

Summary
Although ARF can suppress tumor growth by activating p53 function, the mechanisms by which it suppresses tumor growth independently of p53 are not well understood. Here, we identified ARF as a key regulator of nuclear factor-E2-related factor 2 (NRF2) through complex purification. ARF inhibits the ability of NRF2 to transcriptionally activate its target genes, including SLC7A11, a component of the cystine/glutamate antiporter that regulates reactive oxygen species (ROS)-induced ferroptosis. As a consequence, ARF expression sensitizes cells to ferroptosis in a p53-independent manner while ARF depletion induces NRF2 activation and promotes cancer cell survival in response to oxidative stress. Moreover, the ability of ARF to induce p53-independent tumor growth suppression in mouse xenograft models is significantly abrogated upon NRF2 overexpression. These results demonstrate that NRF2 is a major target of p53-independent tumor suppression by ARF and also suggest that the ARF-NRF2 interaction acts as a new checkpoint for oxidative stress responses.
Keywords: ARF, NRF2, p53, ferroptosis, ROS, oxidative stress, transcriptional regulation
In brief
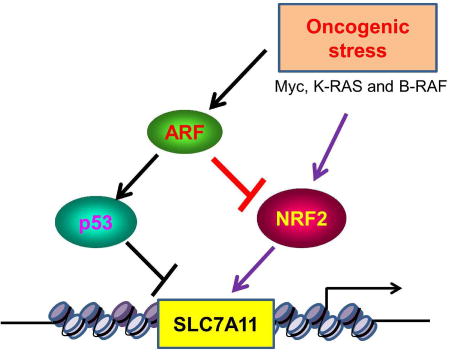
Chen et al., identified ARF as a key regulator of NRF2-mediated activation of SLC7A11, a component of the cystine/glutamate antiporter that regulates reactive oxygen species (ROS)-induced ferroptosis. They showed that the ARF-NRF2 interaction is critical for p53-independent ferroptosis and tumor suppression induced by ARF.
Introduction
Paradoxical results have emerged from recent studies into the effects of oxidative responses in cancer. Although it was popularly thought that antioxidants protect against cancer, accumulating evidence suggests that these agents can actually increase cancer risk (Gorrini et al., 2013; Jaramillo and Zhang, 2013). In particular, genomic analyses of human cancers have uncovered a high frequency of mutations in genes for factors that activate an endogenous antioxidant program (Hayes and McMahon, 2009), such as the NRF2 (nuclear factor erythroid 2-related factor) transcription factor and its repressor, the E3 ligase Keap1 (Kelch-like ECH-associated protein 1). In addition, studies of NRF2-mutant mouse models have demonstrated that the malignantly activated forms of certain oncoproteins, including Myc, K-RAS and B-RAF, can promote tumor cell proliferation in part by stimulating NRF2-mediated expression of endogenous antioxidants and reducing ROS levels (Bauer et al., 2011; Chio et al., 2016; DeNicola et al., 2011; Tao et al., 2014). While these findings underscore the role of oxidative stress responses in tumor suppression, the mechanisms by which these responses promote tumor development remain unclear.
Ferroptosis is a regulated form of non-apoptotic cell death driven by the accumulation of lipid-based reactive oxygen species (ROS), particularly lipid hydroperoxides (Gao et al, 2015; Xie et al., 2016; Yang and Stockwell, 2016). Although cell-cycle arrest, senescence and apoptosis serve as critical barriers to cancer development, accumulating evidence suggests that loss of p53-dependent cell cycle arrest, apoptosis, and senescence is not sufficient to abrogate the tumor suppression activity of p53. We recently showed that the p53 tumor suppressor sensitizes cells to ferroptosis by repressing transcription of the SLC7A11 gene, which encodes a key component of the cystine/glutamate antiporter (Jiang et al., 2015; Wang et al., 2016; Jennis et al., 2016). Cystine uptake is critical for glutathione synthesis to buffer reactive oxygen species (ROS). Although the precise mechanism by which SLC7A11 modulates ferroptosis needs to be further elucidated, suppression of SLC7A11 expression results in intracellular cysteine depletion, which makes the cells incapable of defending oxidative stress and susceptible to ferroptotic cell death. In addition, p53-mediated ferroptosis appears to act as a barrier to cancer development since it can suppress tumor formation independent of p53-mediated cell cycle arrest, senescence and apoptosis (Jiang et al., 2015; Wang et al., 2016). Of note, SLC7A11 is highly expressed in human tumors (Jiang et al., 2015), and its expression is induced by NRF2 in human cancer cells (Suzuki et al., 2013; Ye et al., 2014). Since several studies showed that activation of NRF2 is critical for tumor growth, the precise mechanism by which NRF2 regulates SLC7A11 clearly needs further elucidation. Here, through biochemical purification, we identified ARF as a key regulator of NRF2. ARF is well established as a tumor suppressor critical for p53 activation upon oncogenic stress; however, we found that ARF directly interacts with NRF2 both in vitro and in vivo. ARF has no obvious effect on NRF2 stability but NRF2-mediated transcriptional activation is inhibited upon ARF induction. Moreover, expression of SLC7A11 is severely suppressed by ARF in p53-null cells, which sensitizes the cells to ferroptosis in a p53-independent manner. In contrast, loss of ARF induces NRF2 activation and promotes cancer cell survival upon oxidative stress. Moreover, ARF is able to induce tumor growth suppression in a p53-independent manner in mouse xenograft models but this activity can be significantly abrogated upon NRF2 overexpression. These results indicate that NRF2 activity is tightly regulated by ARF and reveal that ARF modulates ferroptotic responses in both a p53-dependent and a p53-independent manner.
Results
Identification of ARF as a new binding protein of NRF2
To elucidate the mechanisms by which NRF2 modulates SLC7A11 expression and ferroptosis, we sought to isolate NRF2-associated protein complexes from human cancer cells. To this end, cell extracts from a p53-null H1299 lung carcinoma cell line that stably expresses a human NRF2 protein with N-terminal Flag and HA epitopes (FH-NRF2) were subjected to two-step affinity chromatography, as previously described (Chen et al., 2010; Dai et al., 2011). When the affinity-purified NRF2-associated proteins were analyzed by liquid chromatography mass spectrometry/mass spectrometry (LC–MS/MS), we identified several known NRF2-binding proteins, including CBP, p300, KEAP1, MafG, MafK, and MafF (Figure S1A). In addition, Mass Spectrometric (MS) analysis of a protein band (with the apparent size at ~14 kD molecular weight) revealed peptide sequences matching the ARF tumor suppressor (Figure 1A). ARF (known as p14ARF in human and p19ARF in mouse) was initially identified as the product of an alternative reading frame within the Ink4a/ARF tumor suppressor locus (Reed et al., 2014; Sherr, 2006; Wang et al., 2008). A major function of ARF is to activate the p53 pathway in response to oncogenic stress by inhibiting the enzymatic activity of certain ubiquitin E3 ligases (i.e., Mdm2 and ARF-BP1) that target p53 for proteasomal degradation (Forys et al., 2014; Reed et al., 2014; Sherr, 2006; Chen et al., 2005). ARF can also act to suppress tumor growth in a p53-independent manner by the mechanisms that are not completely understood (Eischen and Boyd, 2012; Forys et al., 2014; Itahana and Zhang, 2008; Muniz et al., 2011).
Figure 1. Identification of ARF as a component of the NRF2 protein complexes.

A. Coomassie blue staining of affinity-purified protein complexes from nuclear extracts of a Flag-HA-NRF2/H1299 stable cell line (lane 2) and the parental H1299 cell line (lane 1). Specific NRF2-interacting protein bands were analyzed by mass spectrometry.
B. Human 293 cells were co-transfected with expression vectors encoding NRF2-V5 and/or Flag-ARF, as indicated. Lysates were immunoprecipitated with FLAG/M2 beads, and the fractionated proteins were immunoblotted with antibody specific to the V5 (for NRF2, upper panel) or the Flag (for ARF, lower panel) epitope.
C. Co-immunoprecipitation of ARF with NRF2 from H1299 cells. Whole cell extracts (lane 1) or immunoprecipitates generated with the NRF2 antibody (lane 3) or a control IgG (lane 2) were immunoblotted with anti-ARF (lower) or anti-NRF2 (upper) antibody.
D. Co-immunoprecipitation of NRF2 with ARF from H1299 cells. Whole cell extracts (lane 1) and immunoprecipitates generated with the ARF antibody (lane 3) or a control IgG (lane 2) were immunoblotted with anti-ARF (upper) or anti-NRF2 (lower) antibody.
E. Western blot analysis of an in vitro GST pull-down assays of highly purified FHNRF2 protein incubated with GST-ARF (lane 3) or GST alone (lane 2).
F. Western blot analysis of an in vitro GST pull-down assays of highly purified FHNRF2 protein incubated with GST-ARF (1–64) (lane 3), GST-ARF (65–132) (lane 4) or GST alone (lane 2).
G. H1299 cells were transfected with the SLC7A11-Luc reporter construct together with expression vectors encoding NRF2 and differing amounts of ARF.
H. H1299 cells were transfected with the SLC7A11-Luc reporter construct together with expression vectors encoding NRF2 and either full-length HA-ARF, HAARF(1–64), or HA-ARF(65–132).
See also Figure S1.
ARF interacts with NRF2 both in vitro and in vivo
To validate the interaction between NRF2 and ARF in human cells, we transfected 293 cells with an NRF2 expression vector in the presence or absence of a vector encoding Flag-tagged ARF. As shown in Figure 1B, NRF2 was readily detected in the immunoprecipitated complexes of Flag-ARF. To evaluate this interaction under more physiological conditions, we also performed co-immunoprecipitation assays with endogenous proteins from H1299 cells. As shown in Figure 1C, western blot analysis revealed that the endogenous ARF protein was co-precipitated by the NRF2-specific antibody but not by the IgG control antibody. Conversely, the endogenous NRF2 protein was co-precipitated by the ARF-specific monoclonal antibody but not by the IgG control antibody (Figure 1D). To ascertain whether NRF2 and ARF interact directly, we performed in vitro GST pull-down assays by incubating a GST-fusion protein containing full-length ARF with purified Flag-HA-tagged NRF2. As shown in Figure 1E, NRF2 strongly bound GST-ARF but not GST alone. More specifically, a GST-fusion protein harboring the N-terminal (amino acids 1–64), but not the C-terminal (65–132), domain of ARF also bound NRF2 (Figure 1F). These data demonstrate that ARF is a bona fide binding partner of NRF2.
ARF inhibits the ability of NRF2 to transcriptionally activate its target genes, including SLC7A11
Since ARF expression did not appreciably affect the protein levels of NRF2 (Figure S1B) and had no effect on Keap1-mediated ubiquitination of NRF2 (Figure S1C), we examined whether ARF modulates NRF2-dependent transcriptional activity. To this end, we co-transfected H1299 cells with expression vectors encoding either NRF2 alone, or NRF2 and ARF together, along with a luciferase reporter harboring the promoter sequences of SLC7A11, a known transcriptional target of NRF2 (Ye et al., 2014). As expected, NRF2 expression strongly induced activation of the SLC7A11 reporter (lane 2, Figure 1G). However, co-expression of NRF2 with differing amounts of ARF led to a dosage-dependent repression of the SLC7A11 reporter (Figure 1G), suggesting that ARF is able to suppress the transcriptional activity of NRF2. Consistent with the binding data (Figure 1F), the N-terminal domain of ARF (lane 3, Figure 1H), but not its C-terminal domain (lane 4 vs. lane 3, Figure 1H) although expressing at the similar levels (Figure S1D, S1E), retained the ability to repress NRF2 transcriptional activation.
Further mapping indicate that the 14 amino-terminal residues of ARF is able to directly interact with NRF2 (Figure S2A) whereas ARFΔ14, a truncated polypeptide that lacks the 14 amino-terminal residues of ARF, failed to bind NRF2 (lane 8 vs. lane 6, Figure 2A). Notably, loss of the these resides of ARF (ARFΔ14) also significantly abrogated its ability to suppress NRF2-mediated transcriptional activation (lanes 5, 6 vs. lanes 3, 4, Figure 2B; Figure S2B). To corroborate these findings, we established a tet-on SaoS2 human osteosarcoma p53-null cell line in which ARF expression can be induced by tetracycline. Although ARF induction had no obvious effect on expression of endogenous NRF2 (Figure 2C), the expression levels of SLC7A11 (Figure 2C), as well as several other known NRF2 transcriptional targets (NQO1, GSR and PRDX1; Figure S2C), were significantly reduced upon ARF induction. To examine the role of endogenous ARF in modulating NRF2 function, we also tested whether these NRF2 targets are affected by RNAi-mediated ARF depletion in SaoS2 cells. While the levels of NRF2 protein were unaffected by ARF depletion, the expression levels of SLC7A11, as well as other NRF2 transcriptional targets, were induced upon ARF depletion (Figure 2D; Figure S2D). Taken together, these data demonstrate that ARF acts as a repressor of NRF2-mediated transcriptional activation.
Figure 2. ARF regulates the transcriptional activity of NRF2 and promotes ferroptosis.

A. U2OS cells were co-transfected with expression vectors encoding FH-NRF2 and either full-length ARF or ARF(Δ14), which lacks amino acids 1–14. Lysates were immunoprecipitated with FLAG-M2 beads, and the fractionated proteins were immunoblotted with HA-specific antibody (to detect FH-NRF2, upper panel) or ARF-specific antibody (lower panel).
B. H1299 cells were transfected with the SLC7A11-Luc reporter construct, together with expression vectors encoding NRF2 and full-length ARF or ARFΔ14.
C. Extracts of ARF-inducible SaoS2 cells treated with doxycycline were immunoblotted with antibodies specific for NRF2, SLC7A11, ARF and Actin.
D. Extracts of SaoS2 cells treated with an ARF-specific RNAi (lane 2) or a control RNAi (lane 1) were immunoblotted with antibodies specific for NRF2, SLC7A11, ARF and Actin.
E. Extracts of ARF-inducible H1299 cells treated with doxycycline as indicated were immunoblotted with antibodies specific for NRF2, SLC7A11, ARF and Actin.
F. Representative phase-contrast images of ferroptotic cell death induced by ROS (TBH treatment) in ARF-inducible H1299 cells treated with or without doxycycline and/or Ferrostatin-1 (Ferr-1) as indicated.
G. Quantification of ROS-induced cell death from Figure 2F (error bars, s.d. from three technical replicates).
H. The percent cell death induced by ROS was determined for H1299 cells transfected with an empty expression vector or with expression vectors encoding ARF alone or ARF and NRF2 together (error bars, s.d. from three technical replicates).
See also Figure S2.
The ARF-NRF2 interaction is critical for ferroptosis in p53-null cancer cells
Our previous study showed that p533KR, an acetylation-defective mutant that fails to mediate p53-dependent cell cycle arrest, apoptosis and senescence, still retains the ability to induce ferroptosis through downregulation of SLC7A11 expression (Jiang et al; 2015; Wang et al., 2016). To test the role of ARF in modulating ferroptotic responses upon ROS treatment, we established a tet-on p53-null H1299 cell line in which ARF expression can be induced by tetracycline. Consistent with the above data, SLC7A11 levels were drastically reduced at various time points after ARF induction (Figure 2E, Figure S2E). Next, we examined whether ROS-mediated ferroptosis is also regulated upon ARF induction. In these experiments, ROS treatment was induced by tert-Butyl hydroperoxide (TBH), as described previously (Hughes et al., 2014; Jiang et al., 2015; Wang et al., 2016; Wang et al., 2012). As shown in Figure 2F, no obvious cell death was observed upon either ARF induction alone or ROS treatment alone. Significantly, however, the combination of ARF induction and ROS treatment induced high levels of cell death that was specifically inhibited by the ferroptosis inhibitor Ferrostatin-1(Dixon et al., 2012) (Figure 2F, 2G). Similar data were also obtained when ARF was induced in p53-null SaoS2 human osteosarcoma cells (Figure S2F and S2G). Conversely, RNAi-mediated knockdown of endogenous ARF protected SaoS2 cells from ROS-induced cell death (Figure S2H). To further validate whether the ARF-NRF2 interaction is crucial in the ferroptotic response, we examined whether ARF-mediated ferroptosis can be rescued by ectopic expression of NRF2. To this end, we transfected p53-null H1299 cells with expression vectors encoding either ARF alone or ARF and NRF2-V5 together (Figure S3A), and then tested for ferroptosis upon ROS treatment. Notably, ARF-mediated ferroptosis was largely abrogated by co-expression of NRF2 (Figure 2H). Similar data were also obtained when SLC7A11 was co-expressed (Figure S3B and S3C). Together, these data indicate that ARF is able to induce ferroptosis in a p53-independent manner by suppressing NRF2-mediated transcriptional activity.
ARF can regulate ferroptotic responses in both a p53-dependent and a p53-independent manner
It is well accepted that ARF-mediated tumor suppression acts, at least in part, through p53 activation. To further elucidate the role of ARF in modulating ferroptotic responses, we examined whether ARF induction can promote ferroptosis through both p53-dependent and p53-independent processes. To this end, we used a derivative of the U2OS human osteosarcoma cell line in which ARF expression is controlled by an IPTG-inducible promoter (Abida and Gu, 2008). As expected, IPTG treatment resulted in a significant induction of ARF and activation of the p53 pathway (lane 3 vs. lane 1, Figure 3A). Since SLC7A11 is a transcriptional repression target of p53, RNAi-mediated knockdown of endogenous p53 led to upregulation of SLC7A11 expression (lane 2 vs. lane 1, Figure 3A); conversely, downregulation of SLC7A11 expression was observed upon ARF induction (lane 3 vs. lane 1). To ascertain whether ARF regulates SLC7A11 transcription in a p53-independent manner, we induced ARF expression in these cells after RNAi-mediated knockdown of endogenous p53. As shown in Figure 3A, SLC7A11 expression was strongly repressed by ARF induction, even in the absence of p53 and p21 (lane 4 vs. lane 2, Figure 3A). Moreover, as shown in Figure3B and 3C, ROS-driven ferroptosis was markedly increased upon ARF induction (by IPTG) in cells that express activated p53 (panel 2 vs. panel 1, Figure 3B, 3C). Although knockdown of p53 diminished the basal levels of ferroptosis (panel 3), these levels were again increased upon ARF induction in p53-depleted cells (panel 4). This observation is consistent with our previous studies in which Erastin-induced ferroptosis still occurred in p53-null MEFs, though to a lesser degree than in wild type cells (Jiang et al., 2015). Therefore, we examined p53flox/flox MEFs and p53flox/floxARFflox/flox MEFs to ascertain whether deletion of the ARF gene can further reduce cellular sensitivity to ferroptosis. As expected, infection with Ad-Cre ablated p53 expression in p53flox/flox MEFs and both p53 and ARF expression in p53flox/floxARFflox/flox MEFs (Figure 3D). While low levels of ferroptosis (~28%) were observed in the p53-null p53flox/flox MEFs (Ad-Cre) cells (Figure S3D), high levels (>75%) were observed in control p53flox/flox MEFs (Ad-GFP) and the control p53flox/floxARFflox/flox MEFs (Ad-GFP) (Figure S3E). Notably, much lower levels (~16%) were detected in p53/ARF double-null cells (Ad-Cre) compared to the levels in p53-null cells (Ad-Cre) under the same conditions (Figure 3E). Taken together, these data establish that ARF can regulate ferroptotic responses in both a p53-dependent and a p53-independent manner.
Figure 3. ARF can induce both p53-dependent and p53-independent ferroptosis.

A. ARF-inducible U2OS cells were treated with control RNAi (lane 1), p53 RNAi (lane 2), control RNAi + IPTG (lane 3), or p53 RNAi + IPTG (lane 4) as indicated, and cell extracts were immunoblotted with antibodies specific for NRF2, SLC7A11, p53, p21, ARF and Actin.
B. Representative phase-contrast images of ARF-inducible U2OS cells treated with ROS and either IPTG (to induce ARF expression) and/or Ferrostatin-1 (Ferr-1), as indicated.
C. Quantification of ROS-induced cell death of the cells shown in 3B (error bars, s.d from three technical replicates).
D. Extracts of p53fl/fl and ARFfl/fl/p53fl/fl MEFs treated with either the Ad-Cre or the control Ad-GFP virus were immunoblotted with antibodies specific for p53 and ARF.
E. ARF is critical for p53-independent ferroptosis. Quantification of ferroptotic cell death in p53fl/fl and ARFfl/fl/p53fl/fl MEFs treated with the Ad-Cre virus with or without Erastin as indicated (error bars, s.d. from three technical replicates).
F. 293 cells were transfected with expression vectors encoding Flag-NRF2, CBP-HA, and/or ARF/ARFΔ14 as indicated. Lysates were immunoprecipitated with FLAG-M2 beads, and the fractionated proteins were immunoblotted with the acetylated specific antibody to acetylated NRF2 (upper), the Flag antibody (for Flag-NRF2; middle). ARF or ARFΔ14 in crude lysates were analyzed by western blotting with anti-ARF antibody (lower).
G. ChIP-qPCR analysis of NRF2 binding on the endogenous SLC7A11 promoter. H1299 cells were transfected with expression vectors encoding FH-NRF2 and either full-length ARF or ARF(Δ14), which lacks amino acids 1–14. See also Figure S3.
ARF inhibits CBP-dependent NRF2 acetylation and NRF2 binding to its cognate transcriptional promoter
Since ARF expression has minimal effect on NRF2 protein stability, we explored other possible mechanisms by which ARF might suppress NRF2 transcriptional activity. It is well established that the transcriptional coactivator CBP promotes NRF2-mediated transcriptional activation by binding and acetylating the NRF2 polypeptide (Sun et al., 2009). Therefore, we examined whether ARF impairs either the interaction between CBP and NRF2 and/or CBP-mediated acetylation of NRF2. As shown in Figure S3F, co-immunoprecipitation assays revealed that the CBP/NRF2 interaction was not dramatically affected by ARF expression (lane 3 vs lane 2). Interestingly, however, the levels of CBP-dependent NRF2 acetylation were markedly reduced upon expression of full-length ARF but not ARFΔ14, the truncated ARF polypeptide that fails to bind NRF2 (lane 3 vs. lane 2, Figure 3F). Moreover, since acetylation of NRF2 is critical for its sequence-specific DNA binding (Sun et al., 2009), we examined whether ARF modulates NRF2 binding on the SLC7A11 promoter in vivo. Indeed, ChIP analysis revealed that full-length ARF (lane 3 vs. lane 2, Figure 3G), but not the truncated ARFΔ14 polypeptide (lane 4, Figure 3G), repressed NRF2 loading on the endogenous SLC7A11 promoter. Taken together, these data indicate that ARF represses NRF2-mediated transactivation by inhibiting CBP-dependent NRF2 acetylation and NRF2 binding to its cognate transcriptional promoters.
The ARF-NRF2 interaction is crucial for p53-independent tumor growth repression by ARF
Previous studies have shown that ARF also promotes apoptosis in response to more potent oxidative reagents such as H2O2 (Damalas et al., 2011; Liontos et al., 2012). Therefore, we examined whether the ARF-NRF2 interaction is also involved in H2O2-induced apoptosis using the ARF-tet-on p53-null H1299 cell line. As shown in Figure4A and 4B, although modest levels of cell death (~10%) occurred in the absence of ARF expression (tet-off) (panel 1), a dramatic increase (>70%) was seen upon ARF induction (tet-on) (panel 2). To confirm the mode of H2O2-induced cell death, we treated cells with Z-VAD-FMK, a specific inhibitor of apoptosis. Surprisingly, Z-VAD-FMK only imparted a partial rescue of cell death (pane 3, Figure 4A, 4B), while cell death was also partly suppressed by Ferrostatin-1 (Ferr-1) (panel 4), a specific inhibitor of ferroptosis (Dixon et al., 2012). Notably, combined treatment with both Z-VAD-FMK and Ferrostatin-1 completely rescued cell death (pane 5, Figure4A and 4B). These data indicate that ARF promotes both apoptosis and ferroptosis in respond to H2O2 treatment. More importantly, further analyses indicate that both modes of cell death induced by ARF are largely abrogated by ectopic expression of NRF2 (Figure 4C).
Figure 4. The ARF-NRF2 interaction is critical for ARF-mediated tumor suppression independent of p53.

A. Representative phase-contrast images of H2O2-treated ARF-inducible H1299 cells cultured in the presence or absence of doxycycline, and/or the presence of Ferrostatin-1 (Ferr-1) and/or Z-VAD-FMK, as indicated.
B. Quantification of H2O2-induced cell death from the samples shown in Figure 4A (error bars, s.d from three technical replicates).
C. Quantification of H2O2-induced cell death from H1299 cells co-transfected with an empty expression vector or with expression vectors encoding ARF alone or ARF and NRF2 together.
D. Xenograft tumors from tet-on ARF-inducible H1299 cells transfected with an empty expression vector or an expression vector encoding NRF2.
E. Tumor weight was determined from Figure 4D (error bars, SEM from six tumors). Independent experiments were repeated three times and representative data are shown.
F. Western blot analysis of ARF, NRF2 and SLC7A11 protein levels in tet-on ARFinducible H1299 cells with or without NRF2 overexpression.
See also Figure S4.
Finally, since NRF2 protein is aberrantly accumulated in many types of human cancer (Hayes and McMahon, 2009; Suzuki et al., 2013; Tao et al., 2014), we examined whether NRF2 overexpression affects ARF-mediated tumor growth suppression in xenograft tumor models. Indeed, the growth of p53-null H1299 xenografts was significantly repressed upon tetracycline-induced ARF expression (panel 3 vs. panel 1, Figure4D and 4E), indicating that ARF is capable of inducing tumor growth suppression in a p53-independent manner. Moreover, this tumor growth repression was largely abrogated by NRF2 overexpression (panel 4 vs. panel 3, Figure4D and 4E). Western blot analysis confirmed that the ARF-mediated downregulation of SLC7A11 expression (lane 3 vs. lane 1, Figure 4F) is significantly ablated upon overexpression of NRF2 (lane 4 vs. lane 3, Figure 4F). Notably, the up-regulation of Ptgs2 has recently been identified to be a potential molecular marker of ferroptosis (Yang et al., 2014). We therefore examined Ptgs2 levels in xenograft tumors. Indeed, Ptgs2 was found to be significantly up-regulated in the tumors when ARF is induced (Figure S4A), suggesting that ferroptosis is involved in tumor suppression. In contrast, upregulation of Ptgs2 was suppressed when NRF2 was co-expressed in tumors (Figure S4A), well-correlated with promoting the tumor growth by NRF2 expression (Figure 4E). These data demonstrate that the ARF-NRF2 interaction is crucial for p53-independent tumor growth repression by ARF.
Discussion
The emerging role of oxidative stress responses in metabolism, ferroptosis and tumor suppression has been a topic of great interest (Jiang et al., 2015; Gao et al., 2015; Jennis et al., 2016). Several studies indicate that deregulation of NRF2 is critically involved in tumorigenesis (Hayes and McMahon, 2009; Jaramillo and Zhang, 2013). Nevertheless, the precise mechanisms by which NRF2 is regulated in human cancers need further elucidation. Through biochemical purification, we have identified ARF as a bona fide regulator of NRF2. Interestingly, we found that the NRF2-ARF interaction plays a key role in p53-independent ferroptotic responses in human cancer cells. In particular, our results show that (i) ARF interacts with NRF2 both in vitro and in vivo; (ii) ARF inhibits the ability of NRF2 to transcriptionally activate its target genes, including SLC7A11; (iii) ARF can regulate ferroptotic responses in both a p53-dependent and a p53-independent manner; (iv) The ARF-NRF2 interaction is critical for ferroptosis in p53-null cancer cells; (v) ARF inhibits CBP-dependent NRF2 acetylation and NRF2 binding to its cognate transcriptional promoter; (vi) The ARF-NRF2 interaction is crucial for p53-independent tumor growth repression by ARF. Thus, these results reveal a new mechanism by which the ARF-NRF2 interaction controls cancer cell growth in vivo.
ARF was originally identified as an alternative transcript of the Ink4a tumor suppressor locus that encodes p16INK4a, an inhibitor of cyclin dependent kinases (Sherr, 2006). Alterations affecting this locus, which frequently disrupt p16INK4a and p14ARF, are common in human cancers. The ARF tumor suppressor acts as a key factor to suppress tumor growth upon hyperproliferative signals such as those emanating from the Ras and Myc oncoproteins (Eischen et al., 2009). Numerous studies indicate that ARF suppresses aberrant cell growth in response to oncogene activation by activating the p53 pathway. Although it is well established that ARF and p53 function in a linear pathway of tumor suppression (Reed et al., 2014;Sherr, 2006; Wang et al., 2008), accumulating evidence indicates that ARF also has p53-independent tumor-suppressor activities (Eischen et al., 2012; Forys et al., 2014; Itahana et al., 2008; Muniz et al., 2011). This notion is further supported by recent genomic analyses showing that ARF is inactivated in human cancers containing mutated p53 (Cancer Genome Atlas Research Network, 2012; O’Dell et al., 2012). Our data demonstrate that NRF2 acts as an important target of ARF and that ARF-mediated repression of NRF2 is critical for both the apoptotic and ferroptotic responses under oxidative stress in p53-null cells. Notably, ARF is able to induce tumor growth suppression in a p53-independent manner in mouse xenografts, but this activity can be significantly abrogated by NRF2 overexpression. Thus, our data demonstrate that the ARF-NRF2 interaction regulates not only normal cellular responses to oxidative stress, but also plays a key role in the p53-independent tumor suppression activity of ARF.
Research into effects of oxidative responses in cancer is fraught with controversy and conflicting results. Although it was popularly believed that antioxidants protect against cancer, accumulating evidence suggests that antioxidants actually increase cancer risks (Jaramillo and Zhang, 2013; Chio et al., 2016). On the other hand, cell cycle arrest, apoptosis, and senescence each serve as important mechanisms to prevent inappropriate expansion of cells with malignant potential, recent studies indicate that oxidative stress induced ferroptosis also contributes significantly to the tumor suppression activity (Jiang et al., 2015; Yang et al., 2014). Indeed, high levels of reactive oxygen species (ROS) are generally toxic for normal cell growth, cancer cells appear to bypass normal responses to cellular stress, allowing for sustained growth even in unfavorable environments. It is well established that the levels of ARF are induced upon oncogenic stress (Sherr, 2006). Interestingly, recent studies indicate that several malignantly-activated oncoproteins, including Myc, K-Ras and B-RAF, can protect cancer cells from the toxic effects of oxidative stress by inducing transcription of NRF2 (DeNicola et al., 2011). These studies underscore the role of oxidative responses in tumorigenesis and also suggest that oxidative stress responses are directly linked with oncogenic stress. Since our data indicate that ARF-mediated effects on NRF2 can be detected in both human cancer cells and normal MEFs, it is very likely that ARF is capable of modulating NRF2 activities under both normal conditions and oncogenic stressed conditions.
Moreover, using three independent cancer databases (Bladder, Head and Neck, and Uterine) (Figure S4B, 4C and 4D), we found that SLC7A11 mRNA expression is significantly downregulated in patients with higher expression levels of ARF compared with patients with lower levels of ARF (p <0.05). Further, we show that patients expressing lower levels of ARF have a significantly shorter overall survival compared to patients bearing tumors expressing higher levels of ARF (p = 0.0005) (Figure S4E). Although further investigation is required to examine whether mutations of KEAP1 or NFE2L2 (NRF2) have any effects on ARF-mediated regulation of NRF2, ARF expression has no obvious effect on NRF2 stability and ARF does not interfere with the KEAP1-mediated ubiquitination of NRF2 (Figure S1B and S1C). Moreover, we found that ARF interacts with the Neh1 and Neh3 domains of NRF2, separated from the Neh2 domain, the KEAP1-binding region of NRF2 (Figure S1F, S1G and S1H). It is very likely that ARF-mediated regulation of NRF2 is independent of mutations of KEAP1 or NRF2 in human cancers. Together, our finding indicate that the functional interaction between NRF2 and ARF is critical for ferroptotic responses in both normal and cancer cells, and may act as a novel checkpoint on the oxidative stress responses that is crucial for ARF-mediated tumor suppression.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the followings:
KEY RESOURCE TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell culture and stable lines
Plasmids
METHODS DETAILS
Purification of NRF2-complexes from human cells
Ablation of endogenous ARF by RNAi in SaoS2 cells
RNA extraction, quantitative RT–PCR
Chromatin immunoprecipitation assay
Mouse xenograft
Cell death assay
Mass Spectrometry Assay
Generation of p53-deficient or ARF and p53-deficient MEFs
QUANTIFICATION AND STATISTICAL ANALYSIS
DATA AND SOFTWARE AVAIABILITY
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to the corresponding author, Dr. Wei Gu (wg8@columbia.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell culture and stable lines
All the cell lines were obtained from ATCC and have been proven to be negative for mycoplasma contamination. No cell lines used in this work were listed in the ICLAC database. The cell lines were freshly thawed from the purchased seed cells and were cultured for no more than 2 months. The morphology of cell lines was checked every week and compared with the ATCC cell line image to avoid cross-contamination or misuse of cell lines. Cells were maintained in 37°C incubator with 5% CO2. DMEM media was used by supplementing with 10% FBS, 100 units/ml penicillin and 100 mg/ml streptomycin. To generate ARF inducible stable line, its cDNA was cloned into tet-on pTRIPZ inducible expression vector (Thermo Open Biosystems). The H1299 or SaoS2 cells was transfected with Lipofectamine 2000 (Invitrogen), followed by selection and maintenance with puromycin (1 mg/ml) in DMEM medium containing 10% tetracycline-free FBS. To induce the expression of ARF in H1299 or SaoS2 cells, 0.1 mg/ml of doxycycline was added to the culture medium. To induce the expression of ARF in a U2OS derivative, NARF6 cells, 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) was used. NARF6 cells have been described previously (Abida and Gu, 2008).
Plasmids
cDNA of NRF2 was generously gifted from Dr. Zhang (University of Arizona). For Flag-HA-NRF2, full length NRF2 was subcloned into PCIN4-Flag-HA vector. For NRF2-V5, or Flag-NRF2 construct, full length NRF2 were amplified by PCR without a stop codon or with introducing a Flag sequence to the N terminus, and then cloned into pcDna3.1/v5-His-Topo vector (Invitrogen). To prepare GST-NRF2 mutant constructs, cDNA sequences corresponding to different regions were amplified by PCR from wild type NRF2 construct, and subcloned into pGEX-2T. For GST-ARF 1–14 construct, quick change site-directed mutagenesis kit was used (Stratagene). For HA-ARF, HA-ARF (1–64) and HA-ARF (65–132) constructs, cDNA of ARF were amplified by PCR with introducing HA sequence to the N terminus, and then cloned into pcDna3.1/v5-His-Topo vector (Invitrogen). All the other constructs of ARF have been described previously (Chen et al., 2005).
METHOD DETAILS
Purification of NRF2-complexes from human cells
The epitope-tagging strategy to isolate NRF2-containing protein complexes from human H1299 cells was performed as previously described with some modifications (Chen et al., 2005; Dai et al., 2011). In brief, to obtain an Flag-HA-NRF2 expressing cell line, p53 null H1299 cells were transfected with pCIN4-Flag-HA-NRF2 and selected for 2 weeks in 1 mg/ml G418 (GIBCO). The tagged NRF2 protein levels were detected by western blot analysis. Cells were grown in DMEM with 10% fetal bovine serum plus 0.5 mg/ml G418, and harvested near confluence. The cell pellet was resuspended in buffer A (10 mM HEPES pH 7.9, 0.1 mM EDTA, 10 mM KCl, 0.5 mM PMSF, 1 mM DTT and protein inhibitor mixture [Sigma]). 10% NP 40 (Fluka) was added till a final concentration of 0.5% after the cells were allowed to swell on ice for 15 min. The homogenate is centrifuged for 10 min at 4,000 rpm after the tube is vigorously vortexed for 1 min. The nuclear pellet was resuspended in ice-cold buffer C (20 mM Herpes pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM DTT and protein inhibitor mixture) and the tube was vigorously rocked at 4°C for 45 min. Buffer D (20 mM HEPES [pH 7.9], 1 mM EDTA) was used to dilute the nuclear extract to the 100 mM final NaCl concentration, and ultra-centrifuged 25,000 rpm for 2 hr at 4°C. Filtered with 0.45 µm syringe filters (NALGENE), and then the supernatants were used as nuclear extracts for M2 immunoprecipitations by anti-FLAG antibody-conjugated agarose (Sigma). The bound polypeptides were eluted with the 10 µg/ml FLAG peptide in BC100 solution and were further affinity purified by anti-HA antibody-conjugated agarose (Sigma). The final elutes from the HA-beads with 0.1% trifluoroacetic acid (TFA, Sigma) were resolved by SDS-PAGE on a 4%–20% gradient gel (Novex) for silver staining or colloidal-blue staining analysis. Specific bands were cut and subjected to mass-spectrometry peptide sequencing.
Ablation of endogenous ARF by RNAi in SaoS2
The RNAi-mediated ablation of endogenous ARF was performed essentially as previously described (Chen et al., 2010). A 21-nucleotide siRNA duplex with 3′dTdT overhangs corresponding to ARF mRNA (GAUCAUCAGUCACCGAAGGUU) was synthesized (Dharmacon). Cells were transfected using lipofectamine 3000 following manufacturer's protocol (Invitrogen) three times with 24–48 hr intervals. After three consecutive transfections, cells were harvested for western blot analysis and mRNA analysis.
RNA extraction, quantitative RT–PCR
Total RNA was isolated using TRIzol (Invitrogen) according to the manufacturer’s protocol. One microgram of total RNA was reverse transcribed by the SuperScript IV First-Strand Synthesis SuperMix (Invitrogen) following manufacturer’s protocol. Quantitative PCR was done using a 7500 Fast Real-Time PCR System (Applied Biosystems) with standard protocol. For quantitative RT–PCR primers: human SLC7A11 forward, ATGCAGTGGCAGTGACCTTT, reverse, GGCAACAAAGATCGGAACTG; human NQO1 forward ATGTATGACAAAGGACCCTTCC, reverse, TCCCTTGCAGAGAGTACATGG human PRDX1 forward, AGGCCTTCCAGTTCACTGAC, reverse, CAGGCTTGATGGTATCACTGC; human HPRT forward, TATGGCGACCCGCAGCCCT, reverse, CATCTCGAGCAAGACGTTCAG; human GSR forward, ATGATCAGCACCAACTGCAC, reverse CGACAAAGTCTTTTTAACCTCCTT; human Ptgs2 forward TCACCGTAAATATGATTTAAGTCCAC, reverse CTTCACGCATCAGTTTTTCAAG.
Chromatin immunoprecipitation assay
The assay was performed as described previously with minor modifications (Jiang et al., 2015). Briefly, cells were crosslinked with 1% formaldehyde for 10 min at 25°C and neutralized by glycine to a final concentration of 0.125 M. Cells were harvested after being washed twice with cold PBS, and suspended in cold lysis buffer (10 mM Tris-Cl, pH8.0, 85 mM KCl, 5 mM EDTA 0.25% triton, 0.5% NP40 and proteinase inhibitor mixture). After 10 min incubation at 4°, nuclei were harvested and re-suspended in CHIP lysis buffer (50 mM Tris-HCl pH 8.0, 5 mM EDTA, 1% SDS and protease inhibitor mixture). After sonication, the lysates were centrifuged, the supernatants were added with magnetic beads coated with specific antibodies or IgG control and incubated overnight. Beads were washed seven times with washing buffer (50 mM HEPES, pH7.5, 500 mM HCl, 1 mM EDTA, 0.7% Na-deoxycholate and 1% NP40) and once with TE buffer before the protein–DNA complex was eluted with 1% SDS and 0.1 M NaHCO3. After reverse crosslinking overnight at 55°C, DNA was extracted and quantitative PCR performed. SLC7A11 primer: forward TTACTACTTCTGGATTGGCTA, reverse CTTGTATTTAAGCGCCTGCC.
Mouse xenograft
Pooled stable cell line was derived from H1299 tet-on ARF cells by transfecting either empty vector or vector overexpressing NRF2. Cells were selected by G418 (1 mg /ml) for 2 weeks and then re-transfected the same vectors again. After additional treatment with or without doxycycline (1.0 mg/ml) for 60 h, 1.0× 106 of cells were then mixed with Matrigel (BD Biosciences) at 1:1 ratio (volume) and injected subcutaneously into nude mice (NU/NU, Charles River). The mice were fed with the food containing doxycycline hyclate (Harlan, 625mg/kg) or control food. Seven weeks after injection, the mice were killed and the weight of the tumors was measured. The experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Columbia University. The tumors of the experiments did not exceed the limit for tumour burden (10% of total bodyweight or 2 cm in diameter).
Cell death assay
For cell death assays involving ARF activation, ARF was preactivated for 48 h by either doxycycline (tet-on stable line cells) or IPTG followed by treatment with either TBH or H2O2. Cells were counted with a haemocytomer using a standard protocol. Cells with blue staining were considered as dead cells. Cell death was further quantitated by propidium iodide staining followed by FACS analysis.
Mass Spectrometry Assay
The protein complex was separated by SDS-PAGE and stained with GelCode Blue reagent (Pierce, 24592). The visible band was cut and digested with trypsin and then subjected to liquid chromatography (LC) MS/MS analysis.
Generation of p53-deficient or p53 and ARF deficient MEFs
p53flox/flox, and p53flox/floxARFflox/flox MEFs are generously gifted from Dr. Weber (Washington University in St. Louis). P53 alone or both ARF and p53 conditional knockout MEFs were transduced with Ad-CMV-Cre (Vector Biolabs) for 3 days, in order to undergo Cre-mediated deletion of the floxed p53 or ARF allele. The elimination of p53 or ARF protein in MEFs was confirmed by immunoblotting as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results were shown as the means ± s.d. except in Fig. 4E. Difference was determined by using a two-tailed, unpaired Student’s t test in all figures. All statistical analysis was performed by using GraphPad Prism software. p < 0.05 was denoted as statistically significant.
DATA AND SOFTWARE AVAILABILITY
Raw western blotting data for this paper have been deposited to Mendeley data and are available at http://dx.doi.org/10.17632/9x3z42pvrk.1
Supplementary Material
Highlights.
ARF interacts with NRF2 both in vitro and in vivo
ARF inhibits the ability of NRF2 to activate its target genes.
The ARF-NRF2 interaction is critical for ferroptosis in p53-null cells
NRF2 is a major target for p53-independent tumor suppression by ARF.
Acknowledgments
We thank Dr. Donna Zhang, Dr. Jason Weber to provide critical reagents for this study. This work was supported by the National Cancer Institute of the National Institutes of Health under Award 5R01CA169246, 5RO1CA190477, 5RO1CA085533 and 5RO1CA216884 to W.G, and 5R01-CA172272 to R. B. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: Conception and experimental design: D.C. and W.G. Methodology and data acquisition: D.C., O.T., B. C., L. E. and Y. C. Analysis and interpretation of data: D.C., and W.G. Manuscript writing: D.C., R.B. and W.G.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Research. 2008;68:352–357. doi: 10.1158/0008-5472.CAN-07-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AK, Cho HY, Miller-Degraff L, Walker C, Helms K, Fostel J, Yamamoto M, Kleeberger SR. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS One. 2011;6:e26590. doi: 10.1371/journal.pone.0026590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–83. doi: 10.1016/j.cell.2005.03.037. 2005. [DOI] [PubMed] [Google Scholar]
- Chen D, Shan J, Zhu WG, Qin J, Gu W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature. 2010;464:624–627. doi: 10.1038/nature08820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio II, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Öhlund D, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell. 2016;166:963–76. doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Tang Y, Jung SY, Qin J, Aaronson SA, Gu W. Differential effects on p53-mediated cell cycle arrest vs. apoptosis by p90. Proc Natl Acad Sci U S A. 2011;108:18937–42. doi: 10.1073/pnas.1110988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damalas A, Velimezi G, Kalaitzakis A, Liontos M, Papavassiliou AG, Gorgoulis V, Angelidis C. Loss of p14(ARF) confers resistance to heat shock- and oxidative stress-mediated cell death by upregulating β-catenin. Int J Cancer. 2011;128:1989–95. doi: 10.1002/ijc.25510. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Boyd K. Decreased Mdm2 expression inhibits tumor development and extends survival independent of Arf and dependent on p53. PLoS One. 2012;7:e46148. doi: 10.1371/journal.pone.0046148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forys JT, Kuzmicki CE, Saporita AJ, Winkeler CL, Maggi LB, Jr, Weber JD. ARF and p53 coordinate tumor suppression of an oncogenic IFN-β-STAT1-ISG15 signaling axis. Cell Rep. 2014;7:514–26. doi: 10.1016/j.celrep.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Molecular cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–47. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Hughes RH, Silva VA, Ahmed I, Shreiber DI, Morrison B., 3rd Neuroprotection by genipin against reactive oxygen and reactive nitrogen species-mediated injury in organotypic hippocampal slice cultures. Brain research. 2014;1543:308–314. doi: 10.1016/j.brainres.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Itahana K, Zhang Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell. 2008;13:542–53. doi: 10.1016/j.ccr.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes & development. 2016;30:918–930. doi: 10.1101/gad.275891.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liontos M, Pateras IS, Evangelou K, Gorgoulis VG. The tumor suppressor gene ARF as a sensor of oxidative stress. Curr Mol Med. 2012;12:704–15. doi: 10.2174/156652412800792633. [DOI] [PubMed] [Google Scholar]
- Muniz VP, Barnes JM, Paliwal S, Zhang X, Tang X, Chen S, Zamba KD, Cullen JJ, Meyerholz DK, Meyers S, et al. The ARF tumor suppressor inhibits tumor cell colonization independent of p53 in a novel mouse model of pancreatic ductal adenocarcinoma metastasis. Mol Cancer Res. 2011;9:867–77. doi: 10.1158/1541-7786.MCR-10-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N, et al. Kras (G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72:1557–67. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SM, Hagen J, Tompkins VS, Thies K, Quelle FW, Quelle DE. Nuclear interactor of ARF and Mdm2 regulates multiple pathways to activate p53. Cell Cycle. 2014;13:1288–98. doi: 10.4161/cc.28202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29:2658–72. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci. 2013;34:340–6. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, Zhang DD. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74:7430–41. doi: 10.1158/0008-5472.CAN-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Lushnikova T, Odvody J, Greiner TC, Jones SN, Eischen CM. Elevated Mdm2 expression induces chromosomal instability and confers a survival and growth advantage to B cells. Oncogene. 2008;27:1590–8. doi: 10.1038/sj.onc.1210788. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016;17:366–373. doi: 10.1016/j.celrep.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26:165–76. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, Ohyama C, Itoh K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol. 2014;34:3421–34. doi: 10.1128/MCB.00221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.