

Abstract
T-cell Acute Lymphoblastic Leukemia (T-ALL) is an aggressive malignancy of thymocytes. Using a transgenic screen in zebrafish, thymocyte selection-associated high mobility box protein (TOX) was uncovered as a collaborating oncogenic driver that accelerated T-ALL onset by expanding the initiating pool of transformed clones and elevating genomic instability. TOX is highly expressed in a majority of human T-ALL and is required for proliferation and continued xenograft growth in mice. Using a wide array of functional analyses, we uncovered that TOX binds directly to KU70/80 and suppresses recruitment of this complex to DNA breaks to inhibit Non-Homologous End Joining repair (NHEJ). Impaired NHEJ is well known to cause genomic instability, including development of T cell malignancies in Ku70 and Ku80 deficient mice. Collectively, our work has uncovered important roles for TOX in regulating NHEJ by elevating genomic instability during leukemia initiation and sustaining leukemic cell proliferation following transformation.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) affects thousands of children and adults each year in the United States (1). There are many molecular subtypes of T-ALL, with those expressing the master transcriptional regulators TAL1 and LMO1/LMO2 comprising 60% of new diagnoses (2). Despite their wide differences in transcription factor activation and arrest at different stages of T cell development (1,3), T-ALL subtypes also commonly rely on similar oncogenic pathways for transformation. For example, MYC is now widely recognized as the dominant oncogenic driver in human T-ALL and is often activated downstream of NOTCH1 in a variety of T-ALL subtypes (4). Additional oncogenic drivers also likely collaborate with these potent oncogenes to elevate proliferation, alter tumor initiation, and expand early transformed T cell pools. For example, T-ALLs often harbor genomic deletion of p16INK4A, focal amplification of cMYB, and aberrant activation of TAL1 by genomic deletion of the SIL locus (1,2). Chromosomal translocations are also common in T-ALL and often result in mis-expression of oncogenic transcription factors or creation of novel oncogenic gene fusions that drive transformation and growth (2,5). Finally, recent studies have uncovered novel mechanisms to deregulate oncogenes in T-ALL through acquired small genomic insertions and deletions that drive high transcript expression and are acquired through aberrant DNA repair (6,7). Despite the high number of genomic aberrations found in human T-ALL and the supposition that these are driven by errant DNA repair, it has yet to be fully established how these genetic lesions are acquired and what, if any, DNA repair pathways may be altered in human T-ALL.
Non-homologous End Joining (NHEJ) is important for restoring genomic integrity to cells and provides a rapid and robust mechanism to re-ligate broken DNA strands to protect cells from undergoing apoptosis. Following the creation of a double-strand break (DSB), NHEJ repair can be initiated by recruitment of dimeric KU70/KU80 to sites of DNA damage (8). Other NHEJ factors are then recruited to the DNA lesion including DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, XRCC4, Ligase IV and XLF/Cernunnos (9). These proteins facilitate repair by ligating the DNA ends and restoring DNA integrity. Not unexpectedly, impaired NHEJ often results in DNA translocations, inversions, and deletions that are characteristic of cancer (10). In fact, mice deficient in Ku70 or Ku80 have elevated genomic instability and develop T cell malignancies (11,12). Yet, KU70/KU80 and other NHEJ repair factors are not commonly deleted, mutationally inactivated, or hyper-methylated in human T cell lymphoma or T-ALL, leading investigators to conclude that these pathways may not be dominant oncogenic drivers of T-ALL nor have important roles in regulating aberrant DNA changes seen at leukemia initiation. To date, mechanisms that regulate elevated genomic instability and/or alter NHEJ repair in human T-ALL have not been established.
Using a transgenic screen in zebrafish, we have identified thymocyte selection-associated high mobility group box protein (TOX) as a collaborating oncogene that synergized with MYC and intracellular NOTCH1 to initiate early onset T-ALL, to expand the number of transformed clones, and to elevate genomic instability. TOX contains a single HMG box motif and has been suggested to be a transcription factor based solely by its homology with other HMG containing proteins. Yet, TOX shares remarkable conservation in protein sequence within its HMG box domain with well-known chromatin remodeling proteins that lack DNA binding specificity. Importantly, TOX regulates various aspects of T cell development (13–19) and is also genomically amplified in a subset of mouse and human T-ALL (20). Work presented here has uncovered that TOX is transcriptionally activated by well-known T-ALL oncogenic transcription factors, including TAL1 and LMO1/2. Functional studies revealed that TOX regulates both proliferation and NHEJ in human T-ALL and unexpectedly, these functions do not require binding of TOX to the chromatin. Rather, the HMG box of TOX binds directly to and inhibits KU70/KU80, impairing its recruitment to sites of DNA damage. Thus, one function of TOX is to lock human T-ALL cells in a state of dampened NHEJ repair. In total, our results provide a plausible cellular mechanism for elevated genomic DNA aberrations observed in human T-ALL and reveal additional roles for TOX in regulating proliferation after leukemic cell transformation.
RESULTS
TOX accelerates onset and malignant transformation in zebrafish MYC-induced T-ALL
To identify genes that collaborate with MYC to accelerate time to T-ALL onset, we completed a transgenic screen in zebrafish (Fig. 1a). Importantly, the zebrafish transgenic MYC-induced T-ALL model provides a robust and powerful tool to assess oncogenic drivers required to initiate T-ALL (21–24). This model shares remarkably similar molecular mechanisms of transformation with those found in mouse and human (21,23–25). Moreover, zebrafish T-ALL also harbor similar genomic DNA amplifications and deletions when compared with both human and mouse T-ALL (26,27), supporting conserved roles for genomic instability in driving T-ALL onset. In total, 27 genes were included in our screen based on being highly expressed, amplified, and/or mutationally activated in human T-ALL (Supplementary Table S1 and Supplementary Fig. S1a). Fish with fluorescent labeled thymocytes were followed for disease progression after 21 days of life, with leukemia being defined by >50% of the animal being overtaken by fluorescent-labeled T-ALL cells (24,26). From this analysis, 8 genes were identified that significantly shortened time to leukemia onset and increased disease penetrance (p<0.05, Log-rank statistic, Fig. 1b). As has been previously found using mouse models of T-ALL, BMI1 (28), HIF1α (29), IL7R mutations (30), and GFI1 (31) were identified as collaborating oncogenes in our screen, validating that similar genes and pathways drive leukemogenesis in zebrafish, mouse, and human (Fig. 1b). Our work also uncovered thymocyte selection-associated high mobility group box protein (TOX) as a novel collaborating oncogene that synergized with both MYC and activated intracellular NOTCH1 to induce T-ALL (Fig. 1b,c and Supplementary Fig. S1b). Importantly, TOX alone was insufficient to transform thymocytes over the 150 days of observation.
Figure 1. TOX collaborates with MYC to accelerate T-ALL onset.
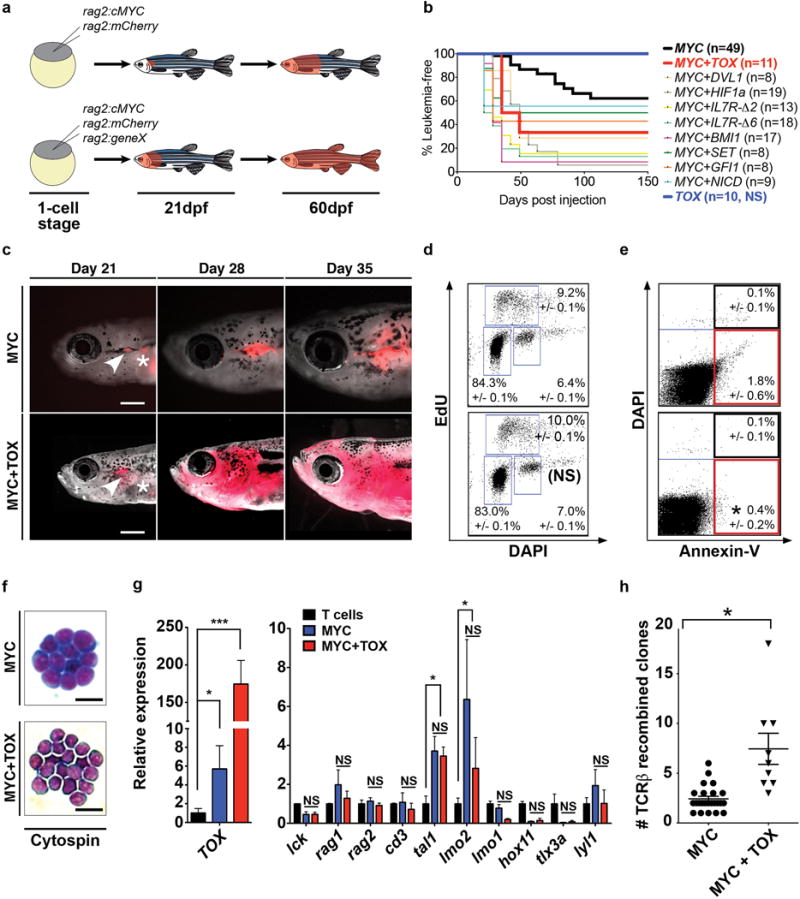
(a) Schematic of transgenic screen. (b) Kaplan-Meier analysis (p<0.05, Log-Rank Statistic). Leukemic fish have >50% of their body overtaken by T-ALL cells. Number of animals analyzed per genotype is shown in parenthesis. (c) Images of T-ALL transgenic fish at 21, 28 and 35 days post-fertilization (dpf). Asterisks denote auto-fluorescence. Arrowheads show leukemias initiating in the thymus. Scale bar equals 2mm. (d) EdU proliferation analysis of zebrafish T-ALL. Not significant (NS). +/− STD noted. (e) Annexin-V apoptosis staining as assessed by flow cytometry. Asterisk denotes p<0.05, Two-tailed Student’s t-test. +/− STD noted. ≥9 primary T-ALLs were analyzed per genotype in d,e. (f) Cytospin showing lymphoblast morphology (n>5 leukemias/genotype analyzed). Scale bar is 20μm. (g) RT-qPCR gene expression comparing MYC and MYC+TOX expressing T-ALL (n=5 per genotype, run in triplicate) with sorted thymocytes isolated from rag2:GFP transgenic fish (purity >95%, viability >95%). *: p<0.05; ***: p<0.001, Two-tailed Student’s t-test. Not significant (NS). Error bars denote standard error of the mean. (h) Number of TCRβ clones per primary leukemia (MYC+TOX: n=9 animals; MYC: n=24 animals). Asterisk indicates p<0.05, Two-tailed Student’s t-test. Error bars denote standard error of the mean.
To define a role for TOX in modulating T-ALL initiation and growth, we next undertook a cellular and molecular characterization of MYC and MYC+TOX expressing T-ALL. Both MYC and MYC+TOX- expressing T-ALL arose exclusively within the thymus (Fig. 1c), had similar lymphoblast morphology (Fig. 1f), and expressed T-cell markers, including higher TOX expression in double transgenic animals (Fig. 1g). Zebrafish MYC and MYC+TOX expressing T-ALLs expressed both tal1/scl and lmo2, confirming that TOX did not alter the subtype of T-ALL induced in zebrafish (Fig. 1g). EdU labeling experiments showed that proliferation rates also did not differ between MYC- or MYC+TOX-expressing T-ALL (p=0.34, Student’s t-test, n>9 T-ALL assessed per genotype, Fig. 1d). However, TOX-expressing T-ALL had slightly fewer apoptotic cells when assessed by Annexin-V staining (Fig. 1e, p=0.012, ANOVA test). Finally, clonal analysis uncovered that MYC+TOX expressing T-ALLs had a dramatic 3-fold increase in TCRβ-recombined clones (MYC+TOX, 7.4+/−1.5 clones compared to MYC, 2.4+/−0.3; p=0.0001, Mann Whitney test, Fig. 1h and Supplementary Table S2). We conclude that TOX has prominent roles in transforming early thymic precursor cells and acts by expanding the overall pool of transformed clones at leukemia initiation.
TOX induces genomic instability in zebrafish T-ALL and MEFs
Human T-ALLs often harbor genomic deletions and amplifications indicative of genome instability and can be assessed clinically by quantitative changes in DNA content following propidium iodide (PI) staining and flow cytometry (32). This same methodology has been previously adapted to zebrafish T-ALL and is a rapid assay to identify leukemias with genomic copy number changes (25). As previously reported, 17% of zebrafish MYC-induced T-ALL had increased DNA content (n=13 of 75) (25). By contrast, 47% of MYC+TOX expressing T-ALLs had altered DNA content (n= 8 of 17, p=0.013, Fisher Exact test, Fig. 2a,b). TOX expressing T-ALLs also comprised a substantial fraction of leukemias that were hypodiploid, contrasting starkly with zebrafish MYC induced T-ALL where hypodiploidy has never been observed (p=0.006, Fisher Exact test). Together, these results show that TOX is a critical driver of genomic instability in zebrafish Myc-induced T-ALL.
Figure 2. TOX promotes genomic instability.
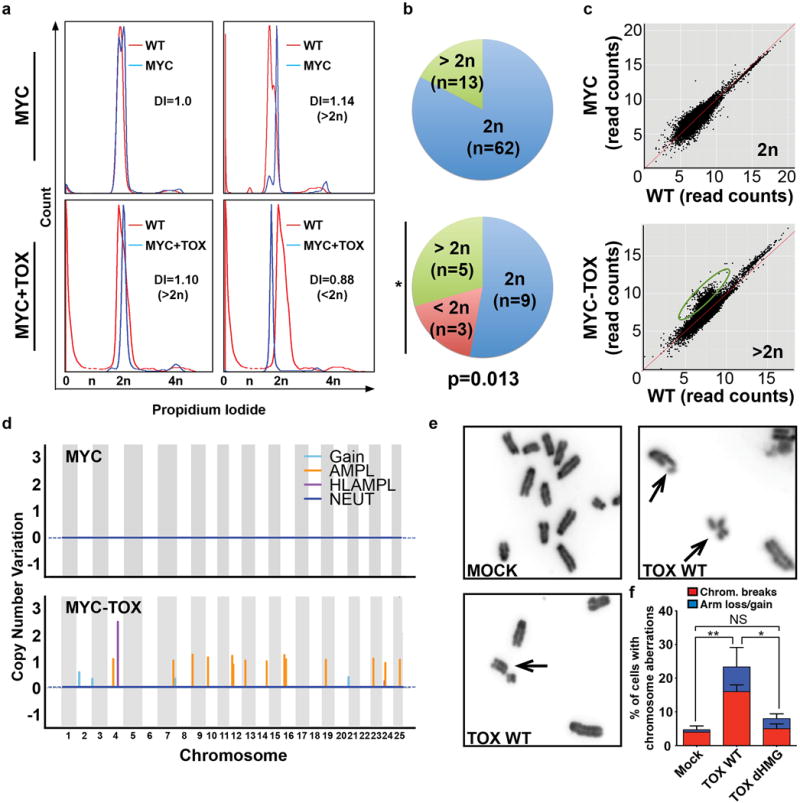
(a) Flow cytometry analysis showing DNA content of zebrafish T-ALL (blue) compared to normal blood-derived DNA (red). Representative leukemias are shown with changes in DNA index noted (DI). 2n has a DNA index of 1. (b) Quantization of DNA content. Asterisk denotes p=0.013, Fisher Exact Test. (c) Genomic DNA alterations identified by whole genome sequencing. The log-log scatter plot represents read counts within non-overlapping window size of 10 kb across the genome comparing leukemia and control tissue from the same, representative animal. Amplifications found in the MYC+TOX leukemia are denoted by the green circle. (d) Manhattan plot representing the copy number variation across the genome of a representative MYC (top) and MYC+TOX (bottom) expressing T-ALL. Regions of significant gain, amplification (AMPL), high level amplification (HLAMPL), and neutral 2N copy number (NEUT) noted. (e-f) Analysis of metaphase spreads from MEF cells infected with control (MOCK), full-length Wild-type TOX (TOX WT), or TOX that lacks the HMG box domain (TOX dHMG). Arrows denote chromosome abnormalities. (f) Quantification of cells with genomic abnormalities. >50 nuclei were counted per condition and replicated three independent times. Error bars denote standard deviation. *, p<0.05 and **, p<0.005, Student’s t-test. Not significant (NS).
To validate that TOX-expressing T-ALLs have elevated genomic instability and harbored alterations in genomic DNA, low-pass whole genome sequencing (WGS) was completed using Illumina Hiseq (0.5–1.0X genome coverage). Low-pass WGS can effectively detect deletions and amplifications akin to array comparative genomic hybridization (array CGH). When compared to matched control tissue isolated from the same leukemic fish, all three MYC+TOX leukemias showed large-scale genomic variations including deletions, gains, and amplifications (Fig. 2c,d and Supplementary Table S3). No recurrent lesions were identified in all three of the TOX+MYC expressing T-ALLs (Supplementary Table S3), indicating that TOX does collaborate with a specified set of amplifications or deletions to drive transformation, but rather TOX is likely a general modulator of genomic instability. Three Myc-induced T-ALLs were also analyzed by WGS, confirming DNA content changes identified by FACs and overall less DNA copy number aberrations when compared with TOX expressing T-ALLs (Fig. 2d and Supplemental Table S3). Taken together, these results independently confirm that TOX imparts elevated genomic instability to zebrafish T-ALL cells.
To further assess a role for TOX in regulating genomic instability, TOX was transfected into mouse embryonic fibroblasts (MEF) cells and chromosomal abnormalities were assessed by metaphase spread (Fig. 2e,f). This analysis revealed that TOX significantly elevated genomic instability that was indicative of impaired NHEJ including elevation in unrepaired chromosome breaks and loss/gains of chromosome arms (p=0.005, Two Tailed Student’s t-test; Fig. 2f). MEF cells that expressed a mutant form of TOX that lacked the HMG box, as a result of deletion of amino acids 261–339, also did not exhibit elevated genomic instability when compared with control treated cells (p=0.15, Student’s t-test). These data further support a role for TOX in elevating genomic instability and suggest prominent roles for the HMG box domain in regulating this process.
TOX is expressed in a majority of human T-ALL and is regulated by the TAL1/MYB transcriptional complex
To determine the extent of TOX expression in human T-ALL, we next analyzed transcript expression in a wide array of human cancer cell lines and uncovered that TOX was highly expressed in human T-ALL (Fig. 3a). This observation was extended to primary T-ALL patient samples, showing that TOX transcripts were highly expressed in human primary T-ALL when compared to normal marrow, irrespective of molecular subtype classification (n=157 of 165 primary T-ALL express high transcript levels of TOX, Fig. 3b and Supplementary Fig. S2a,b). TOX protein expression was also assessed by Western blot analysis and showed that TOX was expressed in both human T-ALL cell lines and primary patient samples (n=10, Fig. 3c and Supplementary Fig. S2c,d). FACS analysis confirmed high expression of TOX at single cell resolution in a large fraction of primary human T-ALLs (Fig. 3d,e). By contrast, TOX protein was detected at lower levels in human thymocytes and B-ALL (n=5).
Figure 3. TOX is highly expressed in human T-ALL and transcriptionally regulated in a subset of leukemias by the TAL1/MYB complex.
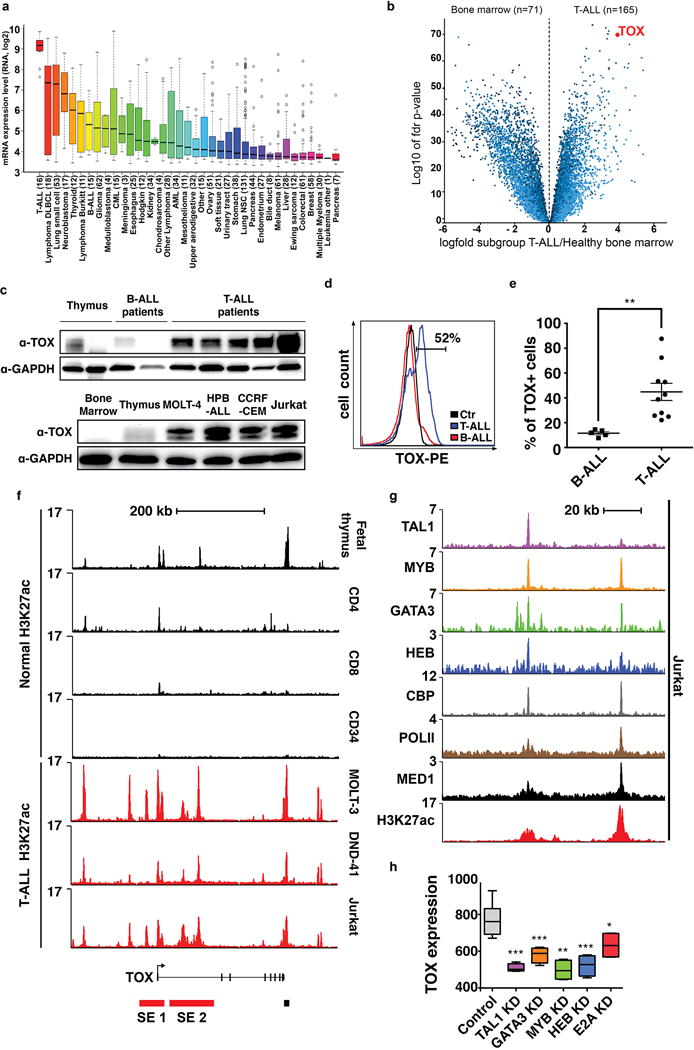
(a) TOX microarray gene expression from the Cancer Cell Line Encyclopedia. Box-and-whisker plots denote median TOX expression (black line), the inter-quartile range (box) and 1.5× the inter-quartile range (bars). (b) Volcano plot comparing gene expression between human T-ALL patient samples and bone marrow. (c) Western blot analysis of primary human leukemia (top) and T-ALL cell lines (bottom). The same thymus sample was run in lane 1, top and lane 2, bottom. (d) Flow cytometry analysis showing TOX expression in primary human T-ALL compared with B-ALL. Control is T-ALL cells stained with conjugated IgG control antibody (ctr). (e) Quantification showing the percentage of TOX+ cells found in each patient sample (right panel, **, p=0.006, Two-tailed Student’s t-test). Mean and standard error of the mean are denoted. (f-h) TOX is associated with two distinct, H3K27 acetylated super-enhancers (SE) in human T-ALL. (f) ChIP sequencing of human T cells, CD34+ progenitor cells from the marrow (black peaks), and T-ALL cell lines (red peaks). Super-enhancer 1 (SE 1) is found in MOLT-3 and Jurkat, while SE 2 is found in all three human T-ALL cell lines. (g) Magnified view of SE 2 in Jurkat T-ALL cells showing super-enhancer occupancy by the TAL1/MYB complex. (h) TOX gene expression in Jurkat cells following knockdown of T-ALL transcription factors found within the H3K27 acetylated super-enhancer (***, p<0.001; **, p<0.01; *, p<0.05, data set from GSE29179; +/− standard error of the mean denoted).\
Given that TOX is highly expressed in a large fraction of human T-ALL and yet only amplified in a small subset of human leukemias (20), we next investigated how TOX might be transcriptionally regulated in malignant T cells. High levels of H3K27ac occupancy are associated with recruitment of mediator and master transcription factors, which define stretch/super enhancers that ultimately drive high transcript expression (33,34). Genomic analysis uncovered elevated H3K27ac occupancy near the TOX locus in human T-ALLs but not immature and mature T-cell subsets or CD34+ marrow cells (Fig. 3f). Super-enhancer analysis revealed two prominent genomic regions that drive high transcript expression in human T-ALL cells, but were not found in normal thymocytes or mature T cell subsets (Supplementary Table S4). Further analysis of H3K27ac peaks revealed that super-enhancer #2 contained both the mediator complex and master T-ALL transcription factors including TAL1, MYB and GATA3 (Fig. 3g). Knockdown of these factors and additional members of the TAL1 complex including HEB and E2A, led to significant reduction in TOX transcript expression in human T-ALL cells (Fig. 3h). We conclude that high TOX transcription is regulated, at least in part, by super-enhancer regulation in human T-ALL.
TOX regulates human T-ALL growth, proliferation, and apoptosis
To assess the consequences of TOX loss of function, HPB-ALL, CCRF-CEM, and MOLT-4 human T-ALL cells were transduced with shRNAs against TOX and compared with scramble control shRNA. Following stable knockdown of TOX using independent shRNAs, all three T-ALL cell lines had reduced viability and overall growth when assessed by Cell Titer-Glo (Fig. 4a,b). EdU proliferation analysis showed that shTOX knockdown T-ALL cells exhibited a marked disruption of cell cycle, with arrest of cells in S-phase and overall reductions in cycling G2/M cells (Fig. 4c). TOX shRNA knockdown cells also had higher levels of apoptosis when assessed by Annexin-V/PI staining when compared with control cells (p<0.02, Student’s t-test, Fig. 4d and Supplementary Figure S3). These results show that TOX has major roles in regulating human T-ALL cell growth and leukemia maintenance, impacting both cell cycle and apoptosis.
Figure 4. TOX is required for continued human T-ALL growth and maintenance.
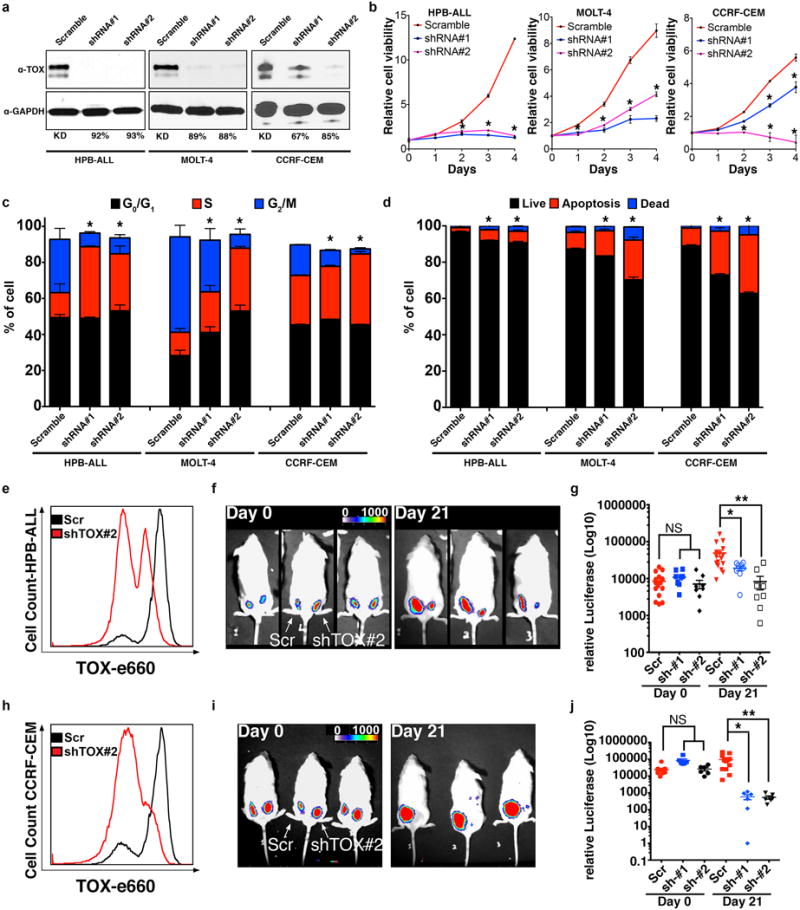
(a) Western blot analysis following stable shRNA knockdown in human T-ALL cells. Percent knockdown noted. (b) Cell viability following knockdown as assessed by Cell TiterGlo. (c) EdU proliferation analysis. (d) Annexin-V staining. Asterisks in b-d denote p<0.05, Student’s t-test. Xenograft studies performed with human HPB-ALL (e-g) and CCRF-CEM (h-j). (e,h) Flow cytometry analysis showing efficiency of TOX knockdown. (f,i) Luciferase bioluminescent imaging of representative animals engrafted at 0 days compared with 21 days. Scramble shRNA control (left flank) or shRNA-TOX #2 (right flank). (g,j) Quantification of xenograft growth using two independent shRNAs. Not significant (NS), p<0.05 (*), p<0.01 (**), and p<0.001 (***), ANOVA test.
To extend our findings to the in vivo setting, human HPB-ALL and CCRF-CEM knockdown cells were transduced with luciferase and assessed for growth in NOD/Scid/IL2Rγ null mice. TOX knockdown cells exhibited significantly reduced xenograft growth by 21 days when compared with control shRNA treated cells (Fig. 4e–j). Together, these data show that TOX also has important roles in regulating continued growth and maintenance of human T-ALL cells in vivo.
TOX binds directly with KU70/KU80 through the HMG box domain
To develop hypotheses about how TOX might be regulating transformation, we sought to identify TOX binding partners. Specifically, endogenous TOX antibody pull-down experiments were performed in human HPB-ALL, MOLT4, and Jurkat T-ALL cells and interacting factors identified through liquid chromatography–mass spectrometry (LC-MS/MS). Following separation of proteins on an SDS-page gel, prominent protein bands were detected in the TOX pull down cell lysates from HPB-ALL cells but not the IgG control cells (Supplementary Fig. S4). These bands were excised and analyzed by LC-MS/MS. From this analysis, KU70 and KU80 were identified as TOX binding factors (Supplementary Table S5a). Additional LC-MS/MS experiments performed in MOLT4 and Jurkat cells confirmed the KU70/KU80 interaction with TOX using analysis of total protein eluates obtained following endogenous TOX immunoprecipitation (Supplementary Table 5b). In these experiments, KU70 and KU80 exhibited a remarkable >22-fold enrichment over IgG control pull down cells, independently verifying TOX interaction with KU70/KU80.
To confirm Ku70/KU80 protein interactions identified by mass spectrometry, TOX immunoprecipitation was performed in the presence and the absence of DNase I and Western blot analysis completed with KU70 and KU80 specific antibodies in all three human T-ALL cell lines. DNase I treatment ensured that interactions of TOX and KU70/KU80 did not result from binding common genomic DNA fragments (Fig. 5a). As was seen by the IP-mass spectrometry experiments, TOX interacted with KU70/KU80 in human HPB-ALL, CCRF-CEM and MOLT-4 human T-ALL cell lines (Fig. 5a and Supplementary Fig. S5a). Reciprocal immunoprecipitation using the KU70 antibody confirmed specificity of the interaction (Fig. 5a, right panel). Consistent with our LC-MS/MS experiments, Western blot analysis also revealed that late acting NHEJ factors including XRCC4 and DNA Ligase IV did not pull down with TOX (Fig. 5b), showing that the interaction of TOX with KU70/KU80 was limited to the dimeric initiating complex that is first recruited to DNA double strand breaks (DSBs). In vitro binding assays using purified protein and performed in the absence of DNA confirmed a direct interaction between TOX, KU70, and KU80 (Fig. 5d and Supplementary Fig. S5b). By contrast, HMG deleted TOX was unable to bind KU70/KU80 in in vitro binding assays (Fig. 5d), confirming a requirement for this domain in regulating the TOX protein:protein interaction with dimeric KU70/KU80.
Figure 5. TOX binds directly to the KU70/KU80 and inhibits Non-Homologous End Joining by suppressing recruitment of KU70/KU80 to sites of DNA damage.
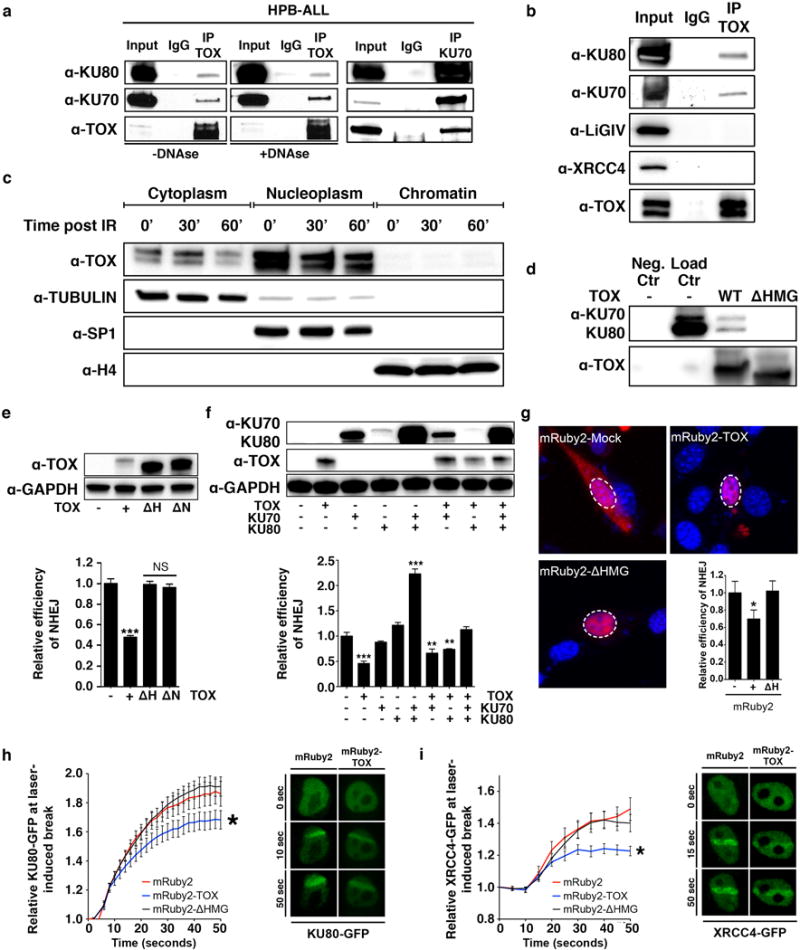
(a) TOX immunoprecipitation followed by Western blot analysis in HPB-ALL cells in the presence or absence of DNAseI treatment. The right panel shows the reciprocal immunoprecipitation using anti-KU70. (b) TOX immunoprecipitation followed by Western blot analysis for members of the NHEJ pathway. (c) Western blot analysis following cell fractionation in non-irradiated and 3Gy-irradiated HPB-ALL cells. TUBULIN, SP1 and Histone H4 (H4) are controls for assessing cytoplasmic, nucleoplasmic and chromatin fractions, respectively. (d) In vitro binding of TOX (WT) or HMG-box (ΔHMG) deletion mutant with KU70/KU80 followed by Western blot analysis. Negative control was KU70/KU80 added to beads and then purified in the absence of TOX (Neg. Ctr). Loading control contains only purified KU70/KU80 proteins (Load Ctr). (e) Western Blot analysis of 3T3 cells following transfection with full-length TOX (+), ΔHMG (ΔH), or ΔNLS (ΔN). Below is the quantification of the NHEJ assay (NS: Not significant; ***, p<0.0001, Two-tailed Student’s t-test comparing experimental samples with control, +/− STD noted). (f) Western Blot analysis of 3T3 cells following transfection with full-length TOX, KU70 and/or KU80. Below is shown the quantification of the NHEJ assay (**, p<0.001, ***, p<0.0001, Two-tailed Student’s t-test, +/− STD noted). (g) Confocal imaging of 3T3 cells transfected with mRuby2-fused constructs. Dashed lines denote nucleus as assessed by Hoechst stain (n>100 cells/construct analyzed). The lower right panel shows the fluorescence-based NHEJ assay completed in 3T3 (*, p=0.03, Two-tailed Student’s t-test). +/− STD noted. (h,i) Quantitative assessment of recruitment of KU80-GFP (h) or XRCC4-GFP (i) to sites of UV laser-induced DNA damage in 3T3 cells. Asterisks denote significant differences (p<0.05, Two-tailed Student’s t-test). +/− STD noted. Representative fluorescent images of cells following laser-induced damage are shown to the right.
Endogenous antibody pull down and in vitro binding assays showed the HMG box motif was responsible for protein:protein interactions with KU70 and KU80 and did not require DNA binding; thus, we wondered if, like other HMG group box containing proteins, TOX bound chromatin. Cell fractionation studies were performed in human HPB-ALL T-ALL cells and identified that TOX was highly expressed in the nuclear fraction and to a lesser degree to the cytoplasm; yet TOX did not bind to chromatin (Fig. 5c). Irradiation of cells induces large amounts of DNA damage and facilitates recruitment of repair enzymes to DNA breaks. Despite high dose irradiation of human T-ALL cells, TOX was still not recruited to chromatin even after 60 minutes following 3Gy irradiation treatment (Fig. 5c and Supplementary Fig. S5c). We conclude that TOX binds directly to KU70/KU80 through its HMG box domain and that this protein:protein interaction does not require DNA binding.
TOX inhibits DNA repair by suppressing recruitment of NHEJ factors to DSBs
Given the prominent roles for KU70/KU80 in regulating NHEJ and subsequently genomic instability (11,12) and that TOX over expression in zebrafish T-ALL and MEFs lead to genomic instability, we hypothesized that TOX might be an inhibitor of KU70/KU80 function and thus affect NHEJ repair. Full-length TOX or deletion mutants that lack the NLS or HMG box domain were stably expressed in 3T3 fibroblast cells (Fig. 5e and Supplementary Fig. S6a). Cells were then transfected with linearized plasmid that contained a DNA DSB between the promoter and the GFP coding sequence, where GFP is expressed only following successful DNA repair. Cells expressing full-length TOX exhibited a remarkable 54+/−4.5% reduction in DNA repair when compared with control cells (Two tailed Student’s t-test, p=0.0004, +/− STD, Fig. 5e), while 3T3 cells expressing TOX deletion mutants that lack the HMG box or nuclear localization domain did not have altered NHEJ. Epistasis experiments revealed that when both KU70 and KU80 were expressed together, NHEJ was fully restored to TOX expressing cells (Fig. 5f). By contrast, expression of either KU70 or KU80 alone was not sufficient to restore NHEJ in TOX expressing cells (Fig. 5f). Suppressed NHEJ was also observed in 3T3 cells transfected with red fluorescent-protein fusions of mRuby2 with full-length TOX but not when fused with deletion mutants that lack the HMG box domain (Fig. 5g). Imaging studies verified that both full-length TOX and HMG box deleted TOX were predominantly localized to the nucleus (Fig. 5g) while the NLS mutant was localized to the cytoplasm (Supplementary Fig. 6b).
To directly assess if TOX modulates recruitment of KU70/KU80 to the sites of DNA repair, 3T3 fibroblasts were transfected with KU80-GFP and mRuby2, mRuby2-TOX or mRuby2-ΔHMG, subjected to UV laser microirradiation-induced DNA damage, and assessed for KU80-GFP recruitment to DSBs (Fig. 5h). Cells expressing full-length TOX exhibited significantly reduced recruitment of KU80-GFP to DNA breaks (p<0.05, Student’s t-test), while cells that expressed mRuby2-ΔHMG efficiently recruited KU80-GFP to sites of DNA damage with similar kinetics to control cells. mRuby2-TOX and fusions with HMG deleted TOX were not recruited to microirradiation induced breaks and remained nuclear localized throughout these experiments. Impaired recruitment of NHEJ pathway regulators to sites of DNA damage was independently confirmed using XRCC4-GFP (p<0.05, Student’s t-test, Fig. 5i). These experiments show that TOX sequesters KU70/KU80 away from sites of active DNA repair.
TOX loss of function in human T-ALL leads to elevated NHEJ repair
We hypothesized that TOX might lock human T-ALL cells in a state of dampened DNA repair, with the prediction that loss of TOX should elevate NHEJ repair. Stable knockdown or shRNA expressing control cells were transfected with linearized plasmid that contained a DNA DSB between the promoter and the mCherry coding sequence, with fluorescence only being detected following successful DNA repair. Plasmid based repair assays showed that HPB-ALL, CCRF-CEM, and MOLT4 T-ALL cells all exhibited enhanced NHEJ repair following stable TOX knock down using independent shRNAs (Fig. 6a–f and Supplementary Fig. S7; p<0.05, Student’s t-test). Similar results were obtained using stable integration of the Traffic Light Reporter (TLR). The TLR assay reads out NHEJ repair via RFP expression following induction of a DSB using tamoxifen-inducible I-SceI restriction enzyme (35). These experiments showed that NHEJ repair was greatly increased following TOX knockdown in CCRF-CEM cells, exhibiting a striking increase in NHEJ following TOX-depletion (p=0.0001, Two-tail Student’s t-test, Fig. 6g,h).
Figure 6. TOX loss-of-function increases DNA repair and accelerates the time to break resolution in human T-ALL cells.
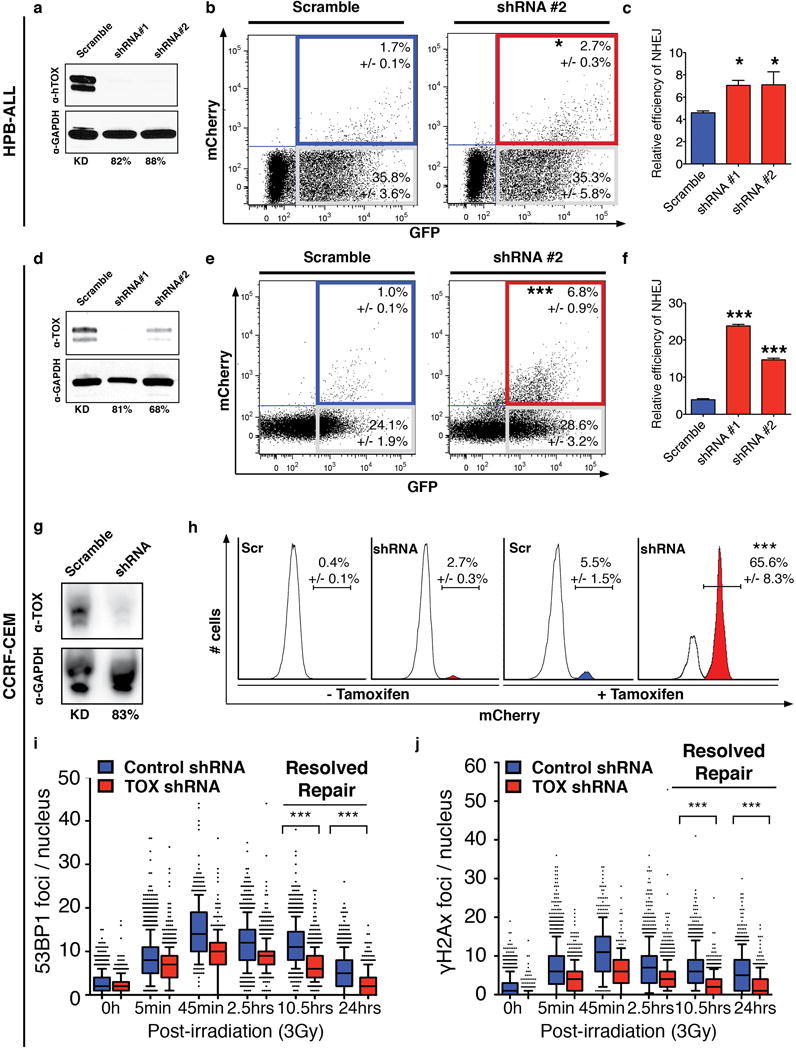
(a–f) Transient repair assays in HPB-ALL (a–c) and CCRF-CEM (d–f) cells that have shRNA knockdown. (a,d) Western blot analysis with percent knockdown noted. (b,e) Flow cytometry analysis of knockdown and control cells following transient fluorescence-based NHEJ assay. (c,f) Graphical summary of data. *, p<0.05; ***, p<0.001 when compared with control shRNA treated cells, Two-tailed Student’s t-test. (g) Western blot analysis of TOX expression in CCRF-CEM cells with stable TOX shRNA #2 or scramble control shRNA knockdown used in the Traffic Light Reporter assay. (h) Flow cytometry analysis of CCRF-CEM cells stably integrated with the Traffic Light Reporter. Cells were nucleofected with Tamoxifen-inducible I-SceI meganuclease and induced as noted in the figure. Percent of cells that undergo NHEJ repair and express mCherry are noted (+/−1STD). ***, p=0.0001, Two-tailed Student’s t-test. (i,j) Kinetics of the DNA repair in CCRF-CEM control and stable knockdown down T-ALL cells (TOX shRNA #2). The number of 53BP1 (i) and γH2A.X (j) foci per nucleus following 3Gγ irradiation are denoted. Each point represents data from a single cell and the black bars denote the median foci per cell (***, p<0.0001, Two-tailed Student’s t-test, >150 cells counted/condition). Box-and-whisker plots denote expression with the median 75% of samples and bars 90%.
To independently confirm a role for TOX in modulating DNA DSB repair, we next assessed the kinetics and localization of 53BP1 and γH2A.X in irradiated CCRF-CEM and HPB-ALL T-ALL cells following stable knockdown of TOX. 53BP1 and γH2A.X are recruited to DNA following induction of DSBs and their kinetics can be measured to quantify resolution of the breaks following γ-irradiation (36). Using this strategy, we found that TOX knockdown led to significantly faster DNA break resolution in human T-ALL cells following 3Gy irradiation (Fig. 6i,j; Supplementary Fig. S8; p<0.001, Student’s t-test). The overall numbers of foci were also reduced at many early time points reflecting ongoing and faster repair in TOX depleted cells following irradiation-induced DSB damage. Similar results were also seen in irradiated HPB-ALL cells (Supplementary Fig. S8; p<0.001, Student’s t-test). Together, our data indicate that TOX negatively regulates the NHEJ pathway in human T-ALL and that human T-ALLs are locked in a state of dampened DSB DNA repair (Supplementary Fig. S9).
DISCUSSION
Our work has uncovered TOX as a novel collaborating oncogenic driver in T-ALL with important roles in both leukemia initiation and maintenance. TOX is amplified in a small subset of mouse and human T-ALL and was included in our screen based on these results (20). Yet, our work uncovered that most human T-ALLs express high levels of TOX and that this high transcript expression largely results from super enhancer regulation by well-known T-ALL transcription factors including TAL1 and LMO1/2. Thus, TOX does not commonly meet the criteria for being a classically-defined oncogene which are activated by translocation, amplification or activating mutations, but rather TOX is expressed in developing T-ALL cells by oncogenic transcription factors, locking leukemia cells both in a state of heightened proliferation and dampened DNA repair. Importantly, TOX also had unexpected roles in regulating leukemia initiation by expanding the pool of initiating clones and elevating genomic instability in the zebrafish T-ALL model. Experiments completed in MEF cells confirmed roles for TOX in inducing genomic instability and generating chromosomal aberrations. Finally, studies of human T-ALL and xenograft studies revealed additional roles for TOX in regulating continued leukemia maintenance by specifically regulating cell cycle proliferation and apoptosis. These results are in keeping with known roles for TOX in regulating cell cycle in primary Sezary cells and Cutaneous T-cell Lymphoma where TOX knockdown led to cell cycle arrest and secondarily cell death (37). Collectively, our work has identified TOX as a new collaborating oncogenic driver in T-ALL and which likely exerts important and diverse functional effects in a large fraction of human T-ALLs.
Because TOX had been previously shown to regulate proliferation in primary Sezary cells and Cutaneous T-cell Lymphoma (37), our studies focused on uncovering additional roles for TOX in regulating DNA repair pathways. Remarkably, our endogenous TOX pull-downs and subsequent LC-MS mass spectrometry analysis uncovered potent interactions between TOX and KU70/KU80, prompting further investigation into roles for TOX in regulating DNA repair and NHEJ. Loss of NHEJ repair genes in mice are well-known to cause elevated genomic instability and cancer. For example, KU70 and KU80 deficiency in mice results in genomic instability and predisposition to T cell malignancy (11,12). Yet, genetic loss of KU70 or KU80 or disruption of other NHEJ factors has not been implicated as a major tumor suppressor pathway in human T-ALL. Rather our work has shown that TOX acts as a negative regulator of KU70/KU80 function to alter NHEJ repair and to elevate genomic instability. Remarkably, TOX remains highly expressed in fully malignant human T-ALL and locks leukemia cells in a state of dampened NHEJ repair, an observation confirmed using a wide array of DNA repair assays performed directly in human T-ALL cells. Even more remarkable is that TOX performs this function by directly binding KU70/KU80 and does not require binding to chromatinized DNA. Perturbations in NHEJ result in the creation of deletions, amplifications, transversions, and translocations that impart growth advantages to early transformed cells and drives cancer initiation. Thus, it is not surprising that mutations in DNA repair proteins can alter NHEJ and are predisposing to cancer. Our work strongly suggests that TOX may be such a factor in T-ALL.
Our results also suggest the existence of a new class of HMG group box proteins that function without binding chromatinized DNA. Indeed, endogenous chromatin immunoprecipitation studies and DNA-free in vitro binding assays show that TOX does not bind to chromatin. Rather, the HMG box domain of TOX is required for protein:protein associations with KU70/KU80 and functionally impairs recruitment of NHEJ factors to breaks. TOX contains a single highly conserved HMG box motif that is structurally distinct from both class I and II HMG box group containing proteins (38). Class I HMG box group proteins are transcription factors and contain a single HMG box that binds DNA in a sequence-specific manner. These transcription factors include the TCF/LEF1 and SOX family members and have important roles in regulating a wide array of developmental and cancer processes (39). By contrast, class II group proteins contain two DNA-binding motifs, bind chromatin in a sequence agnostic manner, and have important roles in modifying DNA structure. For example, HMGB1 modulates nucleosome structure and alters transcription by looping chromatinized DNA to facilitate interaction of transcription factors with enhancer elements (40). HMGB1 also regulates a multitude of DNA repair pathways (41–43). For example, HMGB1 can enhance in vitro DNA ligase activity, by bringing DNA ends into close proximity (44). Yet, to date, functional roles for HMGB1 and other class II HMG group box proteins in regulating DNA bending and repair necessarily require binding to chromatin.
Together, our data suggest that TOX may represent the first of a novel class of HMG proteins that function, at least in part, by binding regulatory proteins through their HMG box and regulating NHEJ repair. Given the prominent role TOX has in T cell development and its coordinated regulation coincident with TCRβ and TCRα rearrangement in mice, it is also possible that TOX may have roles in development including regulation Recombination-Activating Gene (RAG)-mediated recombination. Moreover, TOX is highly and specifically expressed in human T cell malignancies while its related family members TOX2, TOX3, and TOX4 are differentially regulated in a variety of human cancers including Breast cancer, Ewing sarcoma, Multiple Myeloma and Small Cell Lung cancer (45–47). Many of these tumors are well known to have elevated genomic instability and harbor characteristic lesions that are associated with impaired NHEJ. It will be important to assess whether these closely related TOX family members share molecular functions in regulating cell proliferation, transformation and genomic instability in a wider range of human cancers.
Materials and Methods
Transgenic DNA expression constructs
DNA constructs used to generate transgenic zebrafish included rag2:mCherry (23), rag2:MYC (21) and rag2:NICD (24). All genes, except FNBP1, GFI1, GITR, BMI1 and TOX, used in this screen were obtained from hORFeome (48). IL7R constructs were created using site-directed mutagenesis. Full-length ORFs for human FNBP1, GFI1 and GITR were obtained by PCR-amplification of cDNA derived from human T-ALL cells. Zebrafish TOX was amplified from the cDNA of 1 to 5-day-old Tu/AB zebrafish and subcloned into pENTR gateway system (Life Technologies). PCR primer sequences can be found in Table S6. All genes were transferred into the rag2 promoter destination vector using LR clonase II (Life Technologies).
Creation of zebrafish T-ALL
Plasmids were linearized with NotI or XhoI and purified. Mosaic transgenic animals were generated as previously described (22). 40 ng/μL rag2:mCherry was mixed with 40 ng/μL of rag2:Myc or rag2:NICD and 40 ng/μL rag2:experimental gene and micro-injected into one-cell stage Tu/AB embryos. Animals were scored for fluorescent-labeled thymi at 21 and 28 days of life and then followed weekly for disease onset. Leukemic fish are defined by >50% of their body being infiltrated with fluorescent T-ALL cells as previously described (24,26). Zebrafish experiments were approved under animal protocol 2011N000127 (MGH).
Clonality, proliferation, apoptosis and DNA content
Proliferation was assessed using the Click-IT EdU kit (Life Technologies) according to the manufacturer protocol. A pulse of EdU was performed following cell harvest and completed for 1h at 28.5°C. Zebrafish T-ALL cells were assessed for apoptosis using DAPI and Alexa Fluor 647-conjugated AnnexinV (Life Technologies). Numbers indicate the mean of ≥9 biological replicates +/− the s.e.m. For DNA content, 1×106 zebrafish T-ALL cells were permeabilized in 70% ice-cold Ethanol for >2hrs at −20°C. Cells were then incubated in 500μL of a solution containing 50μg/mL PI, 1mg/mL PureLink RNase and PBS-1% BSA for 30 minutes at 25°C. EdU-stained cells, AnnexinV-stained cells and DNA content were then analyzed using LSR II or Fortessa Flow Cytometer (BD Biosciences). The DNA index quantifies differences in total DNA in the test cell population in relation to that in normal diploid cells, comparing maximal G0/G1 peak heights between samples when assessed by FACs. A DNA index of 1.0 is indicative of normal diploid cells.
The zebrafish TCRB recombination assay was first described by Blackburn et al. (24). Briefly, RNA was extracted from FACS-sorted T-ALL cells, made into complementary DNA, and PCR was performed utilizing each Vb and Cb primer (51 variable spanning PCR primers and 2 constant regions). A semi-nested PCR was completed using 1 μl of the PCR product and resolved on a 2% agarose gel. From this analysis, we can quantify the overall numbers of clones contained within the leukemia (24,26).
RT-qPCR was performed on bulk leukemias (n=5 fish/genotype) and compared with FACs sorted rag2:GFP+ thymocytes (n=2 samples). Samples were run in triplicate, with error bars representing the s.e.m of compiled data from all replicates and experimental samples.
Genomic DNA sequencing of zebrafish T-ALL
Zebrafish genomic DNA was extracted from T-ALL cells using a solution containing 10mM Tris-EDTA, 0.2% SDS, 200mM NaCl and 200μg/mL Proteinase K following incubation at 50°C for >16hrs. The next day, the Proteinase K was heat-inactivated at 95°C for 10min. The gDNA extract was then purified using genomic DNA clean and concentrator (Zymo). Purified zebrafish gDNA was sheared and made into a library by the MGH Next generation sequencing core. Next-generation sequencing of zebrafish gDNA from MYC, MYC+TOX and WT samples was performed using Illumina HiSeq Instrument. Following alignment to the Zv9 genome using BWA (49), analysis of large-scale genomic variants using HMMCopy (50). In addition, a direct comparison of mutant and WT coverage was performed by calculating read coverage within non-overlapping 10 Kb windows across the genome. The log-log scatter plots of the resulting read counts from leukemias were compared to the corresponding WT tissue isolated from the same fish.
Metaphase spread on mouse MEF cells
MEF cells were transfected with control, full-length TOX, and dHMG box containing TOX. After 48h post transfection, MEF cells were incubated with 100ng/μL KaryoMax Colcemid for 90min (Life Technologies), washed and incubated for 18 min with 0.075M KCl. 1mL of a 3:1 Methanol:glacial acetic acid (fixative solution) was added to the chromosome preparation and centrifuged 10min at 400g, 4°C. The pellet was washed 2 times with 3mL of fixative solution and resuspended in 1mL of fixative solution. Chromosomes were spread on microscope slides and dried 10min at room temperature. DAPI Fluoromount-G (Southern Biotech) was added to each slide and imaged. At least 50 nuclei are counted per condition and per experiment. Experiments were replicated 3 independent times.
Gene expression analysis of TOX in human T-ALL
TOX transcript expression was assessed in human cell lines using the Cancer Cell Line Encyclopedia (45) (GSE36139) and in human T-ALL patient samples using the program R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). Data sets included normalized gene expression for healthy bone marrow (GSE13159) and human leukemia samples comprising the “Mixed Leukemia – MILE – 2004” (GSE13159) and “Tumor ALL (T) - Meijerink – 124” datasets (GSE26713). Statistical analysis and visualization was completed using Prism6.0. Whiskers plots have been utilized to visualize the data and are represented using the Tukey method. Primary human samples used in this work were collected under the IRB DFCI 05-001.
Protein lysates were extracted using RIPA buffer, quantified using Coomassie Plus Bradford protein assay (Pierce), and analyzed by Western blot analysis. Specifically, denaturated proteins were loaded on a 4–20% gradient gel (Biorad) and then transferred to a PVDF membrane (Biorad). Antibody staining was visualized using Amersham ECL Prime reagent (GE Healthcare) followed by analysis using a Fluor-S MultiImager (Biorad). Band intensity was quantified using the ImageJ software package. Human T-ALL cells were assessed by Western blot analysis using primary antibodies for α-TOX (1:1000 dilution, eBioscience), α-GAPDH (1:2500, #2118, Cell Signaling Technology), α-TAL1 (1:200, BTL73, EMD Millipore), α-LMO2 (1:200, sc-65736, Santa Cruz Biotechnology) and α-cleaved NOTCH1 (1:1000, #4147, Cell Signaling Technology). The secondary antibodies used were HRP-conjugated anti-rat IgG (1:2000, #7077, Cell Signaling Biotechnology), HRP-conjugated anti-rabbit IgG (1:2500, #7074, Cell Signaling Biotechnology) and HRP-conjugated anti-mouse IgG (1:5000, G21040, Life Technologies).
Mouse xenografts and luciferase imaging
Human T-ALL cells were luciferized using the pLKO.1-luc-mKate (gift from Drs. Matthijssens and Van Vlierberghe, Ghent University, Belgium) and equal numbers of viable cells were injected into the flanks of six-week-old NOD/SCID/IL2rg null female mice (1×106 of viable cells per 200 μl). Mice were anesthetized by isofluorane and leukemia growth was monitored by bioluminescence imaging following subcutaneous injection into the loose tissue over the neck of 75mg/kg D-luciferin (Perkin Elmer) in 100 μl of PBS. Comparisons of leukemia size used an ANOVA test and Student’s T-test comparisons between control and treated cells. Mouse experiments were approved under animal protocol 2013N000038 (MGH).
ChIP-Seq analysis
Reads were aligned to the hg19 version of the human genome using bowtie (51) with parameters –k2 –m 2 –best –sam and –l set to read length. Wiggle files to display the density of reads relative to genomic loci were created with MACS (52) with parameters –w –S –space=50 –nomodel –shiftsize=200 –keep-dup=1. The wiggle files were subsequently normalized to the millions of unique positions covered by reads. Wiggle files were displayed in the UCSC genome browser. Array file name used in this study are indicated in Table S6.
Super-enhancer identification
Super-enhancers were identified in two stages as described previously (34,53). First, constituent enhancers were defined as peaks of H3K27ac using MACS with two sets of parameters to account for focal amplifications: -p 1e-9–keep-dup=1 and –p 1e-9 –keep-dup=all. Second, the union of these sets of peaks was used as input for ROSE, which stitches proximal enhancer constituents and ranks stitched enhancers by signal. ROSE was run with parameters –s 12500 and –t 1000 to stitch constituents within 12.5kb of each other and exclude constituents fully contained within +/−1kb from promoters of RefSeq genes.
Stitched enhancers were each assigned to the single expressed RefSeq gene locus whose transcription start site is most proximal to the center of the stitched enhancer. Genes were considered expressed if they were in the top 2/3 of all TSS when ranking by promoter H3K27ac density. RPM-normalized promoter H3K27ac density was calculated in 1kb windows center on TSSs using bamToGFF with parameters –e 200 –m 1 –r –d.
Immunoprecipitation and Western blot analysis
Immunoprecipitation was performed using 300μg of protein lysate along with 4μg of TOX antibody (GeneTex), KU70 antibody (ab3108, Abcam) or control rabbit IgG (Santa Cruz Biotechnology). Immunoprecipitation was completed in the presence of 30–40μL of Dynabeads Protein G (Life Technologies) and incubated for 16h at 4°C in Cell Lysis Buffer (Cell Signaling Technology). Beads were then washed 5 times and proteins were eluted using denaturating Laemmli, at 95°C for 10min. Denaturated proteins were loaded on a 4–20% gradient gel (Biorad) for Western Blot analysis and then transferred to a PVDF membrane (Biorad). Antibody staining was visualized using Amersham ECL Prime reagent (GE Healthcare) followed by analysis using a Fluor-S MultiImager (Biorad). Band intensity was quantified by ImageJ. For DNase I treatment, EDTA- free lysis buffer was used to prepare cell lysates. MgCl2 was added to the final concentration of 0.4 mM. 2.5 μl of 5 mg/ml DNase I were added to 500 μg of cell lysates. The digestion was performed by rotating at room temperature for 10 minutes. EDTA was then added to the final concentration of 5 mM to stop the nuclease reaction.
Primary antibodies were α-TOX (1:1000 dilution, eBioscience), α-KU70 (1:1000, ab92450, Abcam), α-KU80 (1:1000, #2753, Cell Signaling Technology), α-GAPDH (1:2500, #2118, Cell Signaling Technology), α-XRCC4 (1:200, sc-365118, Santa Cruz Biotechnology), and α-LIGASE IV (1:200, sc-271299, Santa Cruz Biotechnology). The secondary antibodies used were the same as described above.
In vitro binding assay
TOX and ΔHMG were subcloned into pT7CFE1-NHis-GST-CHA plasmid using the BamHI and XhoI restriction enzyme sites. 4μg of each construct was then transcribed and translated using the 1-Step Human High-Yield mini IVT kit (Pierce). TOX proteins were purified using the MagneGST Protein Purification System (Promega) and interactions with KU70/KU80 assessed on column. Specifically, 0.5μg–3μg of KU70/KU80 proteins (Abcam) were added to TOX bound beads. Following 2 hours of binding at 25°C, samples were washed 5-times using wash binding buffer supplied from the manufacturer (Pierce). TOX was cleaved from the beads using HRV3C protease (Pierce), purified away from beads, and the elution loaded on a 4–20% gradient gel for Western blot analysis. In vitro binding assays were performed three independent times.
Cell lines and Authentication
NIH3T3 and MEF cells were obtained from ATCC in 2012 and 2015, respectively and used within three months of receipt. Human T-ALL cell lines, MOLT-4, CCRF-CEM, HPB-ALL, JURKAT, DND-41, KE-37, KOPTK1, MOLT13, P12-ICHIKAWA, PEER were a gift from A. Thomas Look in 2012. Human HEK293T cells were ordered from ATCC in 2012. All human cell lines were authenticated at receipt and just prior to use in experiments using Small Tandem Repeat profiling and certified mycoplasma (MycoAlert Plus, Lonza, tested every 6 months).
Cell fractionation
Cell fractionation was completed essentially as described by the manufacturer (Subcellular Protein Fractionation kit, Pierce; 1×107 human T-ALL cells). However, one additional wash was utilized in each cell fraction. Protein fractions were quantified by Bradford assay and 20μg–45μg of proteins were loaded on a 4–20% gradient gel for Western blot analysis. Primary antibodies were α-TOX, α-TUBULIN (1:500, ab4074, Abcam), α-SP1 (1:1000, #9389, Cell Signaling Technology), and α-H4 (1:1000, #2935, Cell Signaling Technology). The secondary antibodies used were the same as described previously. Cell fractionation was performed three times with similar results.
Transient assays for NHEJ in 3T3 cells and human T-ALL cells
For the NHEJ assay, DNA double strand break was made in pEGFP-N1 vector or a pCS2-GW-mCherry by double-digestion with XhoI/BamHI and EcoRI/BamHI respectively, similarly to previous published work (54). When 3T3 cells were 70% confluent, they were transfected with control or TOX expressing vectors with or without KU70 and/or KU80. Cells were also transfected at the same time with linearized NHEJ reporter DNA into a 6-well plate (TransIT-X2 reagent, Mirus Bio). For nucleofection experiments in human T-ALL cells, 5×105 cells were nucleofected with control or TOX expressing vectors along with linearized NHEJ reporter DNA. The Amaxa Cell Line Nucleofector kit L was used for HPB-ALL and CCRF-CEM, while kit V was used for MOLT-4 (program A-030, Lonza). 48hrs/72hrs post transfection/nucleofections, the cells were analyzed by flow cytometry counting the number of GFP or mCherry positive cells and normalized to the number of fluorescent reporter cells. These assays were performed three independent times in triplicate.
Live cell imaging following micro-irradiation induced DNA damage
Human TOX or the HMG box deletion mutant were gateway-cloned into the pDest26 expression vector (Life Technologies). mRuby was then subcloned in frame using SalI and BglII. pmRuby2, pmRuby2-TOX or pmRuby2-ΔHMG constructs were co-transfected into 3T3 cells with KU80-GFP or XRCC4-GFP using electroporation (Neon transfection system, Life Technologies). 8–24 h later, cells were treated with Hoescht 33342 and subjected to 405 nm laser-induced micro-irradiation and imaging as described previously (55). Images were processed and analyzed using Slidebook 5.0 software (Intelligent Imaging Innovations) and Adobe Photoshop CS6. Quantification of fluorescence intensity was measured within laser track regions and normalized to regions outside the track for each time point (> 15 cells assessed for each time point).
Lentivirus infection and Traffic Light Reporter assay
Sequences for each shRNA are provided in Supplementary Table 6. 2μg of pLKO-shTOX or pLKO-scramble (Addgene plasmid 1864) were cotransfected into HEK 293T cells with 2μg pCMV-dR8.91, 0.2μg pVSV-g and TransIT-LT1 reagent (Mirus Bio). Supernatants containing the lentivirus were collected, filtered, and added to T-ALL cell lines in the presence of 4μg/mL polybrene (Millipore). T-ALL cells were spinoculated at 2000g for 90min at 32°C. 2 days post-infection, cells were selected by adding 6μg/mL puromycin (Invivogen). For the Traffic Light reporter assay, 2μg pCVL-TLR-Ef1a-BFP (Addgene plasmid 31481) (35) was cotransfected in addition to the previous plasmids. BFP positive cells were FACs sorted on the Aria Flow Cytometer (BD Biosciences). BFP positive, puromycin-selected cells were then nucleofected with tamoxifen-inducible I-SceI expressing vector (gift from Dr. Mostoslavsky, MGH). 24h post nucleofection, 200nM of Tamoxifen was added to the cells and incubated for 48h before FACs analysis. These experiments were repeated in triplicate and repeated twice.
Tandem mass spectrometry (LC-MS/MS)
Gel separated samples were alkylated with iodoacetamide and digested overnight at pH 8.3 with sequencing grade trypsin (Promega). Peptide mixtures were analyzed by microcapillary liquid chromatography-tandem mass spectrometry (LC-MS/MS) using the EASY-nLC nanoflow HPLC (Thermo Fisher Scientific) with a 75μm inner diameter × 15 cm length C18 capillary column coupled to a hybrid Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). The Elite was operated in data-dependent acquisition (DDA) mode (1 profile FT-MS spectrum followed by 15 centroided IT-MS/MS spectra). The resolution was 60,000 in FT-MS mode and MS/MS spectra were read out at low resolution via CID in the Velos ion trap. The gradient consisted of 3–38% acetonitrile in 0.1% formic acid (FA) at a flow rate of 300 nL/min for 75 min, 38–95% acetonitrile in 0.1% FA for 2 min and held at 95% acetonitrile in 0.1% FA at for 7 min followed by column re-equilibration for 10 min at 3% acetonitrile in 0.1% FA. MS/MS fragmentation spectra were searched for protein identification using the Mascot search engine against the reversed and concatenated SwissProt protein database (v7_2012).
Carbamidomethylation of Cys was set as fixed modification and variable modifications were oxidation of Met and deamidation of Gln and Asn. Precursor ion mass tolerance was set to 12 ppm and fragment ion mass tolerance was set to 0.8 Da. Two missed cleavages were allowed and the minimal length required for a peptide was six amino acids. Relative quantification of proteins was achieved through peptide spectral counting using Scaffold 4 software. The peptide and protein false discovery rates (FDR) were set to 1.5%.
To confirm the results from identifying proteins from selected protein bands from SDS-PAGE gels, the unfractionated sample of affinity enriched proteins was reduced and alkylated in solution basically as described previously (56). Proteins then were precipitated with chloroform-methanol, resuspended in 4 M urea/50 mM HEPES (pH 8.5) and digested using first endoproteinase LysC (Wako) and then sequencing grade trypsin (Promega). The generated peptides were analyzed by LC-MS2 in a 70 min gradient on an Orbitrap Fusion mass spectrometer equipped with an EASY-nLC 1000 autosampler/HPLC pump. The mass spectrometer was operated in a data-dependent mode with a full MS spectrum acquired in the Orbitrap followed by MS2 spectra acquired in the linear ion trap on the most abundant ions detected in the full MS spectrum. MS2 spectra were assigned using a SEQUEST-based (57) proteomics analysis platform (58) by searching against the human Uniprot sequence database. Peptide and protein assignments were filtered to a false discovery rate of < 1 % employing the target-decoy database search strategy (59) and using linear discriminant analysis and posterior error histogram sorting. Peptides with sequences contained in more than one protein sequence from the database were assigned to the protein with most matching peptides (58). Interacting proteins were assigned by the following criteria: 1) at least 10 independent peptides must be found in the TOX pull down samples and 2) there must be at least a 3 fold enrichment over IgG-pull down controls.
Supplementary Material
Statement of Significance.
TOX is a HMG box containing protein that has important roles in T-ALL initiation and maintenance. TOX inhibits the recruitment of KU70/KU80 to DNA breaks thereby inhibiting non-homologous end joining repair. Thus, TOX is likely a dominant oncogenic driver in a large fraction of human T-ALL and enhances genomic instability.
Acknowledgments
We would like to thank Dr. Ravi Mylvaganam for help with flow cytometry and Dr. Virginie Esain for manuscript advice and support.
Financial Support
This work was supported through funding provided to D.M. Langenau from the American Cancer Society, the Leukemia Research Foundation, the MGH Goodman Fellowship, the Live Like Bella Foundation and the Alex’s Lemonade Stand Foundation. Funding support was also garnered from the Harvey Graham Cancer Research Fund and The Terry Fox Foundation (J. Pinder), the Hope Funds for Cancer Research (B.J. Abraham), the William Lawrence and Blanche Hughes Foundation (A.T. Look) and the Boston Children’s Hospital Translational Research Program (A. Gutierrez). We also acknowledge NIH grants 1S10OD010612 (J.M. Asara), 5 P01CA120964 (J.M. Asara), 1K99CA181500 (J.S. Blackburn), CA109901 (A.T. Look and R.A. Young), CA193651 (A. Gutierrez), CA211734 (D.M. Langenau). G. Dellaire is funded by a Discovery Grant (RGPIN 05616) from the Natural Science and Engineering Research Council (NSERC).
Footnotes
The authors have no disclosures or conflicts of interest.
References
- 1.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350:1535–48. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 2.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012;122:3398–406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood. 2017;129:1124–33. doi: 10.1182/blood-2016-09-692582. [DOI] [PubMed] [Google Scholar]
- 5.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–64. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 6.Abraham BJ, Hnisz D, Weintraub AS, Kwiatkowski N, Li CH, Li Z, et al. Small genomic insertions form enhancers that misregulate oncogenes. Nat Commun. 2017;8:14385. doi: 10.1038/ncomms14385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–7. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeggo PA. Identification of genes involved in repair of DNA double-strand breaks in mammalian cells. Radiat Res. 1998;150:S80–91. [PubMed] [Google Scholar]
- 9.Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014;30:326–39. doi: 10.1016/j.tig.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–54. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu Y, Seidl KJ, Rathbun GA, Zhu C, Manis JP, van der Stoep N, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–65. doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- 12.Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, et al. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–5. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- 13.Wilkinson B, Chen JY, Han P, Rufner KM, Goularte OD, Kaye J. TOX: an HMG box protein implicated in the regulation of thymocyte selection. Nat Immunol. 2002;3:272–80. doi: 10.1038/ni767. [DOI] [PubMed] [Google Scholar]
- 14.Aliahmad P, O’Flaherty E, Han P, Goularte OD, Wilkinson B, Satake M, et al. TOX provides a link between calcineurin activation and CD8 lineage commitment. J Exp Med. 2004;199:1089–99. doi: 10.1084/jem.20040051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aliahmad P, Kaye J. Development of all CD4 T lineages requires nuclear factor TOX. J Exp Med. 2008;205:245–56. doi: 10.1084/jem.20071944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol. 2010;11:945–52. doi: 10.1038/ni.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aliahmad P, Kadavallore A, de la Torre B, Kappes D, Kaye J. TOX is required for development of the CD4 T cell lineage gene program. J Immunol. 2011;187:5931–40. doi: 10.4049/jimmunol.1101474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seehus CR, Aliahmad P, de la Torre B, Iliev ID, Spurka L, Funari VA, et al. The development of innate lymphoid cells requires TOX-dependent generation of a common innate lymphoid cell progenitor. Nat Immunol. 2015;16:599–608. doi: 10.1038/ni.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yun S, Lee SH, Yoon SR, Kim MS, Piao ZH, Myung PK, et al. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol Lett. 2011;136:29–36. doi: 10.1016/j.imlet.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–71. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, et al. Myc-induced T cell leukemia in transgenic zebrafish. Science. 2003;299:887–90. doi: 10.1126/science.1080280. [DOI] [PubMed] [Google Scholar]
- 22.Langenau DM, Keefe MD, Storer NY, Jette CA, Smith AC, Ceol CJ, et al. Co-injection strategies to modify radiation sensitivity and tumor initiation in transgenic Zebrafish. Oncogene. 2008;27:4242–8. doi: 10.1038/onc.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith AC, Raimondi AR, Salthouse CD, Ignatius MS, Blackburn JS, Mizgirev IV, et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood. 2010;115:3296–303. doi: 10.1182/blood-2009-10-246488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blackburn JS, Liu S, Raiser DM, Martinez SA, Feng H, Meeker ND, et al. Notch signaling expands a pre-malignant pool of T-cell acute lymphoblastic leukemia clones without affecting leukemia-propagating cell frequency. Leukemia. 2012;26:2069–78. doi: 10.1038/leu.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langenau DM, Feng H, Berghmans S, Kanki JP, Kutok JL, Look AT. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2005;102:6068–73. doi: 10.1073/pnas.0408708102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackburn JS, Liu S, Wilder JL, Dobrinski KP, Lobbardi R, Moore FE, et al. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell. 2014;25:366–78. doi: 10.1016/j.ccr.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frazer JK, Batchelor LA, Bradley DF, Brown KH, Dobrinski KP, Lee C, et al. Genomic amplification of an endogenous retrovirus in zebrafish T-cell malignancies. Adv Hematol. 2012;2012:627920. doi: 10.1155/2012/627920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haupt Y, Bath ML, Harris AW, Adams JM. bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene. 1993;8:3161–4. [PubMed] [Google Scholar]
- 29.Giambra V, Jenkins CE, Lam SH, Hoofd C, Belmonte M, Wang X, et al. Leukemia stem cells in T-ALL require active Hif1alpha and Wnt signaling. Blood. 2015;125:3917–27. doi: 10.1182/blood-2014-10-609370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Treanor LM, Zhou S, Janke L, Churchman ML, Ma Z, Lu T, et al. Interleukin-7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J Exp Med. 2014;211:701–13. doi: 10.1084/jem.20122727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zornig M, Schmidt T, Karsunky H, Grzeschiczek A, Moroy T. Zinc finger protein GFI-1 cooperates with myc and pim-1 in T-cell lymphomagenesis by reducing the requirements for IL-2. Oncogene. 1996;12:1789–801. [PubMed] [Google Scholar]
- 32.Look AT, Melvin SL, Williams DL, Brodeur GM, Dahl GV, Kalwinsky DK, et al. Aneuploidy and percentage of S-phase cells determined by flow cytometry correlate with cell phenotype in childhood acute leukemia. Blood. 1982;60:959–67. [PubMed] [Google Scholar]
- 33.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, et al. Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods. 2011;8:671–6. doi: 10.1038/nmeth.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell. 2013;51:454–68. doi: 10.1016/j.molcel.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y, Su MW, Jiang X, Zhou Y. Evidence of an oncogenic role of aberrant TOX activation in cutaneous T-cell lymphoma. Blood. 2015;125:1435–43. doi: 10.1182/blood-2014-05-571778. [DOI] [PubMed] [Google Scholar]
- 38.O’Flaherty E, Kaye J. TOX defines a conserved subfamily of HMG-box proteins. BMC Genomics. 2003;4:13. doi: 10.1186/1471-2164-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5:504–14. doi: 10.1016/j.stem.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 40.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101–13. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 41.Lange SS, Mitchell DL, Vasquez KM. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci U S A. 2008;105:10320–5. doi: 10.1073/pnas.0803181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prasad R, Liu Y, Deterding LJ, Poltoratsky VP, Kedar PS, Horton JK, et al. HMGB1 is a cofactor in mammalian base excision repair. Mol Cell. 2007;27:829–41. doi: 10.1016/j.molcel.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan F, Gu L, Guo S, Wang C, Li GM. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J Biol Chem. 2004;279:20935–40. doi: 10.1074/jbc.M401931200. [DOI] [PubMed] [Google Scholar]
- 44.Stros M, Cherny D, Jovin TM. HMG1 protein stimulates DNA end joining by promoting association of DNA molecules via their ends. Eur J Biochem. 2000;267:4088–97. doi: 10.1046/j.1432-1327.2000.01450.x. [DOI] [PubMed] [Google Scholar]
- 45.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tessema M, Yingling CM, Grimes MJ, Thomas CL, Liu Y, Leng S, et al. Differential epigenetic regulation of TOX subfamily high mobility group box genes in lung and breast cancers. PLoS One. 2012;7:e34850. doi: 10.1371/journal.pone.0034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung W, Kwabi-Addo B, Ittmann M, Jelinek J, Shen L, Yu Y, et al. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS One. 2008;3:e2079. doi: 10.1371/journal.pone.0002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamesch P, Li N, Milstein S, Fan C, Hao T, Szabo G, et al. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89:307–15. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012;22:1995–2007. doi: 10.1101/gr.137570.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26:909–22. doi: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc Natl Acad Sci U S A. 2004;101:7624–9. doi: 10.1073/pnas.0400726101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrin C, McDonald D, Attwood KM, Rodrigue A, Ghosh S, Mirzayans R, et al. A requirement for polymerized actin in DNA double-strand break repair. Nucleus. 2012;3:384–95. doi: 10.4161/nucl.21055. [DOI] [PubMed] [Google Scholar]
- 56.Edwards A, Haas W. Multiplexed Quantitative Proteomics for High-Throughput Comprehensive Proteome Comparisons of Human Cell Lines. Methods Mol Biol. 2016;1394:1–13. doi: 10.1007/978-1-4939-3341-9_1. [DOI] [PubMed] [Google Scholar]
- 57.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 58.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–89. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.