

Abstract
Background
Real time PCR (rtPCR) is a quantitative assay to determine the relative DNA copy number in a sample versus a reference. The method is the standard for the analysis of the output data generated by an rtPCR experiment. We developed an alternative based on fitting a robust regression to the rtPCR signal. This new data analysis tool reduces potential biases and does not require all of the compared DNA fragments to have the same PCR efficiency.
Results
Comparing the two methods when analysing 96 identical PCR preparations showed similar distributions of the estimated copy numbers. Estimating the efficiency with the method, however, required a dilution series, which is not necessary for the robust regression method. We used rtPCR to quantify mitochondrial DNA (mtDNA) copy numbers in three different tissues types: breast, colon and prostate. For each type, normal tissue and a tumor from the same three patients were analysed. This gives a total of six samples. The mitochondrial copy number is estimated to lie between 200 and 300 copies per cell. Similar results are obtained when using the robust regression or the method. Confidence ratios were slightly narrower for the robust regression. The new data analysis method has been implemented as an R package.
Electronic supplementary material
The online version of this article (10.1186/s13104-017-2913-1) contains supplementary material, which is available to authorized users.
Keywords: rtPCR, Robust regression, Mitochondrial DNA
Background
Mitochondria are the organelle responsible for most of the energy production in eukaryotic cells. Each mitochondrion carries several copies of mitochondrial DNA, which is composed of a single circular chromosome of 16569 base pairs (hg38, GRCh38, Dec. 2013). It encodes for 22 tRNA, 13 protein subunits and two ribosomal RNA subunits. There are currently few accurate measurements of mtDNA copy number in cells [1–5], even though this number affects the symptoms of mitochondrial diseases [6–9]. Better measurements of mtDNA copy numbers would improve the understanding of mtDNA mutagenesis [10–12] as well as the process through which mutations become homoplasmic. Mitochondrial mutations also appear to be involved in cancer development [13–17], and aging [18–21]. Furthermore, most tumors are thought to rely on glycolysis rather than oxidative phosphorylation for the majority of their energy, a process that could be related to mtDNA copy number. The standard method for quantifying DNA copy number is real time PCR (rtPCR) [22–24]. Most methods rely on amplifying a mitochondrial and a nuclear fragment in separate reactions, with the template from the same sample [16, 24]. Although there has been much development in the data analysis algorithms applied to rtPCR output, some challenges remainx [25, 26].
Materials and methods
Tissue and DNA extraction
Anonymous surgical discards were obtained after standardised informed consent. Tissue was stored at the surgical department at − 70 °C until DNA extraction. Normal and tumor tissue was obtained from three different patients with three different tumor types (breast, prostate and colon). The normal tissue was taken at a distance of 10–15 cm from the location of the tumor. A few milligrams were taken from each sample and had their DNA extracted.
DNA extraction
Samples were digested with proteinase K for 4 h at 57 °C in 300 l of digestion Buffer (Qiagen, Hilden, Germany) according to manufacturer’s instructions. DNA was extracted from them using the Qiagen MagAttract DNA Mini-M48 Kit with a dedicated automatic solution also provided by Qiagen. The result is a DNA solution containing approximately 50 ng of DNA per l .
Primers
Primers were designed using the rtPCR primer design tool of IDT (integrated DNA technologies). The nuclear and mitochondrial primer pairs were designed for simultaneous amplification. Table 1 shows the primer pairs. PCR conditions were optimised by testing various annealing temperatures, reaction volumes, and reagent concentrations. The objective was to use the same conditions for both primers pairs. The mitochondrial primer was chosen so that it could not amplify in the nuclear genome and vice versa.
Table 1.
Primer sequences used to amplify nuclear and mitochondrial DNA sequences
| Primer | Sequence (5′ to 3′) | Genome region |
|---|---|---|
| Mitochondrial forward | ACA CCC TCC TAG CCT TAC TAC | chrM: 10087–10192 |
| Mitochondrial reverse | GAT ATA GGG TCG AAG CCG C | |
| Nuclear forward | AGG GTA TCT GGG CTC TGG | chr11: 2170993–2171170 |
| Nuclear reverse | GGC TGA AAA GCT CCC GAT TAT |
rtPCR condition
Real time PCR was performed using a BioRad CFX connect Real-time PCR detection System. The PCR recipe was 2× Perfecta SYBR Green SuperMix for iQ (QuantaBio, Beverly, MA, USA, WHR: 733-1249), 0.2 M of each primer, for a final volume of 20 l . The PCR temperature cycling used: initial denaturing at 94 °C for 4 min, followed by 45 cycles of denaturing at 94 °C for 30 s, annealing at 60 °C for 30 s and extension at 72 °C for 1 min.
Experiment
For both the mitochondrial and the nuclear primers, 96 replicas (a whole plate) of the same identical rtPCR were produced. A 2 ml PCR mix was created (as described the section “rtPCR condition”), to which 4 l of extracted DNA was added. The mix was spread on a PCR plate adding 20 l of it into each well. Serial dilutions for both primers were used to estimate PCR efficiency with the method. The initial rtPCR mix was serially diluted into rtPCR mix without DNA, by a factor of 5, for six steps. There were 16 replicas for each dilution leading to a total of 16 × 6 = 96 reactions. The use of dilutions reveals changes in PCR efficiency and gives an indication of precision.
The DNA copy numbers were estimated for each tissue based on four different rtPCR reactions: nuclear DNA, mitochondrial DNA, nuclear DNA diluted by 10, and mitochondrial DNA diluted by 10. Each rtPCR reaction was replicated 24 times, giving a total number of 4 × 24 = 96 (a complete 96 well plate) reactions.
Data analysis
The data analysis algorithm is available in an R package developed specifically for the analysis of rtPCR results. The package, together with the codes used to generate the graphs and tables are included in the Additional file 1. By fitting a robust linear regression line to the base two logarithm of the signal against the cycle number (c), the efficiency (slope of the regression line) and intercept (I) associated with each rtPCR reaction is estimated. The fitting proceeds by finding the middle point as the couple (), where is closest to middle between the maximal and minimal signal . Forcing passage of the fitted line through the middle point ensures that the line fits the exponential phase of the signal.
The relative copy number between two experiments is defined as which is estimated by taking the difference in the average intercept computed over replicated reactions,
The average intercept is assumed to follow a Normal distribution, which is justified by inspection of the results from 96 replicas. The 95% confidence interval for can therefore be estimated as:
where is the 97.5% quantile of the t distribution, is the t distribution with degrees of freedom, and and are the number of replicas for A and B respectively and the variances are estimated from the replicated values. The resulting confidence interval for the relative copy number is
which shows as a confidence ratio (C.R.). The C.R tells us that the interval captures the actual copy number with a probability of . The boundaries for the confidence interval of the actual relative concentration, can be calculated by multiplying and dividing the estimated relative concentration by the C.R. Baseline noise in a rtPCR reaction is estimated by taking the highest point for which the first derivative of the signal as a function of the cycle number is negative. The threshold to calculate the value is chosen by taking the highest value for the baseline in an experiment. When relative concentrations are calculated between two samples, the same threshold is used to calculate the value for both.
Findings and discussion
Problem
The phases of an rtPCR reaction are:
-
Phase I:
Lag phase: The signal is too low for the detector, only the noise is visible.
-
Phase II:
Exponential phase: Signal grows exponentially with the number of cycles.
-
Phase III:
Saturation phase: Signal increases sub-exponentially, or not at all as the PCR reaction saturates.
The dynamics of the PCR reaction can only be observed during phase II, during which the signal can be modelled by the exponential function . In this equation, N is the number of DNA copies at the start of the experiment, S is the signal, is an unknown constant relating the copy number to the signal intensity and c is the cycle number. The constant is related to parameters such as detection efficiency or fluorescence per base pair. It is assumed that is constant and does not depend on the sample.
The standard algorithm to analyse rtPCR is the method. A signal threshold T is chosen, a little above the noise level. The value is defined as the cycle number at which the signal crosses the threshold. It is calculated by taking a linear interpolation between the first signal value above the threshold and the one immediately below, then taking as the value at which the line intersects the chosen threshold. If there are two samples, A and B, for which rtPCR signal has been obtained this yields an equation relating the initial copy numbers of the two samples.
Assuming equal efficiency for both reactions, , the equation becomes
where is the difference in the values. The method has a few clear flaws, which have already been pointed out and demonstrated by Karlen et al. [25]. The first one is the assumption of equal efficiency which is essential to this method. If the fragments used are not the same, as is the case for the quantification of mtDNA, the reaction needs to be optimised to have equal efficiency. If PCR efficiency depends on initial DNA concentration, as some results suggests [24], this would introduce errors in the measurements.
Proposed solution
The objective of rtPCR is to measure the relative initial copy number between two samples. Taking the logarithm in the equation used for the method leads to the equation we fit to the exponentially increasing signal,
The slope is related to the efficiency and is the intercept or the value of the signal extrapolated to the start of the reaction at . We propose to estimate the values of the intercept and the slope by fitting a regression line to several consecutive pairs (c, S) chosen from the exponential phase of the reaction. If we have intercepts for two samples A and B we obtain
which requires only a constant value of , but gives correct results even when the efficiencies for A and B are different. The slope of the regression gives an estimate of the efficiency for a single reaction without having to perform dilutions. Accuracy can be increased by replicating the reactions several times. Thus, it is possible to compare samples with different efficiencies, which reduces the difficulties in optimising the PCR reactions and improves precision.
In our analyses, we used the robust line fitter that minimises the median of the squared residuals, whereas the least squares estimator minimises the mean of the squared residuals. The line passing through the mid-point has equation and to determine the value of , we fix it such that the median over all measured couples (c, S) of is smallest. Taking the median means that the line can tolerate up to one half of the measured couples not to be near the regression line, which is the case for the phases I and III. The minimisation has to be done numerically and the package supplied in the Additional file 1 will perform the necessary computations.
Results
Figure 1 shows the results of repeating the same reaction 96 times. The efficiency and intercept, calculated using the robust regression, as well as the values are shown. In all three cases, the values group together with five outliers. These outliers are not PCR failures. They represent genuine variation in PCR performance on an identical rtPCR mix. These results justify the use of a normal approximation.
Fig. 1.
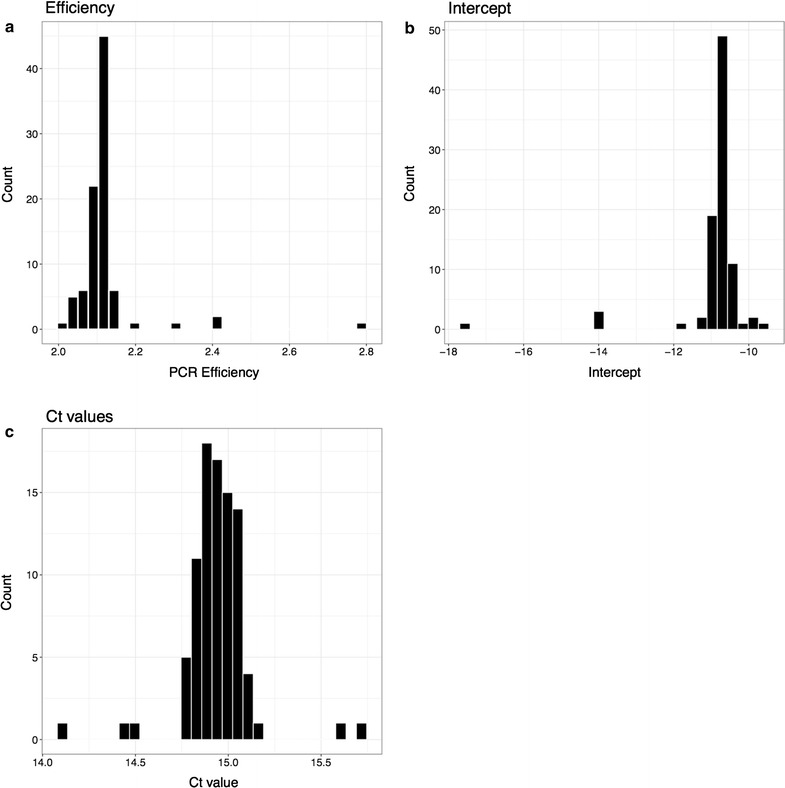
Histograms showing the distribution of efficiency, intercept and . Data shown from 94 replicas. For each reaction, the robust regression method is used to compute the efficiency and intercept
Figure 2 shows the result of the dilution series in which the relative concentrations relative to the initial sample are known. For both nuclear and mitochondrial DNA, the relative concentrations were estimated with the algorithm as well as the new robust regression method. The vertical axis is the logarithm to base five of the relative concentration and since we dilute by a factor of five, the points should lie on a line with slope .
Fig. 2.
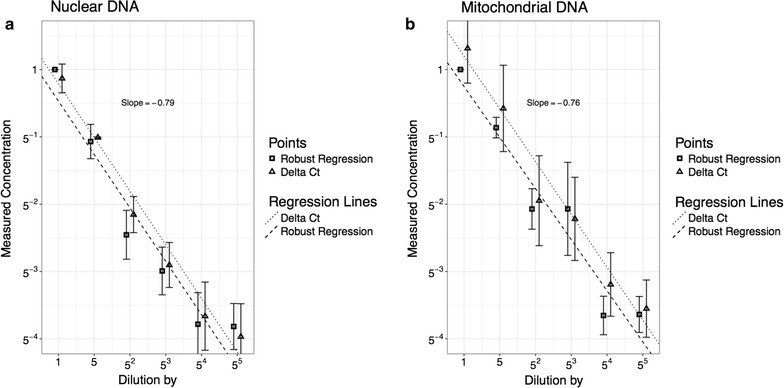
Dilution series: DNA concentration in the PCR mix is serialy diluted by a factor 5, six times. The DNA concentration is measured using the robust regression method and the method. Each concentration is prepared in 16 replicas. The same procedure is reproduced for both nuclear and mitochondrial DNA. Regression lines are fitted using the least squared method. The data has been shifted slightly left and right of the true value for illustration purposes
It can be seen that the robust regression gives, in both cases, a slightly better slope than the method. This difference does not appear to be significant. The dilution series can be used to estimate the efficiency of the PCR using the method. If the efficiency is assume identical in all samples then:
The logarithm of the dilution factor, is linearly related to the , and the slope is the log of the efficiency. Results are shown in Fig. 3.
Fig. 3.
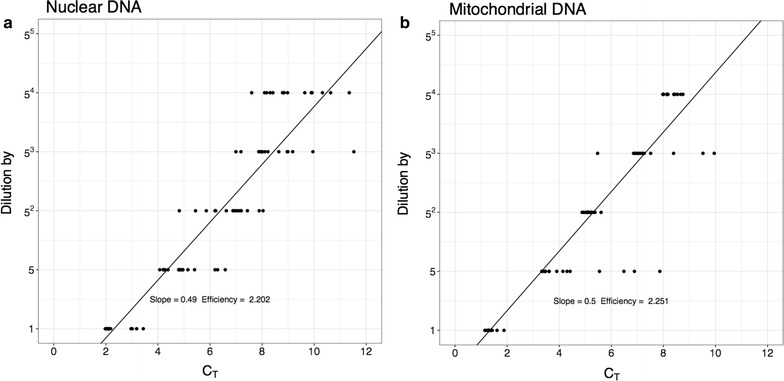
Calculating the efficiency using the method. The measured values for each dilution are plotted. The slope of the fitted line can be used to estimate the efficiency
Table 2 shows the estimates of mtDNA copy numbers based on the relative concentration of mitochondrial DNA compared to nuclear DNA. The numbers range from 100 to 150 for all samples, which represent half the total number of mtDNA copies per cell. The confidence ratios are around 1.3. The ratio between the measured concentrations of mtDNA and diluted mtDNA should be 10. Taking into account the confidence ratios associated with the measurement, the diluted samples have indeed a copy number 10 times below their un-diluted counter-parts. Observing a C.R. of 1.3 for a mitochondrial copy number of 200, corresponds to having a 95% confidence interval between 150 and 260. This precision was achieved with 24 replicas. The C.R. decays very slowly as a function of the number of samples. Using a robust regression to analyse rtPCR data presents major advantages over the method. First, it does not make the assumption of identical PCR efficiency between two samples. This reduces potential biases and allows for the comparison of fragments/samples with clearly different efficiencies. It also allows the estimation of PCR efficiency without performing dilution series. If the efficiency depends on the initial copy number, it would be an additional source of bias for the algorithm. Figure 3 shows efficiency calculations for the dilutions series. For comparison, the prostate tumor tissue is analysed using the method (shown in Table 3). The results are higher than those estimated using the robust regression. They are, however, coherent if the larger confidence ratio is taken into account. Karlen et al. [25] also shows that the method performs well in the case of identical efficiencies, but may be a bad choice in other circumstances. The robust regression method offers an alternative way to analyse rtPCR data which has important advantages.
Table 2.
Relative concentration of samples for each tissue type
| Tissue | Sample type | Efficiency | Concentration | Relative error |
|---|---|---|---|---|
| Breast tumor | ||||
| mtDNA | 2.08 | 129.3 | 1.29 | |
| mtDNA/10 | 2.13 | 6.87 | 1.47 | |
| Nuclear | 2.02 | 1 | 1 | |
| Nuclear/10 | 1.99 | 0.16 | 1.4 | |
| Breast normal | ||||
| mtDNA | 2.19 | 132.13 | 1.4 | |
| mtDNA/10 | 2.02 | 38.54 | 1.36 | |
| Nuclear | 2.03 | 1 | 1 | |
| Nuclear/10 | 2.02 | 0.09 | 1.38 | |
| Colon tumor | ||||
| mtDNA | 2.13 | 132.72 | 1.35 | |
| mtDNA/10 | 2.1 | 12.95 | 1.32 | |
| Nuclear | 2.01 | 1 | 1 | |
| Nuclear/10 | 2.01 | 0.09 | 1.45 | |
| Colon normal | ||||
| mtDNA | 2.11 | 219.19 | 1.5 | |
| mtDNA/10 | 2.11 | 20.82 | 1.56 | |
| Nuclear | 2.05 | 1 | 1 | |
| Nuclear/10 | 1.99 | 0.2 | 1.51 | |
| Prostate tumor | ||||
| mtDNA | 2.17 | 134.14 | 1.41 | |
| mtDNA/10 | 2.11 | 15.79 | 1.39 | |
| Nuclear | 2.04 | 1 | 1 | |
| Nuclear/10 | 2 | 0.16 | 1.47 | |
| Prostate normal | ||||
| mtDNA | 2.12 | 172.65 | 1.49 | |
| mtDNA/10 | 2.08 | 22.21 | 1.47 | |
| Nuclear | 2.02 | 1 | 1 | |
| Nuclear/10 | 2.01 | 0.13 | 1.55 | |
The nuclear DNA concentration is taken as reference, and is therefore 1. The relative error (confidence ratio) is also 1 as there is no uncertainty associated with it. The other concentrations are relative to nuclear DNA and the relative error associated with it. The values for the efficiency are the average taken over the replicas
Table 3.
Concentrations calculated using the method in prostate tumor sample
| Concentration | Relative error | |
|---|---|---|
| mtDNA | 288.33 | 1.52 |
| mtDNA/10 | 23.45 | 1.44 |
| Nuclear | 1.00 | 1.00 |
| Nuclear/10 | 0.11 | 1.52 |
The confidence ratio is estimated using the t-interval
Limitations
The analysis method proposed here is limited to the analysis of rtPCR results. It can be used with any standard rtPCR output data and represents an improvement from the method. However, a large number of replicas is still needed to achieve low C.R.
Authors' contributions
Experimental design by PR and POE and DW. Experiment realisation by PR and POE. Data analysis by PR and SM. Enzyme was provided by DW. All authors contributed to writing the article. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge Edvin Hoovig for allowing the use of rtPCR equipment and advice on experimental design.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data and software tools used are freely available and can be obtained by contacting the corresponding author. The R package used to analyse the data are submitted as Additional file 1.
Consent to publish
Not applicable
Ethics approval and consent to participate
According to Norwegian Law, technical and methodological development work that uses anonymised biological material does not require approval from research ethics committees (Web page, last access November 2016).
Funding
Not applicable
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- rtPCR
real time polymerase chain reaction
- DNA
deoxyribonucleic acid
- mtDNA
mitochondrial DNA
- tRNA
transcription ribonucleic acid
Additional file
Additional file 1. rtPCR package for R. The R package containing the software tools to perform robust regression analysis of rtPCR data. The package is in binary format and can be installed into R.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13104-017-2913-1) contains supplementary material, which is available to authorized users.
Contributor Information
Paulo Refinetti, Email: paulo.refinetti@epfl.ch.
David Warren, Email: david.warren@rr-research.no.
Stephan Morgenthaler, Email: stephan.morgenthaler@epfl.ch.
Per O. Ekstrøm, Email: Per.Olaf.Ekstrom@rr-research.no
References
- 1.Tseng L-M, Yin P-H, Chi C-W, Hsu C-Y, Wu C-W, Lee L-M, Wei Y-H, Lee H-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosom Cancer. 2006;45(7):629–638. doi: 10.1002/gcc.20326. [DOI] [PubMed] [Google Scholar]
- 2.Alonso A, Martín P, Albarrán C, García P. Specific quantification of human genomes from low copy number DNA samples in forensic and ancient DNA studies. Croat Med J. 2003;44:273. [PubMed] [Google Scholar]
- 3.Meissner C, Mohamed SA, Klueter H, Hamann K, von Wurmb N, Oehmichen M. Quantification of mitochondrial DNA in human blood cells using an automated detection system. Forensic Sci Int. 2000;113(1–3):109–112. doi: 10.1016/S0379-0738(00)00249-8. [DOI] [PubMed] [Google Scholar]
- 4.von Wurmb-Schwark N, Higuchi R, Fenech AP, Elfstroem C, Meissner C, Oehmichen M, Cortopassi GA. Quantification of human mitochondrial DNA in a real time PCR. Forensic Sci Int. 2002;126(1):34–39. doi: 10.1016/S0379-0738(02)00026-9. [DOI] [PubMed] [Google Scholar]
- 5.Yin PH, Lee HC, Chau GY, Wu YT, Li SH, Lui WY, Wei YH, Liu TY, Chi CW. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. Br J Cancer. 2004;90:2390. doi: 10.1038/sj.bjc.6601838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeviani M. Mitochondrial disorders. Brain. 2004;127(10):2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- 7.Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, Chinnery PF, Turnbull DM. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63(1):35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 8.Shoffner JM, Wallace DC. Oxidative phosphorylation diseases and mitochondrial DNA mutations: diagnosis and treatment. Annu Rev Nutr. 1994;14:535. doi: 10.1146/annurev.nu.14.070194.002535. [DOI] [PubMed] [Google Scholar]
- 9.Brown M, Starikovskaya E, Derbeneva O, Hosseini S, Allen J, Mikhailovskaya I, Sukernik R, Wallace D. The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup J. Hum Genet. 2002;110(2):130–138. doi: 10.1007/s00439-001-0660-8. [DOI] [PubMed] [Google Scholar]
- 10.Coller HA, Khrapko K, Bodyak ND, Nekhaeva E, Herrero-Jimenez P, Thilly WG. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet. 2001;28(2):147–150. doi: 10.1038/88859. [DOI] [PubMed] [Google Scholar]
- 11.Taylor RW, Barron MJ, Borthwick GM, Gospel A, Chinnery PF, Samuels DC, Taylor GA, Plusa SM, Needham SJ, Greaves LC, Kirkwood TBL, Turnbull DM. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Investig. 2003;112(9):1351–1360. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marcelino LA, Thilly WG. Mitochondrial mutagenesis in human cells and tissues. Mutat Res/DNA Repair. 1999;434(3):177–203. doi: 10.1016/S0921-8777(99)00028-2. [DOI] [PubMed] [Google Scholar]
- 13.Lam ET, Bracci PM, Holly EA, Chu C, Poon A, Wan E, White K, Kwok P-Y, Pawlikowska L, Tranah GJ. Mitochondrial DNA sequence variation and risk of pancreatic cancer. Cancer Res. 2012;72(3):686–695. doi: 10.1158/0008-5472.CAN-11-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1(1):9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25(34):4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 16.Grady JP, Murphy JL, Blakely EL, Haller RG, Taylor RW, Turnbull DM, Tuppen HAL. Accurate measurement of mitochondrial DNA deletion level and copy number differences in human skeletal muscle. PloS ONE. 2014;9(12):114462. doi: 10.1371/journal.pone.0114462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25(34):4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 18.Kujoth GC. Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res. 2006;66(15):7386–7389. doi: 10.1158/0008-5472.CAN-05-4670. [DOI] [PubMed] [Google Scholar]
- 19.Greaves LC, Turnbull DM. Mitochondrial DNA mutations and ageing. Biochim Biophys Acta. 2009;1790(10):1015–1020. doi: 10.1016/j.bbagen.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 20.Lee H-C, Chang C-M, Chi C-W. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res Rev. 2010;9:47–58. doi: 10.1016/j.arr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Melova S, Schneider JA, Coskun PE, Bennett DA, Wallace DC. Mitochondrial DNA rearrangements in aging human brain and in situ PCR of mtDNA. Neurobiol Aging. 1999;20(5):565–571. doi: 10.1016/S0197-4580(99)00092-5. [DOI] [PubMed] [Google Scholar]
- 22.Nicklas JA, Brooks EM, Hunter TC. Development of a quantitative PCR (TaqMan) assay for relative mitochondrial DNA copy number and the common mitochondrial DNA deletion in the rat. Environ Mol Mutagen. 2004;44:313. doi: 10.1002/em.20050. [DOI] [PubMed] [Google Scholar]
- 23.Andreu AL, Martinez R, Marti R, García-Arumí E. Quantification of mitochondrial DNA copy number: pre-analytical factors. Mitochondrion. 2009;9(4):242–246. doi: 10.1016/j.mito.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez-Jimenez N, Castellanos-Rubio A, Plaza-Izurieta L, Gutierrez G, Irastorza I, Castaño L, Vitoria JC, Bilbao JR. Accuracy in copy number calling by qPCR and PRT: a matter of DNA. PloS ONE. 2011;6(12):28910. doi: 10.1371/journal.pone.0028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karlen Y, McNair A, Perseguers S, Mazza C, Mermod N. Statistical significance of quantitative PCR. BMC Bioinform. 2007;8(1):131. doi: 10.1186/1471-2105-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tichopad A. Standardized determination of real-time PCR efficiency from a single reaction set-up. Nucleic Acids Res. 2003;31(20):122. doi: 10.1093/nar/gng122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and software tools used are freely available and can be obtained by contacting the corresponding author. The R package used to analyse the data are submitted as Additional file 1.