

Summary
Context
TERT promoter mutations have been associated with adverse prognosis in papillary thyroid carcinomas (PTCs).
Objective
We investigated the association between TERT promoter mutations and survival from PTC.
Design
Retrospective observational cohort study.
Patients
Eighty consecutive patients with PTC who underwent surgery between 1990 and 2003.
Measurements
TERT promoter was genotyped in DNA from 80 archival PTCs by Sanger sequencing. Median follow‐up was 106 months (range 1–270). Outcomes analysis was stratified according to disease and overall survival status. For each parameter, relative risk (RR) adjusted for age at first surgery and gender was estimated. Both univariate and multivariate analyses were performed using logistic regression, Kaplan–Meier survival analysis and Cox regression models.
Results
PTCs from 11 patients (14%) contained either C228T or C250T TERT promoter mutation. TERT mutations were significantly associated with adverse prognostic features such as older age (P = 0·002), male gender (P = 0·01) and Stage IV disease (P = 0·03). Four patients died from PTC during follow‐up: 3 patients with TERT mutations (27%) and one without (1·5%). Disease‐related mortality rate with or without TERT mutations was 33·7 vs 1·6 per 1000 patient‐years respectively, that is 10 (95% CI = 1·0–104·1, P = 0·05) fold higher, after adjustment for age at first surgery and gender. The combination of TERT promoter mutation and BRAFV 600E significantly increased disease‐related death risk (P = 0·002). TERT mutations increased expression of a reporter gene in thyroid cells containing BRAFV 600E.
Conclusions
TERT promoter mutations are a major indicator of death due to PTCs. Conversely, absence of TERT mutations portends better survival.
Background
Telomerase reactivation, either via the chromosome end‐replicating enzyme telomerase reverse transcriptase (TERT) or alternative lengthening of telomeres, is detected in up to 80% of malignant tumours.1, 2 Somatic mutations in the TERT promoter were first identified in melanoma3, 4 and have been observed at high frequency in multiple cancer types, including those of the thyroid, central nervous system and bladder.5, 6, 7 These mutations occur at two hotspots, chr5:1,295,228C>T (‘C228T’) and chr5:1,295,250C>T (‘C250T’), located, respectively, −124 and −146 bp upstream from the ATG start site. Both mutations create a putative consensus binding site (GGAA) for ETS transcription factors3 and correlate with increased TERT expression.5, 6
In thyroid cancer, TERT promoter mutations were initially noted to occur relatively infrequently in tumours with a better prognosis (papillary and follicular thyroid cancers) and with increasing frequency in progressively worse prognostic subtypes (i.e. poorly differentiated and anaplastic thyroid cancers).8, 9 Subsequent work demonstrated that within papillary (PTC) and follicular (FTC) thyroid cancer types, TERT promoter mutations were correlated to adverse prognostic features such as older age, larger tumour size and male gender.10, 11, 12, 13, 14 Some10, 11, 15 but not all9, 13, 14, 16 studies have found an interaction between the presence of TERT promoter mutations and oncogenic BRAF V600E in predicting recurrence. Two retrospective studies to date have found that TERT promoter mutations decrease survival rates in PTC and FTC.13, 14 TERT promoter mutations have also been identified in papillary microcarcinomas17 and Hurthle cell cancers18 but not in medullary thyroid cancers.8, 9, 10, 19 It has been possible to detect TERT promoter mutations in fine‐needle aspiration samples, suggesting that their pre‐operative detection may contribute to appropriate surgical assessment.20, 21, 22
The importance of these findings to the clinician is double: firstly, the presence of a biomarker that reliably identifies aggressive biological behaviour would justify stronger therapeutic approaches; conversely, if absence of the biomarker is associated with indolent behaviour, then such patients could be more cautiously monitored and treated accordingly.
We sought to address these issues using a retrospective cohort of PTC cases that we have been following for a median of 9 years. We tested the following hypotheses: (a) the prevalence of TERT promoter mutations in this Australian cohort; (b) that TERT promoter mutations are associated with adverse prognostic features in these PTCs; (c) that TERT promoter mutations are associated with disease‐free and/or overall survival; and (d) that TERT promoter mutations interact with BRAF V600E to influence survival.
Materials and methods
Study participants
This study involved 80 consecutive patients with PTC who underwent initial surgery between the years 1990 and 2003.23 Patients were identified from a prospectively maintained endocrine surgical database after approval from the Northern Sydney Local Health District Human Research Ethics Committee (LNR 1312‐417M).
Median clinical follow‐up was 106 months (range 1–270 months), and outcomes analyses were stratified according to disease status.
Disease‐free survival was defined as a negative clinical and radiological examination (i.e. high‐resolution neck ultrasonography and/or whole‐body radioactive iodine scan) in the presence of a serum thyroglobulin level <1 μg/l and undetectable thyroglobulin antibodies (<40 kIU/l). Deceased patients were classified as a disease‐related death or, if their death was unrelated to PTC, classified according to their disease status at the time of death.23
Nodal recurrence was defined as reappearance of PTC within the neck after completion of initial treatment (total thyroidectomy and radioactive iodine remnant ablation). Tumour recurrence was confirmed on the basis of a positive fine‐needle aspiration cytology, abnormal sonographic appearance or positive radioiodine imaging.23
Tissue samples
FFPE tumour blocks from initial surgery for each patient were obtained from the archives of the Department of Anatomical Pathology at the Royal North Shore Hospital. Fresh sections were cut from archived blocks and independently reviewed by a single pathologist to confirm the diagnosis.24 The single paraffin block with the largest dimension of tumour regardless of the ratio of neoplastic to non‐neoplastic tissue was chosen for genomic DNA extraction for subsequent Sanger sequencing.
Genomic DNA extraction and PCR
From each paraffin block confirmed to contain tumour, a ribbon of 10 × 10 μm sections was placed in an Eppendorf container with care being taken to prevent tissue cross‐contamination. Genomic DNA was extracted from the FFPE sections using a QIAamp DNA FFPE Tissue Kit; (Qiagen, Hilden, Germany), according to the manufacturer's instructions. TERT promoter was amplified with HotStartTaq Polymerase Plus (Qiagen) and using the nested PCR methodology utilized by Horn et al.3 Briefly, the first‐round PCR amplicon was amplified using forward 5′‐ACGAACGTGGCCAGCGGCAG and reverse 5′‐CTGGCGTCCCTGCACCCTGG primers with an annealing temperature of 62 °C. Then, the second‐round PCR amplicon was amplified using forward 5′‐CAGCGCTGCCTGAAACTC and reverse 5′‐GTCCTGCCCCTTCACCTT primers with an annealing temperature of 55 °C. The reaction for both rounds of PCR contained Qiagen's 20% Q‐solution. All PCR runs included a standard no DNA template control, which was negative in all cases.
Sanger sequencing
PCR products were purified using Wizard SV Gel and PCR Clean‐up System (Promega, Sydney, NSW, Australia) according to the manufacturer's instructions. Each sample was sequenced using forward and reverse primers on an ABI PRISM 3700 platform (Applied Biosystems, Foster City, CA, USA) (service provided by Australian Genome Research Facility, Sydney, Australia). All samples were genotyped at least twice by two different scientists who utilized their own PCR reagents. Positive controls were included with each sequencing run: normal thyroid (wild type)‐ and cancer cell (e.g. the C228T‐positive SW1736 cell‐line)‐derived genomic DNA that yielded the expected TERT promoter sequence in each case.
Cell culture studies
A region of the TERT promoter encompassing −391 to +83 bp (relative to the TSS) was cloned into pGL3‐basic (Promega) and termed TERT‐LUC. Site‐directed mutagenesis was then performed to generate mutations corresponding to C228T (TERT228‐LUC) and C250T (TERT250‐LUC). These vectors were transfected in triplicate into SW1736 cells (derived from an anaplastic cancer containing BRAF V600E but not RAS mutations and exhibiting low p53 expression although without TP53 mutation9, 25) and treated with either vehicle or the MEK inhibitor U0126. After 48 h, cells were lysed and luciferase activity was measured as previously described.26 Renilla luciferase was used as an internal transfection control to normalize the data.
Statistical analyses
To analyse the association between clinicopathological and molecular factors with TERT promoter mutations in PTCs, we used logistic regression, Fisher's exact test, Cochran–Mantel–Haenszel test stratified for age at first surgery (as a categorical variable) and gender, considering TERT promoter mutations as the dependent variable, and each clinicopathological or molecular factor involved in the univariate or multivariate model as independent variables. Both univariate and multivariate survival analyses were performed using Cox regression model and for each variable adjusting for age at first surgery (as a continuous variable) and gender. In univariate analyses, the variables whose p‐values were <0·05 were retained in multivariate analyses. Tumour size was included in the model as a continuous variable as only a few PTC samples had microscopic deposits (i.e. <10 mm). We used Kaplan–Meier survival curves to present either overall survival (considering only disease‐related deaths) or disease‐free survival (where ‘disease’ was defined as persistent or recurrent nodal or distant metastases, or disease‐related death), according to different clinicopathological and molecular features, such as age at first surgery, gender, tumour size, stage, BRAF V600E and TERT (C228T or C250T) via the logrank test. Statistical significance was set at P < 0·05. Statistical analyses were performed using SAS software version 9·3 (SAS Institute, Inc., Cary, NC, USA) and StatXact version 11.0.0 Cytel Inc (Cambridge, MA, USA).
Results
The study cohort has been previously described23; of the 100 subjects in our previous report, 20 FFPE samples failed to amplify the TERT promoter and were excluded from this analysis. Our cohort therefore consisted of 80 patients (14 men) with mean age 47·3 years at first surgery and a median duration of follow‐up of 106 months (range 1–270). Overall, 59 (74%) were disease‐free at the end of the follow‐up period, 9 had persistent or recurrent disease (11%), 8 had died of PTC‐unrelated causes (10%), and 4 (5%) had died from PTC. If those patients who had died from unrelated causes were reclassified according to their disease status at the time of death, then 66 of 80 (83%) were disease‐free whereas 14 of 80 (18%) had persistent or recurrent disease or had died from PTC. Baseline characteristics of the cohort according to the presence or absence of TERT promoter mutations are presented in Table 1. A total of 11 of 80 patients (14%) had PTCs containing TERT promoter mutations: 8 had the C228T mutation and 4 had the C250T mutation (one tumour had both mutations). TERT promoter mutations were significantly more frequent in PTC with adverse prognostic features such as older age (P = 0·002; Fisher's exact test), male gender (P = 0·01; chi‐squared test, two‐sided), and Stage IV disease (P = 0·03, Cochran–Mantel–Haenszel test, two‐sided). No significant heterogeneity according to age at the first surgery and gender categories was evident for an association between TERT promoter mutations and tumour size (P‐value for heterogeneity of odds‐ratio = 1), that is the lack of association between TERT promoter mutations and tumour size (P = 0·8, Cochran–Mantel–Haenszel test) may be considered as true whatever age at first surgery and gender. BRAF V600E was present in 58 PTCs (73%). Eight patients (10%) had both TERT promoter mutation and BRAF V600E (Table 1).
Table 1.
Summary of clinicopathological or molecular associations with TERT promoter mutations in papillary thyroid cancers
| PTC N | TERT (C228T or C250T) N (%) | Relative risk (95% CI) | P‐Value | |
|---|---|---|---|---|
| Total number | 80 | 11 (13·8) | ||
| Age at first surgerya | ||||
| Mean ± SD | 47·3 ± 17·6 | 62·6 ± 15·3 | 0·002 | |
| ≤45 | 43 | 1 (2·3) | 1 (Referent) | |
| >45 | 37 | 10 (27·0) | 15·2 (1·9–674·6) | |
| Gender | ||||
| Male | 14 | 5 (35·7) | 1 (Referent) | 0·01 |
| Female | 66 | 6 (9·1) | 0·2 (0·05–0·7) | |
| Histological subtypeb , c | ||||
| Classical | 58 | 8 (13·8) | 1 (referent) | 0·2 |
| Follicular | 16 | 0 (0·0) | NCR | |
| Tall cell | 5 | 3 (60·0) | 6·6 (0·8–53·9) | |
| Tumour size (cm)b , c | ||||
| Mean ± SD | 1·1 ± 0·5 | 1·3 ± 0·6 | 0·8 | |
| <1 | 6 | 1 (16·7) | 3·1 (0·2–40·4) | |
| 1 – 4 | 63 | 6 (9·5) | 1 (Referent) | |
| >4 | 11 | 4 (36·4) | 2·2 (0·4–12·2) | |
| Lymph nodes with cancerb , c | ||||
| No | 51 | 5 (9·8) | 1 (Referent) | 0·1 |
| Yes | 29 | 6 (20·7) | 3·4 (0·8–15·6) | |
| Stageb , c | ||||
| I+II+III | 73 | 7 (9·6) | 1 (Referent) | 0·03 |
| IV | 7 | 4 (57·1) | 9·1 (1·3–64·4) | |
| BRAF statusb , c | ||||
| No | 22 | 3 (13·6) | 1 (Referent) | 0·3 |
| Yes | 58 | 8 (13·8) | 2·7 (0·3–23·3) | |
NCR, no convergence reached.
Fisher's exact test.
Adjusted for age at the first surgery as a categorical variable (≤45 or >45) and gender using logistic regression.
Cochran–Mantel–Haenszel test stratified for age at the first surgery on class and gender.
Disease‐related mortality according to TERT promoter mutations is presented in Table 2. Four patients (5%) died from their PTC during follow‐up (mean 50 months, median 42 months, range 2–113 months): 3 of 11 subjects (27%) with TERT promoter mutations died, whereas only 1 of 69 (1·4%) with wild‐type TERT sequence died. Hazard ratio for disease‐related death associated with TERT promoter mutations was 10·0 (95% confidence intervals = 1·0–104·1, P = 0·05) after adjusting for age at the first surgery as a categorical variable (≤45 or >45) and gender.
Table 2.
Papillary thyroid cancer‐related mortality and hazard ratios for patients with TERT‐mutated vs TERT wild‐type sequence
| Disease‐related mortality, n (%) | Disease‐related mortality rate (Deaths per 1000 Person‐Years) (95% CI) | Hazard ratio (95% CI) | ||||||
|---|---|---|---|---|---|---|---|---|
| Overall | TERT Mutated | TERT a wt | TERT Mutated | TERT a wt | Unadjusted | P‐Valueb | Adjustedc | P‐Valueb |
| 4/80 (5·0) | 3/11 (27·3) | 1/69 (1·5) | 33·7 (8·1–532·0) | 1·6 (0·0–14·1) | 19·7 (2·0− 189·5) | 0·0003 | 10·0 (1·0–104·1) | 0·05 |
TERT wt: TERT wild‐type sequence.
Exact Logrank test.
Adjusted for age at the first surgery as a categorical variable (≤45 or >45) and gender using Cox regression.
The presence of TERT promoter mutations (C228T or C250T) (logrank P = 0·0003) was significantly associated with decreased overall survival due to PTC‐related death (Fig. 1a) and also with decreased disease‐free survival (Fig. 1b).
Figure 1.
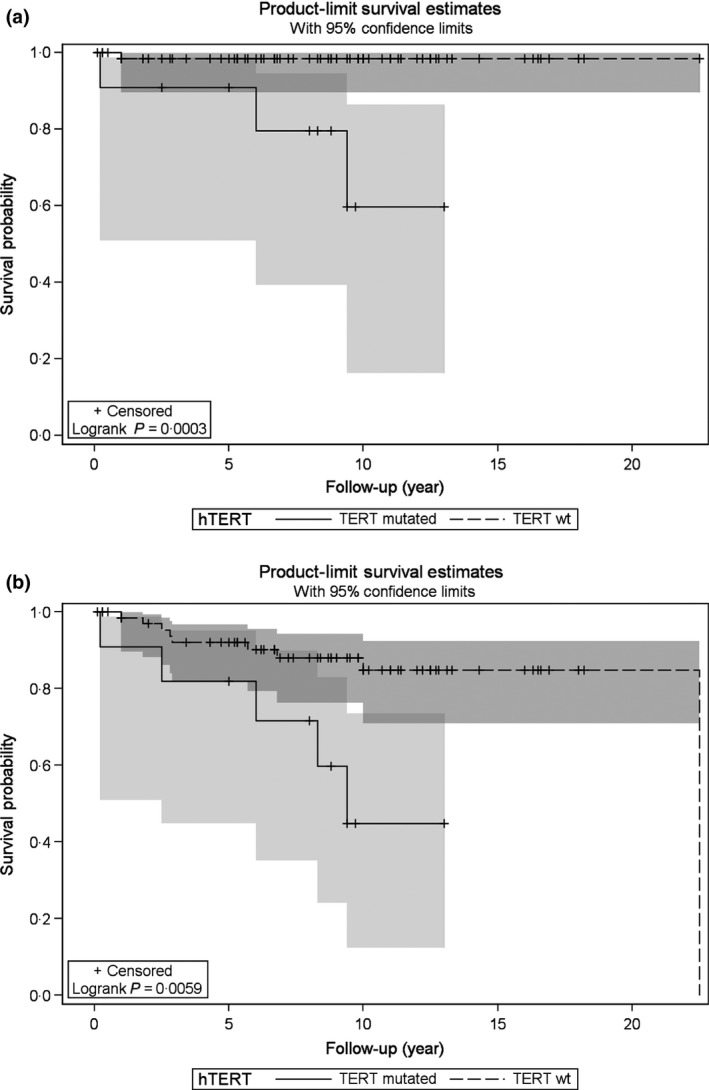
Kaplan–Meier Survival Curves of PTC‐Overall Survival (a) and PTC‐Disease‐free Survival (b) by TERT Status. Survival of subjects with TERT mutations (either C228T or C250T) is shown by the continuous line whereas survival of subjects with wild‐type TERT sequence is shown by the dotted line.
Of 20 patients with either Stage III or IV disease at presentation (13 Stage III, 7 Stage IV), TERT promoter mutations were present in 7, of whom 3 died (43%) compared with one death in 13 patients (8%) with wild‐type TERT promoter sequence.
Examining the possible interaction between TERT promoter mutations and BRAF V600E, the mortality rate for subjects whose tumours were wild type for both genotypes was 0/18; for TERT‐mutated but wild‐type BRAF 0/3; for BRAF V600E but wild‐type TERT 1/51 (2%); and for TERT mutated and BRAF V600E together mortality was 3/8 (38%). The relative risk of disease‐related death in patients with both TERT mutated and BRAF V600E mutated together was 29·4 (Cox’ model with profile‐likelihood 95% confidence limits = 3·8–594·6), as compared to the other patients (P = 0·002) (Table 3).
Table 3.
Hazard ratios of disease‐related death associated with TERT promoter mutations and BRAF V600E
| Presence n (%) | Hazard ratio (95% CI)e | P‐Valuef | |
|---|---|---|---|
| TERT wt & BRAF wta | 0/18 (0) | NCR | NCR |
| TERT mutated & BRAF wtb | 0/3 (0) | NCR | NCR |
| TERT wt & BRAF mutatedc | 1/51 (2·0) | 0·2 (0·01–1·4) | 0·08 |
| TERT mutated & BRAF mutatedd | 3/8 (37·5) | 29·4 (3·8–594·6) | 0·002 |
NCR, no convergence reached.
TERT wild‐type sequence and BRAF wild‐type sequence compared with the other 3 groups (b+c+d).
TERT‐mutated but BRAF wild‐type sequence compared with the other 3 groups (a+c+d).
TERT wild‐type sequence but BRAF mutated compared with the other 3 groups (a+b+d).
TERT mutated and BRAF mutated compared with the other 3 groups (a+b+c).
Cox' model with profile‐likelihood confidence limits.
Logrank exact test.
We therefore explored a possible interaction in vitro using reporter gene containing either the wild‐type or mutated TERT promoter. As shown in Fig. 2, TERT promoter constructs containing either C228T or C250T mutations exhibited increased transcriptional activation when transfected in SW1736 cells (endogenously containing BRAF V600E): TERTC228T‐Luc showed 4·05 ± 0·19‐fold increase (P = 0·001), and TERTC250T‐LUC showed 2·6 ± 0·26‐fold increase (P = 0·008) relative to TERT‐LUC. The addition of MEK inhibitor U0126 resulted in significant reduction of promoter activity for all constructs (Fig. 2).
Figure 2.
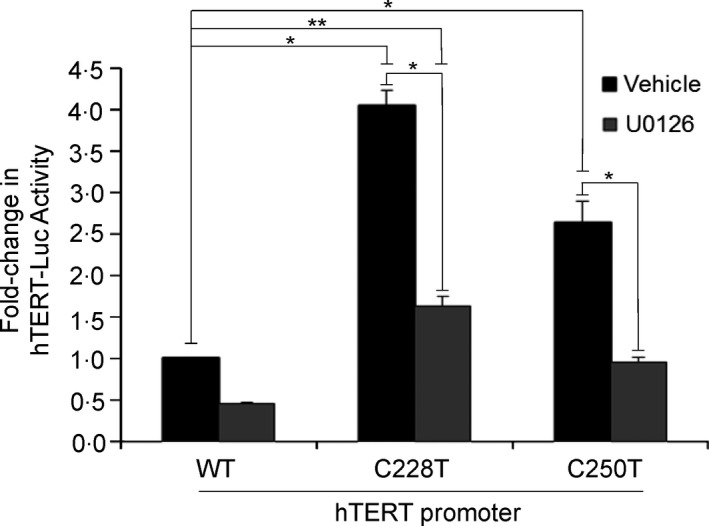
The effect of TERT promoter mutations on reporter gene activity. SW1736 cells were transfected (in triplicate) with TERT‐LUC, TERTC228T‐LUC or TERTC250T‐LUC as shown and treated with vehicle or the MEK inhibitor U0126. Luciferase activity was measured in cell lysates 48 h after transfection and normalised for Renilla luciferase as a transfection control. Data shown are mean ± SD from three experiments. *P < 0·01.
We also considered the association between TERT promoter mutations and the composite end‐point of disease persistence or recurrence, or disease‐related death. Fourteen patients were not disease‐free at the end of follow‐up (Table 4). In univariate analysis, TERT promoter mutations (HR = 4·3, 95% CI = 1·4–13·3, P = 0·01), older age at initial surgery (HR = 1·05, 95% CI = 1·01–1·08, P = 0·01), larger tumour size (HR = 1·04, 95% CI = 1·01–1·06, P = 0·01) and Stage IV disease (HR = 14·0, 95% CI = 4·4–43·9, P < 0·0001) were all significantly associated with a lower disease‐free survival. However, in multivariate analysis, only Stage IV disease (HR = 16·5, 95% CI = 2·9–93·5, P = 0·002) remained significant (Table 4).
Table 4.
Hazard ratios for persistent or recurrent disease or disease‐related death
| Presence (n = 14) | Univariate analysis | Multivariate analysisa | |||
|---|---|---|---|---|---|
| Hazard ratio (95% CI) | P‐Value | Hazard ratio (95% CI) | P‐Value | ||
| Age at the first surgeryb | |||||
| Gender | 14 | 1·05 (1·01–1·08) | 0·01 | 1·00 (0·96–1·04) | 0·9 |
| Male | 3 | 1 (Referent) | 0·5 | 1 (Referent) | 0·5 |
| Female | 11 | 0·6 (0·2–2·3) | 0·6 (0·1–2·5) | ||
| Tumour Size (cm) c | 14 | 1·04 (1·01–1·06) | 0·01 | 1·0 (1·0–1·1) | 0·1 |
| Stage | |||||
| I, II, III | 8 | 1 (Referent) | <0·0001 | 1 (Referent) | 0·002 |
| IV | 6 | 14·0 (4·4–43·9) | 16·5 (2·9–93·5) | ||
| TERT (C228T or C250T) | |||||
| No | 9 | 1 (Referent) | 0·01 | 1 (Referent) | 0·6 |
| Yes | 5 | 4·3 (1·4–13·3) | 0·6 (0·1–3·1) | ||
Adjusted for age at the first surgery as a continuous variable and gender using Cox regression.
Age at the first surgery was included in the model as a continuous variable.
Tumour size was included in the model as a continuous variable.
Discussion
In the current study, we observed that TERT promoter mutations (either C228T or C250T) were significantly associated with adverse prognostic features such as older age, male gender and Stage IV disease. TERT promoter mutations were significantly associated with disease‐related mortality (33·7 deaths per 1000 patient‐years compared with 1·6 per 1000 patient‐years for wild‐type TERT) and the hazard ratio for disease‐related death was 10·0 (95% CI = 1·0–104·1, P = 0·05) after adjustment for age at the first surgery and gender. In Stage III or IV disease, mortality was 43% in patients with TERT mutations compared with 8% in those with wild‐type TERT. The presence of both a TERT promoter mutation and BRAF V600E significantly increased disease‐related death risk (P = 0·002) compared with TERT wild‐type sequence and BRAF wild‐type sequence, TERT‐mutated but BRAF wild‐type sequence, or TERT wild‐type sequence but BRAF mutated. We also found in vitro evidence that TERT mutations increased transcriptional activity of a reporter gene in thyroid cells containing BRAF V600E and that this effect was reversed by treatment with a MEK inhibitor.
Our prevalence of 14% for TERT mutations in PTC is consistent with other reports; globally, an average of 11·5% PTCs harbour TERT mutations (Table S1). The associations with older age, male gender and Stage IV disease are also consistent with previous reports.8, 9, 10, 11, 12, 13, 14 In our study, the presence of TERT mutations was associated with the composite outcome of disease recurrence, persistence or death in univariate analysis, but was no longer significant after accounting for tumour size and Stage IV disease. In larger studies,11, 13 TERT promoter mutations have remained significantly associated with disease persistence in multivariate analyses.
Our data are broadly consistent with two other studies that have reported TERT‐associated mortality data to date13, 14 (Table S1). Melo and colleagues13 studied 332 patients with PTC, and found that TERT promoter mutations were present in 25 (7·5%); after a mean of 7·8 year follow‐up, 2 of 19 patients with TERT promoter mutations had died (10·5%) compared to 3 of 265 patients (1·1%) with wild‐type TERT promoter sequence.13 Mortality rate was very similar in our study (33·7 vs 1·6 per 1000 person‐years for PTCs with and without TERT promoter mutations, respectively) compared with that of Melo et al. (13·64 vs 1·36 deaths per 1000 person‐years).13 In a study of high‐risk (i.e. already persistent/recurrent) PTC, George and colleagues14 found the C228T TERT promoter mutation in 77 of 242 cases (31·8%); after median follow‐up of 112 months, 19 of 77 patients with TERT mutation had died (24·7%) compared to 10 of 165 (6·1%) with wild‐type TERT.
Our in vitro data indicating that TERT promoter mutations increased reporter gene expression by 2‐ to 4‐fold in SW1736 thyroid cells are consistent with studies that show similar stimulation of promoter activity in melanoma, urothelial and hepatoma cell lines4, 27 and in BCPAP and K2 thyroid cell lines.14 We then examined whether MEK inhibition could rescue the effect of these mutations on promoter activity, our rationale being that TERT promoter mutations are activating by virtue of creating new binding sites for MAPK‐responsive transcription factors.3 We chose SW1736 cells for this study, because they are known to endogenously contain BRAF V600E.9 We found that treatment with the MEK inhibitor U0126 did significantly reduce the activity of reporter genes containing either TERT C228T or C250T mutations. This observation provides a basis for potential treatment of TERT‐mutated thyroid cancers using inhibitors of the MAPK pathway.
It is well known that some patients with metastatic PTC will have a very indolent course even without aggressive treatment approaches.28 We found that in patients presenting with Stage III or IV disease, the presence of TERT mutations increased mortality but conversely, the absence of TERT mutations was associated with generally good prognosis (>70% survival after 8 years). If corroborated in larger studies or by meta‐analysis, this would be clinically important: patients with TERT‐mutated advanced disease could be triaged early to aggressive treatment with kinase inhibitors whereas patients with TERT wild‐type disease could be treated more cautiously.
In conclusion, TERT promoter mutations (C228T or C250T) are a major indicator of death due to PTC. Meta‐analysis will be needed to confirm that TERT promoter mutations interact with BRAF V600E to increase PTC‐related mortality.
Funding
MB and RCB were supported by NHMRC project grant 1061941. YR is supported by the Fondation de France and a grant from 2014 Inserm – University of Sydney Research Mobility Funding Scheme.
Conflict of interest
All authors have declared that there is no conflict of interest exists in relation to this article.
Supporting information
Table S1. TERT promoter mutations in thyroid cancer samples.
Acknowledgements
We sincerely acknowledge 2014 Inserm – University of Sydney Research Mobility Funding Scheme for funding a part of this research project.
References
- 1. Kim, N.W. , Piatyszek, M.A. , Prowse, K.R. et al (1994) Specific association of human telomerase activity with immortal cells and cancer. Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 2. Shay, J.W. & Bacchetti, S. (1997) A survey of telomerase activity in human cancer. European Journal of Cancer, 33, 787–791. [DOI] [PubMed] [Google Scholar]
- 3. Horn, S. , Figl, A. , Rachakonda, P.S. et al (2013) TERT promoter mutations in familial and sporadic melanoma. Science, 339, 959–961. [DOI] [PubMed] [Google Scholar]
- 4. Huang, F.W. , Hodis, E. , Xu, M.J. et al (2013) Highly recurrent TERT promoter mutations in human melanoma. Science, 339, 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vinagre, J. , Almeida, A. , Pópulo, H. et al (2013) Frequency of TERT promoter mutations in human cancers. Nature Communications, 4, 2185. [DOI] [PubMed] [Google Scholar]
- 6. Fredrikson, N.J. , Ny, L. , Nilsson, J.A. et al (2014) Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nature Genetics, 46, 1258–1263. [DOI] [PubMed] [Google Scholar]
- 7. Weinhold, N. , Jacobsen, A. , Schultz, N. et al (2014) Genome‐wide analysis of non‐coding regulatory mutations in cancer. Nature Genetics, 46, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu, X. , Bishop, J. , Shan, Y. et al (2013) Highly prevalent TERT promoter mutations in aggressive thyroid cancer. Endocrine Related Cancer, 20, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landa, I. , Ganly, I. , Chan, T.A. et al (2013) Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. Journal of Clinical Endocrinology and Metabolism, 98, E1562–E1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu, X. , Qu, S. , Liu, R. et al (2014) TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. Journal of Clinical Endocrinology and Metabolism, 99, E1130–E1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xing, M. , Liu, R. , Liu, X. et al (2014) BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. Journal of Clinical Oncology, 32, 2718–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang, N. , Liu, T. , Sofiadis, A. et al (2014) TERT promoter mutation as an early genetic event activating telomerase in follicular thyroid adenoma (FTA) and atypical FTA. Cancer, 120, 2965–2979. [DOI] [PubMed] [Google Scholar]
- 13. Melo, M. , da Rocha, A.G. , Vinagre, J. et al (2014) TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. Journal of Clinical Endocrinology and Metabolism, 99, E754–E765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. George, J.R. , Henderson, Y.C. , Williams, M.D. et al (2015) Association of TERT promoter mutation, but not BRAF mutation, with increased mortality in PTC. Journal of Clinical Endocrinology and Metabolism, 100, E1550–E1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gandolfi, G. , Ragazzi, M. , Frasoldati, A. et al (2015) TERT promoter mutations are associated with distant metastases in papillary thyroid carcinoma. European Journal of Endocrinology, 172, 403–413. [DOI] [PubMed] [Google Scholar]
- 16. Muzza, M. , Colombo, C. , Rossi, S. et al (2015) Telomerase in differentiated thyroid cancer: promoter mutations, expression and localization. Molecular and Cellular Endocrinology, 399, 288–295. [DOI] [PubMed] [Google Scholar]
- 17. de Biase, D. , Gandolfi, G. , Ragazzi, M. et al (2015) TERT promoter mutations in papillary thyroid microcarcinomas. Thyroid, 25, 1013–1019. [DOI] [PubMed] [Google Scholar]
- 18. Chindris, A.M. , Casler, J.D. , Bernet, V.J. et al (2015) Clinical and molecular features of Hurthle cell carcinoma of the thyroid. Journal of Clinical Endocrinology and Metabolism, 100, 55–62. [DOI] [PubMed] [Google Scholar]
- 19. Wang, N. , Xu, D. , Sofiadis, A. et al (2014) Telomerase‐dependent and independent telomere maintenance and its clinical implications in medullary thyroid carcinoma. Journal of Clinical Endocrinology and Metabolism, 99, E1571–E1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nikiforov, Y.E. , Carty, S.E. , Chiosea, S.I. et al (2014) Highly accurate diagnosis of cancer in thyroid nodules with follicular neoplasm/suspicious for a follicular neoplasm cytology by ThyroSeq v2 next‐generation sequencing assay. Cancer, 120, 3627–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu, R. & Xing, M. (2014) Diagnostic and prognostic TERT promoter mutations in thyroid fine‐needle aspiration biopsy. Endocrine Related Cancer, 21, 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crescenzi, A. , Trimboli, P. , Modica, D.C. et al (2015) Preoperative assessment of TERT promoter mutation on thyroid core needle biopsies supports diagnosis of malignancy and addresses surgical strategy. Hormone and Metabolism Research, [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 23. O'Neill, C.J. , Bullock, M. , Chou, A. et al (2010) BRAF(V600E) mutation is associated with an increased risk of nodal recurrence requiring reoperative surgery in patients with papillary thyroid cancer. Surgery, 148, 1139–1145. [DOI] [PubMed] [Google Scholar]
- 24. Nikiforov, Y.E.B. , Paul, W. & Thompson, L.D. (2012) Diagnostic Pathology and Molecular Genetics of the Thyroid: A Comprehensive Guide for Practicing Thyroid Pathology. Baltimore, MD: Lippincott Williams & Wilkins (LWW). [Google Scholar]
- 25. Blagosklonny, M.V. , Giannakakou, P. , Wojtowicz, M. et al (1998) Effects of p53‐expressing adenovirus on the chemosensitivity and differentiation of anaplastic thyroid cancer cells. Journal of Clinical Endocrinology and Metabolism, 83, 2516–2522. [DOI] [PubMed] [Google Scholar]
- 26. Bullock, M. , Duncan, E.L. , O'Neill, C. et al (2012) Association of FOXE1 polyalanine repeat region with papillary thyroid cancer. Journal of Clinical Endocrinology and Metabolism, 97, E1814–E1819. [DOI] [PubMed] [Google Scholar]
- 27. Rachakondaa, P.S. , Hosena, I. , de Verdierb, P.J. et al (2013) TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proceedings of the National Academy of Sciences of the United States of America, 110, 17426–17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin, J.D. , Hsueh, C. & Chao, T.C. (2015) Long‐term follow‐up of the therapeutic outcomes for papillary thyroid carcinoma with distant metastases. Medicine (Baltimore), 94, e1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. TERT promoter mutations in thyroid cancer samples.