

Abstract
Targeted therapies promise to increase the safety and efficacy of treatments against diseases ranging from cancer to viral infections. However, the vast majority of targeted therapeutics relies on the recognition of extracellular biomarkers, which are rarely restricted to diseased cells and are thus prone to severe and sometimes-fatal off-target toxicities. In contrast, intracellular antigens present a diverse yet underutilized repertoire of disease markers. Here, we report a protein-based therapeutic platform—termed Cytoplasmic Oncoprotein VErifier and Response Trigger (COVERT)—which enables the interrogation of intracellular proteases to trigger targeted cytotoxicity. COVERT molecules consist of the cytotoxic protein granzyme B (GrB) fused to an inhibitory N-terminal peptide, which can be removed by researcher-specified proteases to activate GrB function. We demonstrate that fusion of a small ubiquitin-like modifier 1 (SUMO1) protein to GrB yields a SUMO-GrB molecule that is specifically activated by the cancer-associated sentrin-specific protease 1 (SENP1). SUMO-GrB selectively triggers apoptotic phenotypes in HEK293T cells that overexpress SENP1, and it is highly sensitive to different SENP1 levels across cell lines. We further demonstrate the rational design of additional COVERT molecules responsive to enterokinase (EK) and tobacco etch virus protease (TEVp), highlighting the COVERT platform’s modularity and adaptability to diverse protease targets. As an initial step toward engineering COVERT-T cells for adoptive T-cell therapy, we verified that primary human T cells can express, package, traffic, and deliver engineered GrB molecules in response to antigen stimulation. Our findings set the foundation for future intracellular-antigen–responsive therapeutics that can complement surface-targeted therapies.
Keywords: granzyme B, targeted therapy, adoptive T-cell therapy, mammalian synthetic biology, rational protein engineering
Graphical Abstract
INTRODUCTION
Precision medicine is an emerging cancer therapy paradigm that utilizes patient-specific genetic information to identify disease-specific molecular targets for therapeutic intervention1,2. Unlike conventional, indiscriminately toxic treatments, targeted therapies recognize aberrantly expressed biomarkers to operate against molecularly defined, diseased cell populations. There has been a long-standing interest in developing anti-tumor “magic bullets” by coupling tumor-targeting moieties to potent cytotoxic molecules3. In particular, monoclonal antibodies bind to unique epitopes with high specificity and are frequently conjugated to small-molecule chemotherapeutics, either through direct chemical linkage in antibody-drug conjugates or by surface presentation on drug-delivery vehicles4. Alternatively, non-selective toxins can be designed to function as prodrugs that activate upon cleavage by tumor-secreted proteases, thereby confining toxicity to cells in the local microenvironment5. While mechanistically diverse, these “smart drugs” share a heavy reliance on the recognition of extracellular signals to achieve specific targeting, even though surface-marker expression and soluble-factor secretion are rarely restricted to tumor cells. Consequently, dose-limiting, off-tumor toxicities are often manifest in therapies directed against extracellular antigen targets6–8.
Despite the lack of tumor-exclusive extracellular biomarkers, contemporary tumor-targeting strategies continue to rely on the recognition of surface-bound antigens. This design focus has persisted through the advent of adoptive T-cell therapy, in which T cells are redirected to attack target cells via engineered receptor-antigen interactions on the cell surface9. Notably, T cells expressing chimeric antigen receptors (CARs) specific for the pan–B-cell marker CD19 have demonstrated robust clinical efficacy in patients with relapsed B-cell malignancies10–13. However, due to natural CD19 expression on non-malignant B cells, sustained B-cell aplasia inevitably accompanies tumor eradication in responding patients10–13. Although B-cell aplasia is a clinically manageable condition, T cells engineered to target other tumor-associated antigens such as carcinoembryonic antigen (CEA) and HER2 have triggered severe dose-limiting or fatal toxicities in response to expression of the targeted molecules on normal tissue14,15. Therefore, the lack of disease-exclusive surface targets presents a recurrent critical barrier to clinical practice.
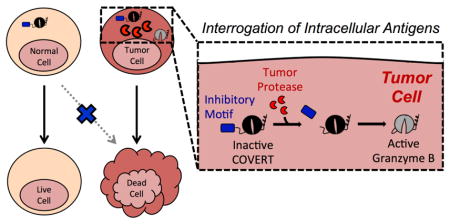
Meanwhile, the completion of the Human Genome Project and parallel advances in sequencing technology have enabled high-throughput, high-resolution analyses of cancer omics data, revealing a plethora of unexploited, intracellular disease signatures16–18. Yet, in stark contrast to the multitude of innovative therapeutic modalities that target extracellular antigens, therapies responsive to intracellular antigens remain few and underdeveloped. Small-molecule drugs can permeate cell membranes to inhibit intracellular enzymes, but they generally display limited surface area and depend on hydrophobic contact for target binding, making them prone to non-specific interactions19,20. We hypothesize that the vast structural and biochemical diversity of proteins can be harnessed to achieve fine-tuned ligand specificity, allowing protein-based therapeutics to react to distinct physiological stimuli. Here, we report a novel protein-based therapeutic platform, termed Cytoplasmic Oncoprotein VErifier and Response Trigger (COVERT), which enables interrogation of intracellular tumor antigens—in particular, proteases—with the use of conditionally active granzyme B (GrB) molecules.
GrB is the initiator of multiple pro-apoptotic pathways and serves as the principle cytotoxic molecule deployed by T cells and natural killer (NK) cells to eliminate target cells21,22. Endogenous GrB is produced as a zymogen bearing an N-terminal Gly-Glu dipeptide that prevents the formation of a functional catalytic triad23. Upon packaging into lytic granules inside the immune cell, GrB is processed by the dipeptidyl peptidase cathepsin C (CatC), which cleaves off GrB’s Gly-Glu dipeptide and frees the newly N-terminal Ile16 residue to insert into the interior of the molecule and form a salt bridge with Asp194. The resulting conformational change enables the simultaneous generation of an oxyanion hole and maturation of the active-site S1 pocket24,25. Since endogenous GrB is activated prior to its release from the lytic granules of T cells and NK cells, it indiscriminately kills any target cell it enters and does not independently ascertain the identity of the target cell. Instead, target-cell identification is established exclusively at the cell surface via receptor-antigen interactions, whose specificities are subject to the limitations outlined above. Given its cytotoxic potential, GrB has been the subject of numerous attempts to engineer targeted therapeutics26,27. The vast majority of these designs consist of C-terminal fusions to binding domains that recognize surface-bound receptors or antigens. As a result, the specificity and therapeutic application of these engineered GrB molecules remain limited by the lack of disease-specific extracellular markers.
We hypothesized that engineering GrB to be a conditionally active molecule that is delivered into target cells in a dormant form but becomes activated after detecting a confirmatory, disease-identifying signal inside the target cell could significantly increase the specificity and safety profile of cell-based immunotherapy. We designed COVERT molecules to achieve this sense-and-respond function by fusing GrB to an N-terminal inhibitory peptide, which can be selectively removed by a cognate protease to activate GrB’s catalytic function. As initial proof of concept, we focused on intracellular proteases as input signals to activate the engineered GrB. Intracellular proteases exert precise control over cellular signaling processes by rapidly enabling or inhibiting the biological activity of cleaved protein substrates, and they play essential roles in post-translational modification processes28. For example, sentrin-specific proteases (SENPs) work in concert with small ubiquitin-like modifier (SUMO) ligases to dynamically regulate the covalent attachment and detachment of SUMO-peptide modifications to protein substrates, and this interplay is responsible for fine-tuning the structure, activity, and subcellular localization of the substrate proteins29,30. Dysregulation of the intricate balance between post-translational–modification states is frequently implicated in tumorigenesis, making intracellular proteases compelling disease targets31,32. In particular, SENP1 has been shown to be upregulated in a variety of tumors, including prostate, pancreatic, and oncocytic thyroid cancers33–35.
Here, we report the development of synthetic COVERT molecules that can be produced, packaged, trafficked, and delivered by human T cells in the same manner as endogenous GrB, but which require the presence of specific proteases such as SENP1 inside target cells for their activation. We demonstrate that fusion of a SUMO1 peptide to the N-terminus of GrB yields a COVERT molecule that is specifically activated by the presence of SENP1. We further demonstrate that the modular COVERT architecture can accommodate a variety of different N-terminal fusion partners recognized by different proteases, and we confirm dose-responsive induction of GrB activity for each version of COVERT molecules tested. As a step toward engineering tumor-targeting T cells with high specificity and low off-tumor toxicity, we demonstrate that human T cells expressing CD19 CARs can be engineered to produce and deploy modified GrB molecules, highlighting the compatibility of the COVERT platform with adoptive T-cell therapy.
RESULTS AND DISCUSSION
Engineering protease-responsive GrB switches
Similar to most trypsin-like serine proteases, GrB activation is strictly dependent on the generation of a free N-terminal Ile16 residue, which initiates the conformational change that creates an oxyanion hole and enables substrate access to the S1 pocket of the enzyme25 (Figure 1a). Taking advantage of this sequence and structural requirement, we developed a protease-responsive COVERT architecture by engineering synthetic GrB zymogens that are only activated upon the removal of inhibitory N-terminal residues by specific proteases, with SENP1 being the protease of interest in our first demonstration (Figure 1b). SENP1 naturally removes SUMO1 modifications from substrate proteins by cleaving the SUMO1 peptide after Gly9736. By replacing the Gly-Glu dipeptide of wild-type GrB with the first 97 residues of SUMO1, we generate SUMO-GrB molecules that remain an inactive zymogen inside the lytic granule since they can no longer be cleaved by CatC to reveal an N-terminal Ile16. Instead, only an encounter with SENP1 can result in proper cleavage between Gly97 of the SUMO1 peptide and Ile16 of GrB, thereby triggering GrB activation.
Figure 1.

N-terminal fusion of SUMO1 to mature GrB yields a SENP1-activated zymogen. (a) Crystal structure of mature GrB (PDB #1IAU) shows the requisite salt bridge interaction between the N-terminal Ile16 (blue) and Asp194 (green), which forms the oxyanion hole between the nitrogen atoms of Gly193, Asp194, and Ser195, and activates the catalytic triad of His57, Asp102, and Ser195 (red). (b) Replacement of the wild-type GrB propeptide with SUMO1 renders SUMO-GrB inactive as a synthetic zymogen that cannot be properly processed by cathepsin C (CatC). Encounter with SENP1 and removal of SUMO1 generates a free Ile16 N-terminus, which enables SUMO-GrB to undergo the conformational change necessary to achieve an active state.
We first confirmed that SUMO-GrB can be properly expressed by human cells and processed by SENP1 into mature GrB. A secreted, His-tagged version of SUMO-GrB was purified from the supernatant of transiently transfected HEK293T cells (which do not endogenously express GrB), and western blot verified that SUMO-GrB is specifically and efficiently cleaved upon co-incubation with lysate taken from SENP1-overexpressing cells (Figure 2a). Human GrB is 32 kDa when glycosylated37, and genetic fusion with the SUMO1 peptide results in expression of full-length SUMO-GrB as a 43-kDa protein. Western blot stained with an anti-GrB antibody indicated that a 32-kDa fragment was released in the presence of SENP1, consistent with the expected processing of SUMO-GrB into mature GrB (Figure 2a).
Figure 2.

SUMO-GrB is efficiently processed and activated by SENP1 in a dose-responsive manner. (a) Cleavage of purified SUMO-GrB following co-incubation with mock or SENP1-overexpressing HEK293T lysates. Percent mature GrB was calculated by normalizing the intensity of the 32-kDa mature GrB band by the total intensity of the 43-kDa SUMO-GrB and the mature GrB bands on the western blot. Purified mature GrB was included as a control, which was unaffected by the presence of SENP1. (b) Enzymatic activity of GrB molecules as quantified by Ac-IEPD-pNA cleavage. Purified SUMO-GrB was co-incubated with mock or SENP1-overexpressing HEK293T lysates as in (a). Percent GrB activity was calculated as the rate of Ac-IEPD-pNA cleavage normalized to the rate of cleavage by mature GrB. (c) SENP1 dose-responsive activation of purified SUMO-GrB following co-incubation with lysates from a panel of seven human cell lines. SENP1 protein level in each cell line was quantified via western blot, and GrB activity was quantified via the Ac-IEPD-pNA cleavage assay. GrB activity and SENP1 protein level for each sample were normalized to those of RWPE-1 to enable clear visualization of the data spread. The red dotted line indicates the SENP1 expression level of HEK293T cells transiently transfected with SENP1-encoding plasmids. Western blots corresponding to x-axis values are shown in Figure S1, Supporting Information. (d) Intracellular cleavage of SUMO-GrB by SENP1. HEK293T cells were transiently transfected with SUMO-GrB plus plasmids encoding either TagBFP-T2A-SENP1 or TagBFP alone. Cell lysates were collected 24 hours post transfection and probed for GrB by western blot. Total GrB refers to the sum of the 43-kDa SUMO-GrB and 32-kDa mature GrB bands, and % mature GrB was calculated as described in (a). (e) Enzymatic activity of SUMO-GrB–transfected cell lysates as quantified by Ac-IEPD-pNA cleavage. Twenty-five μg of the same cell lysates as shown in (d) were reacted with 200 μM Ac-IEPD-pNA. The rate of Ac-IEPD-pNA cleavage was normalized by the amount of total GrB in each lysate based on western blot results shown in (d). All plotted values indicate the mean of triplicate samples and error bars represent ±1 standard deviation (s.d.). * p < 5E–3; ***p < 5E–5; ****p < 5E–12.
To quantify the functional activation of SUMO-GrB, we utilized an N-acetyl-Ile-Glu-Pro-Asp-paranitroanilide (Ac-IEPD-pNA) tetrapeptide substrate, which releases a chromogenic paranitroaniline group upon cleavage by GrB. Purified SUMO-GrB was co-incubated with SENP1-transfected or mock-transfected HEK293T lysates, and the rate of Ac-IEPD-pNA substrate cleavage by GrB was measured as absorbance at 405 nm over time (Figure 2b). SUMO-GrB was nearly catalytically inert in the complete absence of SENP1, confirming the inhibitory nature of an N-terminal fusion architecture. Basal SENP1 levels in HEK293T induced statistically significant but limited activation of SUMO-GrB. In comparison, SENP1 overexpression resulted in a sizable and significant increase in SUMO-GrB’s enzymatic activity (Figure 2b). (Western blots indicated that HEK293T cells transfected with SENP1-encoding plasmids expressed 1.8X to 3.2X more SENP1 compared to untransfected HEK293T cells, with the extent of overexpression correlating to transfection efficiency (red dotted line in Figure 2c; Figure S1, Supporting information).) Using the Ac-IEPD-pNA cleavage assay, we also verified that an S183A mutant of GrB is catalytically inactive and can serve as a negative control in subsequent experiments (Figure 2b).
We next evaluated the sensitivity of SUMO-GrB to endogenous SENP1 expression levels found in different cell lines. The Ac-IEPD-pNA cleavage assay was performed on SUMO-GrB co-incubated with lysates from a panel of seven human cell lines (Jurkat, H9, Raji, HEK293T, PC-3, RWPE-1, and MCF7), and SENP1 protein levels in each cell line were separately quantified by western blot. The results indicated a strong linear correlation between SUMO-GrB activation and SENP1 expression levels, demonstrating a robust SENP1-dose dependent response (Figure 2c and Figure S1, Supporting Information). Strikingly, SUMO-GrB was sensitive to relatively modest fold-differences in SENP1 expression, highlighting its ability to quantitatively differentiate endogenous levels of SENP1 found in different cell types.
To confirm that SENP1-mediated cleavage and activation of SUMO-GrB can also occur in the intracellular environment, we transfected HEK293T cells to express SUMO-GrB with and without SENP1. Western blot results indicate that cells transfected with SUMO-GrB alone contained significant amounts of both SUMO-GrB and mature GrB (Figure 2d), consistent with the fact that HEK293T cells express a basal level of endogenous SENP138 (Figure 2c). In contrast, the vast majority of GrB content in cells co-transfected with SUMO-GrB and SENP1 was in the mature form (Figure 2d), confirming SENP1-dependent cleavage of the SUMO peptide inside transfected cells. To verify that the cleaved GrB was functionally active, we performed Ac-IEPD-pNA cleavage assays using the same cell lysates as used in the western blots. We observed significantly higher enzymatic activity in cells that were transfected with SENP1, after normalizing by the amount of total GrB (SUMO-GrB plus mature GrB) present in each sample (Figure 2e). These results confirm SENP1-specific activation of SUMO-GrB in the intracellular environment. It was noted that HEK293T cells co-transfected with SUMO-GrB and SENP1 contained significantly less total GrB compared to cells transfected with SUMO-GrB alone, suggesting that the activation of SUMO-GrB by SENP1 may have led to toxicities that compromised the cells’ health and ability to produce transgenic proteins at high levels. This hypothesis is supported by the observation that HEK293T cells transfected with mature GrB yielded even lower levels of total GrB expression (Figure S2, Supporting Information). We next sought to confirm whether the presence of active GrB indeed results in cytotoxicity.
SUMO-GrB selectively triggers apoptosis of SENP1-overexpressing cells
The effect of GrB expression in HEK293T cells was first established using wild-type GrB as a positive control. Surprisingly, the results indicated a lack of overt cytotoxicity based on high transfection efficiency (i.e., no depletion of transfected cells due to toxicity of transgenic construct), as well as lack of staining by the viability dye 7-AAD and the apoptosis marker Annexin V (Figure S3, Supporting Information). Although GrB is the main cytotoxic molecule with which T cells trigger target-cell apoptosis, T cells typically release GrB together with other effector molecules such as perforin, a pore-forming protein, during degranulation39. The absence of these accessory proteins may account for the drastic reduction in cytotoxicity observed in cells transfected with GrB alone. Nevertheless, closer inspection of GrB-transfected cells revealed a marked change in cell physiology as evidenced by the appearance of a distinct cell population exhibiting low forward scatter (FSC), which is characteristic of late-apoptotic cells with reduced cell size40,41 (Figure 3a and Figure S4, Supporting Information). Furthermore, we observed a loss of cell adherence resulting from GrB expression, suggesting compromised integrity of GrB-transfected cells (Figure S5a, Supporting Information).
Figure 3.

SUMO-GrB selectively triggers apoptosis of SENP1-overexpressing HEK293T cells. HEK293T cells were transiently transfected to co-express either TagBFP or TagBFP-T2A-SENP1 plus wild-type GrB, SUMO-GrB, or GrB S183A, which is an inactive GrB mutant. (a) Side scatter (SSC) vs. forward scatter (FSC) plot of transfected (TagBFP+) and 7-AAD– cell populations (see Figure 3—supplementary figure 2 for gating strategy). Marked reduction in FSC was observed among cells expressing wild-type GrB regardless of SENP1 presence, as well as among cells expressing SUMO-GrB specifically in the presence of SENP1. Plots shown are representative of two independent experiments, each with triplicate samples. (b) Quantification of the % viability among samples shown in (a), **p < 5E–4. All plotted values indicate the mean of triplicate samples and error bars represent ±1 s.d.
Next, HEK293T cells were transiently transfected to express SUMO-GrB plus either the fluorescent protein TagBFP or SENP1 fused to TagBFP via a 2A peptide, and the physiology of transfected (TagBFP+) cells was analyzed by flow cytometry. SUMO-GrB–expressing cells showed significant reduction in FSC only in the presence of SENP1 (Figure 3), indicating SENP1-induced apoptosis. Concurrent loss of cell attachment was also observed in SUMO-GrB–expressing cells in the presence of SENP1, albeit to a lesser extent compared to cells transfected with wild-type GrB (Figure S5b, Supporting Information). Importantly, control samples transfected with GrB S183A showed no change in FSC or cell-detachment patterns regardless of the presence of SENP1, confirming that SENP1 does not have any intrinsic toxicity (Figure S5c, Supporting Information). Taken together, these results indicate that SUMO-GrB selectively triggers cell apoptosis in a SENP1-specific manner.
The COVERT architecture is modular and compatible with diverse proteases
Given the evidence supporting the predicted mechanism of SUMO-GrB activation, we hypothesized that the basic COVERT architecture could be systematically adjusted to accommodate different N-terminal fusion partners and, consequently, different protease specificities. To explore the potential for extending the COVERT platform to additional protease targets, we designed COVERT molecules that are specifically activated by enterokinase (EK) and tobacco etch virus protease (TEVp). Since EK cleaves at the end of the Asp-Asp-Asp-Asp-Lys (DDDDK) motif, it is readily compatible with the COVERT architecture—i.e., a direct N-terminal fusion of DDDDK to Ile16 of GrB generates an EK-responsive GrB zymogen, termed DDDDK-GrB.
In contrast, TEVp cleaves its canonical recognition sequence Glu-Asn-Leu-Tyr-Phe-Gln-Gly (ENLYFQG) between the P1-Gln and P1′-Gly residues, such that an N-terminal fusion of the entire ENLYFQ↓G sequence would leave an N-terminal Gly residue attached to GrB upon cleavage by TEVp, thus rendering the wild-type TEVp cleavage sequence incompatible with COVERT activation. However, prior studies have demonstrated enzymatic promiscuity by TEVp, which can process sequences containing alternative, non-proline residues in the P1′ position, albeit with reduced efficiency42. Therefore, we rationalized that fusing the first six residues of the TEVp cleavage sequence in front of Ile16 of GrB would constitute a ENLYFQ↓I cleavage site in the resulting COVERT molecule, termed ENLYFQ-GrB, which enables activation in response to TEVp-mediated cleavage.
The conditional activation of DDDDK-GrB and ENLYFQ-GrB were characterized by quantifying their rates of Ac-IEPD-pNA cleavage before and after exposure to their cognate proteases. Secreted, His-tagged DDDDK-GrB and ENLYFQ-GrB were purified from the supernatant of transiently transfected HEK293T cells and co-incubated with increasing amounts of purified EK and purified TEVp, respectively. The results showed efficient, dose-dependent activation of DDDDK-GrB by EK, with a linear, 647-fold increase in GrB activity (R2 = 0.994) over an 80-nM range of EK input concentrations (Figure 4a). As anticipated, ENLYFQ-GrB was also activated by TEVp in a dose-responsive manner, showing linear correlation between GrB activity and TEVp concentration up to 150 nM (R2 = 0.944) (Figure 4b). However, the rates of activation, calculated as GrB activity per mole of target-protease input, indicate that EK is 7.52-fold more efficient than TEVp at activating its cognate COVERT molecule (Figure 4c). This finding is consistent with reports indicating reduced cleavage activity of TEVp against the non-canonical ENLYFQ↓I target sequence42.
Figure 4.

N-terminal fusion architecture can accommodate diverse inhibitory motifs to yield unique COVERT molecules that are specifically activated by cognate proteases. (a,b) DDDDK-GrB and ENLYFQ-GrB were purified from the supernatant of transiently transfected HEK293T cells and co-incubated with EK and TEVp, respectively. The resulting GrB activity of (a) DDDDK-GrB and (b) ENLYFQ-GrB against Ac-IEPD-pNA substrates was quantified by the rate of increase in absorbance at 405 nm. (c) GrB activity per mole of target protease (i.e. EK or TEVp) was calculated to compare the relative rate of COVERT activation. All values indicate the mean of triplicate samples and error bars represent ±1 s.d.
To further investigate the versatility of the COVERT platform, ENLYFQ-GrB was evaluated for its ability to selectively mediate cytotoxicity against HEK293T cells expressing TEVp. In contrast to SENP1, TEVp is not mammalian in origin, and its forced expression mimics a state of viral infection. Transient expression of GrB in HEK293T cells resulted in marked changes in cell physiology regardless of whether the cells co-expressed TEVp (Figure 5a). In contrast, ENLYFQ-GrB elicits the appearance of the low-FSC population only in the presence of TEVp (Figure 5b). This population is again absent in cells transfected with GrB S183A (Figure 5c), indicating that the cytotoxicity in the ENLYFQ-GrB plus TEVp sample can be attributed to selective COVERT activation rather than any potential toxicity directly resulting from TEVp expression. Along with the SUMO-GrB data, these results support the modularity and versatility of the COVERT platform for targeting proteolytic markers of a variety of disease states.
Figure 5.

Selective induction of apoptosis in TEVp-overexpressing HEK293T cells by ENLYFQ-GrB. HEK293T cells were transiently transfected to co-express either mCherry or TEVp (each tagged with mCherry) plus (a) wild-type GrB, (b) ENLYFQ-GrB, or (c) GrB S183A. Co-expression of ENLYFQ-GrB and TEVp resulted in the appearance of a small, but significant, population of cells exhibiting decreased FSC, signifying the specific induction of GrB-mediated cytotoxicity. Contour plots show the SSC vs. FSC scatter of transfected (mCherry+) and DRAQ7– cell populations. Plots shown are representative of triplicate samples.
GrB-based cytotoxic switches are mechanistically compatible with adoptive T-cell therapy
The ability to selectively trigger cell apoptosis in response to intracellular antigen expression would substantially expand the pool of targetable disease markers for adoptive T-cell therapy. As a step toward engineering T cells that deploy COVERT molecules to sense and respond to intracellular disease targets, we first verified that engineered GrB molecules are efficiently expressed and properly stored by human T cells. As a highly potent cytotoxic molecule, wild-type GrB is naturally packaged and stored in lytic granules, both to prevent self-killing and to enable trafficking and subsequent release into the immunological synapse43. To confirm that engineered GrB molecules are compatible with endogenous packaging mechanisms, human Jurkat T cells were nucleofected to express a recombinant GrB molecule fused to a fluorescent mCherry tag via a short peptide linker, and visualized by confocal microscopy. The images reveal a punctate distribution of GrB-mCherry throughout the cytoplasm, indicating granular sequestration (Figure 6a).
Figure 6.

Engineered GrB-mCherry molecules are efficiently expressed, packaged into lytic granules, and trafficked to the immunological synapse by human T cells. (a) Jurkat T cells nucleofected to express a GrB-mCherry fusion (red) and stained with the nuclear dye DRAQ5 (purple) display a punctate and cytoplasmic distribution of GrB-mCherry, consistent with the packaging and storage of wild-type GrB in lytic granules. (b) Time-lapse confocal microscopy of Jurkat T cells co-expressing a CD19 CAR and GrB-mCherry (red) reveals rapid movement of GrB-mCherry to the immunological synapse upon encounter with Raji target cells (green) that naturally expresses CD19. Time signatures indicate minutes:seconds, with 0:00 set to the time of initial cell-cell contact. (c) Jurkat T cells co-expressing CD19 CAR and GrB-mCherry (red) were pre-loaded with the calcium indicator Fluo-4 and co-incubated with CD19+ Raji cells pre-labeled by Hoechst 33342 staining (blue). Fluo-4 signal (green) indicates T-cell activation triggered by target-cell engagement, which was accompanied by rapid and sustained GrB-mCherry polarization to the immunological synapse in preparation for directional release toward the target cell.
To verify that engineered GrB molecules are efficiently trafficked in response to antigen stimulation, we nucleofected Jurkat T cells to co-express a CD19 CAR and GrB-mCherry, and subsequently co-incubated the Jurkat cells with Raji target cells that naturally express CD19 antigen. Time-lapse images show that GrB-mCherry is present in dispersed lytic granules inside the Jurkat cell prior to target-cell conjugation, and that the granules rapidly traffic to the immunological synapse following target-cell conjugation (Figure 6b and Movie S1, Supporting Information). In a subsequent experiment, Jurkat T cells expressing CD19 CAR and GrB-mCherry were pre-loaded with the calcium indicator Fluo-4, and activated Jurkat cells, identified by Fluo-4 signals, exhibited high levels of sustained GrB-mCherry polarization consistent with directional lytic granule release44 (Figure 6c). These images indicate that GrB molecules can be engineered to interface with the same storage compartments and trafficking mechanisms that human T cells use to deploy wild-type GrB.
Despite clear visuals of granule trafficking to the immunological synapse, no GrB-mCherry signal was detected within target cells by confocal microscopy, possibly because the absence of perforin (Pfn) expression by Jurkat T cells precluded efficient delivery of lytic granule contents into target cells. In contrast to Jurkat T cells, activated human CD8+ T cells naturally express ample amounts of Pfn, which is degranulated alongside GrB, and facilitates GrB entry into the target-cell cytoplasm39. To verify the transfer of recombinant GrB molecules from T cells into target cells, we first transduced primary human CD8+ T cells with a retroviral construct encoding a CD19 CAR and GrB-mCherry. The transduced cells were co-incubated with CD19+ Raji target cells, and analyzed by flow cytometry. The results show a significant increase in the percentage of CD19+/mCherry+ target cells (Figure 7a,b) as well as a concurrent reduction in the mCherry MFI of the T cells following co-incubation (Figure 7c,d), suggesting GrB-mCherry release by T cells and uptake by target cells. Together, these results confirm T cells’ ability to produce, package, and deliver engineered GrB molecules and demonstrate the mechanistic compatibility of the COVERT platform with existing surface-receptor technologies for adoptive T-cell therapy.
Figure 7.

CD19 CAR-T cells successfully delivered engineered GrB-mCherry molecules into CD19-expressing Raji target cells. Primary human CD8+ T cells from two healthy donors were transduced to co-express a CD19 CAR and GrB-mCherry and then co-incubated with CD19+ Raji at 3:1 effector:target ratio. Prior to resuspension and analysis via flow cytometry, samples were stained for CD8 and CD19 to distinguish singlet T cells and target cells from T-cell/target-cell conjugates. (a) GrB-mCherry uptake was detected in CD19+/CD8− gated Raji target cells following co-incubation with CD19 CAR-T cells. (b) % mCherry+ of co-cultured Raji cells was calculated by Overton histogram subtraction, with Raji cells cultured in the absence of T cells serving as the reference. (c,d) Degranulation of GrB-mCherry was reflected in a decrease in median mCherry intensity in CD8+/CD19− gated T cells following co-incubation with Raji target cells. T-cell–only samples were assayed in duplicate and error bars represent the range. All other values indicate the mean of triplicate samples and error bars represent ±1 s.d. *p < 5E–3; ***p < 5E–5.
Conclusion
The lack of disease-exclusive surface targets has been a major obstacle to the development of safely targeted therapies, including adoptive T-cell therapy. In this study, we developed a protein-based therapeutic platform that directly addresses this limitation by enabling the interrogation of intracellular antigen expression prior to unleashing cytotoxicity. By taking advantage of the zymogen behavior of wild-type GrB, we were able to apply an N-terminal fusion strategy to construct synthetic GrB zymogens, termed COVERT molecules, that can be activated by a variety of target proteases, including SENP1, EK, and TEVp. Each evaluated COVERT molecule displayed a robustly dose-dependent and protease-specific activation profile, confirming the modularity of the N-terminal fusion architecture. The ability to systematically engineer COVERT molecules responsive to researcher-specified inputs highlight the versatility to target a wide array of previously unexploited disease markers, including those found in cancers as well as viral infections.
In particular, dysregulation of SENP1 expression is frequently implicated in multiple cancers33–35, and SENP1 overexpression is strongly correlated with poor prognosis of prostate cancer patients45. We demonstrated that SUMO-GrB molecules can be engineered to selectively trigger apoptotic phenotypes in human cells that overexpress SENP1. Importantly, basal SENP1 expression levels result in minimal activation and SUMO-GrB–mediated cytotoxicity, corroborating the promise of such engineered GrB molecules as safe, targeted therapeutics. In addition, we demonstrated that SUMO-GrB is remarkably sensitive to physiological differences in SENP1 expression among a panel of seven human cell lines, suggesting additional utility as a SENP1-detection agent during cancer-biomarker screening of patient samples. Standard screening protocols routinely involve RT-PCR or immunohistochemistry, both of which are labor-intensive and prone to variability in sample preparation or scoring46,47. SUMO-GrB enables rapid quantification of SENP1 levels directly from biological samples, potentially offering shorter turnaround times and increased detection accuracy.
The structural modularity of the COVERT platform suggests that novel diagnostic and therapeutic proteins can be systematically generated for a variety of protease targets. The only requirement for compatibility with the COVERT platform is an N-terminal peptide that can be specifically cleaved by the target protease to reveal an N-terminal Ile16 residue for GrB. Each COVERT molecule we have evaluated thus far exhibits highly efficient production in human cells, suggesting robust protein folding of recombinant GrB. Nevertheless, protease cleavage efficiency is an important parameter to be considered, as it dictates the dynamic range of GrB activation. For example, we observed that the highly efficient EK could elicit a >600-fold induction in DDDDK-GrB activity, whereas TEVp could only achieve a 3.3-fold induction in ENLYFQ-GrB activity due to the limited cleavage efficiency of TEVp against the ENLYFQI motif. Despite this limited dynamic range detected by Ac-IEPD-pNA cleavage assays, we were still able to observe significant changes in cell physiology in the presence versus absence of TEVp, highlighting the importance of cell-based evaluations. The fact that proteases such as TEVp can recognize multiple target peptides with different efficiency levels provides a potential method by which to calibrate the sensitivity of COVERT molecules, a flexibility that may be critical in therapeutic applications.
Beyond establishing the selective activation and cytotoxicity of COVERT molecules, our investigation also provides evidence that engineered GrB molecules can readily interface with pre-existing T-cell delivery mechanisms to gain entry into target cells. However, in order for engineered T cells to specifically target intracellular antigens with COVERT molecules, the endogenous cytotoxic payloads, including wild-type GrB, must be silenced. Herein lies a major technical challenge for COVERT applications in adoptive T-cell therapy. Since granzymes are non-chromogenic intracellular proteins, their elimination does not lead to easily detectable changes that are compatible with currently available cell-sorting strategies. As a result, any gene-editing attempt would need to either achieve sufficiently high knockout efficiency as to eliminate the need for cell sorting, or achieve sufficiently high homologous recombination efficiency as to enable cell sorting based on an inserted marker. At this time, T-cell genome editing technologies are not yet able to achieve this goal. Nevertheless, recent advances in T-cell gene modification as well as high-throughput genome editing of primary human cells suggest that such capabilities will likely become available in the not-too-distant future48,49.
The cytotoxic switch architecture developed in this study sets the foundation for a new class of protein-based therapeutics that complements exciting frontier technologies in targeted therapy. In addition to compatibility with T-cell therapy, the COVERT architecture can also be combined with a variety of C-terminal antibody conjugations previously reported to direct GrB molecules to specific cell types26,27. By including both N- and C-terminal fusions, one could engineer AND-gate molecules that perform the first step of target identification at the cell surface via antibody-mediated protein uptake, followed by a second interrogation step inside the target cell prior to activation of GrB’s enzymatic activity. Such engineered GrB molecules would require both a surface-bound antigen and an intracellular protease to trigger target-cell apoptosis, thereby increasing targeting specificity and the safety profile of GrB-based therapeutics. By enabling biologics and cellular therapies to sense and respond to intracellular tumor antigens, the COVERT platform can serve to bridge the gap between omics-mediated biomarker discovery and the transformative promise of precision medicine.
METHODS
Cell Lines
HEK293T, MCF7, Jurkat E6, and H9 cells were obtained from ATCC (Manassas, VA) in 2011, and RWPE-1 and PC-3 were obtained from ATCC in 2014. ATCC verified the identity of each purchased cell line by short tandem repeat analysis prior to shipment. Raji cells were a generous gift from Dr. Michael C. Jensen (Seattle Children’s Research Institute); the cell line was originally obtained from ATCC in 2003 and was authenticated again by short tandem repeat profiling at the University of Arizona Genetics Core in 2015. RWPE-1 cells were cultured in Keratinocyte serum-free medium (K-SFM) supplemented with 0.05 mg/mL bovine pituitary extract (BPE) and 5 ng/mL recombinant human epidermal growth factor (rhEGF), purchased from Life Technologies (Grand Island, NY). Other cell lines were cultured in F-12K (PC-3), DMEM (HEK293T and MCF7), or RPMI-1640 (Jurkat, H9, and Raji) supplemented with 10% heat-inactivated FBS (HI-FBS). All mammalian cell cultures were maintained at 37°C and 5% CO2.
DNA Constructs
DNA was chemically synthesized as oligonucleotides or gBlocks by Integrated DNA Technologies (Coralville, IA) and assembled using standard molecular cloning techniques. Unless otherwise indicated, all constructs were cloned into the epHIV7 lentiviral expression vector50, although expression was induced by transient DNA transfection in the absence of viral packaging. The QPY variant of human GrB was used in this study, and the S183A mutant was generated by introducing a TCT to GCC codon mutation via isothermal DNA assembly. The DNA sequences for the three COVERT molecules and associated GrB control constructs are included in Text S1, Supporting Information. pFLAG-SENP1 was obtained from Addgene (plasmid 17357), and the SENP1 gene was subsequently cloned as a T2A fusion to TagBFP or mCherry. The TEVp gene was constructed from gBlocks. The CD19 CAR was constructed as previously reported51, and fused to GrB-mCherry via a 2A peptide using isothermal DNA assembly. The MSCV-IRES-EGFP retroviral vector and pHIT60 and RD114 retroviral packaging vectors were generous gifts from Dr. Steven Feldman (National Cancer Institute). EF1α-CD19 CAR-T2A-GrB-mCherry was downstream of the 5′ LTR in the MSCV vector via isothermal DNA assembly, and a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) was subsequently inserted at the ClaI site by restriction-ligation cloning.
Cell Transfection
HEK293T cells were seeded at 2.5 × 104 cells/0.25 mL/well in 48-well plates, 24 hours prior to transfection with 250 ng plasmid DNA and 15 nmol linear polyethylenimine (PEI, 25 kDa). For co-transfection experiments, plasmids were mixed at 1:1 mass ratio. The resulting DNA mixture totaling 250 ng was complexed with PEI, incubated at room temperature for 15 min, and then applied to seeded HEK293T cells. Jurkat T cells (5 × 106) were resuspended in 100 μL of Amaxa Cell Line Nucleofector™ V Solution (Lonza, Walkersville, MD) and electroporated with 5 μg plasmid DNA using Program X-001 of the Nucleofector™ 2b Device (Lonza), according to the manufacturer’s protocol.
Retrovirus Production
HEK293T cells were seeded at 6.5 × 106 cells/10 mL/dish in 10-cm tissue-culture dishes and culture medium was replaced with fresh DMEM plus 10% HI-FBS immediately before transfection with 3.8 μg retroviral construct, 3.8 μg pHIT60, and 2.4 μg RD114 using linear PEI. Sixteen hours post-transfection, cells were washed with 10 mL of phosphate buffered saline (PBS) and cultured in DMEM plus 10% HI-FBS, 20 mM HEPES, and 10 mM sodium butyrate for 8 hours before media change to DMEM plus 10% HI-FBS and 20 mM HEPES (no sodium butyrate). Viral supernatants were harvested on each of the two subsequent days post-media change, and filtered through a 0.45-μm low–protein-binding filter immediately prior to transduction of primary human T cells.
Primary Human T-cell Isolation and Retroviral Transduction
Primary human CD8+ T cells were isolated from healthy donor blood samples obtained from the UCLA Blood & Platelet Center using the RosetteSep CD8+ Human T-cell Enrichment Cocktail (Stemcell Technologies, Vancouver, Canada), according to the manufacturer’s protocol. Freshly isolated T cells were seeded at 1 × 106 cells/mL in T-cell media (RPMI-1640 supplemented with 10% HI-FBS), and stimulated with anti-CD3/CD28 Dynabeads (Life Technologies, Carlsbad, CA) at 1:1 cell:bead ratio. Cultures were supplemented with 50 U/mL IL-2 (Life Technologies) and 10 ng/mL IL-15 (Peprotech, Rocky Hill, NJ) every 48 hours. At 48 and 72 hours post-isolation, T cells (1 × 106) were transduced with 1.8 mL of fresh retroviral supernatant supplemented with 5 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO) via spinfection at 800 x g for 90 min at 32°C. Immediately following each spinfection, 1.8 mL of transduction supernatant was replaced with 1.8 mL of fresh T-cell media.
Ni2+-affinity Protein Purification
HEK293T cells were seeded at 15 × 106 cells/25 mL/flask in T-150 flasks and transfected via the linear PEI method. Culture media were changed to serum-free DMEM 16 hours post-transfection, and supernatants were harvested at 48 and 72 hours post-media change. Supernatants from different collection times were pooled, and His-tagged proteins were batch-bound to Ni-NTA resin (Life Technologies) in binding buffer (500 mM NaCl, 20 mM Tris, pH 8.0) for 1 hour prior to being washed three times with binding buffer supplemented with 20 mM imidazole (Fisher Scientific, Hampton, NH), and then eluted with binding buffer supplemented with 500 mM imidazole through a chromatography column. Eluted proteins were buffer-exchanged into storage buffer (50 nM NaCl, 20 mM Tris pH7.4, 10% glycerol) by successive concentration and resuspension steps in Amicon centrifugal columns (10 kDa; EMD Millipore, Billierica, MA) following manufacturer’s recommendations. Purified proteins were then aliquoted and frozen at −20°C.
Western Blot
Lysates were prepared by incubating cells in lysis buffer (150 mM NaCl, 20 mM Tris pH7.2, 1% (v/v) Triton-X) on ice for 45 min, and clarified of nuclear debris by centrifugation at 20,000 x g for 10 min. The protein concentration of the supernatant was determined via Bradford assay (Bio-Rad, Hercules, CA). For the SUMO-GrB cleavage assay, 30 μg of mock or SENP1-overexpressing HEK293T lysate was co-incubated with 2.5 pmol of purified SUMO-GrB for 2 hours at 37°C. Protein samples were resolved on 4–12% bis-tris SDS-PAGE gels, blotted onto nitrocellulose membranes, and probed with antibodies for GrB (clone 2C5; Santa Cruz Biotech, Dallas, TX) or SENP1 (clone C12; Santa Cruz Biotech), followed by staining with an anti-mouse secondary antibody conjugated to horseradish peroxidase (HRP; Jackson ImmunoResearch, West Grove, PA). Blots were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Ac-IEPD-pNA Activity Assay
SUMO-GrB was activated by co-incubating 30 μg of target cell lysate with 2.5 pmol of purified SUMO-GrB in 50 μL of lysis buffer for 30 min at 37°C. Samples were then diluted with 50 μL of assay buffer (50 mM HEPES pH 7.5, 10% (w/v) sucrose, 0.05% (w/v) CHAPS, 5 mM DTT) containing 200 μM Ac-IEPD-pNA substrate (Enzo Life Sciences, Farmingdale, NY), as described by Ewen et al.52. Purified EK (New England Biolabs, Ipswich, MA) and purified TEVp (prepared in-house) were added at the indicated concentrations to 2.5 pmol of DDDDK-GrB and ENLYFQ-GrB, respectively. Absorbance at 405 nm was measured every minute for 4 hours on an EONC microplate reader (BioTek, Winooski, VT). Activity was calculated by using the LINEST function in Excel to determine the line of best fit for the initial rate (dOD405/min) by the least squares method.
Flow Cytometry
For cytotoxicity experiments, transiently transfected HEK293T cells were harvested for analysis at 24 hours (SUMO-GrB) or 48 hours (ENLYFQ-GrB) post-transfection. Culture media containing any dislodged cells were collected in centrifuge tubes before adherent cells were trypsinized and subsequently collected into the same tubes. The cells were washed twice with PBS prior to staining with 7-AAD (Life Technologies), DRAQ7 (eBioscience, San Diego, CA), or Annexin V-FITC (BioLegend, San Diego, CA), according to manufacturer’s protocols. For GrB-mCherry delivery experiments, primary human T cells expressing CD19 CAR and GrB-mCherry were co-cultured with 20,000 Raji cells at 3:1 effector:target ratio for 45 min. Co-cultures were then suspended by rigorous pipetting and stained with CD8-VioGreen (clone BW135/80; Miltenyi Biotec, San Diego, CA) and CD19-VioBlue (clone LT19; Miltenyi Biotec) prior to data acquisition on a MACSQuant VYB flow cytometer (Miltenyi Biotec). Compensation and data analysis were performed using FlowJo Data Analysis software (TreeStar, Ashland, OR).
Live-cell Confocal Microscopy
Raji target cells were resuspended to 3 × 104 cells/50 μL in imaging media (RPMI-1640 without Phenol Red and L-glutamine supplemented with 10% HI-FBS and 20 mM HEPES) and seeded into 48-well glass plates (MatTek, Ashland, MA) to settle for 15 min. For T-cell activation experiments, Jurkat T cells were pre-loaded with calcium indicator by incubation with 1 μM Fluo-4 (Life Technologies) in imaging media containing 0.02% (w/v) Pluronic F127 (Sigma-Aldrich) at room temperature for 20 min. Cells were then washed extensively to remove excess dye and resuspended in fresh imaging media. Jurkat cells expressing CD19 CAR and GrB-mCherry were resuspended to 2× 104 cells/25 μL and added to each well immediately prior to imaging with a C2+ confocal microscope (Nikon, Melville, NY).
Statistics
Statistical significance was determined via two-tailed, homoscedastic Student’s t-test with a p-value cutoff of 5E–3.
Supplementary Material
Acknowledgments
The authors thank W. Clifford Boldridge and ZeNan L. Chang for technical assistance. This research was supported by the National Institutes of Health (5DP5OD012133; grant to YYC) and the National Science Foundation (1553767; grant to YYC). PH was supported by the Biotechnology Training in Biomedical Sciences and Engineering Program funded by the National Institutes of Health.
Footnotes
Author Contributions
PH, CE, and YYC designed the experiments and analyzed data. PH designed and built the COVERT constructs. PH and CE performed the experiments. PH and YYC wrote the manuscript.
COMPETING INTERESTS
The authors declare competing interest in the form of a patent application whose value may be affected by this publication.
Supporting Information. Figure S1: Western blots of SENP1 expression in various cell lines. Figure S2: Presence of active GrB reduces transgenic protein expression in transfected HEK293T cells. Figure S3: Transient expression of GrB was not overtly toxic to HEK293T cells based on transfection efficiency and viability staining. Figure S4: Gating strategy for analysis of GrB-mediated cytotoxicity in transiently transfected HEK293T cells. Figure S5: Expression of GrB results in altered cell morphology and loss of adherence by HEK293T cells. Movie S1: Live-cell imaging of Jurkat T cell expressing engineered GrB upon encounter with antigen-expressing Raji target cell. Text S1: Annotated DNA sequences of COVERT and control constructs. This material is available free of charge on the ACS Publications website at http://pubs.acs.org.
References
- 1.Collins DC, Sundar R, Lim JSJ, Yap TA. Towards Precision Medicine in the Clinic: From Biomarker Discovery to Novel Therapeutics. Trends Pharmacol Sci. 2017;38:25–40. doi: 10.1016/j.tips.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 2.Garraway LA. Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol. 2013;31:1806–14. doi: 10.1200/JCO.2012.46.8934. [DOI] [PubMed] [Google Scholar]
- 3.Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 4.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 5.Denmeade SR, Isaacs JT. Engineering enzymatically activated “molecular grenades” for cancer. Oncotarget. 2012;3:666–7. doi: 10.18632/oncotarget.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23:1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 7.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA. Finding suitable targets is the major obstacle to cancer gene therapy. Cancer Gene Ther. 2014;21:45–7. doi: 10.1038/cgt.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duong CPM, Yong CSM, Kershaw MH, Slaney CY, Darcy PK. Cancer immunotherapy utilizing gene-modified T cells: From the bench to the clinic. Mol Immunol. 2015;67:46–57. doi: 10.1016/j.molimm.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, Li YF, Ngo LT, Goy A, Feldman T, Spaner DE, Wang ML, Chen CC, Kranick SM, Nath A, Nathan DAN, Morton KE, Toomey MA, Rosenberg SA. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J Clin Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DCG, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and Toxicity Management of 19–28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, Norris AP, Wong CW, Urak RZ, Chang WC, Khaled SK, Siddiqi T, Budde LE, Xu J, Chang B, Gidwaney N, Thomas SH, Cooper LJN, Riddell SR, Brown CE, Jensen MC, Forman SJ. Phase 1 studies of central memory–derived CD19 CAR T–cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–2990. doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–6. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JWC, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 17.Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005;1756:81–2. doi: 10.1016/j.bbcan.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kintzing JR, Filsinger Interrante MV, Cochran JR. Emerging Strategies for Developing Next-Generation Protein Therapeutics for Cancer Treatment. Trends Pharmacol Sci. 2016;37:993–1008. doi: 10.1016/j.tips.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 21.Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme B Directly and Efficiently Cleaves Several Downstream Caspase Substrates: Implications for CTL-Induced Apoptosis. Immunity. 1998;8:451–460. doi: 10.1016/s1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- 22.Pinkoski MJ, Hobman M, Heibein JA, Tomaselli K, Li F, Seth P, Froelich CJ, Bleackley RC. Entry and Trafficking of Granzyme B in Target Cells During Granzyme B-Perforin–Mediated Apoptosis. Blood. 1998;92:1044–1054. [PubMed] [Google Scholar]
- 23.Smyth MJ, McGuire MJ, Thia KY. Expression of recombinant human granzyme B. A processing and activation role for dipeptidyl peptidase I. J Immunol. 1995;154:6299–305. [PubMed] [Google Scholar]
- 24.Trapani JA. Granzymes: a family of lymphocyte granule serine proteases. Genome Biol. 2001;2:REVIEWS3014. doi: 10.1186/gb-2001-2-12-reviews3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rotonda J, Garcia-Calvo M, Bull HG, Geissler WM, McKeever BM, Willoughby CA, Thornberry NA, Becker JW. The three-dimensional structure of human granzyme B compared to caspase-3, key mediators of cell death with cleavage specificity for aspartic acid in P1. Chem Biol. 2001;8:357–68. doi: 10.1016/s1074-5521(01)00018-7. [DOI] [PubMed] [Google Scholar]
- 26.Rosenblum MG, Barth S. Development of novel, highly cytotoxic fusion constructs containing granzyme B: unique mechanisms and functions. Curr Pharm Des. 2009;15:2676–92. doi: 10.2174/138161209788923958. [DOI] [PubMed] [Google Scholar]
- 27.Dälken B, Giesübel U, Knauer SK, Wels WS. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006;13:576–85. doi: 10.1038/sj.cdd.4401773. [DOI] [PubMed] [Google Scholar]
- 28.Bond JS, Butler PE. Intracellular Proteases. Ann Rev Biochem. 1987;56:333–64. doi: 10.1146/annurev.bi.56.070187.002001. [DOI] [PubMed] [Google Scholar]
- 29.Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol. 2012;13:755–766. doi: 10.1038/nrm3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flotho A, Melchior F. Sumoylation: A Regulatory Protein Modification in Health and Disease. Annu Rev Biochem. 2013;82:357–385. doi: 10.1146/annurev-biochem-061909-093311. [DOI] [PubMed] [Google Scholar]
- 31.Bawa-Khalfe T, Yeh ETH. SUMO Losing Balance: SUMO Proteases Disrupt SUMO Homeostasis to Facilitate Cancer Development and Progression. Genes Cancer. 2010;1:748–752. doi: 10.1177/1947601910382555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattoscio D, Chiocca S. SUMO pathway components as possible cancer biomarkers. Futur Oncol. 2015;11:1599–1610. doi: 10.2217/fon.15.41. [DOI] [PubMed] [Google Scholar]
- 33.Cheng J, Bawa T, Lee P, Gong L, Yeh ETH. Role of desumoylation in the development of prostate cancer. Neoplasia. 2006;8:667–76. doi: 10.1593/neo.06445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma C, Wu B, Huang X, Yuan Z, Nong K, Dong B, Bai Y, Zhu H, Wang W, Ai K. SUMO-specific protease 1 regulates pancreatic cancer cell proliferation and invasion by targeting MMP-9. Tumour Biol. 2014;35:12729–35. doi: 10.1007/s13277-014-2598-1. [DOI] [PubMed] [Google Scholar]
- 35.Jacques C, Baris O, Prunier-Mirebeau D, Savagner F, Rodien P, Rohmer V, Franc B, Guyetant S, Malthiery Y, Reynier P. Two-Step Differential Expression Analysis Reveals a New Set of Genes Involved in Thyroid Oncocytic Tumors. J Clin Endocrinol Metab. 2005;90:2314–2320. doi: 10.1210/jc.2004-1337. [DOI] [PubMed] [Google Scholar]
- 36.Bailey D, O’Hare P. Characterization of the localization and proteolytic activity of the SUMO-specific protease, SENP1. J Biol Chem. 2004;279:692–703. doi: 10.1074/jbc.M306195200. [DOI] [PubMed] [Google Scholar]
- 37.Trapani JA, Browne KA, Dawson M, Smyth MJ. Immunopurification of Functional Asp-ase (Natural Killer Cell Granzyme B) Using a Monoclonal Antibody. Biochem Biophys Res Commun. 1993;195:910–920. doi: 10.1006/bbrc.1993.2131. [DOI] [PubMed] [Google Scholar]
- 38.Kolli N, Mikolajczyk J, Drag M, Mukhopadhyay D, Moffatt N, Dasso M, Salvesen G, Wilkinson KD. Distribution and paralogue specificity of mammalian deSUMOylating enzymes. Biochem J. 2010;430:335–44. doi: 10.1042/BJ20100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev. 2010;235:35–54. doi: 10.1111/j.0105-2896.2010.00896.x. [DOI] [PubMed] [Google Scholar]
- 40.Desjardins LM, MacManus JP. An Adherent Cell Model to Study Different Stages of Apoptosis. Exp Cell Res. 1995;216:380–387. doi: 10.1006/excr.1995.1048. [DOI] [PubMed] [Google Scholar]
- 41.Bortner CD, Cidlowski JA. A necessary role for cell shrinkage in apoptosis. Biochem Pharmacol. 1998;56:1549–1559. doi: 10.1016/s0006-2952(98)00225-1. [DOI] [PubMed] [Google Scholar]
- 42.Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1′ specificity of tobacco etch virus protease. Biochem Biophys Res Commun. 2002;294:949–955. doi: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- 43.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol. 2003;3:361–70. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- 44.Dustin ML, Long EO. Cytotoxic immunological synapses. Immunol Rev. 2010;235:24–34. doi: 10.1111/j.0105-2896.2010.00904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li T, Huang S, Dong M, Gui Y, Wu D. Prognostic impact of SUMO-specific protease 1 (SENP1) in prostate cancer patients undergoing radical prostatectomy. Urol Oncol. 2013;31:1539–45. doi: 10.1016/j.urolonc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 46.Dall P, Heider KH, Sinn HP, Skroch-Angel P, Adolf Gün, Kaufmann M, Herrlich P, Ponta H. Comparison of immunohistochemistry and RT-PCR for detection of CD44v-expression, a new prognostic factor in human breast cancer. Int J Cancer. 1995;60:471–477. doi: 10.1002/ijc.2910600408. [DOI] [PubMed] [Google Scholar]
- 47.Bernard PS, Wittwer CT. Real-Time PCR Technology for Cancer Diagnostics. Clin Chem. 2002;48:1178–1185. [PubMed] [Google Scholar]
- 48.Chen YY. Efficient Gene Editing in Primary Human T Cells. Trends Immunol. 2015;36:667–669. doi: 10.1016/j.it.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 49.Wang G, Yang L, Grishin D, Rios X, Ye LY, Hu Y, Li K, Zhang D, Church GM, Pu WT. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat Protoc. 2017;12:88–103. doi: 10.1038/nprot.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yam P, Li S, Wu J, Hu J, Zaia JA, Yee JK. Design of HIV Vectors for Efficient Gene Delivery into Human Hematopoietic Cells. Mol Ther. 2002;5:479–484. doi: 10.1006/mthe.2002.0558. [DOI] [PubMed] [Google Scholar]
- 51.Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bi-specific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4:498–508. doi: 10.1158/2326-6066.CIR-15-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ewen C, Kane KP, Shostak I, Griebel PJ, Bertram EM, Watts TH, Bleackley RC, McElhaney JE. A novel cytotoxicity assay to evaluate antigen-specific CTL responses using a colorimetric substrate for Granzyme B. J Immunol Methods. 2003;276:89–101. doi: 10.1016/s0022-1759(03)00073-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.