

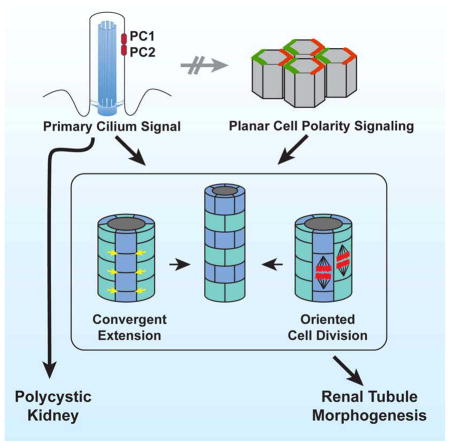
Summary
Oriented cell division (OCD) and convergent extension (CE) shape developing renal tubules, and their disruption has been associated with polycystic kidney disease (PKD) genes, the majority of which encode proteins that localize to primary cilia. Core planar cell polarity (PCP) signaling controls OCD and CE in other contexts, leading to the hypothesis that disruption of PCP signaling interferes with CE and/or OCD to produce PKD. Nonetheless, the contribution of PCP to tubulogenesis and cystogenesis is uncertain, and two major questions remain unanswered. Specifically, the inference that mutation of PKD genes interferes with PCP signaling is untested, and the importance of PCP signaling for cystogenic PKD phenotypes has not been examined. We show that during proliferative stages PCP signaling polarizes renal tubules to control OCD. However, we find that contrary to the prevailing model, PKD mutations do not disrupt PCP signaling, but instead act independently and in parallel with PCP signaling to affect OCD. Indeed, PCP signaling that is normally downregulated once development is completed is retained in cystic adult kidneys. Disrupting PCP signaling results in inaccurate control of tubule diameter, a tightly regulated parameter with important physiological ramifications. However, we show that disruption of PCP signaling is not cystogenic. Our results suggest that regulating tubule diameter is a key function of PCP signaling, but that loss of this control does not induce cysts.
Keywords: Polycystic kidney disease, planar cell polarity, tubule diameter, oriented cell division
Graphical Abstract
Introduction
Polycystic kidney disease (PKD) is a prominent feature of single-gene heritable genetic disorders including Autosomal Dominant PKD, Autosomal Recessive PKD and others (reviewed in [1, 2]). A striking commonality of single-gene PKD’s is that the affected proteins function in or contribute to the structure or function of primary cilia[3, 4]. The importance of primary cilia in the etiology of PKD is well documented. However, the developmental and disease inducing processes affected by primary cilia in the kidney are not well understood.
Two morphogenetic processes, convergent extension (CE) and oriented cell division (OCD), have been proposed to regulate kidney tubule morphogenesis, and their disruption has been associated with development of PKD. CE describes the reorganization of cells to elongate one tissue axis and narrow the orthogonal axis such that, in the renal tubule, CE would reduce tubule diameter[5, 6]. CE has been directly observed by live imaging in the Xenopus nephron[7]. In fixed tissues, CE may be inferred by cellular elongation in the direction of migration, and by this method has been implicated in mouse nephron morphogenesis[8, 9]. Defective inferred CE precedes cystogenesis in Wnt9b[8] and Pkd1[9] mouse mutant collecting ducts, but the appealing hypothesis that impaired CE leads to cyst formation has not been directly examined.
OCD, if aligned with the tubule axis, is expected to maintain tubule diameter while facilitating elongation during proliferative growth, and has been documented during embryonic and postnatal tubule elongation[8, 10–17]. Perturbation of OCD has been associated with development of PKD[8, 10–12, 14, 15], though whether loss of OCD occurs before or after cystogenesis is controversial[14, 15]. Nonetheless, perturbation of OCD is neither necessary nor sufficient to cause cysts, as impaired OCD is observed in Wnt7b and Pkhd1 (causative of ARPKD in humans) mutant mouse kidneys, yet they do not develop cysts[13, 15], and mutation of Ift140, a cilium component, causes cysts without perturbing OCD[16]. Thus, CE and OCD are both integral to normal renal tubule development, but their relationship to PKD is less clear.
PCP signaling controls the polarity of numerous epithelia in both Drosophila and vertebrates, and regulates both OCD and CE in various contexts[18]. The Drosophila wing, in which each cell produces a trichome that emerges from the distal side of the cell, has been an instructive model of PCP signaling, revealing a modular signaling system composed of genetically and biochemically related units[19]. The ‘core’ module acts both to amplify asymmetry, and to coordinate polarization between neighboring cells, producing a local alignment of polarity. Proteins in the core module, including the transmembrane proteins Frizzled (Fz), Flamingo/Starry night (Fmi), Van Gogh/Strabismus (Vang), and the cytosolic/peripheral membrane proteins Dishevelled, Diego and Prickle adopt asymmetric subcellular localizations that predict morphological polarity patterns such as trichome orientation. These proteins communicate at cell boundaries, recruiting one group to the distal side of cells, and the other to the adjacent proximal side through the function of a feedback mechanism, thereby aligning the polarity of adjacent cells[20, 21] (reviewed in [18, 22]).
Downstream of the core module, tissue specific ‘effector’ modules orchestrate morphological responses to the molecular asymmetry produced by core module function. Upstream of the core module, several ‘global’ modules have been proposed to link the direction of core module polarization to tissue axes. One of these, comprising the atypical cadherins Fat (Ft), Dachsous (Ds) and the golgi protein Four-jointed, converts opposing tissue-level expression gradients of Four-jointed and Ds into subcellular asymmetry of intercellular Ft-Ds heterodimers that align PCP with the tissue axes[23]. In addition, Wnt’s and other unknown signals have been implicated as global regulators[24, 25].
In many vertebrate tissues, characteristic features of the PCP signaling mechanism identified in flies are conserved. Yet, in various contexts, features of the mechanism have been adapted and evolved[6, 26], and some PCP genes function in mechanisms other than PCP[18]. Furthermore, while Drosophila has one, or in a few cases, two, paralogs of essential PCP genes, vertebrates typically have three or more paralogs, suggesting redundancy and/or diversification of function.
The contribution of PCP related events, and hence core PCP signaling, to PKD was first proposed based on the observation that OCD is observed along the length of the tubule axis, and is disturbed prior to cystogenesis in a model of PKD[10]. The inference or observation of CE in renal tubule morphogenesis reinforced this idea[7, 8]. However, evidence implicating the PCP signaling mechanism per se in kidney morphogenesis or in PKD is circumstantial. Numerous Wnts are associated with loss of OCD and/or cysts, but assigning function to PCP vs. other pathways is challenging[8, 13, 27, 28]. Mutations in Inversin, a homolog of Drosophila Diego, cause nephronophthisis type 2[29], however, the association of Inversin with cilia and with both canonical Wnt as well as PCP signaling make interpretation difficult (reviewed in[30]). Loss of Fat4, the closest vertebrate ortholog of Drosophila Ft[31], results in disruption of OCD and in very dilated, possibly cystic embryonic tubules[11]. Furthermore, Fat4 and Dchs1 may form a heterodimer pair analogous to Drosophila Ft-Ds[11, 32, 33]. The Fat4 dilated tubule/cystic phenotype was enhanced by Vangl2 heterozygosity, possibly connecting global and core PCP signaling to OCD and cystogenesis. In contrast, Vangl2Lp/Lp mutant animals show tubule branching and glomerular defects but not cysts at embryonic day 18.5 (E18.5)[34]. Confounding a simple interpretation, Fat4 is mostly expressed in the stroma, while Dchs1 is primarily expressed in the metanephric mesenchyme[33].
Studies of additional PCP signaling components have suggested a role for PCP in kidney morphogenesis. Xdd1, a dominant negative Dishevelled derivative, disrupts CE in the frog kidney[7]. However, the association of Dishevelled with basal bodies suggests that basal body docking or other ciliary defects may account for this phenotype[35]. Similarly, knockdown of pk1 in the Zebrafish kidney disorganizes basal bodies and produces cysts. However, genetic interaction with IFT mutants, and the known functions of Pk’s in regulating the cytoskeleton are also consistent with non-PCP mechanisms[24, 36, 37]. Misoriented OCD is observed in Celsr1−/−(ortholog of Fmi) and Celsr1+/−;Vangl2Lp/+ mutant kidneys[17], providing additional evidence for a role of PCP signaling in tubule morphogenesis. Yet, the characteristic subcellular localization of core PCP components that would substantiate conserved PCP signaling function remain to be demonstrated.
Planar polarized behaviors occur in renal tubule morphogenesis, and evidence cited above suggests that they are controlled by PCP signaling. However, two major questions remain unanswered. Specifically, the inference that mutation of PKD genes interferes with PCP signaling is untested, and the importance of PCP signaling for cystogenic PKD phenotypes has not been examined. We therefore directly and functionally interrogated PCP signaling in the mouse kidney.
Results
Core PCP components are required for proper kidney and tubule morphology
To determine whether the core PCP pathway is required for kidney tubule morphogenesis, we analyzed kidney development in mice mutant for Vangl1, Vangl2, Fz3, and Fz6 separately and in combination. Vangl2Lp/Lp homozygotes die at birth. At E18.5, their kidneys were shorter and wider than wildtype littermates, as previously reported[34], and their tubules appeared mildly dilated (Figure 1A–B,C–C″,D–D′). In contrast, Vangl1−/− mice (likely hypomorphs) are viable, fertile, with kidneys macroscopically similar to those of wildtype littermates (not shown). Similar to Vangl1−/−, the kidneys of Fz3−/− and Fz6−/− single mutant mice were not obviously affected (not shown). Fz3−/−;Fz6−/− double mutant mice die perinatally with multiple PCP related phenotypes[38]. Their kidneys were shorter and wider than wildtype littermates and showed dilated tubules comparable to those in Vangl2Lp/Lp kidneys (Figure 1E–E″).
Figure 1. Phenotypes of WT, Vangl2Lp/Lp and Fz3−/−; Fz6−/− mutant kidneys at E18.5.

(A–B) Control and Vangl2Lp/Lp kidneys. (C,C′,D,D′) H&E histology of control and Vangl2Lp/Lp kidneys. (E) Fz3−/−; Fz6−/− kidneys. (E–E′) H&E histology of Fz3−/−; Fz6−/− kidneys. (F–G) Immunofluorescence images of control and Vangl2Lp/Lp kidneys stained for DAPI (blue; nuclei), LTL (green; proximal tubule) and AQP2 (red; collecting duct). (H–I) Median tubule diameters for WT control, Vangl2Lp/Lp and Fz3−/−; Fz6−/− collecting ducts (H) and proximal tubules (I). Images illustrate measurement method (see Experimental Methods). Asterisks indicate significant differences: (H) 22.7 μm and 20.1 μm respectively versus 16.6 for WT control, * = p<0.0001 and (I) 28.0 and 25.4 respectively versus 22.4 for WT control, * = p<0.0001 by Mann-Whitney U test). (J–K) Distribution plots of data from H and I. Scale bars: A,B,E, 1 mm; C,C′,D,D′,E′,E″ 100 μm; F,G, 200 μm; H,I 25 μm. See also Figure S1.
In both Vangl2Lp/Lp and Fz3−/−;Fz6−/− kidneys at E18.5, median tubule diameters of both collecting ducts and proximal tubules were significantly larger compared to wildtype littermates (Figure 1F–I). Enlarged diameters were also observed for Vangl1−/− mutant tubules (Figure S1, Related to Figure 1). Importantly, plots of these measurements revealed a substantially broader distribution in the mutants compared to wildtype (Figure 1J,K; Figure S1, Related to Figure 1). Core PCP components therefore shape the kidney by regulating the diameter of renal tubules. The mutants examined here have not been implicated in ciliogenesis, and we detected no alteration in cilia morphology.
Core PCP components are asymmetrically localized along the proximal-distal tubule axis
We investigated whether core components of the PCP pathway function in tubule epithelia, and whether they function in a conserved PCP signaling mechanism. At E18.5, when each stage of nephrogenesis is present (Figure 2A), Vangl1, Vangl2, Fz3, and Fz6 were detected in all tubule segments, primarily in the lateral membranes of epithelial cells with a diffuse cytosolic presence (Figure 2B–B′, D–E‴; data not shown). This pattern was observed as early as E13.5 and persisted into early postnatal development, after which membrane localization dissipated over several weeks (Figure S2A–I, Related to Figure 2; Figure 7C–C‴). Consistent with this decline, mRNA expression of multiple core PCP genes and related factors decreased between P1 and 16 weeks (Figure S2J, Related to Figure 2). These observations suggest that developing tubule cells participate in PCP signaling, but that tubules in a post-proliferative quiescent state no longer engage in PCP signaling.
Figure 2. Core PCP proteins localize asymmetrically in proximal tubule and collecting duct.

(A) Control kidney stained for phalloidin (blue; actin), LTL (green; proximal tubule) and DBA (red; collecting duct). (B,B′,B″) Collecting duct stained for Vangl1 (green) and E-Cadherin (blue) showing asymmetric proximal localization of Vangl1 relative to E-Cadherin (arrowheads). (C–C″) Collecting duct stained for Vangl1 (green) and Fz6 (red) showing asymmetric proximal localization of Vangl1 and distal localization of Fz6 (arrowheads). (D–D″) Collecting duct showing distal localization of Fz3 (red) relative to β-catenin (blue). (E–E″) Proximal tubule (LTL; red) showing proximal localization of Vangl1 (green) relative to E-Cadherin (blue) (arrowheads). Asymmetry was quantified as an intensity ratio (see STAR methods) and indicated in the white box in associated figure panels and graphically in Figure S3C. Scale bars: A, 500 μm; B, 25 μm; B″,C″,D″,E″, 5 μm. See also Figure S2.
Figure 7. PCP is active in Kif3a mutant cystic kidneys.

(A–A′) H&E stained P30 control (NoCre; Kif3acko/cko) and (B–B′) KspCre; Kif3acko/cko kidneys showing a strongly cystic phenotype. (C–C‴) Control P30 (NoCre; Kif3acko/cko) collecting duct stained for Vangl1 (green), E-Cadherin (blue) and acetylated-α-tubulin (red; cilia). (D–D‴) KspCre; Kif3acko/cko cysts stained for Vangl1 (green), E-Cadherin (blue) and cilia acetylated-α-tubulin; red). Vangl1 signal is strong in cysts (no cilia) but not in the unaffected tubule (cilia present). (E–E″) High magnification images from 3D reconstruction (see Movie S3) showing asymmetric localization of Vangl1 (green) relative to E-Cadherin (blue) in cyst lining cells. Scale bars: A,A′,B,B′, 200 μm; C‴,D″,E″, 5 μm. See also Figure S7.
Using a low calcium dehydration step prior to fixation that induced separation of a fraction of cell-cell junctions, we reliably observed the selective asymmetric accumulation of Vangl1 and Vangl2 along the proximal side and Fz3 and Fz6 along the distal side of cells in both the collecting duct and proximal tubules (Figure 2B–B″,C–C″,D–D″,E–E″). This asymmetric localization is indicative of active PCP signaling and is strongly reminiscent of that first observed in Drosophila, and subsequently in numerous vertebrate tissues.
A characteristic feature of PCP signaling is the dependence of each core protein’s asymmetric localization on the activity of the other core proteins. In Vangl2Lp/Lp mice, in which strong kidney tubule phenotypes are observed (Figure 1), asymmetry of Vangl1, Fz3 and Fz6 localization was eliminated. In addition, their distributions were more punctate, and levels of Vangl1 in particular were decreased (likely due to the dominant negative activity of the Lp allele[39])(Figure 3A–A″,B–B″,C–C″). Similarly, in Fz3−/−;Fz6−/− mice, whose phenotype is comparable to that of Vangl2Lp/Lp mice, asymmetry of Vangl1 and Vangl2 localization was significantly diminished, though not eliminated (Figure 3D–D″; Supplementary Figure S3B–B″, Related to Figure 3; data not shown). This contrasts with Fz3−/−;Fz6+/− and Fz3+/−;Fz6−/− mice, in which the asymmetry of Vangl1 and Vangl2 was not diminished (Supplementary Figure S3A–A″, Related to Figure 3; data not shown). In Vangl1−/− tubules, the asymmetric localization of Vangl2, Fz6, and Fz3 was modestly diminished, perhaps due to the hypomorphic nature of the allele, or to redundancy between Vangl1 and Vangl2 (Supplementary Figure S3B–B″, Related to Figure 3; data not shown). The core components are therefore mutually required for establishment and/or maintenance of their asymmetric localization, indicating that they function in a conserved PCP signaling mechanism to regulate tubule diameter.
Figure 3. Asymmetric localization of core PCP protein localization is lost in PCP mutants.

(A–C) E18.5 Vangl2Lp/Lp collecting ducts (DBA, red) stained for (A) Vangl1 (green) and E-Cadherin (blue), (B) Fz6 (red) and E-Cadherin (blue) or (C) Fz3 (red) and E-Cadherin (blue) showing similar proximal and distal localization of Vangl1, Fz6 and Fz3 (arrowheads). (D) E18.5 Fz3−/−; Fz6−/− collecting duct stained for Vangl1 (green) and E-Cadherin (arrowheads). (E) P1 Vangl1,2DKO, Ksp-Cre collecting duct stained for Fz6 (red) and E-Cadherin (blue). Scale bars: 5 μm. See also Figure S3.
Effects of PCP mutation on postnatal tubules
The mutants examined thus far are perinatally lethal, precluding analyses of potential adult kidney phenotypes. Furthermore, observations from systemic mutants do not rule out the possibility of non-autonomous perturbation of tubule development. We therefore made conditional Vangl1/Vangl2 double knockouts targeted to the collecting ducts from early branching ureteric bud stage (Vl1,2DKO, Ksp-Cre; see methods). Eliminating the only two Vangl paralogs is expected to severely compromise or abolish core PCP signaling. Vl1,2DKO, Ksp-Cre aminals were viable and survived long into adulthood, and recombination in collecting ducts was complete (Supplemental Figure 4A–A″,B–B″). Asymmetry of Fz6 localization was abolished, confirming effectively complete disruption of core PCP signaling (Figure 3E–E″). Macroscopically, Vl1,2DKO, Ksp-Cre kidneys showed no apparent phenotype (Supplemental Figure 4C–D). However, collecting ducts observed in thick sections showed irregular morphology compared to wildtype, consistent with varying duct diameter (Figure 4A–B). We therefore measured collecting duct diameter of postnatal day 1 (P1) Vl1,2DKO, Ksp-Cre animals. Because, to this point, diameter measurements were performed in samples prepared in low calcium dehydration conditions, we first verified that standard fixation (see methods) produced comparable results by repeating measurements on E18.5 Vangl2Lp/Lp kidneys. These samples showed an identical trend, but with larger absolute values due to lack of dehydration (Figure S4E–H, Related to Figure 4). With standard fixation, P1 Vl1,2DKO, Ksp-Cre collecting ducts showed broader diameter distribution compared to wildtype (Figure 4E), much like the E18.5 Vangl2Lp/Lp and Fz3−/−;Fz6−/− mutants, and median diameter was larger than wildtype, but less dramatically so. PCP therefore impacts tubule diameter into the postnatal period, and is required autonomously in ductal epithelial cells.
Figure 4. Phenotype of Vangl1,2 DKO kidneys at P1 and 16 weeks.

(A–B) Longitudinal sections of WT control and Vangl1,2 DKO collecting ducts stained for AQP2 (marking lumens) showing more irregular diameter of the Vangl1,2 DKO ducts along their lengths. Samples are 30 μm thickness. See also Movies S1 and S2. (C–C″) Gross, and H&E sections of WT control and (D–D″) Vangl1,2 DKO mutant kidneys showing absence of cysts. (E–F) Distribution plots of collecting duct diameters of WT and Vangl1,2 DKO at P1 and 16 weeks, measured with standard fixation, showing broader distribution in Vangl1,2 DKO compared to WT. (G–H) Distribution plots of collecting duct diameters showing reduction in overall diameters from E18.5 to 16 weeks in both WT and mutant ducts. Scale bars: A,B,C″,C‴,D″,D‴, 100 μm; C,D, 5 mm; C′,D′, 1 mm. See also Figure S4 and Table S1.
Loss of tubule PCP is not cystogenic
Because disruption of PCP has been proposed to be cystogenic, we aged the Vl1,2DKO, Ksp-Cre mice for up to 50 weeks. Remarkably, and contrary to common conjecture, none of 17 animals aged from 3–50 weeks (Table 1) developed renal cysts (Figure 4C–C‴, D–D‴). Indeed, median tubule diameter continued to decrease into adulthood as it did in wildtype. The median diameter of Vl1,2DKO, Ksp-Cre collecting ducts at 16w decreased compared to P1, yet remained somewhat larger than wildtype, and diameter distribution was broader (Figure 4F). PCP is therefore required to ensure the uniformity of tubule diameters throughout development. During embryogenesis, mutant tubules decrease their diameter less uniformly and reliably than wildtype tubules, leading to both a larger median diameter and a broader diameter distribution. Postnatally, wildtype and mutant tubules continue to decrease in diameter, presumably by a PCP independent process, but the non-uniformity of PCP mutant tubule diameters remains.
PCP signaling controls cellular rearrangement and oriented cell divisions
PCP dependent CE, while only inferred in mice, is hypothesized to underlie narrowing of tubule diameter and concurrent reduction in the number of cells comprising tubule circumference[8, 9]. We therefore measured cell number in circumferential cross-sections of tubules over time. As previously described[8, 9], the cross-sectional cell number in wildtype collecting duct and proximal tubules decreases throughout embryonic development, with the most marked reduction occurring between E16.5 and E18.5 (Figure 5A). However, in Vangl2Lp/Lp mutants, collecting duct cross-sectional cell number decreases more slowly, with the most dramatic difference between mutant and wildtype observed at E17.5, suggesting a maximum perturbation of processes occurring between E16.5 and E17.5. The difference between wildtype and mutant persisted through E18.5. A similar effect was observed in proximal tubules (Figure 5B). In both tubule types, the decline in cell counts in mutants was slowed but not arrested. Thus, either Vangl2Lp/Lp only partially disrupts the relevant mechanism(s), or fully inhibits only a subset of several mechanisms that lead to the reduction in cross-sectional cell number. Due to the difficulty in breeding Fz3−/−;Fz6−/− mice, we were only able to analyze tubules at E18.5. Their cross-sectional cell number was very similar to that of Vangl2Lp/Lp mice (Figure 5A,B).
Figure 5. CE and OCD in WT and Vangl1,2 DKO collecting ducts.

(A–B) Cell number ± SEM per tubule cross-section at the indicated stages in WT control, Vangl2Lp/Lp and Fz3−/−; Fz6−/−collecting ducts and proximal tubules, showing a slowed decrease in mutants compared to WT. Box and whisker format shows the minimum, lower quartile, median, upper quartile and maximum values; + indicates mean. * = p<0.0001 by Mann-Whitney U test. (C–D) Orientation of cell divisions in WT control and in Vl1,2DKO collecting ducts assayed using the histone H3 immunolabeling method[10], quantified at P1. Difference between WT control and Vl1,2DKO is significant at p<0.0001 by Mann-Whitney U test. Representative images showing cell divisions labeled for AQP2 (green; collecting duct), histone H3 (red; dividing nuclei) and DAPI (blue; all nuclei). See also Movies S1, S2 and Figure S5. Scale bars: 10 μm.
Convergent extension is reflected in cellular elongation in the direction of cell intercalation[6]. Unlike previous reports, we observe a bimodal distribution of orientations in wildtype tubules during narrowing, but like Wnt9b−/− mutants that fail to efficiently narrow collecting duct diameters[8], cellular long axes in Vangl2Lp/Lp tubules were shifted toward the tubule axis compared to wildtype (Figure S5, Related to Figure 5). These results support existing evidence that core PCP factors control cellular orientation and CE during the embryonic phase of renal tubule diameter reduction.
PCP signaling controls oriented cell division in tubules
Whereas oriented cell divisions are largely parallel to the tubule axis in wildtype tubules[10], off-axis cell division has been observed in a variety of mutants associated with polycystic kidney disease[10, 11, 13, 15, 16], and is also observed in Pkhd1del4/del4 mouse mutants, which do not develop cysts[15]. We found that in P1 Vl1,2DKO, Ksp-Cre collecting ducts, cell division orientations are significantly off-axis relative to the largely parallel wildtype divisions (Figure 5C,D, Movies S1 and S2). Therefore, PCP signaling regulates renal tubular OCD.
Primary cilia regulate planar polarized behaviors but not core PCP signaling
Many cilium-associated PKD genes, as well as early genetic ablation of primary cilia (through loss of Kif3a) that result in the development of cysts by postnatal day 30 (P30)[40], also show defective CE and OCD. This observation led to the hypothesis that these mutations induce cystogenesis, at least in part, by disrupting PCP signaling. Indeed, results from multiple systems have led to proposals of a mechanistic relationship between PCP signaling and primary cilia, yet no clear understanding of the hypothesized relationship has emerged[41]. We therefore examined the relationship between primary cilia and PCP in renal tubule development.
We determined whether primary cilia contribute to regulation of tubule diameter during embryonic tubulogenesis, a time when PCP signaling contributes to this process. In KspCre;Kif3acko/cko mice at E18.5, cilia were nearly ablated from collecting duct cells, leaving only short ciliary remnants (Supplementary Figure S6B,C, Related to Figure 6). In these kidneys, the median diameter of collecting duct tubules was significantly increased and the distribution broadened compared to wildtype, comparable to Vangl2Lp/Lp and Fz3−/−;Fz6−/− kidneys (Figure 6A,B). Kif3a deletion also had an effect similar to that of the PCP mutants on both the cross-sectional cell number as well as on the distribution of cellular orientations with respect to the tubule axis (Figure 6C and Figure S6A, Related to Figure 6). These similar phenotypes may be explained either by dependence of PCP on primary cilia or vice versa, or by their independent activity on CE and OCD.
Figure 6. PCP in cystic kidneys.

(A) Median collecting duct diameters at E18.5 for WT control, Vangl2Lp/Lp, Fz3−/−; Fz6−/− and KspCre;Kif3acko/cko. Data for WT control, Vangl2Lp/Lp, Fz3−/−; Fz6−/− are from figure 1H. For KspCre;Kif3acko/cko, median = 20.1 μM and * = p<0.0001 compared to wildtype by Mann-Whitney U test. (B) Distribution plots of the measurements from (A). (C) Circumferential cell number for E18.5 collecting ducts for WT control, Vangl2Lp/Lp, Fz3−/−; Fz6−/− and KspCre;Kif3acko/cko. Displayed in box and whisker format showing the minimum, lower quartile, median, upper quartile and maximum values; + indicates mean. * = p<0.0001 by Mann-Whitney U test. Data for WT control, Vangl2Lp/Lp, Fz3−/−; Fz6−/− are from figure 5A. (D–D″) E18.5 collecting duct from KspCre;Kif3acko/cko precystic kidneys showing asymmetrically localized Vangl1 (green); E-Cadherin (blue). (E–E″) P1 Pkhd1del4/del4 collecting duct showing asymmetrically localized Vangl1 (green) and Fz6 (red). Scale bars: 5 μm. See also Figure S6.
We therefore examined PCP signaling in KspCre;Kif3acko/cko mutants by assaying asymmetric PCP factor localization. At E18.5, Vangl1 and Fz6 retain their oppositely oriented asymmetric localization, indicating that core PCP signaling is intact (Figure 6D; not shown). Thus, a reduction of primary cilia length sufficient to cause a tubule diameter defect similar to that seen in the core PCP mutant kidneys showed no effect on core PCP signaling. Primary cilia therefore appear to influence tubule diameter independent of core PCP signaling. However, because ciliary remnants still exist in KspCre;Kif3acko/cko mice at E18.5, these data do not strictly rule out the possibility that renal tubule cilia may be required for generation or maintenance of core PCP factor asymmetric localization.
Pkhd1del4/del4 mutant mice, which do not develop cysts, show off-axis cell division[15], and median tubule diameter is increased and more broadly distributed than wildtype, albeit modestly (Figure S6D; Related to Figure 6). As in Kif3acko/cko, core PCP proteins Vangl1 and Fz6 retain their asymmetric localization in P1 Pkhd1del4/del4 mutant collecting ducts (Figure 6E–E″), indicating that the off-axis cell division in these mutants cannot be attributed to disruption of PCP signaling. Together, these results suggest that PCP signaling is intact when tubule cilia are mutated, and are reminiscent of the findings that Ift88 mutation in the cochlea disrupts sensory hair cell polarity and that Kif3a knockout in ependymal cells disrupts basal body polarity each without altering PCP protein asymmetry[42, 43].
Robust asymmetric localization of core PCP components persists in cyst-lining cells
Several weeks into adulthood, PCP signaling in wildtype ducts is substantially diminished. We therefore determined whether PCP signaling is similarly diminished in P30 Kif3acko/cko mutant collecting ducts, when Kif3a protein is depleted, cilia are severely shortened, and a strongly cystic phenotype is observed as previously reported[40] (Figure 7A,A′,B,B′). Whereas in unaffected P30 proximal tubules, levels of Vangl1 and Fz6 at the lateral cell membrane were significantly diminished from P1 levels (Figure 7C–C‴), In the cyst-lining cells of KspCre;Kif3acko/cko mice, cilia were absent, and strikingly, localization of core PCP components was similar to that observed prenatally. Robust Vangl1 and Fz6 expression co-localized with E-Cadherin along the lateral membrane of tubule cells (Figure 7D–D‴, compare to Figure 7C–C‴; Figure S6F–F″, compare to Figure S6E–E″, Related to Figure 7) and was asymmetric in cyst-lining cells, identical to that observed embryonically (Figure 7E–E″ and Figure S6G–G″, Related to Figure 6; movie 1). Consistent with these observations, Fz3 was previously found to be elevated in cysts, though asymmetric localization was not noted[14]. We were unable to determine the global orientation (proximal-distal axis) of cysts and thus cannot rule out a potential disruption to orientation of polarity with respect to the tubule axis. These results demonstrate that in renal tubule cells, PCP signaling, as revealed by the asymmetric subcellular localization of core PCP proteins, does not depend on primary cilia. Indeed, in adult (P30) kidneys, at a developmental stage at which PCP signaling is no longer active in wildtype, cystic tubule epithelia maintain or restore PCP signaling, perhaps because tubules are inappropriately in an embryonic-like, proliferative state.
Discussion
It has been hypothesized that disruption of PCP, and therefore disruption of CE and OCD, contributes to PKD in a variety of cilium associated mutant conditions. However, we find that contrary to prior conjecture, not only is disruption of PCP signaling not required for cystogenesis, but PCP signaling is retained in adult cysts when it would otherwise be attenuated. We suggest that PCP signaling is part of the developmental program in proliferative renal tubules, and that this program is abnormally activated in proliferating cystic epithelia.
While the core PCP system polarizes cells with respect to their neighbors, global directional cues are also required[18]. In the renal tubules, directional signals remain to be identified. Wnt’s have been proposed as directional signals in Drosophila[25] and in vertebrates[18], though their ability to act as instructive signals is not firmly established. Several Wnt’s required for kidney development[8, 13, 27, 28] should be considered as candidate directional cues. The requirement of Fat4 and Dchs1 for normal OCD suggests the possibility that these provide a directional cue analogous to that proposed for Drosophila Ft-Ds[11]. However, the predominantly non-epithelial expression of these proteins is not easily reconciled with this model. Further work will be required to identify global PCP directional signals in the kidney.
In renal tubules, primary cilium-associated signals are required for OCD (and likely CE)[44]. We find that intact PCP signaling is also required to correctly regulate these events. Our results are most consistent with the model that primary cilia and PCP are independently required for this regulation. PCP is intact in the absence of primary cilia, suggesting that ciliary signals regulating OCD do not act by altering PCP signaling. And while it is formally possible that PCP signaling acts via primary cilia to control OCD, this appears unlikely for several reasons. First, primary cilia appear to be intact in PCP mutants (not shown). Second, PCP regulates OCD in Drosophila tissues that do not have primary cilia. Third, it is difficult to envision a model in which directional information from the PCP system could pass through primary cilia that do not themselves acquire a polarized localization. We therefore propose that PCP provides directional information for OCD and that a primary cilium signal is necessary to enable the response to this directional information.
By conditionally knocking out both Vangl1 and Vangl2 in collecting ducts, an intervention that severely disrupts core PCP signaling, we find that no cysts develop in adults up to one year of age. Therefore, disrupting PCP signaling, which in turn causes off-axis OCD and interferes with CE, does not cause cysts. Similarly, Pkhd1del4/del4 mutants display off-axis OCD but do not become cystic[15]. These results imply that cilium-associated mutations that induce either prenatal or postnatal cysts must do something other than interfere with OCD and CE.
It is likely that proliferative signals fulfill this role. Loss of Kif3a induced conditionally in adult collecting ducts produced cysts only if proliferation was also induced; the induction of cysts by earlier Kif3a loss apparently requires developmentally controlled proliferation[12]. Furthermore, the cystic phenotype of Pkd1 or Pkd2 mutant collecting ducts (encoding polycystin-1 and -2) is suppressed by loss of cilia [45]. Removing polycystins therefore appears to activate a cilium-dependent proliferative signal for robust cystogenesis that is attenuated by disrupting cilia. Blocking PCP does not activate such a signal.
The observation that Fat4 and Dchs1 mutation cause dilations/cysts was suggested to support the idea that impaired PCP signaling is cystogenic[11]. However, whether or not Fat4 and Dchs1 provide PCP directional information in the kidney, the observation that Vl1,2DKO; Ksp-Cre collecting ducts do not produce cysts argues against the model that Fat4 and Dachs1 act solely by disrupting core PCP signaling to induce cysts. We suggest that these mutations activate a proliferative signal. Drosophila ft and ds mutations activate Hippo signaling[46], and Hippo signaling is activated in human ADPKD and ARPKD[47], but Hippo signaling is not activated in Fat4 or Dchs1 associated mouse mutant dilations/cysts[33]. If Fat4 and Dchs1 mutations induce proliferation, this may be indirect, as the earliest indications of abnormal growth were reported to be increased cell death and decreased proliferation[33]. Of note, integrin-β1 is required for the proliferative response to Pkd1 mutation, suggesting another possible proliferative pathway[48]. Taken together, these observations suggest that cilium-based and non-cilium based proliferative signals may be capable of contributing to cystogenesis.
As disrupted OCD is neither necessary nor sufficient for cystogenesis, it is unclear why it is often associated with conditions that cause cysts. One possibility is that disrupting OCD in combination with another signal is one of several distinct mechanisms of cystogenesis. This remains to be explored.
In animals ranging in size from the shrew to the elephant, proximal tubule diameters are essentially invariant[49]. Because mean arterial pressure is independent of size and therefore glomerular filtration pressure is likely also size independent, renal physiology seems to dictate an optimal diameter. One potential consequence of deviation from this optimum is variation in resistance resulting in aberrant nephron pressure. While Vl1,2, DKO median diameters are larger than in wildtype, we also see some points at which diameter is smaller than wildtype, predicting that some nephrons will be subject to abnormally high pressure. Consistent with this, we observe the emergence of Bowman’s space dilations consistent with Type IV Glomerulocystic kidney disease (GCKD)[50] in a fraction of older Vl1,2, DKO animals (Figure S7, Related to Figure 7, and Table S1).
Our results show that PCP regulates OCD and likely CE in the renal tubules, and that it is required to assure uniform tubule diameter. PCP signaling and primary cilium-associated signals appear to be independently required for maintenance of OCD. Although perturbation of OCD is frequently associated with PKD, we find that blocking PCP does not induce cysts, nor is PCP disturbed in cysts induced by disruption of primary cilia. Therefore, the commonly proposed and intuitively pleasing model in which PCP is tightly associated with cystogenesis is not correct.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeff Axelrod (jaxelrod@stanford.edu).
Experimental Model and Subject Details
Animals
Vangl1−/− (previously referred to as Vangl1cko/cko)[51], KspCre;Kif3acko/cko;[40], Fz3−/−;Fz6−/−[38], Vangl2cko/cko[53], Pkhd1del4/del4[15] and Vl2Lp/Lp (JAX) mice and genotyping have been previously described. KspCre; Vangl1cko/cko; Vangl2cko/cko (Vl1,2, DKO, Ksp-Cre) animals were bred from heterozygotes. KspCre drives Cre expression beginning in the early branching ureteric bud stage[55]. Animals were healthy with normal immune status, were not subject to prior procedures, were drug naïve and were reared by standard husbandry and housed in standard cages in the Stanford Animal Care Facility. Approximately equal numbers of animals of either sex were used for all experiments. Specimens were collected at the ages indicated in text and legends. All procedures involving animals were approved by the Stanford University Institutional Animal Care and Use Committee in accordance with established guidelines for animal care.
Method Details
Harvesting, processing and sectioning of kidneys
For dehydration fixation, postnatal and embryonic kidneys (E13.5 – E18.5) were harvested and sub dissected in PBS. Collected kidneys were then placed in 4% paraformaldehyde in PBS for one hour and transferred into a 30% sucrose solution in calcium and magnesium free PBS at 4°C overnight. The following day, the kidneys were submerged in OCT mounting media and frozen in blocks in an ethanol and dry ice bath and could be stored for further use at −80°C. 12 – 20 μ sections through kidneys were made using a Leica CM 3050S cryostat and mounted on charged slides.
For standard fixation, kidneys were harvested and sub dissected in OCT mounting media and frozen in blocks in liquid nitrogen. The samples could be stored for further use at −80°C. 10 – 30 μ sections through kidneys were made using a Leica CM 3050S cryostat and mounted on charged slides. Samples were fixed in 3.7% formaldehyde or 4% paraformaldehyde in PBS for 10 minutes and soaked in 0.2 % triton-X100 in PBS for 5 minutes prior to immunostaining.
For hematoxylin and eosin (H&E) staining, kidneys were fixed in 4% paraformaldehyde in PBS for one hour, then in 70% ethanol for 12 h prior to paraffin-embedding. Paraffin blocks were sectioned at 4 μ thickness and stained with H&E.
Antibodies and reagents
The dilution and sources for primary antibodies used were as follows: rabbit anti-Vangl1 (1:200, Sigma), rabbit anti-Vangl2 (1:200, Santa Cruz), goat anti-Frizzled6 (1:200, R&D systems), goat anti-Frizzled3 (1:100, R&D systems), rat anti-Frizzled3 (1:100, R&D systems), rat anti-E-Cadherin (1:200, Invitrogen) and mouse anti-E-cadherin (1:200, BD Transduction Laboratories), mouse anti-acetylated-α-tubulin (1:500, Sigma), rabbit anti-phospho-Histone H3 (Ser10) (1:200, Millipore) and mouse anti-β-catenin (1:100, BD Transduction Laboratories™). The following secondary antibodies were used at 1:500 and purchased from Molecular Probes (now ThermoFisher): Alexa 488, 594 donkey-anti-rabbit, Alexa 488, 546 donkey-anti-goat, Alexa 594, 633 donkey-anti-rat, Alexa 647 chicken-anti-rat and Streptavidin Alexa 594 conjugate. Phalloidin conjugated to Alexa 633 was used at 1:50. Rhodamine-conjugated Dolichos Biflorus Agglutinin lectin (DBA, 1:50, Vector Laboratories) and goat anti-AQP2 (1:200, Santa Cruz) were used to stain collecting duct. Fluorescein-conjugated Lotus Tetragonolobus lectin (LTL, 1:200, Vector Laboratories) and biotinylated Lotus Tetragonolobus lectin (1:200, Vector Laboratories) were used to stain proximal tubules. DAPI (300nM, Sigma), Sytox Green (1:20,000, Invitrogen) and 7-aminoactinomycin D (7-AAD, 1:40, Invitrogen) were used to stain nuclei.
Immunostaining
Immunostaining was done according to the following steps:
3× 10min wash in PBT (Phosphate Buffered Saline [PBS] + 0.1% Triton X-100)
1 hour incubation in PBT +10% normal horse serum (NHS) or 1% BSA in PBS
Overnight incubation with primary antibodies at required dilution in PBT +10% NHS
Next day, 3× 10min wash in PBT
1 hour incubation with secondary antibodies at required dilution in PBT +10% NHS
3× 10 min wash in PBT
Add mounting media and apply coverslip
Imaging
For immunofluorescence, sections were mounted in ProLong® Gold Antifade Reagent (Invitrogen) or Vectashield® (Vector Laboratories) mounting medium. Specimens were imaged with a Leica SP5 or SP8 confocal microscope, using accompanying acquisition and 3D software. For bright field, kidneys were imaged with a Nikon Eclipse E1000M microscope and acquired using Spot imaging software.
Tubule diameter and cell number measurements
To measure the diameter of kidney tubules, we stained collecting duct (DBA or AQP2) and proximal tubules (LTL), together with E-Cadherin. Branching tubules were avoided and we limited our analysis to tubules that were roughly circular in cross-section[8]. To compensate for obliqueness of section, the shortest diameter was measured using ImageJ or Leica software. For consistency, collecting duct diameter was measured in the cortex and proximal medulla near the corticomedullary junction. Due to non-gaussian distributions, medians were calculated and significance determined by the Mann-Whitney U test. The number of cells within the cross-section of kidney tubules was measured in a similar manner, using (1:20,000, Invitrogen) and 7-aminoactinomycin D (7-AAD, 1:40, Invitrogen) to label nuclei. Nuclei were manually counted in ImageJ. Data are reported as mean ± SEM. Measurements could not be performed in a blinded fashion, as the genotypes are readily recognizable. To address this, for many of the conditions, measurements were performed independently by two different individuals; good concordance between the independent results was achieved.
Measurement of oriented cell division
The method for measuring orientation of mitotic angles was adopted and modified from[10]. Kidney sections (30 μ thick) were stained with anti-phospho-Histone H3 (Ser10) antibody to label the chromosomes of dividing cells in late anaphase and telophase. Sections were co-stained with anti-AQP2 or DBA to label the tubular apical membranes. Z-stack images were acquired by confocal microscopy. The orientation of cell division was determined by measuring the angle between the mitotic spindles of dividing cells and the longitudinal axis of the collecting ducts. Angular distributions were visualized by rose diagram using Oriana software.
Measurement of cell orientation
The orientation of cells was measured in the collecting duct and proximal tubules of kidneys from E15.5 – E18.5 similar to previous reports[8]. For cell orientation analysis, 20 μ kidney sections were stained for DBA, LTL and E-Cadherin. Confocal Z-stacks were captured through entire tubules to ensure that branching tubules were avoided. From these Z-stacks, the outlines of cells, demarcated by E-Cadherin staining, immediately below the apical (luminal) surface were traced using ImageJ (NIH). Cells that shared an edge with the outside of the tubule in our images were excluded from analysis. The software then calculated the length of the long and short axes and the angle of the long axis for each cell. We limited our analyses of cell orientation to cells that possessed a length:width ratio greater than 1.2[8]. The angle of orientation for each cell was calculated as the difference between angle of the long axis of each cell and the angle of the tubule segment from which it is a part of. The data was visualized in histograms by graphing the percentage of events in 10° bins.
Quantitative Real-Time RT-PCR
Total RNA was isolated from 3 P1 kidneys and 3 16w kidneys using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was generated with the SuperScript III 1st Strand cDNA Synthesis Kit (Life Technologies, Carlsbad, CA). Quantitative realtime PCR was performed in triplicate with SYBR GreenER qPCR SuperMix for ABI PRISM (Life Technologies, Carlsbad, CA) in an Applied Biosystems StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA) using the primers in Table S2. PCR was performed for 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds.
Quantification and Statistical Analysis
Quantification of PCP protein asymmetry
Samples prepared by the dehydration method were double labeled with either E-Cadherin or β-Catenin and the PCP protein of interest. At cell-cell junctions that were separated by dehydration, regions encompassing proximal and distal membranes as defined by the E-Cadherin or β-Catenin staining were defined using ImageJ software, and average pixel intensity was recorded. On average, the proximal and distal intensities were approximately equal for E-Cadherin and β-Catenin. The same regions were then used to measure intensities in the PCP channel. PCP intensities were normalized to the corresponding E-Cadherin or β-Catenin measurements, and the ratio of proximal to distal (Vang) or distal to proximal (Fz) of the normalized values was determined. The average intensity ratio (IR) for each PCP protein was then determined and reported in the associated Figure panel and graphically ± SEM in Figure S3. We used a minimum of 10 well imaged and separated proximal-distal junctions, and in most cases more for each condition, except for the E13.5 measurements (n=8 for Vangl1 and n=5 for Fz6).
Statistical Analyses
The Mann-Whitney U test was used to determine significance of difference in median measurements due to the non-Gaussian distribution of data sets.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Vangl1 | Sigma-Aldrich | HPA025235 |
| Rabbit anti-Vangl2 | Millipore | ABN373 |
| Goat anti-Frizzled6 | R&D systems | AF1526 |
| Goat anti-Frizzled3 | R&D systems | AF1001 |
| Rat anti-Frizzled3 | R&D systems | MAB1001 |
| Rat anti-E-Cadherin | Thermo Fisher Scientific | 131900 |
| Mouse anti-E-cadherin | BD Transduction Laboratories™ | 610181 |
| Mouse anti-acetylated-α-tubulin | Sigma-Aldrich | 6-11B-1 |
| Rabbit anti-phospho-Histone H3 (Ser10) | Millipore | 06-570 |
| Mouse anti-β-catenin | BD Transduction Laboratories™ | 610153 |
| Goat anti-AQP2 | Santa Cruz Biotechnology | SC-9882 |
| Alexa 488 donkey-anti-rabbit | Thermo Fisher Scientific | A-21206 |
| Alexa 594 donkey-anti-rabbit | Thermo Fisher Scientific | A-21207 |
| Alexa 488 donkey-anti-goat | Thermo Fisher Scientific | A-11055 |
| Alexa 546 donkey-anti-goat | Thermo Fisher Scientific | A-11056 |
| Alexa 594 donkey-anti-rat | Thermo Fisher Scientific | A-21209 |
| Alexa 633 goat-anti-rat | Thermo Fisher Scientific | A-21094 |
| Alexa 647 chicken-anti-rat | Thermo Fisher Scientific | A-21472 |
| Alexa 647 donkey-anti-mouse | Thermo Fisher Scientific | A-31571 |
| Phalloidin conjugated to Alexa 635 | Thermo Fisher Scientific | A-34054 |
| Streptavidin conjugated to Alexa 488 | Thermo Fisher Scientific | S-32354 |
| Streptavidin conjugated to Alexa 594 | Thermo Fisher Scientific | S-32356 |
| Fluorescein-conjugated Lotus Tetragonolobus lectin (LTL) | Vector Laboratories | FL-1321 |
| Biotinylated Lotus Tetragonolobus lectin (LTL) | Vector Laboratories | B-1325 |
| Rhodamine-conjugated Dolichos Biflorus Agglutinin (DBA) | Vector Laboratories | RL-1032 |
| DAPI | Sigma-Aldrich | D-9542 |
| Sytox Green (1:20,000, Invitrogen) | Thermo Fisher Scientific | S7020 |
| 7-aminoactinomycin D (7-AAD, 1:40, Invitrogen) | Thermo Fisher Scientific | A1310 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Qiagen RNeasy Mini Kit | Qiagen | 74104 |
| SuperScript III 1st Strand cDNA Synthesis Kit | Thermo Fisher Scientific | 18080051 |
| Power SYBR™ Green PCR Master Mix | Thermo Fisher Scientific | 4367659 |
| Applied Biosystems StepOnePlus Real-Time PCR System | Applied Biosystems | 4376592R |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Vangl1cko/cko mouse | Created in our lab [51] | N/A |
| Vangl1−/− (previously referred to as Vangl1CKOΔ /Δ) mouse | Created in our lab [52] | N/A |
| Vangl2Lp/Lp mouse | The Jackson Laboratory | JAX: 000220 |
| Vangl2cko/cko mouse | Gift from Y. Yang [53] | N/A |
| Kif3acko/cko mouse | Gift from J. Helms [40] | N/A |
| Ksp-Cre mouse | Gift from The George M. O’Brien Kidney Center [40] | N/A |
| Fz3−/−;Fz6−/− mouse kidney samples | Gift from H. Chang and J. Nathans [38] | N/A |
| Pkhd1del4/del4 mouse kidney samples | Gift from The George M. O’Brien Kidney Center [54] | N/A |
| Oligonucleotides | ||
| See Table S2 for oligonucleotides for qRT-PCR | Stanford PAN facility | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
Supplementary Material
WT collecting duct stained with AQP2 (green, labeling lumen), anti-phospho-Histone H3 (red, dividing chromosomes) and DAPI (blue, all nuclei).
Vl1,2, DKO collecting duct stained with AQP2 (green, labeling lumen), anti-phospho-Histone H3 (red, dividing chromosomes) and DAPI (blue, all nuclei).
A cyst in a KspCre; Kif3acko/cko kidney labeled for Vangl1 (green) and E-Cadherin (blue) in cyst lining cells, showing asymmetric localization of Vangl1.
Acknowledgments
We thank Jill Helms and Yingzi Yang for mice, Hao Chang and Jeremy Nathans for Fz mutant kidneys, Klara Fekete for help with animal husbandry, Neeraja Kambham and Andrew Connolly for pathology consults, Amarjeet Grewall and Pauline Chu for for H&E staining, Daniel Gray for useful discussions, and Stefan Somlo, Greg Pazour and Axelrod lab members for comments on the manuscript. Work was supported by the Uehara Memorial Foundation Research Fellowship and the JSPS Postdoctoral Fellowships for Research Abroad (KK), NIH 5 T32 CA 9302-34 and National Kidney Foundation Postdoctoral Fellowships (RB), an American Heart Association Scientist Development Grant (ARG), NIH R37 GM059823, NIH R01 GM098582 and the Stanford Child Health Research Institute and the Stanford NIH-NCATS-CTSA (grant no. UL1 TR001085) (JDA) and The George M. O’Brien Kidney Center at Yale (P30 DK079310).
Footnotes
Author Contributions
Conceptualization, K.K., R.B. and J.A.; Methodology, K.K., R.B., E.V. and J.A.; Formal Analysis, K.K.; Investigation, K.K., R.B. and T.V.; Writing – Original Draft, K.K. and J.A.; Writing – Review & Editing, K.K., E.V. and J.A.; Funding Acquisition, K.K. and J.A.; Resources, E.V. and A.G.; Supervision, J.A.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kagan KO, Dufke A, Gembruch U. Renal cystic disease and associated ciliopathies. Curr Opin Obstet Gynecol. 2017 doi: 10.1097/GCO.0000000000000348. [DOI] [PubMed] [Google Scholar]
- 2.Cornec-Le Gall E, Audrezet MP, Le Meur Y, Chen JM, Ferec C. Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum Mutat. 2014;35:1393–1406. doi: 10.1002/humu.22708. [DOI] [PubMed] [Google Scholar]
- 3.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 4.Malicki JJ, Johnson CA. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017;27:126–140. doi: 10.1016/j.tcb.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nigro EA, Castelli M, Boletta A. Role of the Polycystins in Cell Migration, Polarity, and Tissue Morphogenesis. Cells. 2015;4:687–705. doi: 10.3390/cells4040687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallingford JB. Planar cell polarity and the developmental control of cell behavior in vertebrate embryos. Annu Rev Cell Dev Biol. 2012;28:627–653. doi: 10.1146/annurev-cellbio-092910-154208. [DOI] [PubMed] [Google Scholar]
- 7.Lienkamp SS, Liu K, Karner CM, Carroll TJ, Ronneberger O, Wallingford JB, Walz G. Vertebrate kidney tubules elongate using a planar cell polarity-dependent, rosette-based mechanism of convergent extension. Nat Genet. 2012;44:1382–1387. doi: 10.1038/ng.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet. 2009;41:793–799. doi: 10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castelli M, Boca M, Chiaravalli M, Ramalingam H, Rowe I, Distefano G, Carroll T, Boletta A. Polycystin-1 binds Par3/aPKC and controls convergent extension during renal tubular morphogenesis. Nat Commun. 2013;4:2658. doi: 10.1038/ncomms3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38:21–23. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- 11.Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]
- 12.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu J, Carroll TJ, Rajagopal J, Kobayashi A, Ren Q, McMahon AP. A Wnt7b-dependent pathway regulates the orientation of epithelial cell division and establishes the cortico-medullary axis of the mammalian kidney. Development. 2009;136:161–171. doi: 10.1242/dev.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luyten A, Su X, Gondela S, Chen Y, Rompani S, Takakura A, Zhou J. Aberrant regulation of planar cell polarity in polycystic kidney disease. J Am Soc Nephrol. 2010;21:1521–1532. doi: 10.1681/ASN.2010010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol. 2010;21:295–302. doi: 10.1681/ASN.2009060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonassen JA, SanAgustin J, Baker SP, Pazour GJ. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J Am Soc Nephrol. 2012;23:641–651. doi: 10.1681/ASN.2011080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brzoska HL, d’Esposito AM, Kolatsi-Joannou M, Patel V, Igarashi P, Lei Y, Finnell RH, Lythgoe MF, Woolf AS, Papakrivopoulou E, et al. Planar cell polarity genes Celsr1 and Vangl2 are necessary for kidney growth, differentiation, and rostrocaudal patterning. Kidney Int. 2016;90:1274–1284. doi: 10.1016/j.kint.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vladar EK, Antic D, Axelrod JD. Planar cell polarity signaling: The developing cell’s compass. Cold Spring Harb Perspect Biol. 2009;1:a002964. doi: 10.1101/cshperspect.a002964. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tree DR, Ma D, Axelrod JD. A three-tiered mechanism for regulation of planar cell polarity. Semin Cell Dev Biol. 2002;13:217–224. doi: 10.1016/s1084-9521(02)00042-3. [DOI] [PubMed] [Google Scholar]
- 20.Tree DR, Shulman JM, Rousset R, Scott MP, Gubb D, Axelrod JD. Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell. 2002;109:371–381. doi: 10.1016/s0092-8674(02)00715-8. [DOI] [PubMed] [Google Scholar]
- 21.Amonlirdviman K, Khare NA, Tree DR, Chen WS, Axelrod JD, Tomlin CJ. Mathematical modeling of planar cell polarity to understand domineering nonautonomy. Science. 2005;307:423–426. doi: 10.1126/science.1105471. [DOI] [PubMed] [Google Scholar]
- 22.Butler MT, Wallingford JB. Planar cell polarity in development and disease. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matis M, Axelrod JD. Regulation of PCP by the Fat signaling pathway. Genes Dev. 2013;27:2207–2220. doi: 10.1101/gad.228098.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharp KA, Axelrod JD. Prickle isoforms control the direction of tissue polarity by microtubule independent and dependent mechanisms. Biol Open. 2016;5:229–236. doi: 10.1242/bio.016162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu J, Roman AC, Carvajal-Gonzalez JM, Mlodzik M. Wg and Wnt4 provide long-range directional input to planar cell polarity orientation in Drosophila. Nat Cell Biol. 2013;15:1045–1055. doi: 10.1038/ncb2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tissir F, Goffinet AM. Shaping the nervous system: role of the core planar cell polarity genes. Nat Rev Neurosci. 2013;14:525–535. doi: 10.1038/nrn3525. [DOI] [PubMed] [Google Scholar]
- 27.Huang L, Xiao A, Wecker A, McBride DA, Choi SY, Zhou W, Lipschutz JH. A possible zebrafish model of polycystic kidney disease: knockdown of wnt5a causes cysts in zebrafish kidneys. J Vis Exp. 2014 doi: 10.3791/52156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy II, Xu Q, Naillat F, Ali N, Miinalainen I, Samoylenko A, Vainio SJ. Impairment of Wnt11 function leads to kidney tubular abnormalities and secondary glomerular cystogenesis. BMC Dev Biol. 2016;16:30. doi: 10.1186/s12861-016-0131-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Germino GG. Linking cilia to Wnts. Nat Genet. 2005;37:455–457. doi: 10.1038/ng0505-455. [DOI] [PubMed] [Google Scholar]
- 31.Rock R, Schrauth S, Gessler M. Expression of mouse dchs1, fjx1, and fat-j suggests conservation of the planar cell polarity pathway identified in Drosophila. Dev Dyn. 2005;234:747–755. doi: 10.1002/dvdy.20515. [DOI] [PubMed] [Google Scholar]
- 32.Ishiuchi T, Misaki K, Yonemura S, Takeichi M, Tanoue T. Mammalian Fat and Dachsous cadherins regulate apical membrane organization in the embryonic cerebral cortex. J Cell Biol. 2009;185:959–967. doi: 10.1083/jcb.200811030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao Y, Mulvaney J, Zakaria S, Yu T, Morgan KM, Allen S, Basson MA, Francis-West P, Irvine KD. Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development. 2011;138:947–957. doi: 10.1242/dev.057166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yates LL, Papakrivopoulou J, Long DA, Goggolidou P, Connolly JO, Woolf AS, Dean CH. The planar cell polarity gene Vangl2 is required for mammalian kidney-branching morphogenesis and glomerular maturation. Hum Mol Genet. 2010;19:4663–4676. doi: 10.1093/hmg/ddq397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet. 2008;40:871–879. doi: 10.1038/ng.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao Y, Park A, Sun Z. Intraflagellar transport proteins are essential for cilia formation and for planar cell polarity. J Am Soc Nephrol. 2010;21:1326–1333. doi: 10.1681/ASN.2009091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehaideb SN, Iyengar A, Ueda A, lacobucci GJ, Cranston C, Bassuk AG, Gubb D, Axelrod JD, Gunawardena S, Wu CF, et al. prickle modulates microtubule polarity and axonal transport to ameliorate seizures in flies. Proc Natl Acad Sci U S A. 2014;111:11187–11192. doi: 10.1073/pnas.1403357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Guo N, Nathans J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci. 2006;26:2147–2156. doi: 10.1523/JNEUROSCI.4698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin H, Copley CO, Goodrich LV, Deans MR. Comparison of phenotypes between different vangl2 mutants demonstrates dominant effects of the Looptail mutation during hair cell development. PLoS One. 2012;7:e31988. doi: 10.1371/journal.pone.0031988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–213. doi: 10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet. 2008;40:69–77. doi: 10.1038/ng.2007.54. [DOI] [PubMed] [Google Scholar]
- 43.Guirao B, Meunier A, Mortaud S, Aguilar A, Corsi JM, Strehl L, Hirota Y, Desoeuvre A, Boutin C, Han YG, et al. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat Cell Biol. 2010;12:341–350. doi: 10.1038/ncb2040. [DOI] [PubMed] [Google Scholar]
- 44.Costantini F, Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell. 2010;18:698–712. doi: 10.1016/j.devcel.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45:1004–1012. doi: 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma P, McNeill H. Fat and Dachsous cadherins. Prog Mol Biol Transl Sci. 2013;116:215–235. doi: 10.1016/B978-0-12-394311-8.00010-8. [DOI] [PubMed] [Google Scholar]
- 47.Happe H, van der Wal AM, Leonhard WN, Kunnen SJ, Breuning MH, de Heer E, Peters DJ. Altered Hippo signalling in polycystic kidney disease. J Pathol. 2011;224:133–142. doi: 10.1002/path.2856. [DOI] [PubMed] [Google Scholar]
- 48.Lee K, Boctor S, Barisoni LM, Gusella GL. Inactivation of integrin-beta1 prevents the development of polycystic kidney disease after the loss of polycystin-1. J Am Soc Nephrol. 2015;26:888–895. doi: 10.1681/ASN.2013111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calder WA, 3rd, Braun EJ. Scaling of osmotic regulation in mammals and birds. Am J Physiol. 1983;244:R601–606. doi: 10.1152/ajpregu.1983.244.5.R601. [DOI] [PubMed] [Google Scholar]
- 50.Lennerz JK, Spence DC, Iskandar SS, Dehner LP, Liapis H. Glomerulocystic kidney: one hundred-year perspective. Arch Pathol Lab Med. 2010;134:583–605. doi: 10.5858/134.4.583. [DOI] [PubMed] [Google Scholar]
- 51.Vladar EK, Bayly RD, Sangoram AM, Scott MP, Axelrod JD. Microtubules enable the planar cell polarity of airway cilia. Curr Biol. 2012;22:2203–2212. doi: 10.1016/j.cub.2012.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antic D, Stubbs JL, Suyama K, Kintner C, Scott MP, Axelrod JD. Planar cell polarity enables posterior localization of nodal cilia and left-right axis determination during mouse and Xenopus embryogenesis. PLoS One. 2010;5:e8999. doi: 10.1371/journal.pone.0008999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song H, Hu J, Chen W, Elliott G, Andre P, Gao B, Yang Y. Planar cell polarity breaks bilateral symmetry by controlling ciliary positioning. Nature. 2010;466:378–382. doi: 10.1038/nature09129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallagher AR, Esquivel EL, Briere TS, Tian X, Mitobe M, Menezes LF, Markowitz GS, Jain D, Onuchic LF, Somlo S. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am J Pathol. 2008;172:417–429. doi: 10.2353/ajpath.2008.070381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Igarashi P, Shashikant CS, Thomson RB, Whyte DA, Liu-Chen S, Ruddle FH, Aronson PS. Ksp-cadherin gene promoter. II. Kidney-specific activity in transgenic mice. Am J Physiol. 1999;277:F599–610. doi: 10.1152/ajprenal.1999.277.4.F599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
WT collecting duct stained with AQP2 (green, labeling lumen), anti-phospho-Histone H3 (red, dividing chromosomes) and DAPI (blue, all nuclei).
Vl1,2, DKO collecting duct stained with AQP2 (green, labeling lumen), anti-phospho-Histone H3 (red, dividing chromosomes) and DAPI (blue, all nuclei).
A cyst in a KspCre; Kif3acko/cko kidney labeled for Vangl1 (green) and E-Cadherin (blue) in cyst lining cells, showing asymmetric localization of Vangl1.