

Abstract
Inflammasomes are multiprotein complexes that form in the cytoplasm in response to cellular damage and cytosolic pathogen-associated molecules during infection. These complexes play important roles in initiating innate and adaptive immune responses to infectious disease. In addition, inflammasomes are now recognized as important mediators of sterile inflammation in various autoimmune and autoinflammatory diseases. Interestingly, microbiota and infection play critical roles in the development of “sterile inflammation”. Herein, we highlight recent advances in our understanding of the role for inflammasomes in nucleic acid-, nucleosome-, and histone-driven sterile inflammation and discuss knowledge gaps and areas of potential future research.
Keywords: NLRP3, AIM2, Caspase-1, inflammasome, IL-1β, IL-18, autoimmunity, autoinflammation, immunopathology, inflammation
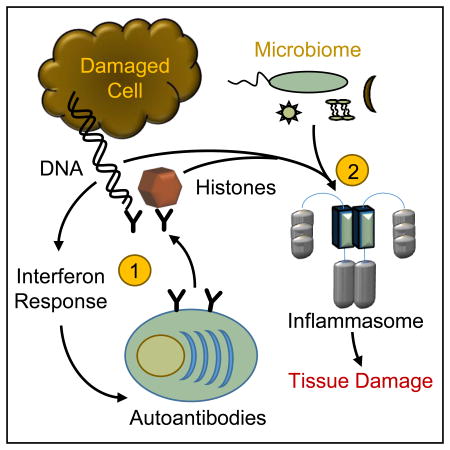
Graphical Abstract
Sterile inflammation associated with self-DNA/nucleosome antigens is initiated by type-I interferon responses and results in autoantibodies against these antigens. The second stage is the development of inflammation leading to tissue damage, which is dependent on autoantibodies and immune priming by both self-antigens and the microbiome. Thus, “sterile inflammation” in this situation is inaccurate, as clinical disease requires the microbiome.
Introduction
Inflammation and immunity are essential to fight infectious diseases, and sensing pathogen-derived nucleic acids is a major mechanism of innate immune cell activation. Pathogens can be detected due to differences in location and structure of host and pathogen nucleic acids. Members of the Toll-like receptor (TLR) family, including TLR-3, TLR-7, TLR-8 and TLR-9, detect nucleic acids in the endosomal compartment, where they survey the contents of vesicles entering the cell. TLRs activate signaling pathways that mediate cytokine secretion, type I interferon responses, and immune cell activation [1]. In the cytoplasm, the Retinoic acid Inducible Gene-I (RIG-I)–like receptor family detects uncapped RNA or long, double-stranded RNA molecules subsequently activating immune cells and triggering an antiviral state [2]. The interferon pathway is similarly activated by a host of DNA or dinucleotide sensors in the cytoplasm that detect infectious agents [3].
The inflammasome is a macromolecular protein complex formed in the cytoplasm in response to pathogen-associated molecular patterns (PAMPs) or cellular damage. Absent in Melanoma 2 (AIM2) oligomerizes in response to cytosolic DNA and binds with the adaptor molecule ASC (Apoptosis-associated Speck-like Protein containing a CARD), which further recruits caspase-1 to form a functional inflammasome [4-7]. Inflammasome activation allows caspase-1 to proteolytically cleave the inactive forms of the cytokines interleukin (IL)-1β and IL-18, resulting in their mature bioactive forms (Figure 1). Inflammasome activation also results in an inflammatory cell death known as pyroptosis via the caspase-1- or caspase-11-mediated proteolytic activation of gasdermin D, where activated gasdermin D forms pores in the cell membrane [8-12].
Figure 1. The AIM2-containing inflammasome recognizes self-DNA.

The PYHIN protein AIM2 is activated in response to DNA in the cytoplasm and interacts with ASC and caspase-1 to form an inflammasome. Inflammasome activation leads to maturation of the proinflammatory cytokines IL-1β and IL-18 and to pyroptotic cell death. Overall, AIM2 activation contributes to sterile inflammatory processes when self-DNA fragments from damaged host cells escape endosomes due to mutations in DNases or endosomal maturation and self-DNA enters the cytoplasm. In order for this inflammatory process to progress, a priming signal is generally required for production of pro-IL-1β. In “sterile inflammation” mediated by AIM2, this priming signal has not been directly determined but may depend on endogenous DNA, other endogenous ligands or on pathogen associated molecular patterns derived from the host microbiota.
Although nucleic acids of pathogens are frequently exposed to endosomal or cytoplasmic sensors, host cell nucleic acids are generally contained in the nucleus or modified (5′ 7-methylguanosine cap of mRNA, 5′ monophosphate of tRNA) to differentiate them from pathogen-associated nucleic acids, thus preventing unwanted inflammation [13, 14]. Under homeostatic conditions, localization and processing of nucleic acids are regulated. Even during cell death by apoptosis, dying cells' DNA is degraded intracellularly. Cellular debris from apoptotic cells are further degraded in phagocytic cells, where DNase II finishes the process of DNA hydrolysis [15]. However, defects in apoptosis or in the removal and degradation of extracellular DNA or apoptotic bodies (as observed in serum amyloid P, noncanonical autophagy, DNase I, or DNase II deficiency) can lead to the persistence of free nucleosomes, histones, or DNA molecules, resulting in inflammation [16-19]. Other forms of cell death, including necrosis, necroptosis, NETosis, and pyroptosis can result in release of self-DNA into the extracellular space, where it can be engulfed and sensed by endosomal or cytoplasmic nucleic acid sensors [20].
Outside the cell, nuclear contents serve as inflammatory stimuli by directly causing damage to the cell membranes of neighbouring cells. Direct membrane damage results from the highly positive charge of histones, which interacts with the phosphate group of phospholipids [21-23]. Thus, instead of activating AIM2, histones activate the NLRP3 inflammasome [24-26]. One could hypothesize that chromatin-mediated inflammation is necessary to initiate proper healing processes during immune responses because self-associated nucleic acids and histones in an extracellular space are indicative of self-damage. However, unrestrained inflammation directed to self-nuclear contents can result in immunopathology and subsequent sequelae associated with autoimmune and autoinflammatory diseases.
In addition to the ability of self-DNA or histones to induce inflammation, the microbiota also play an important role in the development of so-called “sterile inflammation”. For example, depletion or elimination of the microbiota or changes in diet with accompanying changes in microbiota are associated with improved disease outcomes with inflammasome mediated osteomyelitis and gouty arthritis [27, 28]. However, in a mouse model of atherosclerosis, the elimination of microbiota had no effect on disease development [29]. Thus, how microbes interact with the immune system in the development or progression of “sterile inflammation” is an area of current interest.
Herein, we focus our discussion on the known roles that inflammasomes play in the detection of nucleic acids, histones, or nucleosomes during sterile inflammation and on the gaps in our current knowledge. We also highlight recent research demonstrating the importance of pathogen infection or commensal microbiota in the development of diseases associated with sterile inflammation, suggesting a need for change in the current paradigm of sterile inflammation.
Self-DNA–mediated inflammasome activation
Self-tolerance is an essential component of an effective immune response so that pathogens are eliminated but minimal damage is caused to self-tissues. The random nature of immunoglobulin and T-cell receptor recombination that gives rise to the diversity of antibodies and T-cell receptors means that some of these effectors will inevitably react with self-antigens. However, multiple checkpoints have arisen to kill autoreactive T and B cells (negative selection) or prevent their activation (peripheral tolerance, T-regulatory cells). However, persistent immune stimulation coupled with defects in tolerance can lead to autoimmune diseases.
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by severe systemic inflammation, skin rashes (dermatitis), hair loss, cognitive decline, and multi-organ failure [20]. The underlying causes of SLE are not well understood despite decades of research; but the generation of autoantibodies directed at self-DNA and -histones (nucleosomes) is a hallmark of SLE, and these nucleosome-immune complexes induce inflammation through TLR and inflammasome signaling. As discussed above, AIM2 forms an inflammasome in response to cytoplasmic DNA (Figure 1), and polymorphisms or changes in expression of AIM2 are associated with SLE in humans [30, 31]. In SLE-prone mice, impaired degradation of self-DNA immune complexes in the lysosome allows DNA to enter the cytoplasm, where it activates the AIM2 inflammasome in macrophages [32] (Figure 1). Vascular damage is one manifestation of SLE, and expression of AIM2 and IL-18 is elevated in endothelial cells from patients with SLE and in a mouse model of SLE [32, 33]. Inhibition or deletion of caspase-1 increases endothelial cell differentiation in vitro and in mice and reduces the number of autoantibodies to self-DNA, which subsequently protects against vascular damage and glomerulonephritis [33, 34]. AIM2 expression is also positively correlated with autoantibodies in a mouse model of apoptotic DNA–induced SLE (apopDNA mice). Moreover, knocking down AIM2 in apopDNA mice reduces autoantibody levels, immune cell infiltration, and cytokine levels of IL-1β, TNF-α, MCP-1, and IL-6 in the kidney and serum [35]. Similarly, inflammasome activation is important in psoriasis, an autoimmune skin disease caused by extracellular self-DNA [36, 37]. Importantly, keratinocytes can respond to cytosolic DNA, including genomic DNA, in an AIM2-dependent manner [38, 39], and psoriatic lesions contain cytoplasmic DNA, enhanced AIM2 expression and inflammasome activation [38-41].
In SLE, the accumulation of self-DNA due to defects in apoptosis or failure to degrade self-DNA can ultimately lead to AIM2 inflammasome activation. However, some reports demonstrate that autoantibodies to histones or extracellular DNA can exist without clinical signs of autoimmunity [42]. These findings suggest that nucleosome-immune complexes alone are not sufficient for the development of prolonged sterile inflammation and that inflammasome activation alone facilitates disease progression but cannot cause it. In agreement with this postulation, DNaseIIflox/− × Mx1-CreT mice and DNaseII−/− × Ifnar−/− mice accumulate self-DNA in macrophages after phagocytosis of apoptotic cells or erythrocyte precursor nuclei [43]. The authors of this study reported an increase in IL-1β in the joints and IL-18 in the serum of mice lacking DNase II, which suggests involvement of the inflammasome. However, increased IL-1β levels did not precede the onset of clinical disease, demonstrating that inflammasome activation likely enhances disease progression but may not initiate the disease [43]. Subsequently, two groups reported that deletion of AIM2 in DNaseII−/− × Ifnar−/− mice (DNaseII−/− × Ifnar−/− × Aim2−/− mice) results in impaired inflammasome activation and reduced joint inflammation, demonstrating a role for the AIM2 inflammasome in polyarthritis. However, DNaseII−/− × Ifnar−/− × Aim2−/− mice still developed autoantibodies in the absence of clinical disease [44, 45]. Furthermore, deletion of the DNaseII gene alone in mice is embryonically lethal. This lethality can be rescued by deleting genes important in type I interferon signaling (DNaseII−/− × Ifnar−/− or DNaseII−/− × Sting−/− double mutants). However, DNaseII−/− × Aim2−/− mice are not rescued from embryonic lethality [44, 46]. In all, these data support the idea that the AIM2 inflammasome facilitates autoimmune disease progression in response to self-DNA, but interferon signaling is required for the initial inflammatory response. Similarly, examination of SLE mouse models under germ free conditions produces autoantibodies, but germ free conditions reduce clinical signs of disease such as nephritis and lymphoproliferation indicating that microbiota facilitate SLE disease progression but not its initiation [47, 48]. The similarities between germ free mice and Aim2−/− mice with respect to clinical disease may suggest a link between the microbiota and AIM2 activation during SLE disease progression. Although not confirmed, microbiota may provide a priming signal for AIM2 activation (Figure 1).
In contrast to SLE, AIM2 inflammasome activation in response to self-DNA during acute pancreatitis is an essential pathway for disease development [49]. Pancreatitis results from the premature activation of digestive enzymes and subsequent pancreatic tissue damage with release of nuclear material [50]. Importantly, deletion of AIM2 protected against pancreatic cell injury and inflammation [49]. Thus, in autoimmune diseases like SLE, AIM2 plays a supporting role for disease progression, but during autoinflammation like pancreatitis, AIM2 leads the way for disease development.
Although AIM2 inflammasome activation has demonstrated importance in some sterile inflammatory diseases, there is little research on the role of AIM2 in other sterile inflammatory conditions including atherosclerosis, type-I diabetes, or multiple sclerosis. Furthermore, increased AIM2 expression and inflammasome activation have been observed in abdominal aortic aneurisms, dermatitis, venous ulcers, and trauma wounds, though the functional significance of these observations is unknown [40, 51]. Thus, there is still much we do not know about the importance of self-DNA–mediated AIM2 inflammasome activation in the pathology of sterile inflammation.
NLRP3: a sensor of damage signals from nucleic acids and histones
Although AIM2 is activated by cytosolic DNA, NLRP3 is activated by a host of stimuli. NLRP3 stimuli include cellular damage, reactive oxygen species (ROS), and cellular potassium efflux (reviewed in [52]). The NLRP3 inflammasome is also activated in response to a variety of cytosolic nucleic acids and by cellular damage caused by extracellular histones (Figure 2). Pathogen-derived cytosolic nucleic acids or synthetic nucleic acid analogues can activate the NLRP3 inflammasome [53, 54]. Subsequently, it was discovered that damage to mitochondria releases mitochondrial DNA (mtDNA) into the cytoplasm, resulting in NLRP3 inflammasome activation [55-57].
Figure 2. NLRP3 activation by mitochondrial DNA and histones.

NLRP3 inflammasome activation requires two signals in the form of priming the expression of NLRP3 and pro-IL-1β as well as a second damage signal for NLRP3 activation. During sterile inflammation, DNA and histones derived from damaged host cells can prime the NLRP3 inflammasome through TLR-9 or TLR4-mediated increases in NLRP3 and pro-IL-1β expression. The NLRP3 inflammasome can be activated by the presence of cytoplasmic nucleic acids. During sterile inflammation, mitochondrial damage releases mitochondrial DNA (mtDNA) into the cytoplasm where it activates NLRP3. The mechanism of mtDNA-mediated NLRP3 activation is not clear but likely hinges on unknown adaptor proteins or the common signals of potassium efflux or reactive oxygen species generation. NLRP3 activation can also result from histones' ability to damage the cell membrane, but the exact mechanisms are unknown.
Mitochondrial damage and release of mtDNA is an essential part of the autoinflammatory disease caused by mevalonate kinase deficiency. Mutation of mevalonate kinase blocks isoprenoid synthesis and a defect in autophagy arises, resulting in the accumulation of damaged mitochondria. Cytosolic mtDNA subsequently triggers NLRP3 inflammasome activation, resulting in exaggerated production of IL-1β that ultimately contributes to disease [58]. Likewise, ozone-induced lung damage causes mitochondrial damage through oxidative stress. Subsequent release of mtDNA into the cytoplasm triggers NLRP3 inflammasome activation, which leads to lung damage. Treating ozone-exposed mice with the caspase-1 inhibitor YVAD inhibits neutrophil and γδ T-cell infiltration and reduces IL-1β, IL-17, KC, G-CSF, and IP-10 levels. These studies demonstrate that inflammasome activation in response to mtDNA contributes to sterile lung inflammation [59].
During atherosclerosis development, mitochondrial damage triggers NLRP3 inflammasome activation [60]. Cells that are depleted of mtDNA (rho0 cells) have reduced NLRP3 inflammasome activation despite having similar levels of cholesterol accumulation, suggesting that mitochondrial damage is a key trigger of inflammation in atherosclerosis [61]. Mitochondrial damage is associated with a host of additional sterile inflammatory diseases, including Parkinson's disease and Alzheimer's disease [62, 63]. Intriguingly, NLRP3 inflammasome activation is also linked with these diseases [64, 65]. Though likely, a role for cytoplasmic mtDNA in mediating NLRP3 inflammasome activation in these diseases has not been examined. The examples of mtDNA-mediated NLRP3 inflammasome activation show that sensing mtDNA in the incorrect cellular compartment is a common mechanism for the initiation of sterile inflammation. Detection of cytoplasmic mtDNA may have originally evolved as a defence mechanism to prevent outgrowth when the symbiotic relationship between eukaryotic and prokaryotic cells first gave rise to mitochondria. This likely continued to be important during infectious disease as a nonspecific marker of cellular damage. However, the nonspecific nature of this inflammatory signal leading to NLRP3 inflammasome activation has the often-undesirable consequence of causing excessive sterile inflammation, leading to tissue degeneration and clinical disease.
In addition to the NLRP3 response to cytoplasmic mtDNA, histone-mediated NLRP3 inflammasome activation is important in a variety of sterile injury models (Figure 2). Histones activate NLRP3 by inducing ROS, potassium efflux, and calcium influx, possibly resulting from direct damage to the cell membrane caused by interactions of positively charged amino acids in the histones with negatively charged phosphates in phospholipids [24-26]. Importantly, histone H4 can also activate TLR2 and TLR4, thus providing both priming and activation signals necessary for NLRP3 inflammasome formation all in one package [24] (Figure 2). Injecting purified H4 or necrotic cellular debris into the peritoneal cavity of mice causes NLRP3-dependent sterile inflammation [24]. Likewise, liver damage resulting from ischemia/reperfusion is mediated by histones through NLRP3 inflammasome activation [25]. In this instance, TLR9 was required for histone-mediated inflammasome priming [25]. Intratracheal administration of purified histones alone activates the NLRP3 inflammasome and leads to acute lung injury (ALI) [26]. During ALI mediated by C5a or IgG immune complexes, histones in neutrophil extracellular traps (NETs) help drive inflammation in an NLRP3-dependent manner. A positive feedback mechanism of NLRP3 activation induces further histone release through pyroptosis and/or the recruitment of more neutrophils [26]. This report raises the intriguing question of how histone-mediated inflammation is turned off. During infection, elimination of the pathogen in combination with anti-inflammatory cytokines such as IL-10 and TGF-β eventually terminate inflammation. In fact, recent reports demonstrate that IL-10 can dampen NLRP3 inflammasome activation [66-68]. However, the release of histones and the positive feedback loop that could be generated due to an almost-limitless supply of these molecules raise the questions: What mechanisms prevent this from happening, and are these mechanisms defective in autoimmune or autoinflammatory diseases?
NLRP3 inflammasome activation and inflammation can be improved in all three models discussed above by antibody-mediated histone neutralization [24-26]. Thus, it appears that antibodies targeting histones actually help prevent disease. These findings complicate our current understanding of immune complexes in the development of sterile inflammation. DNA immune complexes clearly facilitate SLE progression, as anti-nucleosome immune complexes derived from the serum of patients with lupus can activate the NLRP3 inflammasome when injected in mice. SLE-derived nucleosome-immune complexes facilitate NLRP3 inflammasome activation by upregulating expression of NLRP3 and pro-IL-1β via the TLR4-NF-κB signaling axis [69]. Production of mitochondrial ROS subsequently activates the NLRP3 inflammasome [69]. Also, immune complexes found in patients with SLE consisting of IgG and U1-small nuclear ribonucleoprotein can activate NLRP3 in CD14+ human monocytes [70]. Why antibodies bound to histones can prevent disease but antibodies bound to nucleosomes induce inflammation is unclear. One possibility is that antibodies bound to free histones (not bound to DNA) may have a completely different effect on immune signaling than do antibodies bound to nucleosomes (histones in complex with DNA). Whether the immune complexes formed in each situation are unique and, thus, have different inflammatory outcomes is unknown. Therefore, examining the role of different antibodies, immune complex structures, and immune signaling capacities of these complexes will be essential for understanding sterile inflammation and how to treat it.
Inhibiting inflammasomes as a therapeutic intervention
Based on the important role of DNA-sensing inflammasomes in sterile inflammation, numerous reports have examined the therapeutic potential of targeting inflammasomes in diseases involving sterile inflammation. Citral and epigallocatechin-3-gallate (EGCG), both bioactive compounds derived from traditional Chinese medicine, prevent NLRP3 activation in vivo in mouse models of lupus and improve nephritis by protecting cells from oxidative damage. Both Citral and EGCG treatment increased expression of Nuclear factor (erythroid-derived 2)-like 2 (NRF2) and enhancing expression of antioxidant proteins controlled by NRF2 [71, 72]. Overexpressing the NF-κB inhibitor A20 protein in vivo by using an adenovirus vector (Ad-A20) demonstrates that inhibiting NF-κB signaling impairs NLRP3 expression and IL-1β and autoantibody production [73]. Bay11-7082, an IκBα phosphorylation inhibitor, similarly inhibits the NLRP3 inflammasome and NF-κB activity in SLE mice [74]. Anesthetic isoflurane inhibits the NLRP3 inflammasome in the MRL/lpr SLE mouse model [75]. However, isoflurane is a teratogen; because females are more susceptible to SLE, this treatment is an interesting proof of concept but is not likely to be a viable therapeutic option [76].
The inhibitors above suggest that inhibiting ROS as an NLRP3 activator and inhibiting NLRP3 priming through the NF-κB pathway may have therapeutic benefit. Direct inhibition of caspase-1 during ALI using YVAD inhibited immune cell infiltration and cytokine production and ameliorated lung damage [59]. The NLRP3 inhibitor glibenclamide was examined during SLE and found to partially inhibit inflammasome activation and IL-1β levels [77], likely due to the concurrent involvement of AIM2 in this disease. Psoriasis, ALI, liver damage, and peritonitis were all responsive to IL-1 receptor antagonist treatment, suggesting potential therapeutic interventions here by targeting IL-1β signaling or the inflammasome [24-26, 78]. Overall, blocking inflammasome activation or IL-1β holds promise as a therapeutic treatment during sterile inflammation, but it will also be of interest to know whether inhibition of the interaction of AIM2 with self-DNA is possible as a therapeutic intervention.
Is sterile inflammation truly sterile?
Although autoimmune diseases and trauma are often viewed as sterile events, recent studies demonstrate that most “sterile inflammation” partially depends on the presence of commensal microbes triggering innate immune receptors and “training” the immune system to respond in a biased manner during sterile inflammation. In particular, certain microbes induce a Th17-biased immune response, which is associated with increased susceptibility to autoimmune diseases [79]. Furthermore, in certain instances in which pathogen-associated molecules overlap and demonstrate antigenic similarity to host antigens (a process known as molecular mimicry), self-tolerance to the host antigen can be breached. Indeed, molecular mimicry is associated with rheumatic fever, SLE, type I diabetes, and other autoimmune diseases [80].
Autoinfectome is a term recently coined to describe the history of infectious and commensal microbial encounters that lead up to and facilitate the development of autoimmunity and sterile inflammation [81]. For the diseases discussed in this article, Epstein-Barr virus infection/reactivation is associated with SLE [82-84]. The bacterial amyloid curli in complex with extracellular bacterial DNA is important for bacterial biofilm formation and can accelerate lupus-like disease in mice, perhaps by mimicking nucleosome-immune complexes [85]. Furthermore, alterations in gut microbiota are associated with SLE development [86]. A relative decrease in Firmicutes and increase in Bacteroidetes composition and segmented filamentous bacteria colonization is also associated with SLE [87-90]. These changes in microbial composition result in increased levels of Th17-polarized CD4+ T cells, which contribute to autoimmune disease progression [89, 90]. Importantly, stool samples from patients with SLE induced a higher rate of Th17 polarization than controls when incubated with naïve CD4+ T cells in vitro, suggesting a direct role for the microbiota in shaping the autoimmune-enhancing Th17 response in SLE [90]. The effects of diet on intestinal microbial composition have also been examined in lupus-prone mice and patients with SLE. Seemingly simple changes in diet such as slightly acidic drinking water may prolong disease development or the presence of polyphenols in apples and oranges may alter gut microbiome composition with potential implications on disease development [91, 92]. Staphylococcus aureus is a commensal microorganism found on the skin and anterior nares. Colonization by S. aureus is also associated with higher SLE autoantibody levels and kidney damage [93]. However, some models of SLE performed in germ free mice still develop autoantibodies, suggesting that the initiation of the disease is independent of the microbiota. Instead, the microbiota appears to play a role in clinical disease progression [47, 48]. Clearly, before the microbiome can be utilized for the diagnosis or treatment of disease, there is much research that remains to be done to determine specific bacterial species that can contribute to or ameliorate inflammatory disease. Otherwise, we may be comparing apples and oranges.
In all, there is a strong non-sterile component to the development of autoimmune diseases. We have recently reviewed the role of DNA-sensing inflammasomes, including AIM2, in regulating the gut microbiota, but the effects of microbiota on inflammasome activation in sterile inflammation have not been well studied [94]. The inflammasome is reported to play a role in the microbiota-mediated development of gouty arthritis [28], but as yet, there has been no examination of the role of nucleosome-mediated activation of the inflammasome in arthritis. One report on ALI shows that inflammasome activation is diminished in antibiotic-treated mice [95]. These reports suggest that the microbiota or the autoinfectome are involved in the development of sterile inflammation and further investigation of their importance in inflammasome activation is warranted. Furthermore, understanding how contact with microbes facilitates “sterile inflammation” will help us better understand the complex interaction between genotype, environment, and phenotype.
Conclusions
AIM2 is among the most recently discovered inflammasome adaptors, and our understanding of this protein's importance in sterile inflammation is still in its infancy. As discussed, AIM2 and the mtDNA-sensitive NLRP3 inflammasomes are important drivers of sterile inflammation, but our understanding of the mechanisms involved in particular disease settings needs further research. Especially in the case of non-autoimmune inflammation (wound healing, atherosclerosis, brain trauma), there is a dearth of research on the role of AIM2. In the case of NLRP3, it is still not known how mtDNA activates NLRP3. It is possible that mtDNA binds specific cytoplasmic adaptors that facilitate NLRP3 activation or that cytoplasmic mtDNA merely induces cell damage through currently undefined pathways to induce ROS production or potassium efflux, which then activate NLRP3. Finally, moulding of the immune system by infection or immune stimulation by microbes— be it through shaping the T-helper cell profile and cytokine milieu or through molecular mimicry directly inducing autoimmunity— is increasingly recognized as a requirement for breaking tolerance and transition to a sterile inflammatory state. Thus, as research in the field of sterile inflammation and autoimmunity move forward, we must consider the nonsterile nature of the organisms and environment in which they exist and how this affects the development and progression of sterile inflammation. The examination of sterile inflammation must, therefore, include the study of the infectome and microbiome to present a clear picture of the mechanisms involved and how to effectively treat and diagnose sterile inflammatory diseases. Finally, several studies discussed herein suggest that the inflammasome, IL-1β, and IL-18 are potential therapeutic targets worthy of further examination.
Acknowledgments
We apologize to numerous investigators whose work could not be cited due to space limitations. We thank Prajwal Gurung, R. K. Subbarao Malireddi and Teneema Kuriakose for helpful edits during development of the manuscript. This work was supported by grants from the National Institutes of Health to T-D.K. (Grants AI124346, AR056296, AI101935, and CA163507) and by ALSAC. CL is supported by the Department of Biology, Missouri State University.
Abbreviations
- AIM2
Absent in Melanoma 2
- ALI
Acute lung injury
- apopDNA
Apoptotic DNA–induced SLE
- ASC
Apoptosis-associated Speck-like Protein containing a CARD
- CD
Cluster of differentiation
- DNase
Deoxyribonuclease
- EGCG
epigallocatechin-3-gallate
- G-CSF
Granulocyte-colony stimulating factor
- IFNAR
Interferon-α receptor
- IL
Interleukin
- IP-10
Interferon inducible protein-10
- KC
Keratinocyte chemoattractant
- MCP-1
Monocyte chemoattractant protein 1
- MRL/lpr
Fas (TNF receptor superfamily member 6) mutant mice
- NETs
Neutrophil extracellular traps
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3
Nucleotide-Binding Oligomerization Domain, Leucine Rich Repeat and Pyrin Domain Containing 3
- PAMPs
Pathogen-associated molecular patterns
- RIG-I
Retinoic acid Inducible Gene-I
- ROS
Reactive oxygen species
- SLE
Systemic lupus erythematosus
- STING
Stimulator of interferon genes
- TGF-β
Transforming growth factor-β
- TLR
Toll-like receptor
- TNF-α
Tumor necrosis factor-α
- YVAD
Ac-Tyr-Val-Ala-Asp-Chloromethylketone
Footnotes
Conflicts of Interest: The authors declare no conflicts of interests.
Author Contributions: CL and TDK conceived the ideas. CL, AR, and TDK wrote the paper.
References
- 1.Leifer CA, Medvedev AE. Molecular mechanisms of regulation of Toll-like receptor signaling. J Leukoc Biol. 2016 doi: 10.1189/jlb.2MR0316-117RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan YK, Gack MU. RIG-I-like receptor regulation in virus infection and immunity. Curr Opin Virol. 2015;12:7–14. doi: 10.1016/j.coviro.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dempsey A, Bowie AG. Innate immune recognition of DNA: A recent history. Virology. 2015;479-480:146–52. doi: 10.1016/j.virol.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–8. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–13. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–72. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 7.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–60. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 8.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 10.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 13.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–7. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 14.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 15.Kawane K, Fukuyama H, Kondoh G, Takeda J, Ohsawa Y, Uchiyama Y, Nagata S. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science. 2001;292:1546–9. doi: 10.1126/science.292.5521.1546. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- 17.Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25:177–81. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- 18.Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, Mitchell DA, Cook HT, Butler PJ, Walport MJ, Pepys MB. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med. 1999;5:694–7. doi: 10.1038/9544. [DOI] [PubMed] [Google Scholar]
- 19.Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, Linkermann A, Virgin HW, Green DR. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–9. doi: 10.1038/nature17950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12:716–730. doi: 10.1038/nrrheum.2016.186. [DOI] [PubMed] [Google Scholar]
- 21.Gamberucci A, Fulceri R, Marcolongo P, Pralong WF, Benedetti A. Histones and basic polypeptides activate Ca2+/cation influx in various cell types. Biochem J. 1998;331(Pt 2):623–30. doi: 10.1042/bj3310623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pisetsky DS. Immune activation by histones: plusses and minuses in inflammation. Eur J Immunol. 2013;43:3163–6. doi: 10.1002/eji.201344175. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol. 2013;43:3336–42. doi: 10.1002/eji.201243224. [DOI] [PubMed] [Google Scholar]
- 25.Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, Ding Q, Loughran P, Beer-Stolz D, Billiar TR, Esmon CT, Tsung A. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol. 2013;191:2665–79. doi: 10.4049/jimmunol.1202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, Zetoune FS, Ward PA. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014;192:5974–83. doi: 10.4049/jimmunol.1400368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, Bandi SR, Calabrese CR, Vande Walle L, Lamkanfi M, Kanneganti TD. Dietary modulation of the microbiome affects autoinflammatory disease. Nature. 2014;516:246–9. doi: 10.1038/nature13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vieira AT, Macia L, Galvao I, Martins FS, Canesso MC, Amaral FA, Garcia CC, Maslowski KM, De Leon E, Shim D, Nicoli JR, Harper JL, Teixeira MM, Mackay CR. A Role for Gut Microbiota and the Metabolite-Sensing Receptor GPR43 in a Murine Model of Gout. Arthritis Rheumatol. 2015;67:1646–56. doi: 10.1002/art.39107. [DOI] [PubMed] [Google Scholar]
- 29.Wright SD, Burton C, Hernandez M, Hassing H, Montenegro J, Mundt S, Patel S, Card DJ, Hermanowski-Vosatka A, Bergstrom JD, Sparrow CP, Detmers PA, Chao YS. Infectious agents are not necessary for murine atherogenesis. J Exp Med. 2000;191:1437–42. doi: 10.1084/jem.191.8.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimkong I, Avihingsanon Y, Hirankarn N. Expression profile of HIN200 in leukocytes and renal biopsy of SLE patients by real-time RT-PCR. Lupus. 2009;18:1066–72. doi: 10.1177/0961203309106699. [DOI] [PubMed] [Google Scholar]
- 31.Choubey D. Interferon-inducible Ifi200-family genes as modifiers of lupus susceptibility. Immunol Lett. 2012;147:10–7. doi: 10.1016/j.imlet.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monteith AJ, Kang S, Scott E, Hillman K, Rajfur Z, Jacobson K, Costello MJ, Vilen BJ. Defects in lysosomal maturation facilitate the activation of innate sensors in systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2016;113:E2142–51. doi: 10.1073/pnas.1513943113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol. 2011;187:6143–56. doi: 10.4049/jimmunol.1101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kahlenberg JM, Yalavarthi S, Zhao W, Hodgin JB, Reed TJ, Tsuji NM, Kaplan MJ. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthritis Rheumatol. 2014;66:152–62. doi: 10.1002/art.38225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. AIM2 facilitates the apoptotic DNA-induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J Clin Immunol. 2013;33:925–37. doi: 10.1007/s10875-013-9881-6. [DOI] [PubMed] [Google Scholar]
- 36.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Su B, Nestle FO, Zal T, Mellman I, Schröder JM, Liu YJ, Gilliet M. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–9. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 37.Johansen C, Moeller K, Kragballe K, Iversen L. The activity of caspase-1 is increased in lesional psoriatic epidermis. J Invest Dermatol. 2007;127:2857–64. doi: 10.1038/sj.jid.5700922. [DOI] [PubMed] [Google Scholar]
- 38.Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Goss C, Anz D, Simanski M, Glaser R, Harder J, Hornung V, Gallo RL, Ruzicka T, Besch R, Schauber J. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. 2011;3:82ra38. doi: 10.1126/scitranslmed.3002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kopfnagel V, Wittmann M, Werfel T. Human keratinocytes express AIM2 and respond to dsDNA with IL-1beta secretion. Exp Dermatol. 2011;20:1027–9. doi: 10.1111/j.1600-0625.2011.01382.x. [DOI] [PubMed] [Google Scholar]
- 40.de Koning HD, Bergboer JG, van den Bogaard EH, van Vlijmen-Willems IM, Rodijk-Olthuis D, Simon A, Zeeuwen PL, Schalkwijk J. Strong induction of AIM2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp Dermatol. 2012;21:961–4. doi: 10.1111/exd.12037. [DOI] [PubMed] [Google Scholar]
- 41.Tervaniemi MH, Katayama S, Skoog T, Siitonen HA, Vuola J, Nuutila K, Sormunen R, Johnsson A, Linnarsson S, Suomela S, Kankuri E, Kere J, Elomaa O. NOD-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci Rep. 2016;6:22745. doi: 10.1038/srep22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagele EP, Han M, Acharya NK, DeMarshall C, Kosciuk MC, Nagele RG. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One. 2013;8:e60726. doi: 10.1371/journal.pone.0060726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 44.Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, Marshak-Rothstein A, Gravallese EM. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II-deficient mice. J Immunol. 2015;194:873–7. doi: 10.4049/jimmunol.1402573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jakobs C, Perner S, Hornung V. AIM2 Drives Joint Inflammation in a Self-DNA Triggered Model of Chronic Polyarthritis. PLoS One. 2015;10:e0131702. doi: 10.1371/journal.pone.0131702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–91. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maldonado MA, Kakkanaiah V, MacDonald GC, Chen F, Reap EA, Balish E, Farkas WR, Jennette JC, Madaio MP, Kotzin BL, Cohen PL, Eisenberg RA. The role of environmental antigens in the spontaneous development of autoimmunity in MRL-lpr mice. J Immunol. 1999;162:6322–30. [PubMed] [Google Scholar]
- 48.Unni KK, Holley KE, McDuffie FC, Titus JL. Comparative study of NZB mice under germfree and conventional conditions. J Rheumatol. 1975;2:36–44. [PubMed] [Google Scholar]
- 49.Kang R, Chen R, Xie M, Cao L, Lotze MT, Tang D, Zeh HJ., 3rd The Receptor for Advanced Glycation End Products Activates the AIM2 Inflammasome in Acute Pancreatitis. J Immunol. 2016;196:4331–7. doi: 10.4049/jimmunol.1502340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah AU, Sarwar A, Orabi AI, Gautam S, Grant WM, Park AJ, Liu J, Mistry PK, Jain D, Husain SZ. Protease activation during in vivo pancreatitis is dependent on calcineurin activation. Am J Physiol Gastrointest Liver Physiol. 2009;297:G967–73. doi: 10.1152/ajpgi.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dihlmann S, Erhart P, Mehrabi A, Nickkholgh A, Lasitschka F, Bockler D, Hakimi M. Increased expression and activation of absent in melanoma 2 inflammasome components in lymphocytic infiltrates of abdominal aortic aneurysms. Mol Med. 2014;20:230–7. doi: 10.2119/molmed.2013.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lupfer C, Kanneganti TD. Unsolved Mysteries in NLR Biology. Front Immunol. 2013;4:285. doi: 10.3389/fimmu.2013.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Nunez G. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–6. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 54.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Núñez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–8. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 55.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature immunology. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Burgh R, Nijhuis L, Pervolaraki K, Compeer EB, Jongeneel LH, van Gijn M, Coffer PJ, Murphy MP, Mastroberardino PG, Frenkel J, Boes M. Defects in mitochondrial clearance predispose human monocytes to interleukin-1beta hypersecretion. J Biol Chem. 2014;289:5000–12. doi: 10.1074/jbc.M113.536920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Che L, Jin Y, Zhang C, Lai T, Zhou H, Xia L, Tian B, Zhao Y, Liu J, Wu Y, Wu Y, Du J, Li W, Ying S, Chen Z, Shen H. Ozone-induced IL-17A and neutrophilic airway inflammation is orchestrated by the caspase-1-IL-1 cascade. Sci Rep. 2016;6:18680. doi: 10.1038/srep18680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan H, Li Y, Peng X, Huang D, Gui L, Huang B. Resistance of mitochondrial DNA-depleted cells against oxidized low-density lipoprotein-induced macrophage pyroptosis. Mol Med Rep. 2016;13:4393–9. doi: 10.3892/mmr.2016.5077. [DOI] [PubMed] [Google Scholar]
- 62.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009;5:706–8. doi: 10.4161/auto.5.5.8505. [DOI] [PubMed] [Google Scholar]
- 63.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3:614–5. doi: 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]
- 64.Lu M, Sun XL, Qiao C, Liu Y, Ding JH, Hu G. Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol Aging. 2014;35:421–30. doi: 10.1016/j.neurobiolaging.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 65.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gurung P, Li B, Subbarao Malireddi RK, Lamkanfi M, Geiger TL, Kanneganti TD. Chronic TLR Stimulation Controls NLRP3 Inflammasome Activation through IL-10 Mediated Regulation of NLRP3 Expression and Caspase-8 Activation. Sci Rep. 2015;5:14488. doi: 10.1038/srep14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li B, Gurung P, Malireddi RK, Vogel P, Kanneganti TD, Geiger TL. IL-10 engages macrophages to shift Th17 cytokine dependency and pathogenicity during T-cell-mediated colitis. Nat Commun. 2015;6:6131. doi: 10.1038/ncomms7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao Y, Vent-Schmidt J, McGeough MD, Wong M, Hoffman HM, Steiner TS, Levings MK. Tr1 Cells, but Not Foxp3+ Regulatory T Cells, Suppress NLRP3 Inflammasome Activation via an IL-10-Dependent Mechanism. J Immunol. 2015;195:488–97. doi: 10.4049/jimmunol.1403225. [DOI] [PubMed] [Google Scholar]
- 69.Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S, Zhao J, Yang N. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med. 2016;14:156. doi: 10.1186/s12967-016-0911-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shin MS, Kang Y, Lee N, Kim SH, Kang KS, Lazova R, Kang I. U1-small nuclear ribonucleoprotein activates the NLRP3 inflammasome in human monocytes. J Immunol. 2012;188:4769–75. doi: 10.4049/jimmunol.1103355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ka SM, Lin JC, Lin TJ, Liu FC, Chao LK, Ho CL, Yeh LT, Sytwu HK, Hua KF, Chen A. Citral alleviates an accelerated and severe lupus nephritis model by inhibiting the activation signal of NLRP3 inflammasome and enhancing Nrf2 activation. Arthritis Res Ther. 2015;17:331. doi: 10.1186/s13075-015-0844-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsai PY, Ka SM, Chang JM, Chen HC, Shui HA, Li CY, Hua KF, Chang WL, Huang JJ, Yang SS, Chen A. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic Biol Med. 2011;51:744–54. doi: 10.1016/j.freeradbiomed.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 73.Li M, Shi X, Qian T, Li J, Tian Z, Ni B, Hao F. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-kappaB and NLRP3 inflammasome activation in macrophages of mice. Int J Clin Exp Med. 2015;8:17430–40. [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, Liang Y, Yang N. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-kappaB activation. Int Immunopharmacol. 2013;17:116–22. doi: 10.1016/j.intimp.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 75.Yuan Y, Liu Z. Isoflurane attenuates murine lupus nephritis by inhibiting NLRP3 inflammasome activation. Int J Clin Exp Med. 2015;8:17730–8. [PMC free article] [PubMed] [Google Scholar]
- 76.Wade JG, Stevens WC. Isoflurane: an anesthetic for the eighties? Anesth Analg. 1981;60:666–82. [PubMed] [Google Scholar]
- 77.Herman S, Kny A, Schorn C, Pfatschbacher J, Niederreiter B, Herrmann M, Holmdahl R, Steiner G, Hoffmann MH. Cell death and cytokine production induced by autoimmunogenic hydrocarbon oils. Autoimmunity. 2012;45:602–11. doi: 10.3109/08916934.2012.719948. [DOI] [PubMed] [Google Scholar]
- 78.Viguier M, Guigue P, Pagès C, Smahi A, Bachelez H. Successful treatment of generalized pustular psoriasis with the interleukin-1-receptor antagonist Anakinra: lack of correlation with IL1RN mutations. Ann Intern Med. 2010;153:66–7. doi: 10.7326/0003-4819-153-1-201007060-00030. [DOI] [PubMed] [Google Scholar]
- 79.Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2011;108:11548–53. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sfriso P, Ghirardello A, Botsios C, Tonon M, Zen M, Bassi N, Bassetto F, Doria A. Infections and autoimmunity: the multifaceted relationship. J Leukoc Biol. 2010;87:385–95. doi: 10.1189/jlb.0709517. [DOI] [PubMed] [Google Scholar]
- 81.Bogdanos DP, Smyk DS, Rigopoulou EI, Sakkas LI, Shoenfeld Y. Infectomics and autoinfectomics: a tool to study infectious-induced autoimmunity. Lupus. 2015;24:364–73. doi: 10.1177/0961203314559088. [DOI] [PubMed] [Google Scholar]
- 82.Hanlon P, Avenell A, Aucott L, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and systemic lupus erythematosus. Arthritis Res Ther. 2014;16:R3. doi: 10.1186/ar4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Poole BD, Gross T, Maier S, Harley JB, James JA. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J Autoimmun. 2008;31:362–71. doi: 10.1016/j.jaut.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med. 2005;11:85–9. doi: 10.1038/nm1167. [DOI] [PubMed] [Google Scholar]
- 85.Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, Buttaro B, Caricchio R, Gallucci S, Tukel C. Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity. 2015;42:1171–84. doi: 10.1016/j.immuni.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rojo D, Hevia A, Bargiela R, Lopez P, Cuervo A, Gonzalez S, Suarez A, Sanchez B, Martinez-Martinez M, Milani C, Ventura M, Barbas C, Moya A, Suarez A, Margolles A, Ferrer M. Ranking the impact of human health disorders on gut metabolism: systemic lupus erythematosus and obesity as study cases. Sci Rep. 2015;5:8310. doi: 10.1038/srep08310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, Turroni F, Gonzalez S, Suarez A, Gueimonde M, Ventura M, Sanchez B, Margolles A. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio. 2014;5:e01548–14. doi: 10.1128/mBio.01548-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang H, Liao X, Sparks JB, Luo XM. Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol. 2014;80:7551–60. doi: 10.1128/AEM.02676-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Praet JT, Donovan E, Vanassche I, Drennan MB, Windels F, Dendooven A, Allais L, Cuvelier CA, van de Loo F, Norris PS, Kruglov AA, Nedospasov SA, Rabot S, Tito R, Raes J, Gaboriau-Routhiau V, Cerf-Bensussan N, Van de Wiele T, Eberl G, Ware CF, Elewaut D. Commensal microbiota influence systemic autoimmune responses. EMBO J. 2015;34:466–74. doi: 10.15252/embj.201489966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lopez P, de Paz B, Rodriguez-Carrio J, Hevia A, Sanchez B, Margolles A, Suarez A. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep. 2016;6:24072. doi: 10.1038/srep24072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cuervo A, Hevia A, Lopez P, Suarez A, Sanchez B, Margolles A, Gonzalez S. Association of polyphenols from oranges and apples with specific intestinal microorganisms in systemic lupus erythematosus patients. Nutrients. 2015;7:1301–17. doi: 10.3390/nu7021301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson BM, Gaudreau MC, Al-Gadban MM, Gudi R, Vasu C. Impact of dietary deviation on disease progression and gut microbiome composition in lupus-prone SNF1 mice. Clin Exp Immunol. 2015;181:323–37. doi: 10.1111/cei.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Conti F, Ceccarelli F, Iaiani G, Perricone C, Giordano A, Amori L, Miranda F, Massaro L, Pacucci VA, Truglia S, Girelli G, Fakeri A, Taliani G, Temperoni C, Spinelli FR, Alessandri C, Valesini G. Association between Staphylococcus aureus nasal carriage and disease phenotype in patients affected by systemic lupus erythematosus. Arthritis Res Ther. 2016;18:177. doi: 10.1186/s13075-016-1079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Man SM, Karki R, Kanneganti TD. DNA-sensing inflammasomes: regulation of bacterial host defense and the gut microbiota. Pathog Dis. 2016;74 doi: 10.1093/femspd/ftw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prakash A, Sundar SV, Zhu YG, Tran A, Lee JW, Lowell C, Hellman J. Lung Ischemia-Reperfusion is a Sterile Inflammatory Process Influenced by Commensal Microbiota in Mice. Shock. 2015;44:272–9. doi: 10.1097/SHK.0000000000000415. [DOI] [PMC free article] [PubMed] [Google Scholar]