with 84 ± 5.1% efficiency, based on five replicates. Using the serial dilutions of the positive control, the mean dissociation temperature for 16SSG rDNA qPCR was 72.1 ± 0.2 °C (n = 5).
Quality control of automated DNA extraction and reproducibility
A panel of 37 field samples were subjected to automated DNA extraction, and all the samples were successfully amplified by 16SSG rDNA qPCR, with a mean Ct of 23.3 ± 2.8, suggesting that the DNA extracted by the robot was of good quality and no PCR inhibitors were present (data not shown). The acceptable limit of Ct attesting to the good DNA quality was set at 29, corresponding to mean Ct value of the 37 field samples +2SD. The reproducibility of the whole method (DNA extraction and 16SSG qPCR) was evaluated, and less than 4 Ct variation was found in the same tick lysate extracted in four different repetitions.
Validation of the whole method (automated tick DNA extraction and pCS20 Sol1TqM qPCR)
Limit of detection
The automated DNA extraction followed by pCS20 Sol1TqM qPCR enabled the detection of E. ruminantium from infected cell cultures down to six copies/reaction with Ct = 37.6 ± 1. These results were obtained in two independent assays (data not shown). When the same samples underwent manual extraction and pCS20 nested PCR, the detection limit of E. ruminantium was the same, with six copies/reaction.
Comparison of the performances of automated and manual DNA extraction
Automated and manual DNA extractions were compared in a total of 47 samples screened either by pCS20 Sol1TqM qPCR or nested pCS20 as described in Methods. It is worth noting that only 30% (9/30) of experimentally infected ticks were found to be positive by nested PCR. The relative sensitivity, specificity and accuracy of automated extraction compared to manual extraction was 84.1%, 88% and 86.2%, respectively (Table 3). Kappa agreement between both extraction methods was 72%.
Table 3.
Relative sensitivity and specificity of automated DNA extraction compared with manual DNA extraction based on screening of samples tested either by nested pCS20 PCR or Sol1TqM qPCR. Results (positive and negative E. ruminantium samples, Se, Sp and Acc of the automated extraction method) obtained on tick lysates spiked with E. ruminantium serial dilutions (n = 17) and experimentally infected ticks (n = 30) extracted in parallel by manual and automatic extraction and tested either by nested PCR or pCS20 Sol1TqM qPCR are shown. The kappa test was 72%
| Manual extraction | Total | Se (%) | Sp (%) | Ac (%) | |||
|---|---|---|---|---|---|---|---|
| + | – | ||||||
| Automated extraction | + | 37 | 6 | 43 | 84.1 | 88.0 | 86.2 |
| – | 7 | 44 | 51 | ||||
| Total | 44 | 50 | 94 | ||||
Abbreviations: Se relative sensitivity, Sp relative specificity, Ac accuracy
Six and seven samples were not detected as positive for E. ruminantium with manual and automated extractions, respectively (Table 3). Since the possibility of contamination between samples was excluded by repeated tests, these 13 samples were “true positives”, showing that the two methods of extraction have similar sensitivity. A comparison of Ct values obtained with tick lysates spiked with E. ruminantium serial dilutions and experimentally infected ticks extracted automatically and manually in parallel is shown in Fig. 1. A good correlation was observed (R 2 = 85%) and the Ct values were slightly better under automated DNA extraction (-1.96 Ct, P < 0.001). In conclusion, the performance of the automated DNA extraction method is the same as that of manual extraction for the detection of E. ruminantium by pCS20 Sol1TqM qPCR and nested qPCR.
Fig. 1.
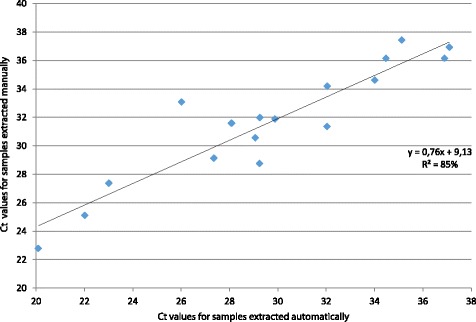
Comparison of Ct values obtained by Sol1TqM qPCR for tick lysates spiked with E. ruminantium serial dilutions and experimentally infected ticks extracted through automated and manual techniques (n = 17)
Relative sensitivity and specificity of the pCS20 Sol1TqM qPCR
Compared to the reference method (pCS20 nested PCR and combined with MLST), the relative sensitivity and specificity of the pCS20 Sol1TqM qPCR was 75.8 and 85.2%, respectively with 80% accuracy (Tables 4, 60 field ticks extracted using manual and automated methods). Out of the 8 false negative and 4 false positive samples, 4 were scored positive and 2 negative, respectively, by the two tests comprising the reference method. Of the 4 false positive samples, the other 2 samples displayed multiple bands by nested PCR and were scored negative by MLST. The Cts obtained for these 4 false positives ranged between 34.2 and 36.7. Out of the 25 true positive and 23 true negative samples, only 19 (76%) were scored positive and 5 (22%) were negative, respectively, by the two tests comprising the reference method. Concerning the true positive samples, out of the 6 remaining samples, 3 were scored positive by nested PCR and negative by MSLT and 3 were scored positive by MLST (corresponding to samples with partial amplification of at least one MLST gene) and negative by nested PCR. The mean Cts for these 6 samples was 37.1 ± 0.6, at the limit of detection. Out of the 23 true negative samples, 18 (78%) displayed multiple bands by nested PCR and were scored negative by MLST.
Table 4.
Relative sensitivity and specificity of pCS20 Sol1TqM qPCR compared with the pCS20 nested PCR and MLST, which were combined as the reference method. Results obtained from 60 field-collected ticks are shown. The kappa test was 60%
| Nested PCR + MLST | Total | Se (%) | Sp (%) | Ac (%) | |||
|---|---|---|---|---|---|---|---|
| + | – | ||||||
| Sol1TqM qPCR | + | 25 | 4 | 29 | 75.8 | 85.2 | 80.0 |
| – | 8 | 23 | 31 | ||||
| Total | 33 | 27 | 60 | ||||
Se relative sensitivity, Sp relative specificity, Ac accuracy
The kappa statistics for the pCS20 Sol1™ qPCR/ pCS20 nested PCR + MLST comparisons were 60%, demonstrating fair to good agreement between the tests.
Relative sensitivity and specificity of the whole method
The relative sensitivity and specificity of the whole method (automated DNA extraction + pCS20 Sol1TqM qPCR) compared with manual extraction + pCS20 nested, were 76.2% and 73.1%, respectively (Table 5), with 7 and 5 additional positive samples detected by the new method and the standard method, respectively. Four out of the 7 false positive samples had a Ct of 37, at the threshold. The 5 false negative samples were clearly positive with the nested pCS20 PCR. The kappa value for this analysis was 49%, demonstrating moderate agreement between the tests.
Table 5.
Relative sensitivity and specificity of automated extraction + pCS20 Sol1TqM qPCR compared with manual extraction + pCS20 nested PCR. Results obtained on 17 E. ruminantium serial dilutions and 30 experimentally infected ticks. The kappa test for this analysis was 49%
| Manual extraction + pCS20 nested PCR | Total | Se (%) | Sp (%) | Ac (%) | |||
|---|---|---|---|---|---|---|---|
| + | – | ||||||
| Automated extraction +Sol1TqM qPCR | + | 16 | 7 | 23 | 76.2 | 73.1 | 74.5 |
| – | 5 | 19 | 24 | ||||
| Total | 21 | 26 | 47 | ||||
Se relative sensitivity, Sp relative specificity, Ac accuracy
Reproducibility of the automated DNA extraction and pCS20 Sol1 ™ qPCR
The reproducibility of the whole method (automated extraction + pCS20 Sol1TqM qPCR) on independent triplicates was high, as demonstrated by the low standard deviation of Ct values: Ct = 33 ± 0.7 (CV = 2.1%) and Ct = 36.8 ± 1.3 (CV = 3.5%) for samples with 60 and 6 E. ruminantium copies/reaction, respectively.
Sample processing time
The total time required to extract DNA/RNA from 96 tick samples automatically was four hours, compared to 2.5 days for manual DNA extraction in our facilities. The first step in the preparation of tick lysates requires manual pipetting of reagents and crushed tick lysates. However, the use of a Tissue Lyser allowed simultaneous processing of 48 samples to obtain tick lysates. In addition, the nested PCR alone takes 1.5 days to perform, whereas the Sol1TqM qPCR can be completed in four hours. Furthermore, the risk of contamination is substantially reduced with the Sol1TqM qPCR.
Discussion
In our conditions, the use of pCS20 CowTqM, described by Steyn et al. [24], was inefficient throughout the present study (PCR efficiency of 69.2%). As we successfully optimized Sol1TqM qPCR in parallel, using the same DNA and reagents, the lack of efficiency cannot be explained by the presence of inhibitors or deficient reagents. Moreover, CowTqM primers and probes hybridized entirely on the Gardel pCS20 gene region, thereby limiting the impact of genetic variability on the result. The reason for unsuccessful implementation of pCS20 CowTqM in our facilities remains unclear.
Both qPCR pCS20 Sol1 using SYBR Green and TaqMan probe chemistries for detection of E. ruminantium were more than 94% efficient. Even though the specificity and sensitivity of Sol1SG qPCR were not evaluated as thoroughly as those of Sol1TqM qPCR, this new qPCR could be a cheaper alternative for the detection of E. ruminantium in laboratories in low income countries. Similar PCR efficiencies obtained for Sol1TqM at 55 °C and 56 °C also confirmed the robustness of the assay.
The limit of detection of the pCS20 Sol1TqM and Sol1SG is three E. ruminantium copies per reaction, which is better than the pCS20 nested PCR (15 copies per reaction in this study, 6 copies in another study [18]), the CowTqM qPCR (14 copies per reaction in the work of Steyn et al. [24], 3000 copies per reaction in our hands). It is also better than the new dual-plex qPCR targeting PME/E. ruminantium (10 copies per reaction) [27] and the new multiple pathogen detection tool enabling the detection of eight Ehrlichia species, including E. ruminantium (5 copies per reaction) [25]. In the present study, the positivity thresholds were set at 37 and 35 cycles for Sol1TqM and Sol1SG qPCRs, respectively. The positivity threshold for Sol1TqM qPCR was confirmed by the results obtained with the whole method (automated extraction and Sol1TqM qPCR) in which three samples with six copies had a mean Ct value(s) of 36.8 ± 1.3. Detection of such a low number of copies is important as it enables the detection of the low bacterial loads as frequently observed in infected ticks. Moreover, pCS20 Sol1TqM qPCR was able to detect E. ruminantium in blood samples of three experimentally infected goats during hyperthermia (data not shown). Thus, pCS20 Sol1TqM can also be recommended for the diagnosis of heartwater from blood samples.
In contrast to CowTqM qPCR, which cross-reacted with E. chaffeensis and E. canis [23], the new pCS20 Sol1TqM qPCR did not cross-react with other tick-borne pathogens including PME. It was also shown to detect 26 different E. ruminantium strains from a wide range of geographic origins including the Caribbean, West, East, and southern Africa. In a previous study, 797 Amblyomma ticks collected in Mozambique and southern Africa were extracted using the whole method (automated DNA extraction and Sol1TqM qPCR), described in this paper [33]. Positive samples obtained with Sol1TqM qPCR were typed by MLST, enabling the identification of genetic groups G1 and G2 including G2A, G2B, G2C, G2D subgroups that covered the wide genetic diversity of E. ruminantium.
The performance of pCS20 Sol1TqM qPCR was tested by comparing the results of detection using African ticks collected in the field and infected tick lysates with those obtained with pCS20 nested PCR and MLST. Among the 25 true positive samples, six samples were scored positive only by nested PCR or by MLST and had high Cts by pCS20 Sol1TqM qPCR. These results confirmed the ability of pCS20 Sol1TqM qPCR to detect low bacterial loads and some strains that could not be amplified by nested PCR or MLST. Moreover, among the true negative samples, 78% of doubtful status (PCR products with multiple bands) identified by nested pCS20 PCR and scored negative by MLST, were also scored negative by Sol1TqM qPCR, showing that Sol1TqM qPCR gives reliable negative results whereas the nested pCS20 gives inconclusive results.
As we observed no cross-reactions with other tick-borne pathogens, including closely related species, and a higher limit of detection, it is possible that the positive results obtained with pCS20 Sol1TqM, which were negative with pCS20 nested PCR and MLST, were truly infected samples.
The specificity and sensitivity for E. ruminantium detection of new PCR method therefore appears to be better than that of nested pCS20 PCR and CowTqM qPCR. Moreover, we demonstrate that this PCR method enabled the detection of a large number of E. ruminantium strains in contrast to CowTqM qPCR which was only able to successfully detect 15 E. ruminantium strains.
Another advantage of our assay is it does not detect PME allowing its use in the USA with a low risk of false positives, avoiding the use of a dual-plex TaqMan qPCR to differentiate PME from E. ruminantium [27]. The recently published new multiple Ehrlichia detection tool [25] is promising since it detects up to eight Ehrlichia species including E. ruminantium. However, it was only tested on five E. ruminantium strains, and not on E. ruminantium positive field samples, so further studies are needed to confirm its possible extensive use on tick samples in the field.
We successfully optimized another qPCR targeting the 16S rDNA tick gene. It can be used for several tick genera or species including Amblyomma, Rhipicephalus and Ixodes [44, 45]. The 16SSG rDNA qPCR is a powerful method for DNA extraction and quality control, DNA quantification and assessment of the presence of PCR inhibitors that complements standard methods using gel migration and nanodrop.
Furthermore, using the 16S qPCR for DNA quality evaluation circumvents the limitations of photometric and fluorometric methods for DNA quality assessment when the viral RNA and DNA Macherey-Nagel kit is used. These limitations are due to the presence of an RNA carrier, which leads to overestimation of the amount of nucleic acids.
We observed a similar performance between the automated and manual extractions of DNA from ticks whatever the PCR used downstream, excepting for samples with low loads of E. ruminantium. For these latter samples, the automated DNA extraction and Sol1TqM qPCR appeared to perform better than conventional methods. We also showed that the whole method (automated DNA extraction and Sol1 qPCRTqM) was highly reproducible. In our laboratory, the processing and testing of 96 samples with the automated method requires only one day of work by one technician whereas with the OIE standard method, it would take three and a half days. Although the first step in the preparation of the tick samples (washing and grinding) before extraction was not automated, it was significantly shortened by the use of a TissueLyser II. We thus conclude that, depending on the number of samples and their bacterial load, DNA extraction methods may be interchangeable.
The commercial kit produced by Macherey-Nagel, which was adapted for automated extraction of tick samples in our experiments, has the advantage of extracting both DNA and RNA from viruses as well as bacteria. This kit was also tested on the avian influenza virus in our laboratory and its virus detection performance was similar (data not shown). The automated DNA extraction method may thus also be useful for the screening of other pathogens, including viruses in ticks, and possibly the genetic characterization of ticks and co-evolution studies.
Conclusions
The whole method, i.e. automated DNA extraction, adapted for tick samples and coupled with the new Sol1 qPCR, is more sensitive, specific and reproducible and reduces the risk of contamination. Using this method, E. ruminantium in ticks is also detected faster than any other existing methods including the OIE reference method based on manual DNA extraction and pCS20 nested PCR. It will be a useful tool for screening a large number of ticks for E. ruminantium. Independently of DNA extraction, the pCS20 Sol1TqM and Sol1SG qPCRs will also be valuable for E. ruminantium detection in ticks or in blood samples from clinically suspicious ruminants. Given there is no cross reaction with the endemic PME or E. chaffeensis, this could be of particular interest in the parts of the American continent that are currently free, such as mainland USA. Likewise, this high-throughput DNA extraction method using a virus RNA/DNA extraction kit validated for tick samples has potential for genetic studies on ticks, and for field screening of other bacteria and viruses in ticks.
Additional files
Text. PCR protocol for the detection of E. ruminantium. Table S1. Primers and probes for pCS20 Sol1TqM, Sol1SG, Cow™ and 16SSG qPCRs. (DOC 48 kb)
Text. Protocol of extraction of tick DNA. (DOCX 12 kb)
Text. Adult ticks from nymphs engorged on experimentally infected goats. (DOCX 11 kb)
Text. Formula for relative sensitivity, specificity and accuracy. (DOCX 12 kb)
Acknowledgements
DNA from A. phagocytophilum, A. platys, E. canis, E. muris, R. felis, R. parkeri was kindly provided by Dr Ulrike Munderloh from the University of Minnesota, Department of Entomology, Minnesota, USA. DNA from A. marginale, B. bovis and B. bigemina was kindly provided by Marisa Farber from INTA (Argentina). PME DNA from 6 A. americanum ticks was kindly provided by Dr Ellen Stromdhal from US Army Public Health Center, USA. We are grateful to the Mozambican Veterinary services, South African National Parks (Kruger National Park) and Zambeze Delta Safaris (Coutada 11 and 12) for support during sampling. We are thankful to Ken Giraud Girard and Rosalie Aprelon who were involved in the production of experimentally infected ticks at CIRAD facilities. We thank Renata Servan de Almeida for her comments and corrections.
Funding
This work was financially supported by CIRAD and the EPIGENESIS project which received funding from the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement No. 31598. FUNDO ABERTO DA UEM 2012–2013 and FUNDO NACIONAL DE INVESTIGAÇÃO Projecto No. 133-Inv/FNI/2012–2013 funded the field trips and the purchase of reagents in Mozambique. This study was partly conducted in the framework of the project MALIN “Surveillance, diagnosis, control and impact of infectious diseases of humans, animals and plants in tropical islands” supported by the European Union in the framework of the European Regional Development Fund (ERDF) and the Regional Council of Guadeloupe.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- Ac
Accuracy
- B
Blood
- CC p
Cell culture passage
- Ct
Cycle threshold
- FN
False negative
- FP
False positive
- MLST
Multi-locus sequence typing
- NTC
Non-template control
- PME
Panola Mountain Ehrlichia
- qPCR
Quantitative polymerase chain reaction
- RLB
Reverse line blot
- Se
Sensitivity
- SG
SYBR green
- Sp
Specificity
- T
Ticks
- Th
Temperature of hybridization
- TN
True negative
- TP
True positive
- TqM
TaqMan
- Undet
Undetermined
- w +
Weak positive.
Authors’ contributions
NC and VP optimized and generated DNA extraction and PCR results. NC, VP, LB, EA, LN and NV interpreted the results and wrote the manuscript. TL, EA, NV and LN designed the project. All authors read and approved the final manuscript.
Ethics approval
Experiments on goats to obtain E. ruminantium infected ticks were performed according to approved World Organization for Animal Health (OIE) international standards, and the CIRAD institution was authorized by the director of veterinary services of Guadeloupe on behalf of the Prefect of Guadeloupe (authorization number: A-971-18-01).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13071-017-2490-0) contains supplementary material, which is available to authorized users.
Contributor Information
Nídia Cangi, Email: nidicangi@yahoo.com.br.
Valérie Pinarello, Email: valerie.pinarello@cirad.fr.
Laure Bournez, Email: laurebournez@gmail.com.
Thierry Lefrançois, Email: thierry.lefrancois@cirad.fr.
Emmanuel Albina, Email: emmanuel.albina@cirad.fr.
Luís Neves, Email: lneves17@gmail.com.
Nathalie Vachiéry, Email: nathalie.vachiery@cirad.fr.
References
- 1.Allsopp BA. Trends in the control of heartwater. Onderstepoort J Vet Res. 2009;76(1):81–88. doi: 10.4102/ojvr.v76i1.69. [DOI] [PubMed] [Google Scholar]
- 2.Walker JB, Olwage A. The tick vectors of Cowdria ruminantium (Ixodoidea, Ixodidae, genus Amblyomma) and their distribution. Onderstepoort J Vet Res. 1987;54(3):353–379. [PubMed] [Google Scholar]
- 3.Stachurski F, Tortosa P, Rahajarison P, Jacquet S, Yssouf A, Huber K. New data regarding distribution of cattle ticks in the south-western Indian Ocean islands. Vet Res. 2013;44:79. doi: 10.1186/1297-9716-44-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Provost A, Bezuidenhout JD. The historical background and global importance of heartwater. Onderstepoort J Vet Res. 1987;54(3):165–169. [PubMed] [Google Scholar]
- 5.Minjauw B. The economic impact of heartwater (Cowdria ruminantium) infection in the SADC region, and its control through the use of new inactivated vaccines. UF/USAID/SADC Heartwater Research Project Report: ILRI; 2000. [Google Scholar]
- 6.Roth JA, Richt JA, Morozov IA (editors). Vaccines and diagnostics for transboundary animal diseases. In: International Symposium, Ames, Iowa, September 2012 Proceedings. Karger; 2013.
- 7.Faburay B, Geysen D, Ceesay A, Marcelino I, Alves PM, Taoufik A, et al. Immunisation of sheep against heartwater in the Gambia using inactivated and attenuated Ehrlichia ruminantium vaccines. Vaccine. 2007;25(46):7939–7947. doi: 10.1016/j.vaccine.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Adakal H, Stachurski F, Konkobo M, Zoungrana S, Meyer DF, Pinarello V, et al. Efficiency of inactivated vaccines against heartwater in Burkina Faso: impact of Ehrlichia ruminantium genetic diversity. Vaccine. 2010;28(29):4573–4580. doi: 10.1016/j.vaccine.2010.04.087. [DOI] [PubMed] [Google Scholar]
- 9.Adakal H, Gavotte L, Stachurski F, Konkobo M, Henri H, Zoungrana S, et al. Clonal origin of emerging populations of Ehrlichia ruminantium in Burkina Faso. Infect Genet Evol. 2010;10(7):903–912. doi: 10.1016/j.meegid.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Esemu SN, Besong WO, Ndip RN, Ndip LM. Prevalence of Ehrlichia ruminantium in adult Amblyomma variegatum collected from cattle in Cameroon. Exp Appl Acarol. 2013;59(3):377–387. doi: 10.1007/s10493-012-9599-9. [DOI] [PubMed] [Google Scholar]
- 11.Molia S, Frebling M, Vachiéry N, Pinarello V, Petitclerc M, Rousteau A, et al. Amblyomma variegatum in cattle in Marie Galante, French Antilles. Prevalence, control measures, and infection by Ehrlichia ruminantium. Vet Parasitol. 2008;153(3–4):338–346. doi: 10.1016/j.vetpar.2008.01.046. [DOI] [PubMed] [Google Scholar]
- 12.Ammazzalorso AD, Zolnik CP, Daniels TJ, Kolokotronis SO. To beat or not to beat a tick: comparison of DNA extraction methods for ticks (Ixodes scapularis) PeerJ. 2015;3:e1147. doi: 10.7717/peerj.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allender MC, Bunick D, Dzhaman E, Burrus L, Maddox C. Development and use of a real-time polymerase chain reaction assay for the detection of Ophidiomyces ophiodiicola in snakes. J Vet Diagn Investig. 2015;27(2):217–220. doi: 10.1177/1040638715573983. [DOI] [PubMed] [Google Scholar]
- 14.Vidergar N, Toplak N, Kuntner M, Streamlining DNA. Barcoding protocols: automated DNA extraction and a new cox1 primer in arachnid systematics. PLoS One. 2014;9(11):e113030. doi: 10.1371/journal.pone.0113030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Perez MA, Gopal H, Adeleke MA, De Luna-Santillana EJ, Gurrola-Reyes JN, Guo X. Detection of Onchocerca volvulus in Latin American black flies for pool screening PCR using high-throughput automated DNA isolation for transmission surveillance. Parasitol Res. 2013;112(11):3925–3931. doi: 10.1007/s00436-013-3583-0. [DOI] [PubMed] [Google Scholar]
- 16.Moriarity JR, Loftis AD, Dasch GA. High-throughput molecular testing of ticks using a liquid-handling robot. J Med Entomol. 2005;42(6):1063–1067. doi: 10.1093/jmedent/42.6.1063. [DOI] [PubMed] [Google Scholar]
- 17.Crowder CD, Rounds MA, Phillipson CA, Picuri JM, Matthews HE, Halverson J, et al. Extraction of total nucleic acids from ticks for the detection of bacterial and viral pathogens. J Med Entomol. 2010;47(1):89–94. doi: 10.1093/jmedent/47.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez D, Vachiéry N, Stachurski F, Kandassamy Y, Raliniaina M, Aprelon R, Gueye A, Nested PCR. For detection and genotyping of Ehrlichia ruminantium: use in genetic diversity analysis. Ann N Y Acad Sci. 2004;1026:106–113. doi: 10.1196/annals.1307.014. [DOI] [PubMed] [Google Scholar]
- 19.Faburay B, Geysen D, Munstermann S, Taoufik A, Postigo M, Jongejan F. Molecular detection of Ehrlichia ruminantium infection in Amblyomma variegatum ticks in the Gambia. Exp Appl Acarol. 2007;42(1):61–74. doi: 10.1007/s10493-007-9073-2. [DOI] [PubMed] [Google Scholar]
- 20.Vachiéry N, Jeffery H, Pegram R, Aprelon R, Pinarello V, Kandassamy RL, et al. Amblyomma variegatum ticks and heartwater on three Caribbean Islands. Ann N Y Acad Sci. 2008;1149:191–195. doi: 10.1196/annals.1428.081. [DOI] [PubMed] [Google Scholar]
- 21.Peixoto CC, Marcelino I, Vachiery N, Bensaid A, Martinez D, Carrondo MJ, Alves PM. Quantification of Ehrlichia ruminantium by real time PCR. Vet Microbiol. 2005;107(3–4):273–278. doi: 10.1016/j.vetmic.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Pruneau L, Emboule L, Gely P, Marcelino I, Mari B, Pinarello V, et al. Global gene expression profiling of Ehrlichia ruminantium at different stages of development. FEMS Immunol Med Microbiol. 2012;64(1):66–73. doi: 10.1111/j.1574-695X.2011.00901.x. [DOI] [PubMed] [Google Scholar]
- 23.Postigo M, Taoufik A, Bell-Sakyi L, de Vries E, Morrison WI, Jongejan F. Differential transcription of the major antigenic protein 1 multigene family of Ehrlichia ruminantium in Amblyomma variegatum ticks. Vet Microbiol. 2007;122(3–4):298–305. doi: 10.1016/j.vetmic.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 24.Steyn HC, Pretorius A, McCrindle CM, Steinmann CM, Van Kleef MA. Quantitative real-time PCR assay for Ehrlichia ruminantium using pCS20. Vet Microbiol. 2008;131(3–4):258–265. doi: 10.1016/j.vetmic.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Kelly P, Guo W, Xu C, Wei1 L, Jongejan F, et al.. Development of a generic Ehrlichia FRET-qPCR and investigation of ehrlichioses in domestic ruminants on five Caribbean islands. Parasit Vectors 2015;8:506. [DOI] [PMC free article] [PubMed]
- 26.Nakao R, Stromdahl EY, Magona JW, Faburay B, Namangala B, Malele I, et al. Development of loop-mediated isothermal amplification (LAMP) assays for rapid detection of Ehrlichia ruminantium. BMC Microbiol. 2010;10:296. doi: 10.1186/1471-2180-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sayler KA, Loftis AD, Mahan SM, Barbet AF. Development of a quantitative PCR sssay for differentiating the agent of heartwater disease, Ehrlichia ruminantium, from the Panola Mountain Ehrlichia. Transbound Emerg Dis. 2015; doi:10.1111/tbed.12339. [DOI] [PubMed]
- 28.Marcelino I, Verissimo C, Sousa MF, Carrondo MJ, Alves PM. Characterization of Ehrlichia ruminantium replication and release kinetics in endothelial cell cultures. Vet Microbiol. 2005;110:87–96. doi: 10.1016/j.vetmic.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Frutos R, Viari A, Ferraz C, Morgat A, Eychenié S, Kandassamy Y et al. Comparative genomics analysis of three strains of Ehrlichia ruminantium reveals an active process of genome size plasticity. J of Bacteriol. 2006;188(8):2533-2542. [DOI] [PMC free article] [PubMed]
- 30.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 31.Raliniaina M, Meyer DF, Pinarello V, Sheikboudou C, Emboulé L, Kandassamy Y et al. Mining the genetic diversity of Ehrlichia ruminantium using map genes family. Vet Parasitol. 2010;167(2-4):187-195. [DOI] [PubMed]
- 32.Pilet H, Vachiery N, Berrich M, Bouchouicha R, Durand B, Pruneau L et al. A new typing technique for the rickettsiales Ehrlichia ruminantium: multiple-locus variable number tandem repeat analysis. J Microbiol Methods. 2012;88(2):205-211. [DOI] [PubMed]
- 33.Cangi N, Gordon JL, Bournez L, Pinarello V, Aprelon R, Huber K, et al. Recombination is a major driving force of denetic diversity in the anaplasmataceae Ehrlichia ruminantium. Front Cell Infect Microbiol. 2016;6:111. [DOI] [PMC free article] [PubMed]
- 34.Mathew JS, Ewing SA, Barker RW, Fox JC, Dawson JE, Warner CK et al. Attempted transmission of Ehrlichia canis by Rhipicephalus sanguineus after passage in celle culture. Am J Vet Res. 1996;57:1594-1598. [PubMed]
- 35.Lynn GE, Oliver JD, Nelson CM, Felsheim RF, Kurtti TJ, Munderloh UG. Tissue distribution of the Ehrlichia muris-like agent in a tick vector. PLoS One. 2015; 10(3):e0122007. [DOI] [PMC free article] [PubMed]
- 36.Pornwiroon W, Pourciau SS, Foil LD, Macaluso KR. Rickettsia felis from cat fleas: isolation and culture in a tick-derived cell line. Appl Environ Microbiol 2006;72:5589-5595. doi:72/8/5589 [pii]. [DOI] [PMC free article] [PubMed]
- 37.Paddock CD, Fournier PE, Sumner JW, Goddard J, Elshenawy Y, Metcalfe MG et al. Isolation of Rickettsia parkeri and identification of a noval spotted fever group Rickettsa sp. from Gold Coast ticks (Amblyomma maculatum) in the United States. Appl Environ Microbiol 2010;76:2689-2696. doi:10.1128/AEM.02737-09. [DOI] [PMC free article] [PubMed]
- 38.Norris DE, Klompen JSH, Keirans JE, Black WC. Population genetics of Ixodes scapularis (Acari: Ixodidae) based on mitochondrial 16S and 12S genes. J Med Entomol. 1996;33:78–89. doi: 10.1093/jmedent/33.1.78. [DOI] [PubMed] [Google Scholar]
- 39.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3 - new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleiss JL, Levin B, Paik MC. The measurement of interrater agreement. In: Fleiss JL, Levin B, Paik MC, editors. Statistical methods for rates and proportions. New Jersey: John Wiley & Sons, Inc.; 2004. pp. 598–626. [Google Scholar]
- 41.Wilcoxon F. Individual comparisons by ranking methods. Biom Bull. 1945;1(6):80–83. doi: 10.2307/3001968. [DOI] [Google Scholar]
- 42.Bournez L, Cangi N, Lancelot R, Pleydell DR, Stachurski F, Bouyer J, et al. Parapatric distribution and sexual competition between two tick species, Amblyomma variegatum and A. hebraeum (Acari, Ixodidae), in Mozambique. Parasit Vectors. 2015;8:504. doi: 10.1186/s13071-015-1116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lalkhen AG, McCluskey A. Clinical tests: sensitivity and specificity. Continuing Educ. In Anaesthesia, critical care. Pain. 2008;8:221–223. [Google Scholar]
- 44.Black WC. 4th, Piesman J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc Natl Acad Sci USA. 1994;91(21):10034–8. [DOI] [PMC free article] [PubMed]
- 45.Mangold JA, Bargues DM, Mas-Coma S. Mitochondrial 16S rDNA sequences and phylogenetic relationships of species of Rhipicephalus and other tick genera among Metastriata (Acari: Ixodidae) Parasitol Res. 1998;84:478–484. doi: 10.1007/s004360050433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Text. PCR protocol for the detection of E. ruminantium. Table S1. Primers and probes for pCS20 Sol1TqM, Sol1SG, Cow™ and 16SSG qPCRs. (DOC 48 kb)
Text. Protocol of extraction of tick DNA. (DOCX 12 kb)
Text. Adult ticks from nymphs engorged on experimentally infected goats. (DOCX 11 kb)
Text. Formula for relative sensitivity, specificity and accuracy. (DOCX 12 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.