

Abstract
Background
As DNA sequencing costs decline, genetic testing options have expanded. Whole exome and whole genome sequencing (WGS) are entering clinical use, posing questions about their incremental value compared with disease-specific multi-gene panels that have been the cornerstone of genetic testing.
Methods and Results
Forty-one patients with hypertrophic cardiomyopathy (HCM) who had undergone targeted HCM genetic testing (either multi-gene panel or familial variant test) were recruited into the MedSeq Project, a clinical trial of WGS. Results from panel genetic testing and WGS were compared. In 20 of 41 participants panel genetic testing identified variants classified as pathogenic, likely pathogenic or uncertain significance (VUS). WGS identified 19 of these 20 variants but the variant detection algorithm missed a pathogenic 18-base pair duplication in MYBPC3 due to low coverage. In 3 individuals, WGS identified variants in genes implicated in cardiomyopathy but not included in panel testing: a pathogenic PTPN11 variant and VUSs in ILK and FLNC. WGS also identified 84 secondary findings (mean=2/person, range= 0–6), which mostly defined carrier status for recessive conditions.
Conclusions
WGS detected nearly all variants identified on panel testing, provided one new diagnostic finding, and allowed interrogation of posited disease genes. Several variants of uncertain clinical utility and numerous secondary genetic findings were also identified. While panel testing and WGS provided similar diagnostic yield, WGS offered the advantage of re-analysis over time to incorporate advances in knowledge, but necessitated expertise in genomic interpretation to appropriately incorporate WGS into clinical care.
Keywords: hypertrophic cardiomyopathy, genetic testing, whole genome sequencing
Journal Subject Terms: Genetics, Functional Genomics, Cardiomyopathy
Introduction
Current consensus guidelines recommend the use of genetic testing to establish a molecular etiology in patients diagnosed with hypertrophic cardiomyopathy (HCM), and to identify at-risk relatives to target for longitudinal clinical screening.1,2 Over the past decade, there has been rapid growth in the availability and utilization of HCM genetic testing.3 With the development of next generation sequencing technology, HCM multi gene-panels have expanded from 5 genes in 2004, when genetic testing was first commercially available, to now over 100 genes. However, expanding panels to include genes beyond the sarcomere genes has not substantially improved diagnostic yield,3 as many of these genes have not been definitively established to cause disease and any variants identified in these genes will be of uncertain significance4. This is a particular limitation when pretest probability for identifying a causal mutation is reduced due to the absence of family history or phenotypic ambiguity.5–7 Furthermore, regardless of panel size, genetic testing does not yield a molecular etiology in 40–70% of HCM patients.3
More recently whole exome and genome sequencing (WES and WGS) have been increasingly utilized for molecular diagnosis.8, 9 Initially reserved for complex clinical presentations, or as second tier tests following negative targeted genetic testing, decreasing price and wider availability now make such technology more accessible, raising the question of whether these comprehensive tests might replace multi-gene panels to determine the molecular etiology of monogenic conditions such as inherited cardiomyopathies. The breadth of sequence analysis afforded from WES/WGS offers great promise for increased diagnostic yield as well as the ability to perpetually reexamine the comprehensive sequence data as knowledge emerges; a key advantage over targeted testing. However, their expansive scope also requires careful consideration, particularly regarding the potential impact of unanticipated secondary findings. The American College of Medical Genetics and Genomics (ACMG) recommends reporting incidentally-identified pathogenic variants in 59 genes considered to be medically actionable.10, 11 Learning about secondary findings from WGS has been cited as both a potential advantage and barrier to its use in clinical medicine.12 In addition, concerns about whether WGS read depth is sufficient to supplant panel testing13 make WGS sensitivity central to the discussion of its use relative to panel testing, although examination of non-exonic regulatory elements and regions with high GC content may be superior with WGS.
In this study, we compared targeted HCM genetic testing, performed by multi-gene panel or familial variant test, to WGS in HCM patients to: 1) examine the difference in diagnostic yield 2) quantify the occurrence of secondary findings from WGS and 3) explore the clinical actions that resulted from additional findings from WGS.
Methods
Study Cohort
The study population for this analysis was drawn from the MedSeq Project, a randomized clinical trial of the incorporation of WGS into clinical practice in adult medicine. The design of this study has been previously reported.14 Briefly, the MedSeq Project cohort included 100 primary care patients and 100 patients with presumptive inherited hypertrophic or dilated cardiomyopathy (DCM). Eligible patients received a study mailing and were approached for participation by telephone or in person during clinic visits. Participants had targeted HCM genetic testing, prior to or concurrent with their enrollment and were randomized 1:1 to undergo family history collection and review of targeted HCM genetic testing, or family history collection, review of targeted HCM genetic testing and WGS.
In this report, we limited the analyses to the 41 HCM patients who underwent WGS. This project was approved by the Partners Human Research Committee and all participants provided informed consent.
Genetic Testing
Targeted HCM genetic testing
Multi-gene panel size ranged from 4–62 genes depending on year of testing (2004–2016) and clinician panel selection. All but two subjects who underwent panel testing had a minimum of 8 sarcomere genes sequenced, including myosin binding protein C (MYBPC3), myosin heavy chain (MYH7), cardiac troponin T (TNNT2), cardiac troponin I (TNNI3), alpha-tropomyosin (TPM1), myosin essential and regulatory light chains (MLY2, MYL3), and cardiac actin (ACTC). The two subjects who had only four sarcomere genes sequenced (MYBPC3, MYH7, TNNT2, and TPM1) had pathogenic variants identified. Variants were classified as pathogenic, likely pathogenic (LP), uncertain significance (VUS), likely benign or benign using the clinical standard of the laboratory at the time of testing.15,16,17 The majority of subjects (32/41) had their targeted testing performed by CLIA-certified Partners Laboratory for Molecular Medicine (LMM), Cambridge, MA (see supplemental material for methodology).
WGS
The WGS methodology and bioinformatic pipeline used in the MedSeq Project have been previously described.16,18,19 Genome sequencing was performed by the CLIA-certified, CAP-accredited Illumina Clinical Services Laboratory (San Diego, CA) using paired-end 100 base pair reads on the Illumina HiSeq platform between 2013–2015. Genomes were sequenced to minimum of 30X mean coverage, with ≥ 95% of bases sequenced to at least 8X coverage. Sequencing data were then transferred to the LMM for analysis and reporting. The medical exome content evolved with current knowledge throughout the study, but included ~4000 genes. Non-coding regions outside clinical regions of interest were not interpreted, unless a previously known pathogenic variant was identified. Single nucleotide variants and small insertions/deletions were identified and assessed. Detection of insertion/deletion variants >10 bp was limited due to the sequencing depth and read length. Larger copy number and structural variants are being investigated separately. See supplemental material for sequence alignment and variant calling information.
Variants were classified using a seven-tier system: benign, likely benign, uncertain significance – favor benign (VUS-FB), uncertain significance (VUS), uncertain significance – favor pathogenic (VUS-FP), likely pathogenic (LP), and pathogenic. Pathogenic, LP, and VUS-FP were reported. VUSs in cardiomyopathy- associated genes were also reported16, 17 WGS results were analyzed independently of targeted HCM panel testing data. Subsequent comparisons of WGS and targeted HCM genetic test results were to assess both the accuracy of WGS and its ability to identify new causal variants.
WGS information reported in the MedSeq Project extended beyond monogenic disease and recessive conditions to include an array of genetic risk information that might impact cardiovascular disease management. The MedSeq Project genome report itself has been described in detail elsewhere.16,20,21 Briefly, it was divided into different categories to report:
Monogenic disease risk, both related and unrelated to the indication for testing (i.e. cardiomyopathy).
Carrier variants for recessive conditions
Selected pharmacogenomic associations
- A cardiac risk report incorporating predictions based on genomic variation:
-
○Predicted fasting lipid profile
-
○Data from genome wide association studies (GWAS) on alleles conferring small to moderate risk for eight common phenotypes: atrial fibrillation, hypertension, QT prolongation, abdominal aortic aneurysm, coronary heart disease, type 2 diabetes, obesity, and platelet aggregation21
-
○
Secondary findings were not limited to the genes defined by the ACMG guidelines.10, 11 Variants were designated as a secondary finding, by consensus, when there was a lack of moderate, strong, or definitive association with cardiomyopathy, but other potential medical significance. Secondary findings were tallied to determine the burden of such findings in each individual and the cohort.
Clinical actions triggered by WGS results
WGS results were disclosed by the patient’s cardiologist after completing a genetics education module. The majority (4/7) of cardiologists had genetics expertise. Physicians completed a post-disclosure survey to indicate whether specific WGS findings resulted in any further action (referrals, additional diagnostic testing, etc.). Medical records were then reviewed a minimum of one year after disclosure to determine the outcome of the recommended actions.
Results
Patient characteristics
Forty-one unrelated participants with HCM underwent WGS and targeted HCM genetic testing (multi-gene panel (n=38) or familial variant testing (n=3)) (Figure). The mean (standard deviation) age was 58 (12) years; 54% were female and 95% were Caucasian (Table 1). A family history of HCM was present in 17/41 (42%) subjects. Participants demonstrated the known clinical heterogeneity in HCM, ranging from those who were asymptomatic to those requiring therapy for advanced heart failure (Table 1).
Figure.
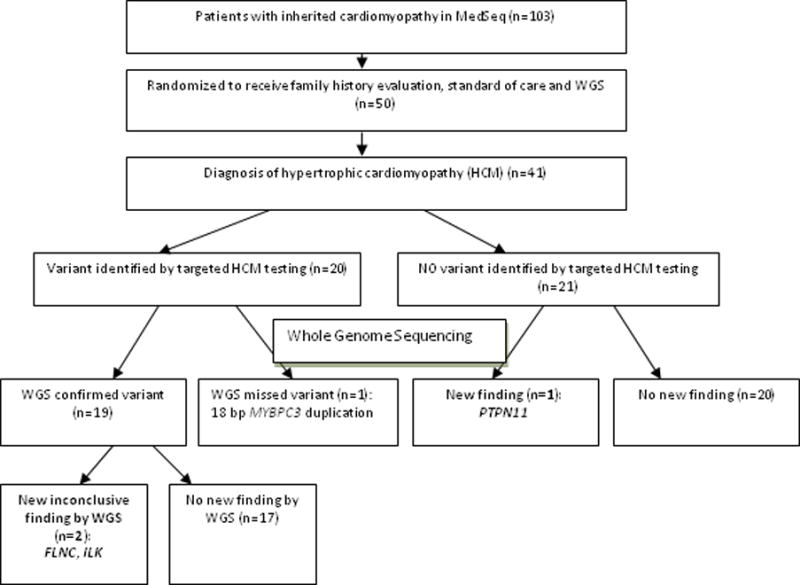
Subject enrollment and cardiomyopathy-related genetic test results
ILK, integrin-linked kinase; FLNC, Filamin-C; PTPN11, Protein Tyrosine Phosphatase, Non-Receptor Type 11
Table 1.
Characteristics of HCM patients participating in MedSeq who underwent multi-gene HCM panel testing and WGS (n=41)
| Mean Age (SD), years | 58 (12) |
| Female, n (%) | 22 (54%) |
| Caucasian, n (%) | 39 (95%) |
| Family history of HCM, n (%) | 17 (42%) |
| Atrial fibrillation, n (%) | 10 (24%) |
| End stage HCM/HF death/transplant | 4 (10%) |
| Sudden cardiac arrest/death | 5/41 (12.2%) |
| Mean Maximal left ventricular wall thickness, mm (SD) | 17.2 (4.3) |
| Mean left ventricular ejection fraction, % (SD) | 62.2 (14.6) |
| New York Heart Association Functional Class | |
| I | 19 (63%) |
| II | 11 (36%) |
| III | 1 (3%) |
| Sarcomere genes implicated by targeted HCM genetic testing | 18/41 (44%) |
| MYBPC3, n (%) | 10 (56%) |
| MYH7, n (%) | 5 (28%) |
| TNNI3, n (%) | 1 (6%) |
| MYL2, n (%) | 1 (6%) |
| ACTN2, n (%) | 1 (6%) |
Age and maximal left ventricular wall thickness are presented as mean and standard deviation (SD), left ventricular ejection fraction as mean percentage and standard deviation (SD) and categorical variables as numbers (n) and percentages. HCM, hypertrophic cardiomyopathy; MYBPC3, Myosin binding protein C; MYH7, Cardiac β-myosin heavy chain; TNNI3, Troponin I; MYL2, Myosin Light Chain 2; ACTN2, Actinin Alpha 2
Monogenic findings related to cardiomyopathy
Table 2 shows the variants reported by targeted HCM genetic testing and by WGS. Twenty subjects (49%) had variants identified by targeted HCM genetic testing (10 pathogenic, 3 L P, and 7 VUS). The majority of positive results (pathogenic or LP, n=13, 32% of the cohort) involved MYBPC3 and MYH7 (54% and 23% of positive results, respectively). Twenty-one subjects (51%) had no variants identified by targeted HCM testing. Nineteen of 20 variants identified by targeted HCM testing were detected by WGS. One variant, an 18-base pair duplication in MYBPC3 (c.3742_3759dup), was initially missed by the WGS variant detection algorithm. As prior genetic testing, using a resequencing array, had identified this variant, the WGS data was manually reviewed. The variant occurred in 1 of 12 reads covering the duplication, which was below the threshold for variant detection in the WGS algorithm. It was confirmed by Sanger sequencing. As such, this variant was missed due to a combination of the duplication size and the reduced coverage of this region by WGS.
Table 2.
Variants reported by panel testing and WGS that may cause, or contribute to, cardiomyopathy
| HYPERTROPHIC CARDIOMYOPATHY TARGETED TEST RESULT | WGS RESULT | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | Gene | DNA variant | Protein Variant | Classification | Gene | DNA variant | Protein Variant | Classification | ExAC allele frequency | Reported in other HCM probands? |
| 1 | MYBPC3 | c.2827C>T | p.Arg943X | P | Same | 1/16138 South Asian, 1/64974 European | Y | |||
| 2 | MYBPC3 | c.772G>A | p.Glu258Lys | P | Same | 3/43348 European | Y | |||
| 3 | MYBPC3 | c.3742_3759dup | p.Cys1253_Arg1254insGlyGlyIleTyrValCys | P | Variant ultimately identified and reported but initially missed by WGS | Absent | Y | |||
| 18 | MYBPC3 | c.772G>A | p.Glu258Lys | P | Same | 3/43348 European | Y | |||
| 19 | MYBPC3 | c.2905C>T | p.Gln969X | P | Same | Absent | Y | |||
| 21 | MYBPC3 | c.103C>T | p.Arg35Trp | VUS | Same | 3/50036 European | ||||
| 27 | MYBPC3 | c.927-9G>A | P | Same | Absent | Y | ||||
| 33 | MYBPC3 | c.2747G>A | p.Trp916X | P | Same | Absent | Y | |||
| 35 | MYBPC3 | c.3771C>A | p.Asn1257Lys | VUS | Same | Absent | Y | |||
| 26 | MYBPC3 | c.3005G>A | p.Arg1002Gln | VUS | Variant identified but did not meet MedSeq WGS reporting standards due to insufficient evidence for pathogenicity | 4/62092 European | Y | |||
| 6 | MYH7 | c.1987C>T | p.Arg663Cys | LP | Same | Absent | Y | |||
| 11 | MYH7 | c.4031G>A | p.Arg1344Gln | VUS | Same | Absent | Y | |||
| ILK | c.211del | p.Leu71CysfsX26 | VUS | Absent | N | |||||
| 15 | MYH7 | c.1357C>T | p.Arg453Cys | P | Same | Absent | Y | |||
| 22 | MYH7 | c.2717A>G | p.Asp906Gly | P | Same | Absent | Y | |||
| 38 | MYH7 | c.2609G>A | p.Arg870His | P | Same | 1/66732 European | Y | |||
| 34 | TNNI3 | c.568G>T | p.Asp190Tyr | LP | Same | Absent | Y | |||
| 41 | MYL2 | c.484G>A | p.Gly162Arg | LP | Same | Absent | Y | |||
| 31 | ACTN2 | c.1839+5G>C | VUS | Same | Absent | N | ||||
| 5 | ABCC9 | c.1982G>A | p.Arg661His | VUS | Same | 1/11498 Latino, 1/66718 European | N | |||
| FLNC | c.2450T>C | p.Ile817Thr | VUS | 1/9640 African, 1/16472 South Asian, 1/65918 European | N | |||||
| 37 | ABCC9 | c.2238-1G>A | VUS | Variant identified but did not meet MedSeq WGS reporting standards due to insufficient evidence for pathogenicity | 118/16384 South Asian, 70/9748 European, 1/9748 African | N | ||||
| 4 | No variant identified* | PTPN11 | c.1403C>T | p.Thr468Met | Pathogenic | 1/6614 European | N | |||
WGS indicates whole genome sequencing; P, Pathogenic; LP, likely pathogenic; MYBPC3, Myosin binding protein C; MYH7, Cardiac β-myosin heavy chain; TNNI3, Troponin I; MYL2, Myosin Light Chain 2; ACTN2, Actinin Alpha 2; ABCC9, ATP Binding Cassette Subfamily C Member 9; ILK, integrin-linked kinase; FLNC, Filamin-C; PTPN11, Protein Tyrosine Phosphatase, Non-Receptor Type 11
targeted genetic testing panel did not include PTPN11
Three patients had findings identified by WGS in genes that were not analyzed in their prior HCM genetic testing. In one subject with prior negative genetic testing, WGS found a pathogenic PTPN11 variant (c.1403C>T) associated with Noonan syndrome with multiple lentigines (NSML), an autosomal dominant condition characterized by lentigines, typical facial features, pulmonic stenosis, and left ventricular hypertrophy (LVH) among other features.24 She was diagnosed with HCM at 20-years-old due to a murmur and symptoms of effort intolerance. She is 5′1″ tall with mild facial dysmorphism, lentigenes on her upper arms, LVH (maximum wall thickness 15mm) and outflow tract obstruction. Family history was negative for HCM or LVH, but one daughter was known to have mild aortic coarctation. Genetic testing in 2009 included 11 genes but did not examine RAS/MAPK pathway genes, often included on current HCM panels. After the initial negative genetic analyses, additional genetic testing and clinical evaluations were deferred due to the family’s lack of interest and the patient’s perception of limited clinical utility. Following the identification of the PTPN11 variant, her two adult children were evaluated. Though neither has pursued testing for the PTPN11 variant, one with aortic coarctation has mild facial dysmorphism and lentigenes, consistent with NSML.
The second new WGS finding was a VUS in the integrin-linked kinase (ILK) gene in a patient with a previously known VUS in MYH7 (p.Arg1344Gln), a definitive HCM gene. Arginine at position 1344 in MYH7 is highly conserved in evolution. Arg1344Gln has been identified in at least three HCM probands, but is also reported in four samples from the gnomAD database.25 ILK participates in the regulation of cardiomyocyte growth and has been implicated in DCM by studies in mice and zebrafish26, but is not known to cause HCM. The patient had a maximum left ventricular wall thickness of 17mm with outflow tract obstruction that led to septal myectomy. Clinical evaluations in three adult children, all who carry the ILK variant and one with the MYH7 variant, were normal.
The third new finding from WGS was a VUS (p.Ile817Thr) in the filamin C (FLNC) gene, found in a patient with a previously identified VUS in ABCC9, which was included on his panel testing but is not known to cause HCM. FLNC variants are primarily associated with adult-onset skeletal myopathy but also occur with cardiomyopathy in some families.27 Recently FLNC missense variants (but not Ile817Thr) were reported in familial HCM with incomplete penetrance.28 FLNC was not previously analyzed in this patient. He is a 51-year-old male with no family history of HCM and no personal or family history of neuromuscular abnormalities.
Secondary genetic findings
Secondary findings, variants identified in several thousand disease genes14 that are unrelated to the patient’s indication for testing, were reported. There is variability in laboratory practices for reporting secondary findings, with some laboratories only reporting findings in genes on the ACMG list.29 The approach for the MedSeq Project was broad in order to assess the utility of WGS, taking into account all possible genetic results with any clinical significance. Hence, variants that are associated with monogenic dominant diseases might identify previously undiagnosed conditions or risk for future disease development, whereas single variants in recessive genes would not cause disease, but variant carriers could incur risk to subsequent generations. In total, 84 secondary variants were identified in 41 subjects (mean=2.05/person, range= 0–6). Monogenic secondary findings and their disease associations are summarized in Table 3. None of the secondary findings reported in the MedSeq Project were in genes on the ACMG list.10, 11
Table 3.
Monogenic secondary findings from WGS
| Subject age, years | Gene | DNA variant | Protein Variant | Classification | Disease association |
|---|---|---|---|---|---|
| 44 | F5 | c.1601G>A | p.Arg534Gln | Risk allele | Factor V Leiden Thrombophilia |
| 63 | EYA4 | c.1739-1G>A | Likely pathogenic | Postlingual deafness | |
| 55 | SQSTM1 | c.1175C>T | p.Pro392Leu | Likely pathogenic | Paget disease of the bone |
| 62 | CHEK2 | c.1100del | p.Thr367MetfsX15 | Pathogenic | CHEK2-related cancer risk |
| 24 | APP | c.2137G>A | p.Ala713Thr | VUS-FP | Late onset Alzheimer disease |
F5 indicates Coagulation Factor 5; EYA4, EYA Transcriptional Coactivator And Phosphatase 4; SQSTM1, Sequestosome 1; CHEK2, Checkpoint Kinase 2; APP, Amyloid Precursor Protein
Five subjects (12%) had a variant in one of the following genes, with variably robust disease associations: coagulation factor 5 (F5; Factor V Leiden), EYA transcriptional coactivator and phosphatase 4 (EYA4), sequestosome 1 (SQSTM1), checkpoint kinase 2 (CHEK2), and amyloid precursor protein (APP). No clinical interventions were initiated based on these secondary findings. Two of these variants may contribute to non-cardiac phenotypes in subjects. Factor V Leiden was present in a 44-year-old who had subclavian vein thrombosis associated with implantable cardioverter-defibrillator implantation. While lead-associated venous thrombosis is a known complication of device implantation and only 10% of individuals with Factor V Leiden typically develop a blood clot, the F5 variant may have been a predisposing factor in this case. The EYA4 variant is predicted to alter splicing, and similar EYA4 variants cause dominant post-lingual deafness. The subject developed hearing loss around age 50 years that he attributed to excessive noise exposure; however, review of his audiology tracings revealed a pattern more consistent with EYA4 mutations30,31 than the 4kHz notch characteristic of noise induced hearing loss.32 He has no family history of hearing loss. Family members have not pursued EYA4 variant testing or audiological evaluations. An EYA4 deletion was associated with hearing loss and DCM in one family33 and in one proband31, but no other EYA4 variants have been identified in DCM patients with or without hearing loss. As such, the authors considered this a secondary finding with likely association to the patient’s hearing loss, but not to HCM.
The other 3 patients with monogenic secondary findings did not exhibit any phenotypic manifestations of the condition, although each condition has reduced penetrance or variable expressivity.34, 35 A likely pathogenic variant in SQSTM1 which causes Paget disease of the bone, a dominant late-onset disorder associated with increased bone turnover34 was identified in a 55-year-old male without history of orthopedic problems; his cortical bone volume has not been objectively assessed. A pathogenic variant in CHEK2, a gene associated with increased risk for various types of cancer36 was found in 62-year-old female without a personal or family history of cancer. She declined a referral to a cancer genetics program but will continue age-appropriate cancer screening. A VUS in APP was identified in a 24-year-old subject with a grandparent who had Alzheimer disease. Although some APP variants are associated with autosomal dominant late-onset Alzheimer disease37, the potential clinical relevance is uncertain.
There were 79 pathogenic/likely pathogenic recessive carrier variants identified; an average of 2 carrier variants/subject (see supplemental material). Hemochromatosis (HFE) carrier variants were most common (16/41 participants; 39%). Approximately 10–15% of people of European ancestry in the United States are heterozygote HFE variant carriers.38 The remaining carrier states represented recessive conditions with widely variable, even unknown, carrier frequencies in the general population. While most participants were beyond their reproductive years, carrier testing in offspring, who each have a 50% chance to carry the variant, would better define risk for future generations.
Clinical actions triggered by WGS findings
In addition to monogenic and recessive carrier variants, MedSeq Project WGS reported GWAS-based risk predictions for selected common, complex cardiovascular phenotypes.20 In 5/41 (12%) patients, physicians offered referrals to other providers (n=2) or ordered further diagnostic testing (n=3) based on WGS findings (Table 4). The three diagnostic tests were prompted by common alleles that suggested an increased risk of either abdominal aortic aneurysms or atrial fibrillation. Follow-up testing was only conducted in a single case. This patient was predicted to have an increased risk for abdominal aortic aneurysm (90th–100th percentile rank of relative risk), however an abdominal ultrasound revealed normal aortic size. Of note, the physician cited the patient’s strong desire for testing as a significant factor in ordering the ultrasound, rather than physician perception of increased risk. For a patient with a predicted increased risk for atrial fibrillation(90th–100th percentile rank of relative risk) ambulatory electrocardiographic monitoring was initially considered, but the cardiologist then opted to examine existing electrocardiographic information from the medical record. The patient continues to be monitored for the development of atrial fibrillation as part of her routine cardiomyopathy care. A patient considering future reproduction was found to have two recessive carrier variants and was, therefore, advised to get preconception genetic counseling. Similar prenatal referrals would likely be more common in a younger cohort.
Table 4.
Clinical actions resulting from WGS findings unrelated to cardiomyopathy
| WGS finding prompting the clinical action | Clinical Testing Ordered | Findings from Clinical Testing |
|---|---|---|
| GWAS predicted increased risk for atrial fibrillation | Ambulatory electrocardiographic monitoring, n=1 | Test not completed. Existing medical information used instead. |
| GWAS predicted increased risk for aortic aneurysm | Abdominal ultrasound, n=2 | No aortic dilatation identified (n=1); Imaging not completed (n=1) |
| CHEK2 variant | Cancer genetics referral, n=1 | Declined by patient |
| Two carrier variants | Preconception genetic counseling recommended, n=1 | Not yet completed |
Discussion
In the MedSeq Project, the diagnostic yield of genetic testing in HCM patients was similar using either targeted/multi-gene panel testing (32%) or WGS (34%). Expanding the scope of genetic testing to interrogate the genome did not trigger substantive additional clinical action for the patients in the study. In this cohort, WGS detected all but one variant (95%) previously identified by multi-gene panels, allaying major concerns about reduced sensitivity and accuracy with WGS. Moreover, the ability to re-analyze the genome sequencing data provides a valuable resource that will be sought as knowledge evolves and new associations between genes and diseases are discovered, allowing WGS to be more dynamic and flexible than panel testing that is inherently constrained to the included genes. However, to achieve the benefit of re-analysis, a realistic workflow is needed to determine how sensitive genomic data would be securely stored and what prompts re-analysis, as well as who would be responsible for testing and communicating results.
While much of the existing literature on the clinical experience using genomic sequencing in inherited cardiomyopathies consists of case reports describing the use of WES for gene discovery in a proband39 or small collections of families with severe complex cardiomyopathies of unknown etiology,27,40 data from small cardiomyopathy cohorts have also been reported. Seidelmann et al41 reported their experience with WES in a variety of inherited cardiovascular conditions, including HCM. In 28 HCM patients, 13/28 (46.4%) had pathogenic or likely pathogenic variants identified; twelve occurred in genes found on current cardiomyopathy panels. Two patients (7.1%) had novel candidate genes identified. Golbus et al42 performed WGS in 11 individuals with nonischemic DCM. WGS confirmed a previously identified variant in 3 subjects, identified possible new causal variants in 6 subjects, was negative in 2 subjects, and identified potential disease modifiers in two families exhibiting variable disease expression. As such, the MedSeq Project is the only study to date that directly compares targeted testing and WGS in HCM patients while also providing new information regarding the largely undescribed consequences of secondary findings from WGS in a disease specific patient population.
Candidate genes and genetic modifiers
The potential for discovering candidate genes or genetic modifiers of disease over time are major drivers for the shift from targeted to comprehensive sequencing. No novel candidate genes for HCM were identified in this study. However this was not anticipated given the small cohort size and the stringent criteria used for clinical variant reporting, illustrating the need for studying larger populations of panel negative patients using different bioinformatic pipelines and deeper analysis of potential candidate genes or candidate pathways to foster gene discovery. Such efforts are underway.
In our cohort, three patients had variants identified in genes that have potential associations with different cardiomyopathies (ILK, EYA4, FLNC).27,33,43 Although none of these genes has a well-established role in the pathogenesis of HCM, it is conceivable that one or more of these variants may contribute to cardiomyopathy in these patients. The EYA4 variant was found in isolation, while the FLNC and ILK variants were each found in the presence of a VUS in a cardiomyopathy-associated gene (ABCC9 and MYBPC3, respectively). Environmental and genetic modifiers are thought to underlie the substantial clinical heterogeneity of HCM and other cardiomyopathies. It is possible that these variants are modifiers, rather than the primary cause of disease. Additional investigation, including more systematic family evaluation, is required to better understand whether any of the identified variants may be playing a primary or modifier role in the cardiomyopathy phenotype.
Secondary findings and clinical implications
The potential to identify secondary findings may be considered an advantage of WGS by some patients and providers. Indeed, most patients and research participants wish to receive all secondary findings when presented with hypothetical scenarios.44,45, 46 However, others may raise concerns about what WES/WGS might find, and whether that information would be helpful, particularly if there is no ability to prevent disease expression. In the MedSeq Project, WGS revealed a secondary finding with disease risk in 12% of patients (5/41). Virtually all patients had carrier variants identified, with an average of two carrier variants per patient. While there are no expected health consequences for the patient there are reproductive implications for the patient and family. It is important to note that given the broad approach to reporting secondary findings in the MedSeq Project, results may not reflect the typical experience in clinical practice.
With the exception of the potential relationship between the EYA4 variant and hearing loss in one patient and Factor V Leiden in a patient with lead-associated venous thrombosis, secondary findings were not associated with demonstrable clinical features and did not lead to new diagnoses or changes in medical management in this cohort. However, as MedSeq participants had relatively short follow-up and limited phenotyping, clinical features may still emerge. Furthermore, the implications and relevance of a secondary finding to a patient may vary based on context; someone starting a family may be more concerned about a carrier variant than those beyond their reproductive years. Moreover, secondary findings may be largely unexpected by family members if pre-test counseling is not appropriately provided. As with all genetic testing, providers should equip patients with information and resources to facilitate family communication about the implications of results. The additional time demands on providers to investigate the potential clinical relevance of new findings and to facilitate family communication may be considered a disadvantage of WGS, which, when coupled with potential increased healthcare utilization, could have important downstream economic impact on the healthcare system.47 However, while more extensive economic analyses of MedSeq Project data are underway, data derived from physician-participant ordering practices following disclosure indicate that WGS results in patients with established cardiomyopathy had limited clinical impact and; therefore, led to few downstream clinical actions.
Currently, clinical testing laboratories rarely report late onset diseases with no treatment or cure as secondary findings.29 By contrast, we took a broader approach to secondary findings and reported an APP variant to a young patient, an endeavor that epitomizes the concerns about presymptomatic testing for adult-onset neurodegenerative conditions, such as Alzheimer disease. Joint practice guidelines on genetic counseling and testing for Alzheimer disease suggest adopting the multidisciplinary genetic testing model employed for Huntington disease, using both neurologic and psychiatric evaluation to minimize adverse psychological outcomes in those considering presymptomatic testing.48 Given the time and expertise required, this model is challenging to deploy for WGS, particularly for diseases exhibiting variable expression or reduced penetrance, again highlighting the importance of thorough pre-test counseling. Standards for pre-test counseling have been proposed; ongoing evaluation of the consent process will be important as WES/WGS use increases.49
Based on this experience using WGS in clinical practice, we highlight the following considerations:
Providers and patients should have reasonable expectations about diagnostic yield and the potential for secondary findings, and knowledge that our understanding of the genomic sequence data will evolve such that results may need to be revisited.
Proper data interpretation is critical and starts with the genetic testing laboratory but often requires careful phenotyping of patients and family members, in specialized clinical programs with the necessary expertise, to allow for deeper understanding of the potential relationship between phenotype and genotype.
Given the importance of detailed pre- and post-test counseling, collaboration with providers with specific expertise in cardiovascular genetics is recommended to help achieve the best outcomes for patients and families.
Limitations
Although this is to date the largest study of WGS in HCM, the cohort was small and predominantly of European ancestry. Similar results may not be attainable in a more ethnically diverse population where population data in variant interpretation is limited. The use of WGS as the primary genetic testing strategy requires ongoing study to guide appropriate use in the clinic. Well-recognized limitations of WGS include insensitivity to copy number variation and variants characterized by multi-nucleotide repeats. Some panel testing is optimized for these in ways that have not yet been applied to WGS.
Conclusions
Clinical WGS in HCM patients has sufficient sensitivity to detect nearly all sarcomere variants identified with multi-gene panels. Indeed, the overall diagnostic yield of WGS in the MedSeq HCM cohort was similar to that achieved from current and historical multi-gene HCM panels. Despite the potential to identify important secondary findings WGS resulted in few clinical actions. While recognizing that these findings underscore the difficulties of translating genomic data into clinically useful information and define targeted panel testing as less laborious and more cost effective, we conclude that the wealth of information garnered from WGS provided valuable insights that will likely grow with continued discovery of disease genes, risk and modifiers. Even in this small cohort, WGS reclassified disease based on precise etiology (e.g., pathogenic PTPN11 variant) rather than a prespecified phenotype (HCM). We suggest that this may be an important and real impact of genomics: a deeper appreciation of the full spectrum of disease biology that improves medical taxonomy and thereby clinical management. Programs positioned at the interface of clinical care and genetics to properly interpret genomic sequence data and precisely phenotype patients and family members will be best positioned to lead these efforts.
Supplementary Material
Clinical Perspective.
Although multi-gene panel genetic testing for hypertrophic cardiomyopathy (HCM) has been available for over a decade, many HCM patients do not have a molecular etiology identified by current testing panels. As whole exome and genome sequencing become more accessible, there has been speculation that these more comprehensive tests may replace multi-gene panel tests as the preferred strategy for determining the molecular etiology in patients with HCM and other inherited cardiomyopathies. However, the efficacy of this approach in the clinical arena has not been carefully assessed. In this study, 41 patients with HCM who had previously undergone genetic testing with either a multi-gene panel or known familial variant test were randomized to receive whole genome sequencing (WGS), allowing direct comparison of the diagnostic yield of multi-gene panels and whole genome sequencing. WGS and multi-gene panel testing had comparable diagnostic yield. We also assessed the incidence and consequences of secondary genetic findings—genetic variation associated with diseases unrelated to the testing indication of cardiomyopathy, but identified from genomic sequencing. Through these efforts, we describe that broadening the scope of sequencing to interrogate the genome did not lead to the discovery of new genes associated with HCM, nor did it lead to substantial downstream clinical action as a result of secondary genetic findings.
Acknowledgments
The authors would like to thank the MedSeq Project participants and acknowledge Sarah Kalia and Danielle Azzariti for their contributions to this work.
Sources of Funding: The MedSeq Project is funded by the National Human Genome Research Institute U01-HG006500.
Footnotes
Clinical Trial Registration: https://clinicaltrials.gov; Unique Identifier: NCT01736566.
Disclosures: RCG receives compensation for speaking or consultation from AIA, GenePeeks, Helix, Illumina, Prudential and Veritas; and is co-founder and advisor to Genome Medical. CES is a founder and owns shares in Myokardia Inc. HKR directs a fee-for service laboratory (Laboratory for Molecular Medicine) and is on the scientific advisory board for Genome Medical. ML and KM and employed by a fee-for service laboratory (Laboratory for Molecular Medicine). CAM receives funding from American Heart Association, Verily and AstraZeneca and consults for Personome.
References
- 1.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:e783–831. doi: 10.1161/CIR.0b013e318223e2bd. [DOI] [PubMed] [Google Scholar]
- 2.Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC) Eur Heart J. 2014;35:2733–2779. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 3.Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–888. doi: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 4.Walsh R, Thomson K, Ware JS, Funke BH, Woodley J, McGuire KJ, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingles J, Burns C, Bagnall RD, Lam L, Yeates L, Sarina T, et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ Cardiovasc Genet. 2017;10:e001620. doi: 10.1161/CIRCGENETICS.116.001620. [DOI] [PubMed] [Google Scholar]
- 6.Murphy SL, Anderson JH, Kapplinger JD, Kruisselbrink TM, Gersh BJ, Ommen SR, et al. Evaluation of the Mayo Clinic Phenotype-Based Genotype Predictor Score in Patients with Clinically Diagnosed Hypertrophic Cardiomyopathy. J Cardiovasc Transl Res. 2016;9:153–161. doi: 10.1007/s12265-016-9681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gruner C, Ivanov J, Care M, Williams L, Moravsky G, Yang H, et al. Toronto hypertrophic cardiomyopathy genotype score for prediction of a positive genotype in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2013;6:19–26. doi: 10.1161/CIRCGENETICS.112.963363. [DOI] [PubMed] [Google Scholar]
- 8.Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014;371:1170. doi: 10.1056/NEJMc1408914. [DOI] [PubMed] [Google Scholar]
- 9.Green R, Rehm H, Kohane I. Clinical Genome Sequencing. In: Ginsburg G, Willard H, editors. Genomic and Personalized Medicine. 2nd. San Diego: Academic Press / Elsevier; 2013. [Google Scholar]
- 10.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 12.Krier JB, Green RC. Management of Incidental Findings in Clinical Genomic Sequencing. Curr Protoc Hum Genet. 2015;879(23):1–16. doi: 10.1002/0471142905.hg0923s87. [DOI] [PubMed] [Google Scholar]
- 13.Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311:1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vassy JL, Lautenbach DM, McLaughlin HM, Kong SW, Christensen KD, Krier J, et al. The MedSeq Project: a randomized trial of integrating whole genome sequencing into clinical medicine. Trials. 2014;15:85. doi: 10.1186/1745-6215-15-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLaughlin HM, Ceyhan-Birsoy O, Christensen KD, Kohane IS, Krier J, Lane WJ, et al. A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med Genet. 2014;15:134. doi: 10.1186/s12881-014-0134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duzkale H, Shen J, McLaughlin H, Alfares A, Kelly MA, Pugh TJ, et al. A systematic approach to assessing the clinical significance of genetic variants. Clin Genet. 2013;84:453–463. doi: 10.1111/cge.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai EA, Shakbatyan R, Evans J, Rossetti P, Graham C, Sharma H, et al. Bioinformatics Workflow for Clinical Whole Genome Sequencing at Partners HealthCare Personalized Medicine. J Pers Med. 2016;6:12. doi: 10.3390/jpm6010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassy JL, McLaughlin HM, McLaughlin HL, MacRae CA, Seidman CE, Lautenbach D, et al. A one-page summary report of genome sequencing for the healthy adult. Public Health Genomics. 2015;18:123–129. doi: 10.1159/000370102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong SW, Lee IH, Leshchiner I, Krier J, Kraft P, Rehm HL, et al. Summarizing polygenic risks for complex diseases in a clinical whole-genome report. Genet Med. 2015;17:536–544. doi: 10.1038/gim.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane WJ, Westhoff CM, Uy JM, Aguad M, Smeland-Wagman R, Kaufman RM, et al. Comprehensive red blood cell and platelet antigen prediction from whole genome sequencing: proof of principle. Transfusion. 2016;56:743–754. doi: 10.1111/trf.13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baronas J, Westhoff CM, Vege S, Mah H, Aguad M, Smeland-Wagman R, et al. RHD Zygosity Determination from Whole Genome Sequencing Data. J Blood Disord Transfus. 2016;7:365. [Google Scholar]
- 24.Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog Pediatr Cardiol. 2015;39:13–19. doi: 10.1016/j.ppedcard.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hannigan GE, Coles JG, Dedhar S. Integrin-linked kinase at the heart of cardiac contractility, repair, and disease. Circ Res. 2007;100:1408–1414. doi: 10.1161/01.RES.0000265233.40455.62. [DOI] [PubMed] [Google Scholar]
- 27.Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum Mutat. 2016;37:269–279. doi: 10.1002/humu.22942. [DOI] [PubMed] [Google Scholar]
- 28.Gomez J, Lorca R, Reguero JR, Moris C, Martin M, Tranche S, et al. Screening of the Filamin C Gene in a Large Cohort of Hypertrophic Cardiomyopathy Patients. Circ Cardiovasc Genet. 2017;10:e001584. doi: 10.1161/CIRCGENETICS.116.001584. [DOI] [PubMed] [Google Scholar]
- 29.O’Daniel JM, McLaughlin HM, Amendola LM, Bale SJ, Berg JS, Bick D, et al. A survey of current practices for genomic sequencing test interpretation and reporting processes in US laboratories. Genet Med. 2017;19:575–582. doi: 10.1038/gim.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hildebrand MS, Coman D, Yang T, Gardner RJ, Rose E, Smith RJ, et al. A novel splice site mutation in EYA4 causes DFNA10 hearing loss. Am J Med Genet A. 2007;143A:1599–1604. doi: 10.1002/ajmg.a.31860. [DOI] [PubMed] [Google Scholar]
- 31.Makishima T, Madeo AC, Brewer CC, Zalewski CK, Butman JA, Sachdev V, et al. Nonsyndromic hearing loss DFNA10 and a novel mutation of EYA4: evidence for correlation of normal cardiac phenotype with truncating mutations of the Eya domain. Am J Med Genet A. 2007;143A:1592–1598. doi: 10.1002/ajmg.a.31793. [DOI] [PubMed] [Google Scholar]
- 32.Walker JJ, Cleveland LM, Davis JL, Seales JS. Audiometry screening and interpretation. Am Fam Physician. 2013;87:41–47. [PubMed] [Google Scholar]
- 33.Schönberger J, Wang L, Shin JT, Kim SD, Depreux FF, Zhu H, et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet. 2005;37:418–422. doi: 10.1038/ng1527. [DOI] [PubMed] [Google Scholar]
- 34.Galson DL, Roodman GD. Pathobiology of Paget’s Disease of Bone. J Bone Metab. 2014;21:85–98. doi: 10.11005/jbm.2014.21.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiovitz S, Korde LA. Genetics of breast cancer: a topic in evolution. Ann Oncol. 2015;26:1291–1299. doi: 10.1093/annonc/mdv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lerner-Ellis J, Khalouei S, Sopik V, Narod SA. Genetic risk assessment and prevention: the role of genetic testing panels in breast cancer. Expert Rev Anticancer Ther. 2015;15:1315–1326. doi: 10.1586/14737140.2015.1090879. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg RN, Lambracht-Washington D, Yu G, Xia W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016;73:867–874. doi: 10.1001/jamaneurol.2016.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinberg KK, Cogswell ME, Chang JC, Caudill SP, McQuillan GM, Bowman BA, et al. Prevalence of C282Y and H63D mutations in the hemochromatosis (HFE) gene in the United States. JAMA. 2001;285:2216–2222. doi: 10.1001/jama.285.17.2216. [DOI] [PubMed] [Google Scholar]
- 39.Long PA, Larsen BT, Evans JM, Olson TM. Exome Sequencing Identifies Pathogenic and Modifier Mutations in a Child With Sporadic Dilated Cardiomyopathy. J Am Heart Assoc. 2015;4:e002443. doi: 10.1161/JAHA.115.002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Almomani R, Verhagen JM, Herkert JC, Brosens E, van Spaendonck-Zwarts KY, Asimaki A, et al. Biallelic Truncating Mutations in ALPK3 Cause Severe Pediatric Cardiomyopathy. J Am Coll Cardiol. 2016;67:515–525. doi: 10.1016/j.jacc.2015.10.093. [DOI] [PubMed] [Google Scholar]
- 41.Seidelmann SB, Smith E, Subrahmanyan L, Dykas D, Abou Ziki MD, Azari B, et al. Application of Whole Exome Sequencing in the Clinical Diagnosis and Management of Inherited Cardiovascular Diseases in Adults. Circ Cardiovasc Genet. 2017;10:e001573. doi: 10.1161/CIRCGENETICS.116.001573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, et al. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:751–759. doi: 10.1161/CIRCGENETICS.113.000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:5326. doi: 10.1038/ncomms6326. [DOI] [PubMed] [Google Scholar]
- 44.Wynn J, Martinez J, Duong J, Chiuzan C, Phelan JC, Fyer A, et al. Research Participants’ Preferences for Hypothetical Secondary Results from Genomic Research. J Genet Couns. 2017;26:841–851. doi: 10.1007/s10897-016-0059-2. [DOI] [PubMed] [Google Scholar]
- 45.Ziniel SI, Savage SK, Huntington N, Amatruda J, Green RC, Weitzman ER, et al. Parents’ preferences for return of results in pediatric genomic research. Public Health Genomics. 2014;17:105–114. doi: 10.1159/000358539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christensen KD, S S, Huntington NL, Weitzman ER, Ziniel SI, Bacon PL, Cacioppo CN, et al. Preferences for the return of individual results from research on pediatric biobank samples. J Emp Res Hum Res Ethics. 2017;12:97–106. doi: 10.1177/1556264617697839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christensen KD, Dukhovny D, Siebert U, Green RC. Assessing the Costs and Cost-Effectiveness of Genomic Sequencing. J Pers Med. 2015;5:470–486. doi: 10.3390/jpm5040470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13:597–605. doi: 10.1097/GIM.0b013e31821d69b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ayuso C, Millán JM, Mancheño M, Dal-Ré R. Informed consent for whole-genome sequencing studies in the clinical setting. Proposed recommendations on essential content and process. Eur J Hum Genet. 2013;21:1054–1059. doi: 10.1038/ejhg.2012.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.