

Abstract
Age‐related macular degeneration (AMD) is the leading cause of central vision loss worldwide. Loss of retinal pigment epithelium (RPE) is a major pathological hallmark in AMD with or without pathological neovascularization. Although activation of the immune system is implicated in disease progression, pathological pathways remain diverse and unclear. Here, we report an unexpected protective role of a pro‐inflammatory cytokine, interleukin‐33 (IL‐33), in ocular angiogenesis. IL‐33 and its receptor (ST2) are expressed constitutively in human and murine retina and choroid. When RPE was activated, IL‐33 expression was markedly elevated in vitro. We found that IL‐33 regulated tissue remodelling by attenuating wound‐healing responses, including reduction in the migration of choroidal fibroblasts and retinal microvascular endothelial cells, and inhibition of collagen gel contraction. In vivo, local administration of recombinant IL‐33 inhibited murine choroidal neovascularization (CNV) formation, a surrogate of human neovascular AMD, and this effect was ST2‐dependent. Collectively, these data demonstrate IL‐33 as a potential immunotherapy and distinguishes pathways for subverting AMD pathology. © 2016 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: IL‐33, AMD, RPE, angiogenesis, wound healing
Introduction
Age‐related macular degeneration (AMD) is the leading cause of visual loss in developed countries, affecting one in ten people older than 50 years (http://www.amdalliance.org). With a global increase in the ageing population, the numbers suffering with this progressive disease are expected to increase in 2050 by 50% 1. AMD is a progressive, polygenic, and multifactorial disease leading to a clinico‐pathological spectrum of cell loss and atrophy and neovascularization. Although current anti‐vascular endothelial growth factor (VEGF) therapies have revolutionized treatment for neovascular (n)AMD, we are treating late in disease, only after the insidious progression of degeneration has ensued. Tailoring therapies to treat early is desirable to overcome the potential amplification of degenerative pathways, including neovascularization, and to offer, in such cases, alternatives for those unresponsive to anti‐VEGF agents 2.
Altered immune responses are recognized as integral to the pathogenesis of AMD 3. The NLRP3 inflammasome has been reported to monitor cell stress through pattern recognition receptors [e.g. Toll‐like receptors (TLRs)], activating inflammatory caspases 4. The activation of the NLRP3 inflammasome is almost certainly a protective response initially, providing a rapid response to danger in order to preserve tissue function and integrity. The corollary is that inflammasome activation may also cause tissue damage. So, why patients convert from early or intermediate AMD (clinically manifest as drusen and RPE changes) to advanced AMD (including choroidal neovascularization) is purported to be as a switch from the ageing homeostatic regulatory response 5 to a pathological inflammatory response. For example, recent studies have shown that the NLRP3 inflammasome, which is capable of liberating active interleukin (IL) 1β and IL‐18 production, can ‘sense’ drusen isolated from human AMD donor eyes. IL‐18, however, has been shown to protect against the development of choroidal neovascularization (CNV) 6.
Given the controversy over the role of inflammasome activation and the role for adjuvant immunotherapy in AMD treatment, we have investigated the role of the type 2 IL‐1 family member IL‐33 7, which is unique as it is active without caspase‐1 cleavage and does not require inflammasome activation for secretion and bioactivity 8. IL‐33 is released following cellular damage 9 and biomechanical overload 10, behaving as an ‘alarmin’ 11 that signals through the heterodimeric receptor consisting of ST2 and IL‐1R accessory protein 12. The membrane form of ST2 (IL1RL1) 13 is expressed on many immune cells 14, 15. An alternative spliced soluble form (sST2) acts as a decoy receptor of IL‐33 16. In experimental autoimmune uveitis (EAU), IL‐33 attenuates disease severity 17. However, IL‐33 triggers an inflammatory response, recruiting monocytes, contributing to photoreceptor loss in a photoxic retinal model of degeneration 18, and this infers a pathogenic role for endogenous IL‐33 and offers an a priori rationale for neutralizing IL‐33 to reduce myeloid cell accumulation as a possible intervention.
On the basis of the emerging role of IL‐33 in inflammatory disorders 19, 20 and the controversy over its role, we hypothesized that IL‐33/ST2 signalling in the absence of overt cell death regulates tissue responses in neovascular AMD disease pathology. We showed that IL‐33 subverts angiogenesis, via direct inhibition of fibroblasts and endothelial cells that express high levels of ST2. We further showed that recombinant IL‐33 protects against CNV development. Our data provide new insights into the regulation of angiogenesis by IL‐33, and are contrary to previous data.
Materials and methods
Cell culture
The murine B6‐RPE07 and human ARPE‐19 cell lines were cultured in DMEM medium supplemented with 10% heat‐inactivated fetal calf serum, 2% l‐glutamine, 1 mm sodium pyruvate, 60 μm 2‐mercaptoethanol, 100 U/ml penicillin, and 100 µg/ml streptomycin. Primary human retinal and cerebral microvascular endothelial cells (HRMECs and HCMECs) were purchased from a commercial source (Innoprot, Derio‐Bizkaia, Spain). HRMECs were isolated from healthy retinal tissue and were cryopreserved at passage 1 and delivered to our laboratory frozen. The vendor had characterized cells by immunofluorescent methods with antibodies directed against VWF/Factor VIII and CD31 (P‐CAM) and by uptake of DiI‐Ac‐LDL. Human choroidal fibroblasts were purchased from SciencCell Research Laboratories (Carlsbad, CA, USA) and cultured in fibroblast medium (SciencCell Research Laboratories).
Mice and in vivo experimental procedures
C57BL/6J mice were purchased from Charles River Laboratories, Margate, UK. ST2 knock‐out (St2−/−) C57BL/6 mice (backcrossed for ten generations) were kindly provided by C Emanueli (School of Clinical Sciences, Bristol Heart Institute, University of Bristol) and were generated as described earlier 20. IL‐33 knock‐out (Il‐33−/−) mice were generated as described earlier 21. Il33−/− mice were backcrossed for six generations to C57/Bl6J mice to remove the rd8 mutation. All mice were used at 8–10 weeks of age and maintained in the animal house facilities of the University of Bristol, according to Home Office Regulations. Treatment of the animals conformed to the Association for Research in Vision and Ophthalmology (ARVO) statement.
CNV was induced by laser photocoagulation in mice; four laser spots (power 200 mW, duration 75 ms, spot size 75 µm) were delivered to the posterior retina using an OculightSlx Krypton Red Laser system (IRIS Medical, Iridex, Mountain View, CA, USA). Local administration of IL‐33 (doses in 2 µl of normal saline) or vehicle control was performed by intravitreal injection using a 33‐gauge hypodermic needle. Optical coherence tomography (OCT) was performed using the Micron IV (Phoenix Research Laboratories, Pleasanton, CA, USA) to evaluate lesion size and retinal thickness.
Tissue preparation and immunofluorescence staining
To prepare RPE/choroid whole‐mounts, enucleated eyes were initially fixed in 2% PFA overnight. Eyes were dissected, and RPE/choroidal tissues were blocked and permeabilized in 5% BSA with 0.3% Triton X‐100 in PBS for 2 h. For evaluation of CNV formation, the neovascular membrane was stained with biotin‐conjugated isolectin B4 (IB4; Sigma‐Aldrich, St Louis, MO, USA; L2140; 1:100), followed by incubation with Rhodamine Red‐X‐labelled streptavidin (Jackson Immuno Research Laboratories, West Grove, PA, USA; 016‐290‐084; 1:400). The CNV volume was measured using a series of Z‐stack images (from the surface to the deepest focal plane) using Volocity® Image Analysis Software 6.0. TMR Red‐dUTP TUNEL (Roche Diagnostics, Burgess Hill, UK) staining of DNA breaks was performed on 10 µm cryosections of enucleated eyes from treated mice, according to the manufacturer's protocol.
Histology and immunohistochemistry
Eyes from treated mice were enucleated at various time points and cryosections prepared. For immunostaining, sections were fixed in acetone and blocked in 5% donkey serum, 3% BSA, and 0.3% Triton X‐100 in PBS. Sections were incubated with the following primary antibodies – anti‐IL‐33 Nessy‐1 (ALX‐804‐840; Enzo Life Sciences, Ltd, Exeter, UK; 1:1000), ST2 (3363; ProSci Inc, Poway, CA, USA; 1:1000), Iba1 (L2140; Sigma, St Louis, MO, USA; 1:100), IL1RAP (ab8110; Abcam, Cambridge, MA, USA; 1:1000), c‐kit (105816; Biolegend, San Diego, CA, USA; 1:200), vimentin (ab92547; Abcam; 1:100) and tryptase (BS7353; bioWORLD, Dublin, OH, USA; 1:400) – overnight at 4°C. Sections were incubated with the appropriate secondary antibody (Life Technologies, Paisley, UK) donkey anti‐rabbit conjugated to AlexaFluor‐488 or AlexaFluor‐594, or donkey anti‐rat AlexaFluor‐594 (Life Technologies; dilution 1:400) or goat anti‐rabbit Cy2 (Abcam). DAPI (Vector Laboratories, Peterborough, UK) was used to show nuclei in sections.
Human ocular samples
Healthy adult (age 18–65 years) human post‐mortem eyes from anonymous donors collected by Moorfields Eye Bank for corneal transplantation were fixed in 10% formalin and embedded in paraffin wax. This work was ethically approved by the NHS Research Ethics Committee (10/H0106/57‐14ETR41).
Recombinant proteins
Recombinant mouse IL‐33 (ALX‐522‐101‐C010) and human IL‐33 (ALX‐522‐098‐C010) were purchased from Enzo Life Sciences Ltd (Exeter, UK). The truncated form of human IL‐33 was generated as described previously 22 and was a kind gift from Professor Seamus Martin (Trinity College Dublin).
Proliferation and functional assays
Cell numbers were assessed using an MTT assay, with absorbance measured at 570 nm. For migration assays, cells were seeded at 1 × 104 cells per culture‐insert and incubated overnight. Inserts were removed before addition of fresh medium with treatment or vehicle. Phase contrast images were taken at 0 h and 4 h post‐treatment using a wide‐field microscope (DM16000; Leica, Wetzlar, Germany). The whole area of gaps was quantified by ImageJ 1.46r (National Institutes of Health, USA). For Boyden chamber assays, cells were cultured in EBM medium with treatment or vehicle for 24 h before the supernatant was transferred to the lower chamber of a 24‐well format Transwell (Corning, UK) for 16 h incubation. The membranes of the upper chamber were fixed with 4% PFA for 20 min, followed by staining with DAPI for cell counting using confocal microscopy.
Cytokine measurements
IL‐6 levels were measured with an ELISA and following the manufacturer's instructions (550799; BD Biosciences, San Jose, CA, USA). For western blots, cells lysates were separated by SDS‐PAGE gel electrophoresis, and proteins were transferred to a nitrocellulose membrane. Following blocking in 5% milk–TBS–Tween‐20, the membrane was subjected to analysis using the anti‐IL‐33 Nessy‐1 antibody (ALX‐804‐840; Enzo Life Sciences; 1:1000), the ST2 antibody (3363; ProSci; 1:1000), and β‐actin antibody (4970; Cell Signaling Technology, Beverley, MA, USA; 1:1000).
Proteins were detected with HRP‐conjugated polyclonal anti‐mouse IgG and visualized using a chemiluminescent method (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, UK). RNA was extracted using an RNeasy Mini Kit (Qiagen, Hamburg, Germany) before cDNA synthesis using the ImProm‐IITM Reverse Transcription System (Promega, Fitchburg, WI, USA). cDNA was amplified using the Power SYBR® Green PCR Master Mix Reagent on a StepOne™ Applied Biosystems Real‐Time PCR System. Relative expression was calculated using Actb and RNA18S as reference transcripts for mouse and human tissues, respectively.
Statistical analysis
Statistical analysis was performed using an unpaired two‐tailed Student's t‐test or ANOVA as indicated for comparison between groups. p < 0.05 was considered significant.
Results
IL‐33 expression is induced in human and murine RPE cells
The retinal pigment epithelium (RPE) is central to retinal homeostasis. We first investigated the expression of IL‐33 and ST2 in established human and murine RPE cell lines 23, 24. Both human (ARPE‐19) and mouse (B6RPE‐07) RPE cells were stimulated with Toll‐like receptor (TLR) agonists, including TLR3 and TLR4 agonists [poly(I:C) and LPS], to simulate activation of the innate immune response and inflammasome 4. Our data show that murine and human RPE cells constitutively expressed IL‐33 and its cognate receptor ST2, at both mRNA and protein levels (Figure 1A–C). TLR activation of RPE cells increased expression of IL‐33 and ST2L (IL1RL1) mRNA in a dose‐dependent manner, although the effect of poly(I:C) was not as profound as LPS stimulation (Figure 1A,B). The effect of TLR agonists on IL‐33 and ST2 induction was confirmed at the protein level by western blot (Figure 1C). These results demonstrate that IL‐33 expression is up‐regulated in the RPE cells following an inflammatory stimulus.
Figure 1.
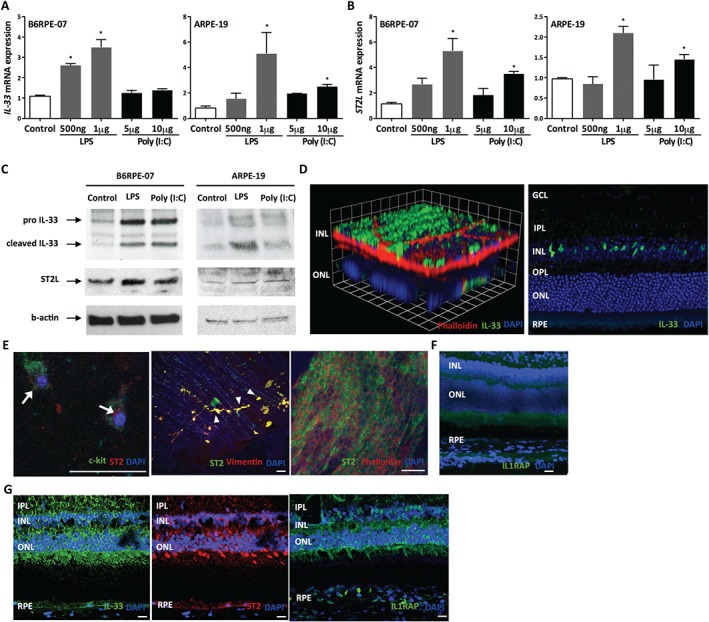
IL‐33 is expressed in murine and human retinas, and is induced in RPE. Expression of IL‐33 (A) and its receptor, ST2L (B), mRNA expression in retinal pigment epithelium (RPE) treated with TLR agonists [LPS, poly(I:C)] as indicated for 24 h (n = 4 per group). (C) Western blot analysis of IL‐33 and ST2 in mouse (B6RPE‐07) and human (ARPE‐19) RPE cells upon activation with TLR agonists. (D) Immunofluorescence staining showing expression of IL‐33 in adult murine retina. GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. Scale bar = 100 µm. (E) ST2 expression in choroidal mast cells (arrows), choroidal fibroblasts (arrowheads), and RPE. (F) Immunofluorescence staining showing expression of IL‐1 receptor accessory protein (IL1RAP) in murine retina. Scale bar =100 µm. (G) Representative images of immunofluorescence staining showing expression of IL‐33, ST2, and IL1RAP in sections of human retina. Scale bar = 100 µm. Data are shown as mean ± SEM. Data are representative of at least three independent experiments with similar results. *p < 0.05. Statistical analysis was performed with Student's t‐test.
IL‐33 and its receptor ST2 are expressed in murine and human retina
To establish expression patterns of IL‐33 and ST2 in vivo, we examined normal eye tissues from naïve C57BL/6J mice. Immunostaining of adult murine retina demonstrated that IL‐33 was expressed by cells in the inner nuclear layer (INL) and RPE, with expression localized to the nuclei of Müller glial cells and retinal microvascular endothelial cells, respectively (Figure 1D). In the developing mouse retina, there is a gradual but marked increase of IL‐33 expression from postnatal day 4 to postnatal day 10; the ciliary body was similarly enriched (see supplementary material, Figure S1A–C).
Immunostaining of choroid from WT animals demonstrated ST2 expression in RPE (phalloidin‐stained), as well as choroidal mast cells (c‐kit+) and choroidal fibroblasts (vimentin+) (Figure 1E). Furthermore, immunohistochemical analysis of murine retina demonstrated that IL‐1 receptor accessory protein (IL1RAP) was expressed by cells in the retina and RPE (Figure 1F). We next sought to determine whether similar expression profiles were present in healthy human ocular tissue. This revealed robust expression of IL‐33, ST2, and IL1RAP in the nerve fibre layer of the inner retina and the RPE (Figure 1G). Furthermore, IL‐33 is known to be stored within red blood cells 25, which may explain the strong IL‐33 staining within choroidal vessels (see supplementary material, Figure S2).
Retinal endothelial cells are a direct target of IL‐33
Although IL‐33 is expressed in human choroidal vascular endothelium, IL‐33 is down‐regulated at the onset of angiogenesis 26, suggesting a homeostatic role of IL‐33 regulation of tissue vasculature. To investigate further, primary human retinal microvascular endothelial cells (HRMECs) were cultured in the presence of IL‐33. First, we found that HRMECs responded to IL‐33, as evidenced by the increased production of downstream cytokine IL‐6, as a marker of their activation, when cultured with IL‐33 (Figure 2A). Furthermore, HRMECs expressed ST2 at high levels, compared with monocytes (Figure 2B), and soluble ST2 was present in the supernatant of HRMECs (see supplementary material, Figure S3A). More importantly, ST2 was almost completely absent in other tissue‐derived endothelial cells, such as human cerebral microvascular endothelial cells (HCMECs) (Figure 2B). As shown in Figure 2A,B, HCMECs do not express IL‐33 receptor and do not respond to IL‐33.
Figure 2.
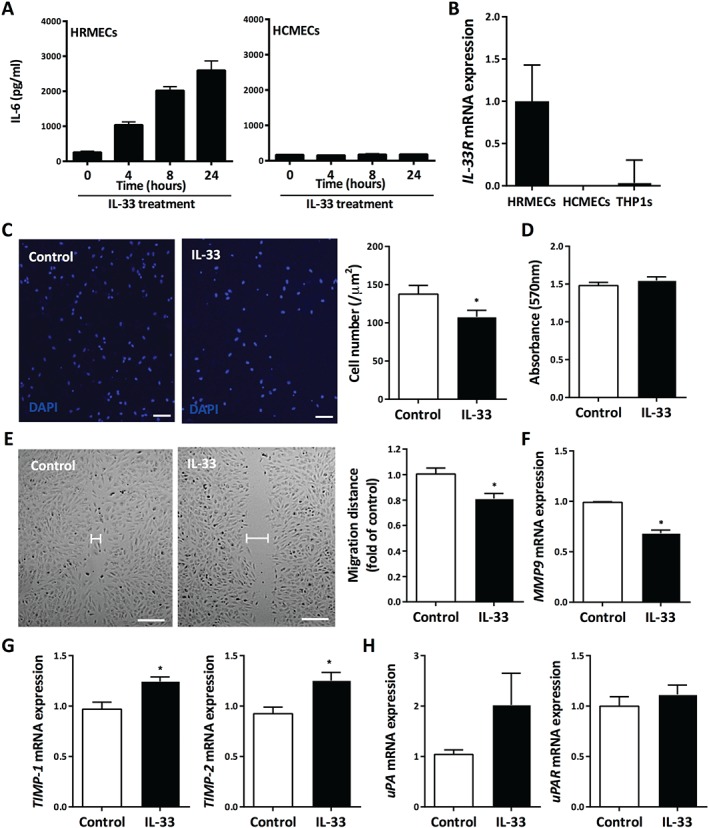
Retinal endothelial cells are a direct target of IL‐33. (A) Production of IL‐6 by HRMECs and HCMECs when treated with IL‐33 for varying amounts of time, measured by ELISA (n = 3 per group). (B) ST2L mRNA expression in HRMECs and HCMECs and human myeloid cell line THP1 (n = 3 per group). (C) The Boyden chamber assay was performed and chemotactic activity was quantified by blind counting of the migrating cells on the lower surface of the membrane in ten high‐power microscope fields per chamber using a × 100 objective. IL‐33 significantly inhibited the migratory ability of HRMECs (scale bar = 50 µm) (n = 4 per group). (D) The MTT assay showed that IL‐33 treatment had no effect on the proliferation of HRMECs (n = 3 per group). (E) IL‐33 inhibited the migration of HRMECs in a wound‐healing assay (n = 5 per group). Scale bar = 100 µm. (F) Matrix metalloproteinase‐9 (MMP9) mRNA expression in HRMECs in response to IL‐33 (n = 3 per group). (G) Expression of tissue inhibitors of metalloproteinases‐1 (TIMP1) and ‐2 (TIMP2) in HRMECs in response to IL‐33 (n = 3 per group). (H) Urokinase‐type plasminogen activator (uPA) and its receptor uPAR expression in HRMECs in response to IL‐33 (n = 3 per group). Data are shown as mean ± SEM. Data are representative of at least two independent experiments with similar results. *p < 0.05. Statistical analysis was performed with Student's t‐test.
Next, we investigated the effect of IL‐33 on the migratory ability of HRMECs. Using a modified Boyden chamber, the chemotactic motility of HRMECs was inhibited when incubated with recombinant human IL‐33, with the maximal effect at 100 ng/ml, although proliferative responses remained unaltered (Figure 2C,D and supplementary material, Figure S3B). The inhibitory effect elicited by IL‐33 treatment was further demonstrated in a wound‐healing assay, in which there was a significant decrease in the migratory ability of HRMECs (Figure 2E). We also assessed the production of matrix metalloproteinase (MMP)‐9, and found that HRMECs produced significantly lower levels of MMP9 in response to IL‐33 stimulation (Figure 2F). Furthermore, there was a small but significant increase in the expression of tissue inhibitors of metalloproteinases‐1 (TIMP1) and ‐2 (TIMP2) upon IL‐33 treatment of HRMECs (Figure 2G). There was no change in the expression of urokinase‐type plasminogen activator uPA (PLAU) or its receptor uPAR (PLAUR) (Figure 2H), or of vascular endothelial growth factor receptor (VEGFR)‐1 (FLT1) or VEGFR2 (KDR) in response to IL‐33 (see supplementary material, Figure S3C–E).
IL‐33 regulates the function of human choroidal fibroblasts
Alteration in choroidal architecture including vascular changes and choroidal stromal thinning accompanying fibrosis is associated with the RPE atrophy and photoreceptor death as hallmarks of AMD 27. As choroidal fibroblasts express ST2, we wished to determine if IL‐33 could attenuate responses as observed with its anti‐fibrotic and anti‐hypertrophic effects in the myocardium 28. Experiments using human choroidal fibroblasts demonstrated that cellular migration was significantly inhibited when incubated with IL‐33 (Figure 3A). Collagen gel contraction assays have been widely used to evaluate the interaction of fibroblasts with a collagenous matrix and their ability to remodel this matrix 29, 30, 31. Treatment of human choroidal fibroblasts with IL‐33 significantly inhibited their ability to contract collagen gels by approximately 25% over the 12 h period examined, with no effect on their proliferation (Figure 3B,C). A number of factors including MMPs 32 are recognized to enhance the migration and contraction of three‐dimensional collagen gels by fibroblasts. In response to IL‐33, the human choroidal fibroblasts expressed significantly lower levels of both MMP2 and MMP9 (Figure 3D,E). Furthermore, we found an increase in TIMP1 and TIMP2 expression upon IL‐33 stimulation of choroidal fibroblasts (Figure 3F), while there was no change in the expression of uPA (PLAU) or uPAR (PLAUR) (Figure 3G).
Figure 3.
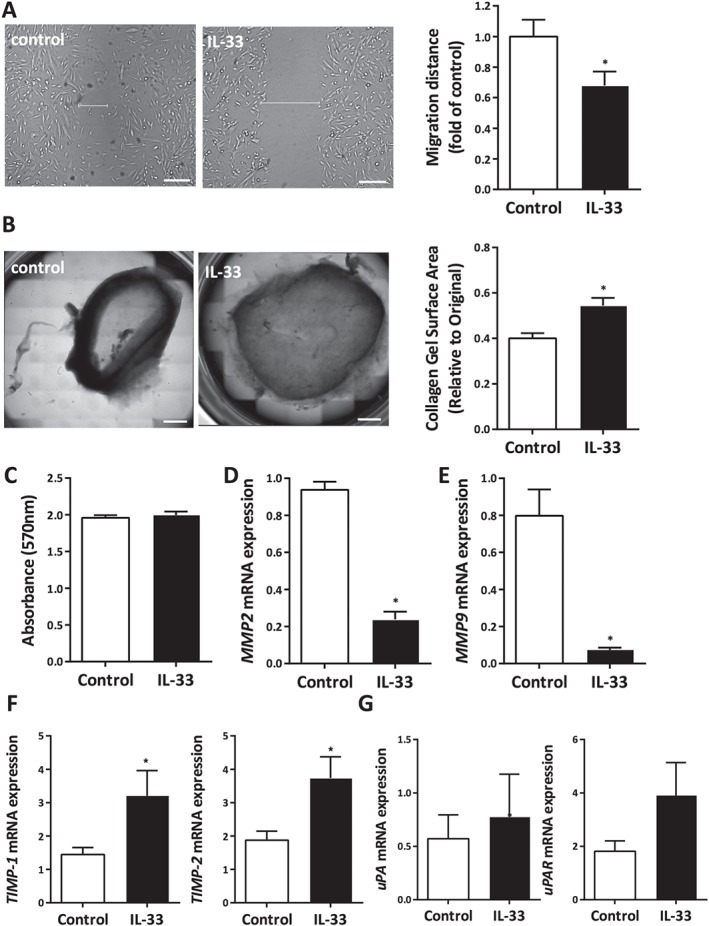
IL‐33 regulates the function of human choroidal fibroblasts. IL‐33 inhibited the ability of human choroidal fibroblasts to (A) migrate in a wound‐healing assay and (B) contract collagen gel (scale bar = 100 µm). (C) The MTT assay showed that IL‐33 treatment had no effect on their proliferation. (D) MMP2 and (E) MMP9 mRNA expression in choroidal fibroblasts upon treatment with IL‐33. (F) TIMP1 and TIMP2 expression in choroidal fibroblasts in response to IL‐33 (n = 3 per group). (G) Expression of uPA and its receptor uPAR in choroidal fibroblasts in response to IL‐33 (n = 3 per group). Data are shown as mean ± SEM. Data are representative of three independent experiments with similar results. *p < 0.05. Statistical analysis was performed with Student's t‐test.
Intravitreal administration of IL‐33 attenuates CNV
To investigate a functional role of IL‐33, we explored its action in angiogenesis in the context of nAMD using the murine model of laser‐induced choroidal neovascularization (CNV). Groups of mice received an intravitreal injection of recombinant mouse IL‐33 immediately following laser‐induced CNV. Seven days post‐laser exposure, in vivo optical coherence tomography (OCT) assessment and ex vivo isolectin‐B4 (IB4) staining of RPE‐choroidal flat mounts demonstrated that CNV volume was significantly reduced in eyes receiving IL‐33 compared with the vehicle control (Figure 4A,B). Furthermore, IB4 staining revealed that two lower doses of IL‐33 (200 pg/µl and 1 ng/µl) were more effective at attenuating CNV development, while the higher dose of 5 ng/µl IL‐33 had no significant effect. This effect is similar to what has been observed previously in a primate model of immunotherapy with AMD 33. Intravitreal IL‐33 at the dose of 200 pg/µl decreased choroidal neovascular lesion (CNV) volume by up to 50% (Figure 4B,C). This observation was manifest in reduced choroidal neovascular lesions and disease severity (sub‐retinal fluid) in IL‐33‐treated eyes, as demonstrated in OCT images (Figure 4A). Furthermore, the reduction in clinical disease was accompanied by significantly decreased c‐kit+ cells, which demonstrated a potentially reduced population of endothelial progenitor cells in the lesions in IL‐33‐treated eyes compared with control eyes (see supplementary material, Figure S4A,B). Resident immune cells of the choroid include macrophages and mast cells 34. In addition to the monocyte/macrophage cell infiltrate following laser treatment, we found that tryptase‐positive mast cells had infiltrated CNV lesions (see supplementary material, Figure S4C).
Figure 4.
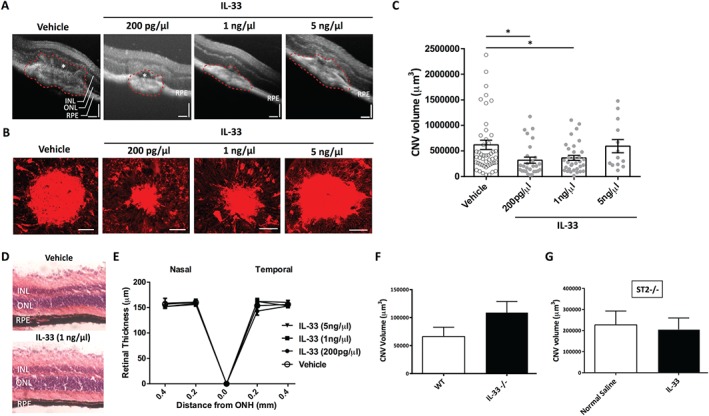
Intravitreal administration of IL‐33 attenuates CNV in an ST2‐dependent manner. (A) Representative OCT images showing choroidal neovascular lesions in control and IL‐33‐treated eyes. INL = inner nuclear layer; ONL = outer nuclear layer; RPE = retina pigment epithelium. The white asterisk denotes the sub‐retinal fluid. Scale bar = 200 µm. (B) Fluorescence IB4 staining of choroidal neovascular (CNV) lesions in all treatment groups. Images are representative of CNV volume in each experimental group. Scale bar = 75 µm. (C) Quantitative analysis of the volume of CNV lesions showed that the two lower doses of IL‐33 significantly attenuated CNV development. IL‐33 did not affect the integrity and thickness of the retina, as shown in histological sections (D) and OCT analysis (E) of eyes that had only intravitreal injection of IL‐33. ONH = optic nerve head. Data are representative of two measurements per retina with four eyes per group. (F) Il33−/− mice had more pronounced lesions but not to a statistically significant level. (G) IL‐33 treatment did not affect the severity of CNV development in St2−/− mice (n = 10 eyes per group). Data are shown as mean ± SEM. Data are representative of at least three independent experiments with similar results. *p < 0.05. Statistical analysis was performed with ANOVA with post‐hoc t‐test.
A recent report has suggested that endogenous release of IL‐33 triggers the recruitment of monocytes contributing to photoreceptor loss 18. To examine this, RPE/choroidal tissue was collected at 3 days post‐laser exposure and intravitreal IL‐33 injection, and immunostained for Iba1, a marker for both microglia and monocytes. In the laser‐induced CNV model, macrophage numbers peak on day 3 following laser damage 35. Our results showed that Iba1+ cell accumulation was no different between vehicle and IL‐33‐treated eyes. Furthermore, a 500‐fold increase to the therapeutic dose of IL‐33 did not influence Iba1+ cell accumulation in RPE/choroid (see supplementary material, Figure S5A). In addition, monocyte recruitment was assessed via flow cytometric analysis of the cell infiltrate from the retinas of mice receiving either the low (200 pg/l) or the high (100 ng/l) doses of IL‐33. IL‐33 treatment did not lead to any significant increase of CD45+/CD11b+/Ly6C+ cells (see supplementary material, Figure S5B).
To examine the safety and tolerability of recombinant IL‐33, groups of mice receiving varying doses of IL‐33 were clinically monitored using OCT at 3, 5, and 7 days post‐injection. Histological and OCT analyses of the retina at day 7 indicated that IL‐33 administration did not alter retinal integrity and thickness, or result in cell death (Figure 4D,E and supplementary material, Figure S6A,B). Exogenous administration of recombinant IL‐33 had no effect on retinal pigment epithelial (RPE) cell viability in vitro (see supplementary material, Figure S6C).
ST2 is required for IL‐33 anti‐angiogenic properties
As exogenous IL‐33 suppresses angiogenesis, we asked if endogenous IL‐33 is protective. To this end, we noted that choroidal lesion volume measurements were more pronounced in Il33−/− mice (Figure 4F). These results open a plausible explanation that endogenous IL‐33 expression may regulate but not significantly attenuate CNV development in this model. To confirm that the IL‐33 effect was direct, we noted that IL‐33 treatment did not alter the severity of CNV development in St2−/− mice (Figure 4G).
A truncated form of human IL‐33 is biologically active in mice
As mast cells are activated by IL‐33 15, murine bone marrow‐derived mast cells (BMMCs) were treated with mouse and human IL‐33, and IL‐6 and IL‐1β mRNA levels were measured to compare their potency. We found that BMMCs responded similarly to both forms of IL‐33 (Figure 5A), suggesting that human IL‐33 is bioactive in mice.
Figure 5.
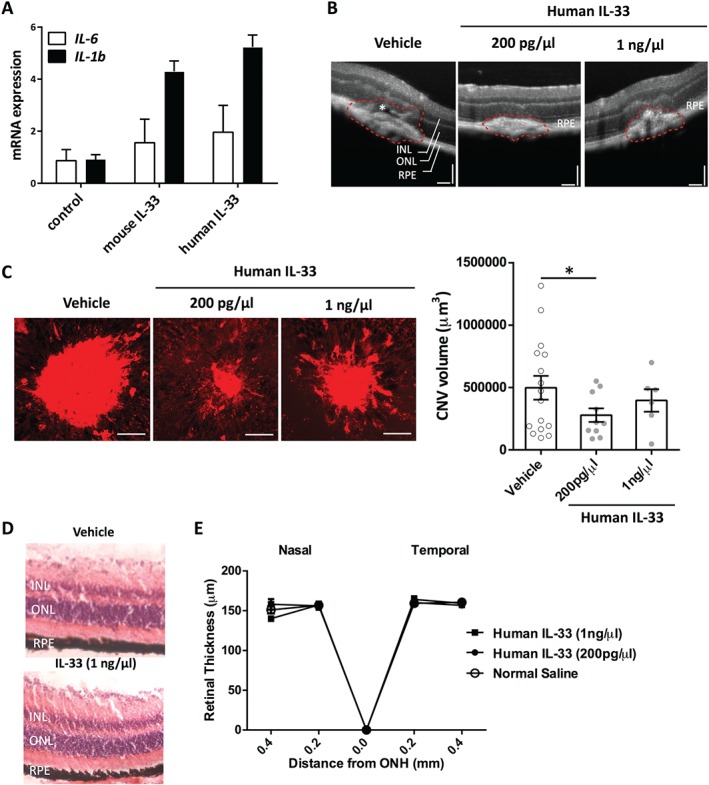
A truncated form of human IL‐33 is biologically active in mice. (A) We used a truncated form of human IL‐33, which is bioactive in mice, as shown by IL‐6 and IL‐1β mRNA levels produced by bone marrow‐derived mast cells (BMMCs) upon treatment with mouse and human IL‐33. (B) Representative images of OCT showing choroidal neovascular lesions in control and human IL‐33‐treated eyes. INL = inner nuclear layer; ONL = outer nuclear layer; RPE = retina pigment epithelium. The white asterisk denotes the sub‐retinal fluid. Scale bar = 200 µm. (C) Immunofluorescence IB4 staining and quantitative analysis of choroidal neovascular lesions in all treatment groups with normal saline or human IL‐33. Images are representative of CNV volume in each experimental group. Scale bar = 75 µm. (D) Representative images of histology in control and eyes treated only with intravitreal injections (n = 4 eyes per group). (E) The truncated form of human IL‐33 did not affect the thickness of the retina, as measured by OCT in eyes that had intravitreal injection of IL‐33. ONH = optic nerve head. Data are representative of two measurements per retina with four eyes per group. Data are shown as mean ± SEM and are representative of at least two independent experiments with similar results. *p < 0.05. Statistical analysis was performed with ANOVA with post‐hoc t‐test.
In vivo, administration of recombinant human IL‐33 reduced angiogenesis, with the lower dose of 200 pg/µl being the most effective (Figure 5B,C). Treatment with human IL‐33 had no effect on human RPE cell viability in vitro (see supplementary material, Figure S6D). Intravitreal administration did not induce cell death or alter retinal integrity or thickness (Figure 5D,E and supplementary material, Figure S6E,F).
Discussion
Our data demonstrate a protective role for IL‐33 in a murine model of injury‐induced CNV. IL‐33 is expressed in the human retina and RPE, and IL‐33 and its cognate receptor are selectively expressed in primary human retinal microvascular endothelial cells. Local administration of recombinant IL‐33 protected against the development of CNV, supporting its immunomodulatory therapeutic potential for nAMD 6, 33, 36. IL‐33 suppression of CNV does not support a primary VEGF dependency because VEGFR levels remained unchanged, but instead supports a direct inhibition of ST2‐positive fibroblasts and endothelial cells.
Current anti‐VEGF therapies arguably do not address major aspects of the underlying pathophysiology, such as circumventing inflammation and wound healing leading to angiogenesis. Furthermore, VEGF's role as a neuronal survival factor cannot be overlooked, underscored by the observation that in patients receiving long‐term anti‐VEGF therapy the neural retina can begin to degenerate 2. These data and others' question the current development of therapeutic targets redressing either complement dysregulation or inhibiting the assumed negative impact of chronic low‐grade inflammation such as inflammasome activation 37. The identification of an anti‐angiogenic role of IL‐33 in CNV extends current understanding of the functional diversity of members of the IL‐1 family in angiogenesis. For example, IL‐1 receptor antagonist treatment significantly inhibits CNV, as there is a direct effect of IL‐1β on choroidal endothelial cell proliferation 38. On the other hand, IL‐18 production following inflammasome activation suppresses CNV and down‐regulates VEGFR signalling 36. IL‐33 treatments directly inhibit the migration of retinal endothelial cells and choroidal fibroblasts without down‐regulation of VEGFR and appear to be potent in reducing angiogenic responses. Conceivably, IL‐33 may synergize with IL‐18 and further amplify an anti‐angiogenic response in both early and late phases of the disease. IL‐33 is bioactive without requiring the NLRP3 inflammasome and caspase cleavage 8. Inflammatory cues strongly induce expression of endogenous IL‐33 39. Indeed, whether IL‐33 is also liberated following intravitreal IL‐4 administration directing a macrophage‐mediated suppression of CNV warrants further investigation 40.
Supporting the diverse constitutive tissue expression of IL‐33 7, 41, the current data show IL‐33 and ST2 expression in human and murine RPE. Many RPE‐derived molecules are central to regulating and maintaining local immune privilege in the eye 42. To support a regulatory behaviour, IL‐33 is constitutively expressed by cells in the inner nuclear layer (INL) of the retina, displaying eye‐specific homeostatic expression similar to others' data 18. Although IL‐33 is released from Müller cells in vitro and in vivo after phototoxic stress 18, and the effect is detrimental, we have shown TLR‐induced up‐regulation of IL‐33 in RPE cells without cell death. This likely represents an attempt to maintain homeostasis and tissue health. In support of our observation, Kakkar et al have reported that IL‐33 is localized simultaneously to both nuclear euchromatin and membrane‐bound cytoplasmic vesicles, and have demonstrated that mechanical strain induced IL‐33 secretion in the absence of cellular necrosis in murine fibroblasts 10. We speculate a similar dynamic inter‐organelle trafficking of IL‐33 in RPE under stress, which induces increased membrane‐bound cytoplasmic vesicles and subsequent IL‐33 release without cell death.
Endogenous intracellular IL‐33 does not alter the development of angiogenesis in the laser‐CNV model. The lack of effect with loss of endogenous IL‐33 can be explained by the low level of its basal expression in normal tissue. We speculate that IL‐33 acts as an in‐built brake and that under stress conditions, as shown in vitro (Figure 1), IL‐33 is up‐regulated, regulating angiogenesis. To support this notion, exogenous recombinant IL‐33 suppresses CNV. Further, we found that the two lower doses of murine IL‐33 were more efficient at reducing CNV development. We speculate that higher doses of IL‐33 up‐regulate ST2 expression, neutralizing IL‐33 activity. Such a possibility is consistent with our in vitro data demonstrating IL‐33‐induced enhancement of the IL‐33/ST2 axis in the RPE. Furthermore, we found no adverse effect of exogenous application of IL‐33, as reported previously 18. Injection of 500‐fold higher than the therapeutic dose of recombinant IL‐33 did not induce cell death or increased monocyte recruitment (see supplementary material, Figure S5).
IL‐33 exhibits unique features and represents a guardian of barriers and a local alarmin 43. In an intact barrier, cellular contacts provide the signals to produce full‐length IL‐33 ‘constitutively’ but neither process nor release it. In these barrier cells, full‐length IL‐33 resides in the nucleus, where it controls gene expression and is possibly involved in maintaining the cells' resting state 43. During injury, some of the barrier cells are destroyed and full‐length IL‐33 is liberated. Depending on the location of the barrier and the cause of perturbation, the bioactivity and effect of extracellular IL‐33 will be different. Although triggers differ, the basic principles of alarming resident sentinel cells via IL‐33 may yet apply. Extracellular IL‐33 coordinates immune defence and repair mechanisms, and elucidating the local effects of IL‐33 enables a rationally based approach to developing pharmacological intervention strategies to target the IL‐33/ST2 axis. IL‐33 is expressed in multiple endothelial cells 26, 28, and exogenous administration of IL‐33 induces the expression of adhesion molecules and inflammatory cytokines 44. The presence of mast cells within CNV lesions infers the possibility of additional sources of IL‐33 and the ability to rapidly process IL‐33 delivering enhanced biological activity, augmenting local immune responses 43. On the other hand, IL‐33 has anti‐hypertrophic and anti‐fibrotic effects 45. Despite data indicating that IL‐33 promotes angiogenesis 46, we believe that this is tissue context‐dependent, in that endothelial IL‐33 receptor expression has tissue specificity. Human retinal endothelial cells express ST2 at high levels, compared with monocytes, and ST2 is almost completely absent in human brain endothelial cells (Figure 2B) which do not respond to IL‐33 treatment. Cytokine pleiotropism is supported because IL‐33 exacerbates autoimmune collagen‐induced arthritis via mast cell degranulation 20, but IL‐33 elicits high levels of IL‐5 and IL‐13 45, driving M2 macrophages and protecting against EAU 17 and atherosclerosis 45. IL‐33 prevents cognitive decline in mouse models of Alzheimer's disease 47 and inhibits atherosclerosis 45, and here IL‐33 also directly inhibited choroidal fibroblast activity, implicating a potentially wider role clinically. These actions add to the increasing evidence and support for immunotherapeutic mediated adjuncts to aid the current treatment paradigm for patients suffering from nAMD 48.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary materials and methods
Supplementary figure legends
Figure S1. Expression pattern and localization of IL‐33 in developing retina
Figure S2. Expression of IL‐33 in adult human ocular tissue
Figure S3. Effect of IL‐33 on retinal endothelial cells
Figure S4. Reduced population of endothelial progenitor cells in IL‐33 eyes with CNV
Figure S5. Recombinant IL‐33 does not increase monocyte recruitment in retina
Figure S6. Safety and tolerability of IL‐33
Supporting information
Supplementary materials and methods
Supplementary figure legends
Figure S1. Expression pattern and localization of IL‐33 in developing retina. (A) P0, P4, P7, P11, P17, and P18 murine retinal sections stained for IL‐33 were analysed under the fluorescence microscope using a 40× objective. IL‐33 is stained green (secondary goat IgG Cy2) and nuclei are stained blue (DAPI). ONL = outer nuclear layer; INL = inner nuclear layer; GCL = ganglion cell layer. Scale bar for images = 100 µm. (B) In the developing mouse retina of P0, P10, P11, P12, P13, and P14, pro‐IL‐33 was detected at 30–32 kDa by western blot analysis. A marked increase in the expression levels of pro‐IL‐33 can be seen from P0 through to P12. IL‐33 expression increases further at P13 and expression levels remain constant from this time point onwards. A lower molecular weight form of IL‐33 was detected at 17 kDa and appears to be differentially regulated. C1 (control 1) represents a P8 cerebrum cell lysate control. β‐Actin was used as a loading control and can be detected equally at 43 kDa in each retinal lysate. (C) Fluorescence microscopy analysis of the expression pattern of IL‐33 was performed in the ciliary margins of the developing retina/eye. P0, P4, P7, P10, P14, and P18 murine retinal sections stained for IL‐33 were analysed under the fluorescence microscope using a 20× objective. IL‐33 is stained green (secondary goat IgG Cy2) and nuclei are stained blue (DAPI). Scale bar for P0 and P4 = 60 µm. Scale bar for P7–P18= 100 µm.
Figure S2. Expression of IL‐33 in adult human ocular tissue. (A) Immunofluorescence staining showing expression of IL‐33, ST2, and IL1RAP in adult human choroid. IL‐33, ST2, and IL1RAP are expressed in RPE and within choroidal vessels (arrows). RPE = retinal pigment epithelium. (B) Human ciliary body was also enriched in IL‐33 and ST2. (C) Negative control for staining of IL‐33, ST2, and IL1RAP (secondary IgG only). GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. Nuclei are stained with DAPI. Scale bar = 100 µm.
Figure S3. Effect of IL‐33 on retinal endothelial cells. (A) Soluble ST2 was present in the supernatant of HRMECs and did not vary between control and IL‐33‐stimulated cells. (B) The Boyden chamber assay was performed and chemotactic activity was quantified by blind counting of the migrating cells on the lower surface of the membrane in ten high‐power microscope fields per chamber using a ×100 objective. IL‐33 significantly inhibited the migratory ability of HRMECs in a dose‐dependent manner (scale bar = 50 µm) (n = 4 per group). (C) IL‐33 at a range of doses did not affect the expression of vascular endothelial growth factor receptor (VEGFR) 2, measured by western blot analysis. (D, E) Expression of VEGFR1 (D) and VEGFR2 (E) by HRMECs when treated with IL‐33 for varying amounts of time, measured by RT‐PCR (n = 3 per group). Data are shown as mean ± SEM. Data are representative of at least two independent experiments with similar results. *p < 0.05. Statistical analysis was performed with Student's t‐test.
Figure S4. Reduced population of endothelial progenitor cells in IL‐33 eyes with CNV. (A) c‐kit staining of choroidal neovascular (CNV) lesion in control and treatment groups. Images are representative of CNV volume in each experimental group. Scale bar = 75 µm. (B) Quantitative analysis of c‐kit+ cells in the area of each choroidal neovascular lesion showed significantly decreased c‐kit+ cells in the two lower doses of IL‐33 treatment groups (n = 15–20 eyes per group). (C) Tryptase staining of CNV lesion in control and treatment (IL‐33 200 pg/µl) groups showed that mast cells are present in the CNV lesion. Scale bar = 75 µm. Data are shown as mean ± SEM. Data are representative of at least two independent experiments with similar results. *p < 0.05. Statistical analysis was performed with Student's t‐test.
Figure S5. Recombinant IL‐33 does not increase monocyte recruitment in retina. (A) Confocal images and semi‐quantitative mean fluorescence intensity (MFI) analysis using ImageJ software for Iba1+ cell accumulation on RPE/choroid whole‐mounts taken on day 3 post‐laser and treatment (n ≥ 8 per group). There was no significant difference in monocyte recruitments between treatment groups and control group. (B) Flow cytometric analysis of retina shows no increase in CD45+/CD11b+ cell infiltration on day 3 post‐IL‐33 treatment compared with control (n = 4 per group). Data are shown as mean ± SEM.
Figure S6. Safety and tolerability of IL‐33. (A) Mouse recombinant IL‐33 did not affect the thickness of the retina, as measured by OCT at days 3 and 5 in eyes that had intravitreal injection of IL‐33. (B) Confocal microscopic analysis did not show TUNEL‐positive apoptotic cells. INL= inner nuclear layer; ONL = outer nuclear layer. The MTT assay showed that IL‐33 at a range of doses had no effect on the viability of mouse (B6RPE‐07) (C) and human (ARPE‐19) (D) RPE cells. Phase contrast microscopy of cells treated with IL‐33 showed a healthy RPE appearance. (E, F) The truncated form of human IL‐33 did not affect the thickness of the retina, as measured by OCT at days 3 and 5 in eyes that had intravitreal injection of IL‐33, and did not induce apoptosis. ONH = optic nerve head. Data are representative of two measurements per retina with four eyes per group. Data are shown as mean ± SEM. Data are representative of at least two independent experiments with similar results. Statistical analysis was performed with Student's t‐test.
Acknowledgements
Research was funded through the National Eye Research Centre, UK (RJ6056). ADD and ST were supported in part through the National Institute for Health Research, UK. Additional support was received from the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and University College London Institute of Ophthalmology. SD is supported by grants from Science Foundation Ireland (SFI), The Health Research Board of Ireland (HRB), and the Bright Focus Foundation. MC was supported by grants from Science Foundation Ireland (SFI), The Health Research Board of Ireland (HRB) and the Bright Focus Foundation. The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR or the Department of Health. We would like to thank Costanza Emanueli (School of Clinical Sciences, Bristol Heart Institute, University of Bristol, Bristol, UK) for providing the ST2 knock‐out (St2−/−) C57BL/6 mice. Additionally, we would like to thank Professor Seamus Martin and Dr Graeme Sullivan for providing human recombinant IL‐33. We would also like to acknowledge the assistance of the Flow Cytometry Facility and the Wolfson Bioimaging Facility at the University of Bristol.
Author contribution statement
ADD conceived the project and supervised all research. ST, DAC, JL, JW, PJG, EO, and MC performed research. ST, DAC, SLD, and MC analysed data. ST, DAC, SLD, MC, and ADD wrote the manuscript.
No conflicts of interest were declared
References
- 1. Rein DB, Wittenborn JS, Zhang X, et al. Forecasting age‐related macular degeneration through the year 2050: the potential impact of new treatments. Arch Ophthalmol 2009; 127: 533–540. [DOI] [PubMed] [Google Scholar]
- 2. Xu L, Mrejen S, Jung JJ, et al. Geographic atrophy in patients receiving anti‐vascular endothelial growth factor for neovascular age‐related macular degeneration. Retina 2015; 35: 176–186. [DOI] [PubMed] [Google Scholar]
- 3. Nussenblatt RB, Lee RW, Chew E, et al. Immune responses in age‐related macular degeneration and a possible long‐term therapeutic strategy for prevention. Am J Ophthalmol 2014; 158: 5–11.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lamkanfi M. Emerging inflammasome effector mechanisms. Nature Rev Immunol 2011; 11: 213–220. [DOI] [PubMed] [Google Scholar]
- 5. Xu H, Chen M, Forrester JV. Para‐inflammation in the aging retina. Prog Retin Eye Res 2009; 28: 348–368. [DOI] [PubMed] [Google Scholar]
- 6. Doyle SL, Ozaki E, Brennan K, et al. IL‐18 attenuates experimental choroidal neovascularization as a potential therapy for wet age‐related macular degeneration. Sci Transl Med 2014; 6: 230ra244. [DOI] [PubMed] [Google Scholar]
- 7. Schmitz J, Owyang A, Oldham E, et al. IL‐33, an interleukin‐1‐like cytokine that signals via the IL‐1 receptor‐related protein ST2 and induces T helper type 2‐associated cytokines. Immunity 2005; 23: 479–490. [DOI] [PubMed] [Google Scholar]
- 8. Cayrol C, Girard JP. The IL‐1‐like cytokine IL‐33 is inactivated after maturation by caspase‐1. Proc Natl Acad Sci U S A 2009; 106: 9021–9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lefrancais E, Roga S, Gautier V, et al. IL‐33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A 2012; 109: 1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kakkar R, Hei H, Dobner S, et al. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem 2012; 287: 6941–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lamkanfi M, Dixit VM. IL‐33 raises alarm. Immunity 2009; 31: 5–7. [DOI] [PubMed] [Google Scholar]
- 12. Chackerian AA, Oldham ER, Murphy EE, et al. IL‐1 receptor accessory protein and ST2 comprise the IL‐33 receptor complex. J Immunol 2007; 179: 2551–2555. [DOI] [PubMed] [Google Scholar]
- 13. Yanagisawa K, Tsukamoto T, Takagi T, et al. Murine ST2 gene is a member of the primary response gene family induced by growth factors. FEBS Lett 1992; 302: 51–53. [DOI] [PubMed] [Google Scholar]
- 14. Xu D, Chan WL, Leung BP, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med 1998; 187: 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allakhverdi Z, Smith DE, Comeau MR, et al. Cutting edge: the ST2 ligand IL‐33 potently activates and drives maturation of human mast cells. J Immunol 2007; 179: 2051–2054. [DOI] [PubMed] [Google Scholar]
- 16. Leung BP, Xu D, Culshaw S, et al. A novel therapy of murine collagen‐induced arthritis with soluble T1/ST2. J Immunol 2004; 173: 145–150. [DOI] [PubMed] [Google Scholar]
- 17. Barbour M, Allan D, Xu H, et al. IL‐33 attenuates the development of experimental autoimmune uveitis. Eur J Immunol 2014; 44: 3320–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xi H, Katschke KJ Jr, Li Y, et al. IL‐33 amplifies an innate immune response in the degenerating retina. J Exp Med 2016; 213: 189–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liew FY, Pitman NI, McInnes IB. Disease‐associated functions of IL‐33: the new kid in the IL‐1 family. Nature Rev Immunol 2010; 10: 103–110. [DOI] [PubMed] [Google Scholar]
- 20. Xu D, Jiang HR, Kewin P, et al. IL‐33 exacerbates antigen‐induced arthritis by activating mast cells. Proc Natl Acad Sci U S A 2008; 105: 10913–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oboki K, Ohno T, Kajiwara N, et al. IL‐33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A 2010; 107: 18581–18586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin‐33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009; 31: 84–98. [DOI] [PubMed] [Google Scholar]
- 23. Chen M, Muckersie E, Robertson M, et al. Characterization of a spontaneous mouse retinal pigment epithelial cell line B6‐RPE07. Invest Ophthalmol Vis Sci 2008; 49: 3699–3706. [DOI] [PubMed] [Google Scholar]
- 24. Dunn KC, Aotaki‐Keen AE, Putkey FR, et al. ARPE‐19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res 1996; 62: 155–169. [DOI] [PubMed] [Google Scholar]
- 25. Wei J, Zhao J, Schrott V, et al. Red blood cells store and release interleukin‐33. J Investig Med 2015; 63: 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuchler AM, Pollheimer J, Balogh J, et al. Nuclear interleukin‐33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol 2008; 173: 1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Friedlander M. Fibrosis and diseases of the eye. J Clin Invest 2007; 117: 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen WY, Hong J, Gannon J, et al. Myocardial pressure overload induces systemic inflammation through endothelial cell IL‐33. Proc Natl Acad Sci U S A 2015; 112: 7249–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bell E, Ivarsson B, Merrill C. Production of a tissue‐like structure by contraction of collagen lattices by human fibroblasts of different proliferative potential in vitro . Proc Natl Acad Sci U S A 1979; 76: 1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guidry C, Grinnell F. Contraction of hydrated collagen gels by fibroblasts: evidence for two mechanisms by which collagen fibrils are stabilized. Coll Relat Res 1987; 6: 515–529. [DOI] [PubMed] [Google Scholar]
- 31. Carver W, Molano I, Reaves TA, et al. Role of the alpha 1 beta 1 integrin complex in collagen gel contraction in vitro by fibroblasts. J Cell Physiol 1995; 165: 425–437. [DOI] [PubMed] [Google Scholar]
- 32. Daniels JT, Cambrey AD, Occleston NL, et al. Matrix metalloproteinase inhibition modulates fibroblast‐mediated matrix contraction and collagen production in vitro . Invest Ophthalmol Vis Sci 2003; 44: 1104–1110. [DOI] [PubMed] [Google Scholar]
- 33. Doyle SL, Lopez FJ, Celkova L, et al. IL‐18 immunotherapy for neovascular AMD: tolerability and efficacy in nonhuman primates. Invest Ophthalmol Vis Sci 2015; 56: 5424–5430. [DOI] [PubMed] [Google Scholar]
- 34. McMenamin PG. The distribution of immune cells in the uveal tract of the normal eye. Eye (Lond) 1997; 11: 183–193. [DOI] [PubMed] [Google Scholar]
- 35. Liu J, Copland DA, Horie S, et al. Myeloid cells expressing VEGF and arginase‐1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PLoS One 2013; 8: e72935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Campbell M, Doyle S, Humphries P. IL‐18: a new player in immunotherapy for age‐related macular degeneration? Expert Rev Clin Immunol 2014; 10: 1273–1275. [DOI] [PubMed] [Google Scholar]
- 37. Doyle SL, Campbell M, Ozaki E, et al. NLRP3 has a protective role in age‐related macular degeneration through the induction of IL‐18 by drusen components. Nature Med 2012; 18: 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lavalette S, Raoul W, Houssier M, et al. Interleukin‐1β inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol 2011; 178: 2416–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pichery M, Mirey E, Mercier P, et al. Endogenous IL‐33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il‐33‐LacZ gene trap reporter strain. J Immunol 2012; 188: 3488–3495. [DOI] [PubMed] [Google Scholar]
- 40. Wu WK, Georgiadis A, Copland DA, et al. IL‐4 regulates specific Arg‐1(+) macrophage sFlt‐1‐mediated inhibition of angiogenesis. Am J Pathol 2015; 185: 2324–2335. [DOI] [PubMed] [Google Scholar]
- 41. Carriere V, Roussel L, Ortega N, et al. IL‐33, the IL‐1‐like cytokine ligand for ST2 receptor, is a chromatin‐associated nuclear factor in vivo . Proc Natl Acad Sci U S A 2007; 104: 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Forrester JV, Xu H, Lambe T, et al. Immune privilege or privileged immunity? Mucosal Immunol 2008; 1: 372–381. [DOI] [PubMed] [Google Scholar]
- 43. Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nature Immunol 2016; 17: 122–131. [DOI] [PubMed] [Google Scholar]
- 44. Demyanets S, Konya V, Kastl SP, et al. Interleukin‐33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol 2011; 31: 2080–2089. [DOI] [PubMed] [Google Scholar]
- 45. Miller AM, Xu D, Asquith DL, et al. IL‐33 reduces the development of atherosclerosis. J Exp Med 2008; 205: 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Choi YS, Choi HJ, Min JK, et al. Interleukin‐33 induces angiogenesis and vascular permeability through ST2/TRAF6‐mediated endothelial nitric oxide production. Blood 2009; 114: 3117–3126. [DOI] [PubMed] [Google Scholar]
- 47. Fu AK, Hung KW, Yuen MY, et al. IL‐33 ameliorates Alzheimer's disease‐like pathology and cognitive decline. Proc Natl Acad Sci U S A 2016; 113: E2705–E2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dick AD. Enhancing inflammation as an adjuvant to neovascular AMD therapy. Invest Ophthalmol Vis Sci 2015; 56: 5431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Supplementary figure legends
Figure S1. Expression pattern and localization of IL‐33 in developing retina. (A) P0, P4, P7, P11, P17, and P18 murine retinal sections stained for IL‐33 were analysed under the fluorescence microscope using a 40× objective. IL‐33 is stained green (secondary goat IgG Cy2) and nuclei are stained blue (DAPI). ONL = outer nuclear layer; INL = inner nuclear layer; GCL = ganglion cell layer. Scale bar for images = 100 µm. (B) In the developing mouse retina of P0, P10, P11, P12, P13, and P14, pro‐IL‐33 was detected at 30–32 kDa by western blot analysis. A marked increase in the expression levels of pro‐IL‐33 can be seen from P0 through to P12. IL‐33 expression increases further at P13 and expression levels remain constant from this time point onwards. A lower molecular weight form of IL‐33 was detected at 17 kDa and appears to be differentially regulated. C1 (control 1) represents a P8 cerebrum cell lysate control. β‐Actin was used as a loading control and can be detected equally at 43 kDa in each retinal lysate. (C) Fluorescence microscopy analysis of the expression pattern of IL‐33 was performed in the ciliary margins of the developing retina/eye. P0, P4, P7, P10, P14, and P18 murine retinal sections stained for IL‐33 were analysed under the fluorescence microscope using a 20× objective. IL‐33 is stained green (secondary goat IgG Cy2) and nuclei are stained blue (DAPI). Scale bar for P0 and P4 = 60 µm. Scale bar for P7–P18= 100 µm.
Figure S2. Expression of IL‐33 in adult human ocular tissue. (A) Immunofluorescence staining showing expression of IL‐33, ST2, and IL1RAP in adult human choroid. IL‐33, ST2, and IL1RAP are expressed in RPE and within choroidal vessels (arrows). RPE = retinal pigment epithelium. (B) Human ciliary body was also enriched in IL‐33 and ST2. (C) Negative control for staining of IL‐33, ST2, and IL1RAP (secondary IgG only). GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. Nuclei are stained with DAPI. Scale bar = 100 µm.
Figure S3. Effect of IL‐33 on retinal endothelial cells. (A) Soluble ST2 was present in the supernatant of HRMECs and did not vary between control and IL‐33‐stimulated cells. (B) The Boyden chamber assay was performed and chemotactic activity was quantified by blind counting of the migrating cells on the lower surface of the membrane in ten high‐power microscope fields per chamber using a ×100 objective. IL‐33 significantly inhibited the migratory ability of HRMECs in a dose‐dependent manner (scale bar = 50 µm) (n = 4 per group). (C) IL‐33 at a range of doses did not affect the expression of vascular endothelial growth factor receptor (VEGFR) 2, measured by western blot analysis. (D, E) Expression of VEGFR1 (D) and VEGFR2 (E) by HRMECs when treated with IL‐33 for varying amounts of time, measured by RT‐PCR (n = 3 per group). Data are shown as mean ± SEM. Data are representative of at least two independent experiments with similar results. *p < 0.05. Statistical analysis was performed with Student's t‐test.
Figure S4. Reduced population of endothelial progenitor cells in IL‐33 eyes with CNV. (A) c‐kit staining of choroidal neovascular (CNV) lesion in control and treatment groups. Images are representative of CNV volume in each experimental group. Scale bar = 75 µm. (B) Quantitative analysis of c‐kit+ cells in the area of each choroidal neovascular lesion showed significantly decreased c‐kit+ cells in the two lower doses of IL‐33 treatment groups (n = 15–20 eyes per group). (C) Tryptase staining of CNV lesion in control and treatment (IL‐33 200 pg/µl) groups showed that mast cells are present in the CNV lesion. Scale bar = 75 µm. Data are shown as mean ± SEM. Data are representative of at least two independent experiments with similar results. *p < 0.05. Statistical analysis was performed with Student's t‐test.
Figure S5. Recombinant IL‐33 does not increase monocyte recruitment in retina. (A) Confocal images and semi‐quantitative mean fluorescence intensity (MFI) analysis using ImageJ software for Iba1+ cell accumulation on RPE/choroid whole‐mounts taken on day 3 post‐laser and treatment (n ≥ 8 per group). There was no significant difference in monocyte recruitments between treatment groups and control group. (B) Flow cytometric analysis of retina shows no increase in CD45+/CD11b+ cell infiltration on day 3 post‐IL‐33 treatment compared with control (n = 4 per group). Data are shown as mean ± SEM.
Figure S6. Safety and tolerability of IL‐33. (A) Mouse recombinant IL‐33 did not affect the thickness of the retina, as measured by OCT at days 3 and 5 in eyes that had intravitreal injection of IL‐33. (B) Confocal microscopic analysis did not show TUNEL‐positive apoptotic cells. INL= inner nuclear layer; ONL = outer nuclear layer. The MTT assay showed that IL‐33 at a range of doses had no effect on the viability of mouse (B6RPE‐07) (C) and human (ARPE‐19) (D) RPE cells. Phase contrast microscopy of cells treated with IL‐33 showed a healthy RPE appearance. (E, F) The truncated form of human IL‐33 did not affect the thickness of the retina, as measured by OCT at days 3 and 5 in eyes that had intravitreal injection of IL‐33, and did not induce apoptosis. ONH = optic nerve head. Data are representative of two measurements per retina with four eyes per group. Data are shown as mean ± SEM. Data are representative of at least two independent experiments with similar results. Statistical analysis was performed with Student's t‐test.