

Abstract
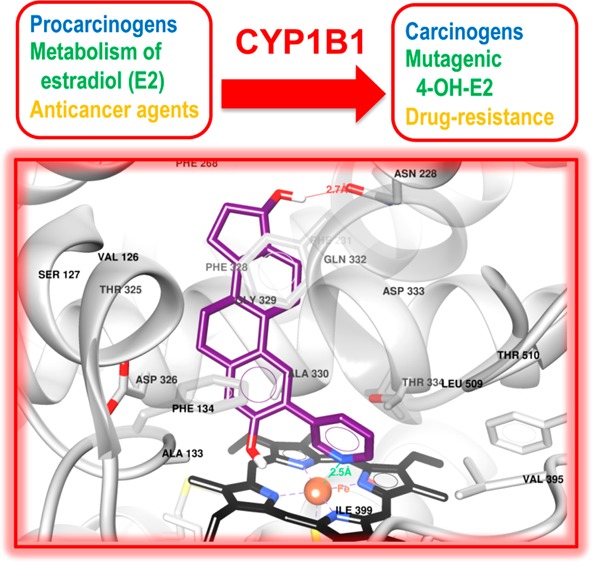
Inhibition of cytochrome P450 (CYP) 1B1 is a promising therapeutic strategy, as such an inhibitor could modulate the bioactivation of procarcinogens while reducing drug resistance. Based on docking studies, the synthesis of 12 estra-1,3,5(10)-triene derivatives containing a pyridin-3-/4-yl moiety at position C2, C3, or C4 was performed, and we measured their inhibitory activity on CYP1B1 using the ethoxyresorufin-O-deethylase (EROD) assay. The position of the nitrogen atom in the aromatic ring has little influence on their inhibition potency, but compounds with a pyridinyl at C2 of the steroid nucleus are more potent CYP1B1 inhibitors than those with a pyridinyl at C3 or C4. Estradiol derivatives (OH at C17β) are also 10-fold more potent inhibitors than estrone derivatives (carbonyl at C17). Thus, 2-(pyridin-3-yl)-estradiol (4a) is the best CYP1B1 inhibitor (IC50 = 0.011 μM) from this series of compounds, and the best steroid inhibitor reported until now. It is also 7.5-fold more potent than the well-known nonsteroidal CYP1B1 inhibitor α-naphthoflavone (IC50 = 0.083 μM).
Keywords: Steroid, estrane, enzyme inhibitor, cytochrome P450, CYP1B1, molecular modeling
Cytochromes P450 (CYPs) form a large family of hemoproteins involved in many reduction and oxidation reactions on both endogenic and xenobiotic compounds of different sizes. Eighteen (18) CYP gene families, including over 50 enzymes, are found in humans.1,2 The CYP1 family comprises three members: CYP1A1, CYP1A2, and CYP1B1. The latter is mainly expressed in extrahepatic mesodermal cells, including steroidogenic tissues (ovaries, testes, and adrenal glands) and steroid-responsive tissues (breast, uterus and prostate).3 CYP1 enzymes have been extensively studied because they are involved in the conversion of many polycyclic aromatic hydrocarbons (PAHs), such as benzo[α]pyrene, into carcinogens.4,5 Moreover, it should be noted that CYP1 enzymes are involved in the modulation of proinflammatory and inflammatory pathways through the metabolism of leukotrienes and eicosanoids.6 These enzymes can also act as tumor suppressors via the metabolism of some flavonoid- and stilbene-type compounds.7−9
Among these three CYP1 enzymes, CYP1B1 represents an interesting therapeutic target, notably because it is involved in the 4-hydroxylation of 17β-estradiol (E2) (Figure 1A), whereas CYP1A1 and CYP1A2 mainly catalyze the 2-hydroxylation of E2.10,11 Unlike 2-hydroxy-E2 and its oxidation product E2-2,3-quinone, 4-hydroxy-E2 can be oxidized into E2-3,4-quinone, a mutagenic compound that can bind DNA covalently, leading to the formation of depurinating adducts.12 In addition, it has been observed that CYP1B1 is overexpressed in a wide variety of human cancers (breast, lung, colon, esophagus, skin, testis, lymph node, and brain), but not in healthy tissue.13 Consequently, CYP1B1 can also be a good marker for the prevention of certain cancers. Finally, it should also be emphasized that CYP1B1 is involved in the metabolism of certain anticancer agents, such as docetaxel, leading to drug-resistance associated with the overexpression of CYP1B1.14−16 Thus, a CYP1B1 inhibitor could be useful within a multitherapy context. Based on these observations, the inhibition of CYP1B1 constitutes a promising therapeutic strategy because it would enable a biological action at three distinct levels: (1) by inhibiting the formation of 4-hydroxy-E2, (2) by inhibiting the bioactivation of procarcinogens, and (3) by reducing drug-resistance.15
Figure 1.

Conversion of estrogenic C18-steroid hormones by CYP1B1 (A) and the structures of the newly synthesized CYP1B1 inhibitors (B). Partial numbering of carbons (left structure) and steroid (A–D) ring identification (right structure) are reported.
Over the past years, several CYP1 inhibitors have been identified within different families, including flavonoids, trans-stilbenes, coumarins, alkaloids, and anthraquinone derivatives,1,17−19 but in the steroid family, only 16α-fluoro-5-androsten-17-one and three methoxyestradiol derivatives were reported as weak CYP1B1 inhibitors.20,21 This is surprising, considering that C18-steroid E2 and its oxidized form at position 17, estrone (E1), are both substrates of several CYPs, including CYP1B1.11
Our research group has expertise in the development of enzyme inhibitors of steroidogenesis having a steroid nucleus (C18-, C19-, or C21-steroids).22−25 In our previous study, we reported the results obtained from the screening of 90 steroids and steroid derivatives on the CYP1B1 using the ethoxyresorufin-O-deethylase (EROD) assay.26 Among the molecules tested, we identified 3-thioestrone (IC50 = 3.4 μM) as an inhibitor of CYP1B1.26 Furthermore, molecular modeling studies have shown that the 3-SH group of this steroid is closer (3.36 Å) to the iron atom of the heme system of CYP1B1 than the 3-OH of E1 and E2.
Based on these observations, we considered introducing a chemical group on the A-ring of a C18-steroid (estra-1,3,5(10)-triene), which could be able to interact with the heme of CYP1B1. Pyridine, an aromatic ring containing a nitrogen atom, is known to generate interactions with the iron of heme systems.27 We therefore synthesized 12 steroid derivatives containing a pyridin-3-/4-yl moiety at position C2, C3, or C4 of E1 and E2 (Figure 1B). Their inhibitory activity against CYP1B1 and docking simulations have also been reported.
Results and Discussion
Docking Simulations and Rational Inhibitor Design
The α-naphthoflavone (ANF) is one of the best CYP1B1 inhibitors reported in literature, and this flavonoid binds the CYP1B1 catalytic site.28 The crystal structure of CYP1B1 (PDB ID: 3PM0) is also known, and it was used in our design of new steroidal CYP1B1 inhibitors. We first divided the fused tricyclic core of ANF as three mimetic parts for the A/B/C rings of the steroidal scaffold. Furthermore, in the cocrystallized structure of ANF with CYP1B1, we observed that the phenyl moiety at C2 was oriented toward the porphyrin and ferrous ion of the heme group. Thus, we believe that replacing this phenyl ring by a pyridine moiety could generate a favorable interaction with the heme group by means of a nitrogen–iron bond. Additionally, in our previous docking studies using E1 and E2 (Supporting Information, Figure A), we observed that the A-ring was oriented toward the heme group. However, it was less close when compared to the phenyl moiety at C2 of ANF. From these observations, we hypothesized that a desired interaction between a pyridine ring and the heme of CYP1B1 could be achieved by adding a pyridine to the A-ring of E1 or E2.
To address this idea, we performed docking simulations using GOLD-5.4 software29,30 and the 3D structure of CYP1B1 (PDB ID: 3PM0). The molecules screened in this study were ANF for comparison purposes, and those represented in Figure 1B.
This screening provided a GOLD score (GS) of 69.8 and a ChemPLP fitness score (CFS) of 95.5 for the best docked conformation of ANF (Table 1). The 2-substituted compounds, such as 3a (GS = 60.6 and CFS = 81.2), fit better into the binding site of CYP1B1 than the corresponding 3-substituted compound 6a (GS = 58.8 and CFS = 49.9) and 4-substituted compound 11a (GS = 45.6 and CFS = 76.0). In fact, the pyridine ring of 3a interacts with the heme group (Figure 2A) through a nitrogen–iron bond. Moreover, the nitrogen atom of 3a is closer to the iron than the nitrogen atom of 6a (2.4 and 3.2 Å, respectively, Figure 2A,B). In addition, we found that the pyridinyl moiety promotes an ideal orientation to perform pi-stacking interactions between the A-ring of the steroid and the side chain of Phe134. On the contrary, the structure of 11a is inverted when compared to 3a and 6a (Figure 2C). Thus, this orientation, suggesting an interaction of the carbonyl at C17 with iron, significantly decreases the interactions into the binding site. However, 4a (Figure 2D), which is an E2 derivative having a pyridin-3-yl at C2, presented the best GS and CFS for this series (62.6 and 86.0, respectively).
Table 1. Docking Scores and Inhibition of CYP1B1 Activity by a Series of E1 and E2 Derivatives.
| compda | X | Y | Z | GSb | CFSb | inhibition (%) at 0.3 μMc | inhibition IC50(μM)c |
|---|---|---|---|---|---|---|---|
| 3a | =O | N | CH | 60.6 | 81.2 | 37.8 ± 1.5 | 0.063 |
| 3b | =O | CH | N | 52.5 | 74.8 | 21.3 ± 3.1 | 0.120 |
| 4a | 17βOH | N | CH | 62.6 | 86.0 | 85.4 ± 0.3 | 0.011 |
| 4b | 17βOH | CH | N | 54.2 | 81.8 | 87.4 ± 0.9 | 0.032 |
| 6a | =O | N | CH | 58.8 | 49.9 | 28.2 ± 2.5 | |
| 6b | =O | CH | N | 56.2 | 49.7 | 6.1 ± 2.2 | |
| 7a | 17βOH | N | CH | 45.7 | 85.7 | 19.6 ± 1.7 | |
| 7b | 17βOH | CH | N | 49.0 | 84.4 | 19.5 ± 0.7 | |
| 11a | =O | N | CH | 45.6 | 76.0 | 14.0 ± 1.8 | |
| 11b | =O | CH | N | 43.2 | 74.8 | 16.9 ± 2.3 | |
| 12a | 17βOH | N | CH | 44.1 | 71.2 | 28.9 ± 1.1 | |
| 12b | 17βOH | CH | N | 42.8 | 71.9 | 20.9 ± 4.5 | |
| E1 | 58.6 | 49.7 | 16.0 ± 4.0 | ||||
| E2 | 48.5 | 53.6 | 12.4 ± 3.0 | ||||
| T-E1 | 69.8 | 13.3 ± 3.3 | |||||
| ANF | 69.8 | 95.5 | 94.0 ± 3.2 | 0.083 |
E1, estrone; E2, 17β-estradiol; T-E1, 3-thioestrone; ANF, α-naphthoflavone.
GS, gold score; CFS, ChemPLP fitness score.
Inhibition (% and IC50) of the transformation of resorufin ethyl ether into resorufin by human CYP1B1 in the presence of NADPH in triplicate (±SD). See Supporting Information (Figure B) for the inhibition curves of 3a, 3b, 4a, 4b, and ANF.
Figure 2.
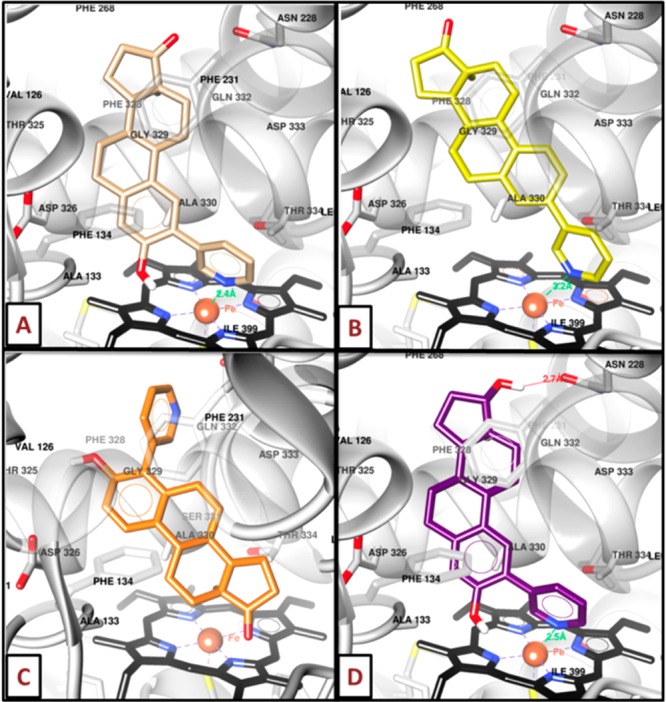
Binding mode of 3a (A), 6a (B), 11a (C), and 4a (D) docked in CYP1B1. Black sticks represent the heme group, whereas the iron atom is highlighted as a red sphere. Images were produced using the UCSF Chimera package.30
The binding mode for this compound showed the same orientation and interactions as 3a. These include a nitrogen–iron bond with the heme as well as a pi-stacking interaction between the A-ring and Phe134. Additionally, the steroidal core of 3a and 4a interacts by means of hydrophobic contacts with Phe268, Ala133, and Phe134. Nevertheless, 4a produced an H-bond between the 17β-OH group and the carbonyl group from the side chain of Asn228. This extra interaction is also performed by the E2 derivatives 4b, 7a, and 7b (data not shown) and is the main reason why we found better GS and CFS values than those of 3b, 6a, and 6b. Hence, the E2 derivatives with a pyridine ring linked to C2 could induce a significant inhibitory effect on CYP1B1.
Chemistry
To confirm our docking results, we performed the synthesis of 12 E1 and E2 derivatives bearing a pyridin-3-/4-yl moiety in C2, C3, or C4 of the estra-1,3,5(10)-triene (Figure 1B). A different synthesis pathway was used to introduce the pyridine ring at each steroid position, and the final step is the reduction of the C17 ketone into a secondary alcohol. Compounds 3a, 3b, 4a, and 4b bearing a pyridinyl moiety at C2 were obtained in two or three steps from 1 (Scheme 1). Compound 1 was previously obtained from E1 in two steps: (1) 2-iodo-E1 was prepared via a selective halogenation of E1 with mercuric acetate and iodine in acetic acid,31 and (2) the 3-OH was protected by a reaction with the methoxymethyl (MOM) chloride in the presence of Cs2CO3. Compounds 2a and 2b were obtained by a Suzuki coupling between 1 and 3- or 4-pyridine boronic acid in the presence of K3PO4 and the catalyst Pd(dppf)Cl2. This reaction was performed in DMF under microwaves (MW). Then, the MOM ether was hydrolyzed with HCl to form 3a and 3b. Finally, 4a and 4b were synthesized from 3a and 3b through a reduction of the ketone with NaBH4.
Scheme 1.

Reagents and conditions: (a) Hg(OAc)2, I2, AcOH, THF, rt, 2 h; (b) MOM-Cl, Cs2CO3, ACN, reflux, 2–3 h; (c) 3- or 4-pyridine boronic acid, Pd(dppf)Cl2, K3PO4, DMF, MW, 120 °C, 2–3 h; (d) 10% HCl aq. in MeOH (1:9), 50 °C, overnight; (e) NaBH4, MeOH/DCM (9:1), 0 °C, 2–3 h.
Steroid derivatives with a pyridinyl at C3 were obtained from 5 in a single step for the oxidized forms 6a and 6b or in two steps for the reduced forms 7a and 7b (Scheme 2). A classical reaction between E1 and triflic anhydride with DMAP as base gave 5 in a high yield of 90%.32 Compounds 6a and 6b were obtained by a Suzuki coupling of 5 with 3- or 4-pyridine boronic acid under the same reaction conditions used for 2a and 2b. Compounds 6a and 6b were then reduced with NaBH4 to give compounds 7a and 7b.
Scheme 2.

Reagents and conditions: (a) Tf2O, 2,6-lutidine, DMAP, DCM, 0 °C to rt; (b) 3- or 4-pyridine boronic acid, Pd(dppf)Cl2, K3PO4, DMF, MW, 120 °C, 2–3 h; (c) NaBH4, MeOH/DCM (9:1), 0 °C, 2–3 h.
Steroid derivatives with a pyridinyl at C4 were synthesized from 4-bromo-E1 (8) in three steps for the oxidized forms 11a and 11b or in four steps for the reduced forms 12a and 12b (Scheme 3). Compound 8 was obtained from E1 by a bromination with Br2 in the presence of powdered iron, acetic acid, and water, as previously reported by Slaunwhite and Neely.33 The MOM derivative 9 was formed by the protection of the 3-OH of 8 with MOM-Cl and Cs2CO3 in refluxing ACN. Thereafter, 10a and 10b were obtained by a Suzuki coupling of 9 with 3- or 4-pyridine boronic acid under different conditions than those previously reported for C2 and C3. Indeed, due to the lower reactivity of the bromine atom compared to the iodine, the methodology used for the synthesis of compounds with the pyridinyl at C2 or C3 was not efficient for the coupling at C4. After investigation, we found alternative conditions for the Suzuki coupling of boronic acids with aromatic compounds bearing a bromine atom. In this methodology, Pd(PPh3)4 was used as the catalyst, aqueous K2CO3 (2 M) as the base, and the reaction was carried out in a mixture of toluene and ethanol. Subsequent deprotection of the MOM ether with HCl led to the formation of 11a and 11b. Compounds 12a and 12b were next synthesized through a reduction of 11a and 11b with NaBH4.
Scheme 3.

Reagents and conditions: (a) Br2, 80% aq. acetic acid, Fe, 20 °C; (b) MOM-Cl, Cs2CO3, ACN, reflux, 2–3 h; (c) 3- or 4-pyridine boronic acid, Pd(PPh3)4, toluene, EtOH, K2CO3 aq. (2 M), reflux, overnight; (d) 10% HCl aq. in MeOH (1:9), 50 °C, overnight; (e) NaBH4, MeOH/DCM (9:1), 0 °C, 2–3 h.
The chemical reactions described above were generally complete, but it is important to mention that a protection of the 3-OH with a MOM group is necessary to subsequently perform the Suzuki coupling. In fact, this protecting group promotes the formation of palladium complexes during this reaction, thus optimizing the coupling of steroid derivatives 1 and 9 with pyridine boronic acid. In this context, it should be noted that Ivanov et al. worked on the optimization of the Suzuki–Miyaura reaction conditions for the synthesis of 2-aryl-E1 and 2,4-diaryl-E1 by using different ligands, and notably sterically encumbered biaryl ligands.34 We were able to obtain E1 derivatives with a pyridinyl at C2 in good yields (approximately 70–80%) under conventional Suzuki coupling reaction conditions through the introduction of the MOM group at C3.
Structure–Activity Relationships against CYP1B1
The inhibitory activity of synthesized compounds 3a–b, 4a–b, 6a–b, 7a–b, 11a–b, and 12a–b against recombinant human CYP1B1 enzyme was evaluated using the standard EROD assay, which is widely used to assess CYP1 activity (Table 1). In this assay performed in the presence of NADPH as cofactor, the transformation of resorufin ethyl ether, used as an enzyme substrate, into resorufin, was measured by recording the fluorescence. ANF, a potent CYP1B1 inhibitor,1 was used as the reference for this assay. As the best steroidal CYP1B1 inhibitor,26 3-thioestrone was included in the protocol to compare its activity with those of our new steroid derivatives. The activity of E1 and E2, two steroidal substrates of CYP1B1, was also evaluated.
The results of the EROD assay clearly highlight 4a and 4b, which have shown a very close inhibitory activity against CYP1B1 (85.4 ± 0.3 and 87.4 ± 0.9%, respectively) to that of ANF (94.0 ± 3.2%) at 0.3 μM. Compounds 4a and 4b bear a pyridin-3-/4-yl, respectively, at C2 of the steroid backbone. We also note that 3a and 3b, which are distinguished from 4a and 4b only by the presence of a ketone instead of an alcohol at C17, have a lower inhibitory activity than their reduced forms. E2 derivatives appear to be better inhibitors than their oxidized forms in most cases, but the difference is particularly pronounced for 3a–b and 4a–b. Compounds 6a–b and 7a–b, with a pyridinyl at C3, show a weaker activity than their counterparts with the pyridinyl in C2. Except 6b, which has the lowest activity among the 12 synthesized compounds, these derivatives at C3 present a similar inhibitory potency (∼20%). Finally, the C4-steroid derivatives 11a–b and 12a–b have an inhibitory activity similar to that of 6a and 7a–b, with the pyridinyl at C3. Compounds 12a–b have a slightly higher inhibitory activity than their oxidized homologues 11a–b. 3-thioestrone is clearly a lower inhibitor in comparison with these pyridine derivatives. Otherwise, both substrates E1 and E2 showed a very weak inhibitory potency toward CYP1B1 (∼15% inhibition), thus demonstrating the role of the pyridine ring.
From screening study results, we selected four steroid derivatives with a pyridine ring at C2 as the best CYP1B1 inhibitors. From their inhibition curves, we determined their IC50 values (Table 1), which allow a better comparison between each inhibitor.
Considering these results, the most important point is that steroid derivatives with a pyridinyl at C2 (3a–b and 4a–b) are the best inhibitors of CYP1B1 activity among the 12 steroids bearing a pyridine moiety. This is quite interesting and confirms the docking results showing the best CFS and GS when the pyridine ring was located at C2, while the introduction of the pyridinyl in C3 or C4 seemed to lessen the affinity of these compounds for the enzyme. Also note that E2 derivatives 4a–b are better CYP1B1 inhibitors than their oxidized E1 homologues 3a–b confirming our docking results. Given that E1 and E2 have shown the same inhibitory activity against CYP1B1, this result suggests that the hydroxy function in C17 is involved in the formation of a H-bond, probably with the Asn228 residue into the catalytic site of CYP1B1 as suggested by our docking simulations. Moreover, it should be stressed that 4a–b appear to be very potent inhibitors of CYP1B1. Indeed, they have shown better inhibitory activities than ANF, a potent and well-known CYP1B1 inhibitor.
Plasmatic Concentration of 4a in Rats
A preliminary assessment of the stability of these new CYP1B1 inhibitors was performed by determination of the plasmatic concentration of 4a as a function of time after a subcutaneous (sc) injection in rats (Figure 3). The concentration of the corresponding oxidized compound 3a was also measured because the secondary alcohol of an E2 derivative is known to be metabolized by the Phase-I metabolism enzymes. Surprisingly, the ketonic form of 4a, compound 3a, was more concentrated in plasma than the alcohol 4a, but the shape of their concentration curves was the same. In fact, following a sc injection of 0.66 mg of 4a in propylene glycol (PG)/DMSO (92:8), the plasmatic concentrations of 4a and 3a rapidly increased to 4.0 and 7.0 ng/mL (at 2 h), respectively, and gradually decreased thereafter. Although the plasmatic concentrations measured and AUC values calculated (12.8, 33.2, and 46.0 ng·h/mL for 4a, 3a, and 4a + 3a, respectively) are relatively acceptable for E2-derivatives, these preliminary results highlight that both 17β-OH and 3-OH of 4a could be protected toward Phase-I and Phase-II metabolism, in order to increase their metabolic stability and, consequently, to increase their plasmatic concentrations.
Figure 3.

Plasmatic concentrations of 2-(pyridin-3-yl)-E2 (4a) and 2-(pyridin-3-yl)-E1 (3a) after a sc injection of 4a (0.66 mg in 500 μL of PG/DMSO (92:8); 2.0 mg/kg) in rats. AUC: area under curve.
Conclusion
In conclusion, we have successfully synthesized a series of estra-1,3,5(10)-triene derivatives bearing a pyridinyl linked to the steroid A-ring (position C2, C3, or C4) and a carbonyl or a hydroxyl group on D-ring (C17). This study constitutes an important advance in the field of CYP1B1 inhibitors, as we have identified the most potent CYP1B1 inhibitors with a steroid scaffold. Indeed, 4a and 4b have shown a better (7.5- and 2.5-fold) inhibitory activity when compared to ANF, a potent nonsteroidal CYP1B1 inhibitor.
A major point of consideration is that the position of the pyridine ring plays a key role for the inhibitory activity of the synthesized steroid derivatives, and the best inhibitors appear to be those with the pyridinyl at C2. Notably, 4a and 4b (IC50 = 0.011 and 0.032 μM, respectively) are 6- and 4-fold more active inhibitors against CYP1B1 activity than their oxidized homologues in C17 (3a and 3b), thus reflecting that 17β-OH contributes to improve the affinity of these compounds for the catalytic site of CYP1B1. However, the difference of inhibitory activity between the reduced form and the oxidized form is not so important for the other derivatives with a pyridinyl in C3 or C4, but they are poor CYP1B1 inhibitors.
The position of the nitrogen atom into the pyridine ring does not strongly alter the inhibitory potency. Indeed, compounds with a pyridin-3-yl moiety (series a) almost have the same inhibitory profile as their analogues with a pyridin-4-yl moiety (series b). However, the measurement of IC50 values of 3a–b and 4a–b showed that 3a and 4a, with a pyridin-3-yl at C2, were 2- and 3-fold more potent CYP1B1 inhibitors than their counterparts 3b and 4b, with a pyridin-4-yl at C2.
The interesting results we obtained open the door to the development of a new generation of steroidal CYP1B1 inhibitors. In fact, based on these SAR results and additional docking studies, the synthesis of a large series of new and diversified estrane derivatives is planned to reveal their inhibitory activity against CYP1B1. Modification at C17 will also be tested to avoid the formation of a less potent ketone derivative, which is the major metabolite observed when the corresponding alcohol was injected in rats.
Acknowledgments
F.C.B. thanks the National Autonomous University of Mexico and the National Council for Sciences and Technology (CONACyT) for the awarded fellowship. Careful reading of this manuscript by Mrs. Micheline Harvey is also greatly acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00265.
Docking methodology, experimental procedures for the synthesis, characterization, and NMR spectra of all compounds, enzymatic EROD assay, plasmatic concentration assay, and additional figures (PDF)
Author Contributions
All authors contributed to writing the manuscript and have given approval to the final version of the manuscript.
Fonds de recherche du Québec–Santé (FRQS) is acknowledged for financial support from the Strategic Program.
The authors declare no competing financial interest.
Supplementary Material
References
- Chun Y. J.; Kim S. Discovery of cytochrome P450 1B1 inhibitors as new promising anti-cancer agents. Med. Res. Rev. 2003, 23, 657–668. 10.1002/med.10050. [DOI] [PubMed] [Google Scholar]
- Nebert D. W.; Wikvall K.; Miller W. L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc., B 2013, 368, 20120431. 10.1098/rstb.2012.0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter T. R.; Tang Y. M.; Hayes C. L.; Wo Y. Y.; Jabs E. W.; Li X.; Yin H.; Cody C. W.; Greenlee W. F. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J. Biol. Chem. 1994, 269, 13092–13099. [PubMed] [Google Scholar]
- Shen Z.; Liu J.; Wells R. L.; Elkind M. M. cDNAcloning, sequence analysis, and induction by aryl hydrocarbons of a murine cytochrome P450 gene, CYP1B1. DNA Cell Biol. 1994, 13, 763–769. 10.1089/dna.1994.13.763. [DOI] [PubMed] [Google Scholar]
- Androutsopoulos V. P.; Tsatsakis A. M.; Spandidos D. A. Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. 10.1186/1471-2407-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divanovic S.; Dalli J.; Jorge-Nebert L. F.; Flick L. M.; Gálvez-Peralta M.; Boespflug N. D.; Stankiewicz T. E.; Fitzgerald J. M.; Somarathna M.; Karp C. L.; Serhan C. N.; Nebert D. W. Contributions of the three CYP1 monooxygenases to pro-Inflammatory and inflammation-resolution lipid mediator pathways. J. Immunol. 2013, 191, 3347–3357. 10.4049/jimmunol.1300699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsopoulos V. P.; Papakyriakou A.; Vourloumis D.; Tsatsakis A. M.; Spandidos D. A. Dietary flavonoids in cancer therapy and prevention: Substrates and inhibitors of cytochrome P450 CYP1 enzymes. Pharmacol. Ther. 2010, 126, 9–20. 10.1016/j.pharmthera.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Androutsopoulos V. P.; Papakyriakou A.; Vourloumis D.; Spandidos D. E. Comparative CYP1A1 and CYP1B1 substrate and inhibitor profile of dietary flavonoids. Bioorg. Med. Chem. 2011, 19, 2842–2849. 10.1016/j.bmc.2011.03.042. [DOI] [PubMed] [Google Scholar]
- Androutsopoulos V. P.; Li N.; Aroo R. R. J. The Methoxylated Flavones Eupatorin and Cirsiliol Induce CYP1 Enzyme Expression in MCF7 Cells. J. Nat. Prod. 2009, 72, 1390–1394. 10.1021/np900051s. [DOI] [PubMed] [Google Scholar]
- Hayes C. L.; Spink D. C.; Spink B. C.; Cao J. Q.; Walker N. J.; Sutter T. R. 17β-Estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 9776–9781. 10.1073/pnas.93.18.9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. J.; Cai M. X.; Thomas P. E.; Conney A. H.; Zhu B. T. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology 2003, 144, 3382–3398. 10.1210/en.2003-0192. [DOI] [PubMed] [Google Scholar]
- Cavalieri E. L.; Stack D. E.; Devanesan P. D.; Todorovic R.; Dwivedy I.; Higginbotham S.; Johansson S. L.; Patil K. D.; Gross M. L.; Gooden J. K.; Ramanathan R.; Cerny R. L.; Rogan E. G. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10937–10942. 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray G. I.; Taylor M. C.; McFadyen M. C.; McKay J. A.; Greenlee W. F.; Burke M. D.; Melvin W. T. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997, 57, 3026–3031. [PubMed] [Google Scholar]
- McFadyen M. C. E.; McLeod H. L.; Jackson F. C.; Melvin W. T.; Doehmer J.; Murray G. I. Cytochrome P450 CYP1B1 protein expression: A novel mechanism of anticancer drug resistance. Biochem. Pharmacol. 2001, 62, 207–212. 10.1016/S0006-2952(01)00643-8. [DOI] [PubMed] [Google Scholar]
- Cui J.; Meng Q.; Zhang X.; Cui Q.; Zhou W.; Li S. Design and synthesis of new α-naphthoflavones as cytochrome P450 (CYP) 1B1 inhibitors to overcome docetaxel-resistance associated with CYP1B1 overexpression. J. Med. Chem. 2015, 58, 3534–3547. 10.1021/acs.jmedchem.5b00265. [DOI] [PubMed] [Google Scholar]
- Rochat B.; Morsman J. M.; Murray G. I.; Figg W. D.; McLeod H. L. Human CYP1B1 and anticancer agent metabolism: mechanism for tumor-specific drug inactivation?. J. Pharmacol. Exp. Ther. 2001, 296, 537–541. [PubMed] [Google Scholar]
- Cui J.; Li S. Inhibitors and prodrugs targeting CYP1: a novel approach in cancer prevention and therapy. Curr. Med. Chem. 2014, 21, 519–552. 10.2174/09298673113206660277. [DOI] [PubMed] [Google Scholar]
- Dong J.; Zhang Q.; Cui Q.; Huang G.; Pan X.; Li S. Flavonoids and naphthoflavonoids: wider roles in the modulation of cytochrome P450 family 1 enzymes. ChemMedChem 2016, 11, 2102–2118. 10.1002/cmdc.201600316. [DOI] [PubMed] [Google Scholar]
- Dutour R.; Poirier D. Inhibitors of cytochrome P450 (CYP) 1B1. Eur. J. Med. Chem. 2017, 135, 296–306. 10.1016/j.ejmech.2017.04.042. [DOI] [PubMed] [Google Scholar]
- Dawling S.; Roodi N.; Parl F. F. Methoxyestrogens exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res. 2003, 63, 3127–3132. [PubMed] [Google Scholar]
- Ciolino H. P.; MacDonald C. J.; Yeh G. C. Inhibition of carcinogen-activating enzymes by 16α-fluoro-5-androsten-17-one. Cancer Res. 2002, 62, 3685–3690. [PubMed] [Google Scholar]
- Poirier D. Contribution to the development of inhibitors of 17β-hydroxysteroid dehydrogenase types 1 and 7: key tools for studying and treatingestrogen-dependent diseases. J. Steroid Biochem. Mol. Biol. 2011, 125, 83–94. 10.1016/j.jsbmb.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Maltais R.; Ayan D.; Trottier A.; Barbeau X.; Lagüe P.; Bouchard J. E.; Poirier D. Discovery of a non-estrogenic irreversible inhibitor of 17β-hydroxysteroid dehydrogenase type 1 from 3-substituted-16β-(m-carbamoylbenzyl)-estradiol derivatives. J. Med. Chem. 2014, 57, 204–222. 10.1021/jm401639v. [DOI] [PubMed] [Google Scholar]
- Maltais R.; Trottier A.; Delhomme A.; Barbeau X.; Lagüe P.; Poirier D. Identification of fused 16β,17β-oxazinone-estradiol derivatives as a new family of non-estrogenic 17β-hydroxysteroid dehydrogenase type 1 inhibitors. Eur. J. Med. Chem. 2015, 93, 470–480. 10.1016/j.ejmech.2015.01.059. [DOI] [PubMed] [Google Scholar]
- Ciobanu L. C.; Boivin R. P.; Luu-The V.; Poirier D. 3β-Sulfamate derivatives of C19 and C21 steroids bearing a t-butylbenzyl or a benzyl group: synthesis and evaluation as non-estrogenic and non-androgenic steroid sulfatase inhibitors. J. Enzyme Inhib. Med. Chem. 2003, 18, 15–26. 10.1080/1475636031000069282. [DOI] [PubMed] [Google Scholar]
- Poirier D.; Roy J.; Cortés-Benítez F.; Dutour R. Targeting cytochrome P450 (CYP) 1B1 with steroid derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 5272–5276. 10.1016/j.bmcl.2016.09.046. [DOI] [PubMed] [Google Scholar]
- DeVore N. M.; Scott E. E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–119. 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.; Savas U.; Stout C. D.; Johnson E. F. Structural Characterization of the complex between α-naphthoflavone and human cytochrome P450 1B1. J. Biol. Chem. 2011, 286, 5736–5743. 10.1074/jbc.M110.204420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a generic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Potter B. V. L.; Reed M. J.; Woo L. W. L.. Steroidal compounds for inhibiting steroid sulphatase. Patent WO2003033518A1, 2003.
- Radu I. I.; Poirier D.; Provencher L. New efficient pathway for the synthesis of 3-aminoestrone. Tetrahedron Lett. 2002, 43, 7617–7619. 10.1016/S0040-4039(02)01823-3. [DOI] [Google Scholar]
- Slaunwhite W. R.; Neely L. Bromination of phenolic steroids. I. Substitution of estrone and 17β-estradiol in ring A. J. Org. Chem. 1962, 27, 1749–1752. 10.1021/jo01052a062. [DOI] [Google Scholar]
- Ivanov A.; Ejaz S. A.; Shah S. J. A.; Ehlers P.; Villinger A.; Frank E.; Schneider G.; Wölfling J.; Rahman Q.; Iqbal J.; Langer P. Synthesis, functionalization and biological activity of arylated derivatives of (+)-estrone. Bioorg. Med. Chem. 2017, 25, 949–962. 10.1016/j.bmc.2016.12.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.