

Abstract
The unique polarization of neurons depends on selective sorting of axonal and somatodendritic cargos to their correct compartments. Axodendritic sorting and filtering occurs within the axon initial segment (AIS). However, the underlying molecular mechanisms responsible for this filter are not well understood. Here, we show that local activation of the neuronal-specific kinase CDK5 is required to maintain AIS integrity, as depletion or inhibition of CDK5 induces disordered microtubule polarity and loss of AIS cytoskeletal structure. Furthermore, CDK5-dependent phosphorylation of the dynein regulator Ndel1 is required for proper re-routing of mislocalized somatodendritic cargo out of the AIS; inhibition of this pathway induces profound mis-sorting defects. While inhibition of the CDK5-Ndel1-Lis1-dynein pathway alters both axonal microtubule polarity and axodendritic sorting, we found that these defects occur on distinct timescales; brief inhibition of dynein disrupts axonal cargo sorting before loss of microtubule polarity becomes evident. Together, these studies identify CDK5 as a master upstream regulator of trafficking in vertebrate neurons, required for both AIS microtubule organization and polarized dynein-dependent sorting of axodendritic cargos, and support an ongoing and essential role for dynein at the AIS.
Keywords: Axon initial segment, polarized transport, dynein, Ndel1, Lis1, CDK5, microtubule polarity
Graphical abstract
Introduction
Neurons are highly polarized cells, with proteins and organelles differentially localized to the axon, dendrites, and soma.1–3 In order to maintain the functional integrity of these compartments, there is polarized sorting and trafficking of newly synthesized proteins. An active or passive barrier has been postulated,4–6 preventing unauthorized trafficking into the axon. The small region that divides the soma from the axon, the axon initial segment (AIS), or alternatively the pre-AIS (PAEZ7), fulfills this barrier role. However, the mechanism or mechanisms by which somatodendritic cargos are excluded while axonal proteins are permitted to pass through the AIS remains unclear.1,8
The AIS is a specialized cellular region located between the cell body and the proximal axon, with a unique structural and functional identity not found in other parts of the neuron. One prominent feature of the AIS is its high concentration of ion channels, particularly voltage-gated sodium channels and cell adhesion molecules.9 These channels are held in place by a distinct and well-organized cytoskeletal architecture composed of actin rings, microtubule bundles, and neurofilaments.10 The master organizer of the AIS, ankyrin G (AnkG), is responsible for establishing the unique cluster of proteins that comprise the AIS.11 Due to its location delineating the soma from the axon, the AIS permits or blocks the transport of cytoplasmic and membrane-bound structures into or out of the axonal compartment.
A variety of mechanisms have been proposed to explain how sorting occurs at the AIS. One early proposal involved a passive diffusion barrier tethered to the cellular membrane, wherein the flow of vesicles both into4 and out of12 the axon would be limited. The identification of a cytoplasmic size-exclusion barrier to passive diffusion supported this model.5 These passive diffusion barriers have been attributed to a putative dense actin cytoskeleton specifically localized to the AIS.4–6 However, recent studies using either superresolution or electron microscopy10,13 do not support the presence of a dense actin meshwork at the AIS, although small patches of actin have been identified.14 A more active barrier maintaining selective transport was proposed, based on earlier observations of the uniform polarity of microtubules within the AIS.15,16 This newer model postulates the localized activation of the minus-end directed microtubule motor dynein returns misplaced somatodendritic cargo to the correct neuronal compartment.17
Dynein is the major retrograde-directed microtubule-based motor protein, required for proper polarization of microtubules, exclusion of Golgi bodies from the axon, and re-routing of somatodendritic cargo to the soma for re-sorting, in addition to its canonical role in retrograde-directed axonal cargo transport.17–19 However, the mechanism by which dynein is locally activated in the AIS, and the resulting regulation of dynein-dependent functions are not fully understood.
One known regulator of dynein activity is cyclin-dependent kinase 5 (CDK5), a kinase expressed in post-mitotic cells including neurons. CDK5 has many known functions in neurons, including dendritic development,20 neuronal migration,21 axonal guidance,22 and organelle transport.23–27 Previous work has shown that CDK5 activates dynein indirectly, through phosphorylation of Ndel1, which controls dynein activity in conjunction with the dynein-binding protein Lis1.25
Phosphorylation of Ndel1 by CDK5 results in the formation of a phospho-Ndel1/Lis1 complex27; the binding of Lis1 to the dynein motor domain within this complex results in steric blockade of the dynein powerstroke.28 This leaves dynein unable to release from the microtubule even when ATP is bound. Strong binding of this phospho-Ndel1/Lis1/dynein complex to the microtubule interrupts processive cargo transport in the mid-axon.27 This Lis1-dependent mechanism is required for the effective initiation of retrograde transport, likely enhancing the initial recruitment of dynein to the microtubule.29 Intriguingly, Ndel1 was recently found to localize preferentially to the AIS.17 We hypothesized that the localized phosphorylation of Ndel1 at the AIS by CDK5 would promote an effective dynein-microtubule interaction that might prove integral to the localized sorting of axonal and somatodendritic cargos.
In this study, we show that cyclin dependent kinase 5 (CDK5) is a key regulator of AIS structure and function. The CDK5 activator p35 localizes to the AIS, and inhibition of CDK5 disrupts AIS integrity. Furthermore, inhibition of CDK5 activity disrupts the filtration barrier for somatodendritic proteins in the AIS, causing mis-localization of Golgi bodies to the axon. We demonstrate that the CDK5-dependent phosphorylation of dynein cofactor Ndel1 and the formation of a Ndel1/Lis1/dynein complex is required for proper microtubule organization in the axon, as well as correct trafficking of somatodendritic cargo out of the AIS and back to the soma. However, acute inhibition of dynein is sufficient to disrupt polarized trafficking without significantly altering microtubule organization, supporting a direct role for dynein in mediating the selectivity of the AIS filter. Taken together, these findings highlight CDK5 activity, and the CDK5-dependent regulation of dynein through Ndel1 and Lis1, as integral for the cytoskeletal integrity and barrier function of the AIS.
Results
CDK5 activator p35 localizes to the AIS of hippocampal neurons
CDK5 displays pan-neuronal expression, but its activity is spatially restricted by regulated binding to its activator, the membrane-bound protein p35.30,31 The localization of p35 within neurons is unknown, as it has primarily been evaluated in non-neuronal cells.30 To determine if the subcellular localization of p35, and thus CDK5 activity, is spatially regulated in the AIS, we used live-cell imaging of 7 days in vitro (DIV) embryonic rat hippocampal neurons expressing RFP-tagged p35 and an AIS marker.
Twenty-four hours after transfection, p35 was clearly visible throughout the neuron, with strong enrichment in the cell body. The tagged p35 could also be visualized along the length of hippocampal dendrites and faintly along the axon (Figure 1A). As determined by co-expression with the AIS-specific voltage-gated sodium channel NaV II–III, p35 accumulated at higher levels in the AIS than along the rest of the proximal axon. We performed a direct comparison of p35 accumulation in ~10µm regions within the AIS, within the axon (>20µm from the distal end of the AIS), and within a secondary dendrite (>10µm from the cell body), and found that p35 levels were significantly higher in the AIS than in either other compartment (Figure 1B, p>0.05). Comparing the relative local intensity of p35 within the AIS, axon, and dendrites revealed a 7-fold increase in p35 expression in the AIS compared to the axon, and a 3-fold increase compared to secondary dendrites (Figure 1C). This recruitment of p35 to the AIS implies high levels of localized CDK5 activity. This led us to hypothesize that local regulation of CDK5 may determine aspects of AIS structure and function.
Figure 1. Reducing CDK5 activity shortens the axon initial segment.

(A) Live-cell fluorescence microscopy of 8 DIV rat hippocampal neurons expressing mCherry-tagged p35 and YFP-tagged NaV II–III. White arrows indicate AIS region.
(B) Quantification of p35 fluorescence intensity from the AIS, axon, and dendrite of 8 DIV hippocampal neurons as in (A).
(C) Comparative intensity of p35 fluorescence of AIS vs. axon and AIS vs. dendrite, data from (B).
(D) STED super-resolution images of 8 DIV rat hippocampal neurons stained for AnkG expression. Neurons transfected with dnCDK5 or p25 as indicated. Pink arrows mark beginning and end of AIS, cell body to the left.
(E) Quantification of the length of AnkG staining in 8 DIV fixed rat hippocampal neurons under STED microscopy, data from (D). Neurons aligned by 75% maximum signal set to 0µm. Grey arrows indicate end of AnkG signal for various CDK5 conditions. Graphs depict means; n ≥ 18 neurons from 5 biological replicates, average of 4 neurons imaged per replicate.
(F) Quantification of the data from (E), each dot represents the length of AnkG signal for one biological replicate.
(G) Fluorescence microscopy images of 7 DIV live rat hippocampal neurons stained transfected with full-length AnkG-GFP and co-transfected with the indicated construct. Pink arrows mark beginning and end of AIS, cell body to the left.
(H) Quantification of length of AnkG fluorescence from 7 DIV live hippocampal neurons transfected with full-length AnkG-GFP. Neurons aligned by 75% maximum signal set to 0µm. Grey arrows indicate end of AnkG signal for various CDK5 conditions. Graphs depict means; n ≥ 21 neurons from 3 or more biological replicates, average of 17 neurons imaged per replicate.
(I) Quantification of the data from (H), each dot represents the length of AnkG signal for one biological replicate.
Scale bars represent 10µm in (A) and 5 µm in (D) and (G). Graphs depict means ± SEM in (B) and means ± standard deviation in (C); n ≥ 13 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test in (B); Students t-test in (C)) are noted on graphs (*p < 0.05, **p < 0.01, ***p < 0.001).
AIS structure is altered by CDK5 inhibition
Abnormal AIS length has been linked to several diseases, including schizophrenia and bipolar disorder.32 The membrane-bound protein AnkG is integral for formation of the AIS and the localized assembly of associated ion channels and cytoskeletal components; it is also frequently used as a marker of AIS length.11,33 Previous work in Drosophila melanogaster suggested that the length of the AIS varies based on CDK5 activity levels.34 Therefore, we sought to determine whether this effect is conserved in mammalian neurons via inhibition or activation of CDK5.
Rat hippocampal neurons were fixed after 7–8 DIV and stained for endogenous AnkG.2 Super-resolution Stimulated Emission Depletion (STED) microscopy was used to determine the distance over which AnkG signal persisted along the axon (Figure 1D). As expected, AnkG localized to the cellular membrane within the AIS. Inhibition of CDK5 by expression of a dominant negative construct (dnCDK5) or the chemical CDK5 inhibitor roscovitine shortened the length of AnkG enrichment as compared to untreated control neurons (Figure 1E; p<0.05). Hyperactivation of CDK5 induced by overexpression of the stress-induced activator p25 caused a slight lengthening of the AIS that was not significantly different from control neurons, but was significantly different from neurons in which CDK5 activity was inhibited either by expression of dnCDK5 or by roscovitine (Figure 1E; p<0.001).
Live-cell imaging of 6–7 DIV hippocampal neurons transfected with full-length GFP-tagged AnkG11 led to similar results. Live cultured neurons exhibited significantly shortened regions of AnkG signal when CDK5 was inhibited by expression of dnCDK5, addition of roscovitine, or depletion of endogenous CDK5 by siRNA (Figure 1G–I; p<0.05). In contrast, co-transfection of the CDK5 activator p25 did not induce a significant lengthening of the AnkG-positive AIS as compared to control neurons, although the effects of p25 were significant as compared to neurons with CDK5 inhibited by dnCDK5, roscovitine, or CDK5 knockdown (Figure 1H–I; p<0.01). Taken together, these observations suggest that CDK5 maintains AIS length, a function that is conserved from insects to mammals.
The CDK5-dependent change in AIS length led us to question whether other structural aspects of the AIS were altered by CDK5 activity. A prominent feature of the AIS and axonal cytoskeleton is parallel actin rings that line the membrane, running perpendicular to the length of the AIS. These parallel rings stretch from the start of the AIS down into the axon and display a characteristic spacing of ~180nm.13,35 Actin rings are predicted to provide structural support and stability to the narrow and otherwise fragile axon.35,36
We assessed the integrity of the actin rings upon CDK5 manipulation using STED super-resolution microscopy on 8 DIV fixed hippocampal neurons stained with fluorescently-tagged phalloidin.35,37 In control neurons, periodic actin rings were observed throughout the AIS (Figure S1A) as previously described.35 Activation of CDK5 by p25 overexpression preserved actin ring structure, but led to smaller gaps between neighboring rings (Figure S1B). In contrast, inactivation of CDK5 by roscovitine or expression of dnCDK5 caused disruption of ring orientation and formation (Figure S1A). Thus, super-resolution microscopy reveals that CDK5 is necessary to preserve actin cytoskeleton organization within the AIS, in addition to modulating AIS length.
Inhibition of CDK5 alters axonal microtubule polarity
Microtubules, like actin, are uniquely organized within the AIS and axon of mammalian neurons. In proximal dendrites, microtubules exhibit mixed polarity: they can be oriented with their plus ends towards the cell body or towards the end of the dendrite.15,16 In contrast, within the AIS and axon, microtubules are found with uniform plus-end-out polarity15,16,38 (Figure 2A). This organization facilitates the polarized activities of the anterograde motor kinesin and retrograde motor dynein, moving cargo either toward or away from the axon terminal, respectively.
Figure 2. Inhibition of CDK5 activity causes mis-polarization of microtubules in the AIS.

(A) Schematic of microtubule polarity in axons. Magnification depicts EB3 tracking with the growing plus-end of axonal microtubules, oriented uniformly plus-end-out in the axon.
(B) Kymographs of EB3 motion in live-cell fluorescence microscopy of 7 DIV rat hippocampal neurons expressing mCherry-EB3. Schematic on far right highlights 10 randomly chosen anterograde-directed EB3 comets in axons (black), and any visible retrograde-directed EB3 comets (purple). Black bracket on kymographs indicate the AIS, which has intrinsic EB3 binding. All visible comets throughout the AIS and axon were used for analysis.
(C) Quantification of retrograde-directed EB3 motility in the axon from 7 DIV rat hippocampal neurons as in (B).
(D) Quantification of density of EB3 comets in the axons of 7 DIV rat hippocampal neurons as in (B).
See methods for the sequence of the siRNA oligonucleotide used for knockdown. Scale bar represents 10µm (horizontal) and 30 seconds (vertical). Graphs depict means ± SEM; n ≥ 14 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (****p < 0.0001), n.s. indicates results not significant.
To determine if microtubule structure is grossly disrupted in the AIS when CDK5 is inhibited, we assessed microtubule localization with super-resolution STED microscopy. Hippocampal neurons were fixed at 8 DIV and stained with α-tubulin (Figure S2A). We observed no changes in microtubule bundle number in the AIS with increased or decreased CDK5 activity (Figure S2B). However, the polarity of microtubules within the bundles, rather than the number of bundles, determines the effectiveness of motor protein-driven transport.38 Thus, we tracked end-binding protein 3 (EB3), which binds to the dynamic plus-ends of microtubules, as well as to the membrane in the AIS (Figure 2A).16 EB3 binds preferentially to the growing end of the microtubule, resulting in a comet-like appearance during live cell imaging, and short angled runs in kymographs of the resulting videos (Figure 2B, control). Consistent with prior studies, we found that in dendrites, EB3 comets appear bi-directional, due to the mixed polarity of microtubules within this compartment (Figure S2C).15,16 However, in the AIS and axon of control neurons, EB3 comets are almost uniformly anterograde-directed in control cells, as expected. We also noted brighter background levels of EB3 within the AIS, denoted by brackets in Figure 2B. This is likely due to the previously noted association of EB3 with the cell membrane at the AIS (Figure 2A).16
Activation of CDK5 by p25 had no effect on microtubule polarity in dendrites or within the AIS and axon. In contrast, inhibition of CDK5 led to a significant increase in the number of observed retrograde-directed EB3 comets in the AIS and axon (Figure 2B; p<0.0001). The number of retrograde-directed EB3 comets, and thus minus-end-out microtubules, increased from <5% in control neurons to ~20% when CDK5 was inhibited by dnCDK5 expression or by depletion of endogenous CDK5 (Figure 2C). The observed change in microtubule orientation occurred without affecting the overall density of EB3 comets within the axon (Figure 2D), indicating that the treatments did not grossly disrupt microtubule dynamics, nor did we observe significant changes in microtubule orientation in dendrites (Figure S2C). This loss of uniform polarity of microtubules has the potential to change the trafficking of cargo within the AIS.
Dynein cofactors Lis1 and phospho-Ndel1 are required for maintenance of proper microtubule polarity in the AIS and axon
CDK5 is not known to directly phosphorylate microtubules or alter their polarity. However, CDK5 can indirectly influence microtubules through control of dynein, the minus-end-directed microtubule motor protein. This regulation depends on phosphorylation of Ndel1 by CDK5. Recently, Kuijpers17 demonstrated that Ndel1 activation of dynein is important for proper sorting of somatodendritic cargo into the correct neuronal compartment. We sought to determine if this Ndel1-dependent sorting mechanism was also regulated by CDK5.
As previously shown,17 we found that wild-type Ndel1 was enriched in the AIS (Figure S3A–B, left). A phospho-deficient Ndel1 5A mutant in which all five CDK5 phosphorylation sites were mutated from serine to alanine residues39,40 also localized primarily to the AIS, although there is some increased localization to the axon distal to the AIS (Figure S3A–B, right). Thus, recruitment of Ndel1 to the AIS is independent of CDK5 phosphorylation.
Ndel1 requires Lis1 to strongly affect dynein motility.41 Together, Lis1 and Ndel1 form a 2:2 complex to bind dynein and cause it to latch onto the microtubule, both enhancing initiation but also disrupting the processivity of dynein-driven transport.27,29,42 To determine if Ndel1, Lis1, and dynein are required to mediate the effects of CDK5-driven changes in microtubule polarity within the AIS and axon, we assessed the movement of mCherry-tagged EB3 in 7 DIV rat hippocampal neurons transfected with either mutant Ndel1 5A or a mutant Lis1 construct containing a point mutation in its dynein binding domain (K147A). Both the Ndel1 and the Lis1 mutant constructs recapitulated the effects of CDK5 inhibition on EB3 comet direction, and thus, microtubule polarity. In the AIS and axons of transfected neurons, retrograde-directed EB3 comets were visible with both the Ndel1 5A and Lis1 K147A mutations (Figure 3A). Quantification of retrograde-directed EB3 comets demonstrated that expression of either the Ndel1 or Lis1 mutants let to a significantly greater percent of minus-end-outward microtubules than observed in control neurons (Figure 3B; p<0.001). Moreover, hyperactivation of CDK5 through expression of p25 was not sufficient to rescue the disrupted microtubule phenotype in neurons co-expressing phospho-deficient Ndel1 (Figure 3A–B).
Figure 3. Dynein cofactors Ndel1 and Lis1 require CDK5 phosphorylation to promote proper polarization of microtubules.

(A) Kymographs of EB3 motion in the axons of 7 DIV rat hippocampal neurons. Schematic on the right highlights 10 randomly chosen anterograde-directed EB3 comets in axons (black), and any visible retrograde-directed EB3 comets (red). All visible comets throughout the AIS and axon were used for analysis.
(B) Quantification of retrograde-directed EB3 motility in the axons of 7 DIV rat hippocampal neurons as in (A).
(C) Schematic depicting CDK5/p35 phosphorylating Ndel1, promoting Ndel1 recruitment of Lis1 to dynein, and leading to interaction of dynein with the microtubule.
Scale bar represents 10µm (horizontal) and 30 seconds (vertical). Graph depicts means ± SEM; n ≥ 21 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on the graph (***p<0.001).
Taken together, these findings suggest that the CDK5-dependent phosphorylation of Ndel1, not CDK5 itself or unphosphorylated Ndel1, is required to maintain the proper polarity of microtubules within the AIS and axon of mammalian neurons. As CDK5 activity acts through Ndel1 and Lis1 to regulate dynein motor function (Figure 3C), we hypothesize that the observed effects of CDK5 disruption are due to dysregulation of dynein activation in the AIS. These results confirm previous observations in Drosophila wherein inhibition of dynein or NudE disrupted axonal microtubule polarity,18,43 but extend these findings by demonstrating the upstream regulatory pathway involved. We next sought to determine if other known functions of dynein in the AIS are affected by CDK5 inhibition.
Polarized trafficking of somatodendritic cargo at the AIS requires CDK5 activity
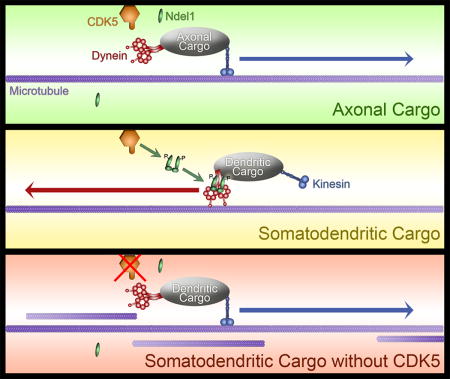
Disruption of the microtubule cytoskeleton in the AIS occurs with inhibition of CDK5. This suggests that altering CDK5 activity may affect sorting of somatodendritic and axon-bound cargos. Somatodendritic cargos such as the transferrin receptor (TfR) preferentially localize to the dendrites and cell body of mature hippocampal neurons, which we confirmed by fluorescence microscopy of tagged TfR (Figure 4A). TfR puncta enter the AIS at a constant rate, but once in the AIS, these cargos stop, reverse direction, and return to the cell body (Figure 4B).44 We hypothesized that CDK5 activates dynein-dependent retrieval of dendritic cargos that enter the AIS, and ensures their return to the dendrites.
Figure 4. CDK5 controls trafficking of somatodendritic TfR puncta.

(A) Live-cell fluorescence imaging of 7 DIV rat hippocampal neuron expressing Halo-tagged TfR and GFP-tagged AnkG.
(B) Schematic of TfR behavior in hippocampal neurons. TfR (red dots) localize to the somatodendritic compartment, and upon entering the AIS (green), rebound (red arrow) into the cell body rather than continue into the axon proper (over the dotted black line).
(C) Kymographs of TfR motion in 7 DIV rat hippocampal neurons expressing indicated CDK5 constructs. Individual runs highlighted to the right: runs shown in pink, axon in light green and AIS, as determined by co-expression of NaV II–III, in dark green. Yellow arrow marks the same run on both the kymograph and the cartoon.
(D) Quantification of the ratio of total TfR density in the axon versus the total to enter the AIS and axon in 7 DIV rat hippocampal neurons transfected with varying CDK5 constructs as in (C).
(E) Quantification of the total density of TfR puncta within both the AIS and axonal compartment in 7 DIV rat hippocampal neurons transfected with varying CDK5 constructs as in (C).
(F) Quantification of the density of TfR puncta in the axon of 7 DIV rat hippocampal neurons transfected with varying CDK5 constructs as in (C).
(G) Quantification of the density of TfR puncta in the AIS of 7 DIV rat hippocampal neurons transfected with varying CDK5 constructs as in (C).
(H) Quantification of the percent of TfR puncta rebounding from the AIS/axon boundary from 7 DIV rat hippocampal neurons transfected with varying CDK5 constructs as in (C).
(I) Quantification of the density of cytoplasmic or surface puncta that make it past the AIS and into the axon as determined by selective ligand addition to 7 DIV rat hippocampal neurons expressing TfR-HTC. Scale bars represent 10µm and 10 seconds. Graphs depict means ± SEM; n ≥ 22 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (*p<0.5, **p < 0.01, ****p < 0.0001).
To determine if CDK5 regulates this sorting, we transfected 6 DIV rat hippocampal neurons with Halo-tagged TfR, an AIS marker, and either the CDK5 activator p25, or the dominant-negative CDK5 inhibitor, dnCDK5.22 In parallel, we depleted CDK5 by siRNA, as previously described.27,45 We then examined the trafficking of TfR into and out of the AIS using live-cell microscopy. When CDK5 was active at basal levels, or when CDK5 activity was elevated by expression of p25, TfR puncta entered the AIS, stopped, and then switched direction to return to the cell body (Figure 4C, S4A). However, when CDK5 was inhibited by dnCDK5, TfR puncta that entered the AIS passed straight through the AIS and into the axon (Figure 4C, S4A). Once in the axon, TfR puncta continued to move in the anterograde direction towards the axon terminal. This pronounced alteration in TfR trafficking led to a significant increase in TfR puncta that enter into the axon (Figure 4D; p<0.0001). Depletion of endogenous CDK5 by siRNA had a similar effect, inducing a significant increase in the ratio of axonal/total TfR puncta (Figure 4D). Live-cell imaging demonstrates that this change was caused by a loss of barrier function at the AIS, as TfR puncta were observed to move processively through the AIS in the absence of CDK5, in contrast to the directional reversals at the AIS that predominated in the control neurons (Figure 4C).
This change in the ability of somatodendritic cargo to aberrantly enter the axon is not the byproduct of an increase in overall cargo density, as the total density of TfR puncta in the AIS and axon combined remained steady despite changes in CDK5 activity (Figure 4E). Rather, an increase in TfR density was only found within the axon, while a concomitant decrease was observed in the AIS region of the cell when CDK5 was inhibited or depleted (Figure 4F–G; p<0.001). This increase in axonal TfR density was due to an overall increase in the percentage of TfR puncta aberrantly continuing past the AIS-axon boundary, rather than rebounding from it to return to the soma (Figure 4H). These changes in TfR trafficking upon inhibition or depletion of CDK5 indicate a critical role for kinase in the regulation of sorting at the AIS, but do not preclude the involvement of other regulatory mechanisms for dynein within the AIS.46,47
TfR is a transmembrane protein that binds to its ligand, transferrin, to deliver iron to the cell following receptor-mediated endocytosis.48 We wanted to determine whether the surface or the cytoplasmic pool of TfR was most strongly affected by changes in CDK5 activity, and thus establish whether the altered pattern of trafficking resulted from changes in membrane or cytoplasmic barrier integrity. To achieve this, we incubated neurons expressing Halo-tagged TfR with a pulse of non-cell-permeable Halo ligand to uniquely tag surface TfR, before chasing with a cell-permeable ligand of a different wavelength to identify cytoplasmic TfR. This permitted us to discriminate between cytoplasmic and surface TfR by color (Figure 4I). Although both the plasma-membrane-associated and the cytoplasmic pools of TfR demonstrated an increased density in the axons of neurons expressing dnCDK5, the cytoplasmic pool was more strongly affected, suggesting that the altered trafficking observed at the AIS was primarily due to a defect in microtubule-based transport rather than the failure of the cell-surface barrier.
Cargo sorting at the AIS is a phospho-Ndel1 and Lis1-dependent process
The observation that cytoplasmic transport of TfR was heavily influenced by changes in CDK5 activity led us to assess if microtubule-based transport by dynein was the target of regulation. As noted above, CDK5 does not directly phosphorylate dynein, but rather phosphorylates the dynein cofactor Ndel1,31,49 and Ndel1 has been recently localized to the AIS17 (see Figure 3C). To determine if the observed defect in sorting of somatodendritic cargo into the axon upon inhibition of CDK5 is due to a loss of Ndel1 phosphorylation, we transfected cells with the phospho-deficient Ndel1 5A construct and observed trafficking of Halo-tagged TfR.
Expression of the Ndel1 5A mutant phenocopied CDK5 inhibition; we observed the mislocalization of TfR into the axon due to altered trafficking at the AIS (Figure 5A). Again, TfR puncta consistently advanced processively through the AIS rather than halting their anterograde movement and switching to retrograde movement back to the soma as observed in control neurons. Furthermore, TfR mislocalization could neither be rescued by CDK5 overactivation nor exacerbated by CDK5 inhibition in neurons expressing Ndel1 5A, suggesting that CDK5 restriction of TfR from the axon is solely mediated by Ndel1 phosphorylation (Figure 5A). As with CDK5 inhibition, Ndel1 5A did not increase the overall density of TfR puncta entering the axonal region, indicating that the changes in axonal TfR observed are not due to higher cargo density (Figure 5B). Rather, expression of the Ndel1 mutant led to an increase in the number of TfR puncta that passed through the AIS and into the axon unimpeded (Figure 5C–D; p<0.0001), and decreased the overall percentage of TfR puncta that rebounded from within the AIS to return towards the soma (Figure 5E; p<0.0001). Together, these data indicate that CDK5 acts through Ndel1 phosphorylation and dynein activation at the AIS to regulate the return of somatodendritic cargo to the soma.
Figure 5. Proper trafficking of somatodendritic cargo depends on dynein cofactors Ndel1 and Lis1.

(A) Kymographs of TfR motion in live-cell fluorescence imaging of 7 DIV rat hippocampal neurons expressing a phosphorylation-deficient 5A Ndel1 mutant or a Lis1 K147A point mutation rendering it unable to bind dynein, and the indicated CDK5 constructs. Individual runs highlighted to the right: runs shown in pink, axon in light green and AIS, as determined by co-expression of NaV II–III, in dark green. Yellow arrow marks the same run on both the kymograph and the cartoon.
(B) Quantification of the additive density of TfR puncta in the axon plus AIS with expression of phospho-deficient Ndel1, dynein-binding Lis1 mutant, and varying CDK5 conditions in 7 DIV rat hippocampal neurons as in (A).
(C) Quantification of the density of TfR puncta in the AIS with varying CDK5 activity and expression of the Ndel1 or Lis1 mutant in 7 DIV rat hippocampal neurons as in (A).
(D) Quantification of the density of TfR puncta in the axon with varying CDK5 activity and expression of the Ndel1 or Lis1 mutant in 7 DIV rat hippocampal neurons as in (A).
(E) Quantification of the percent of TfR puncta rebounding from the AIS/axon boundary with varying CDK5 activity and expression of the Ndel1 or Lis1 mutant in 7 DIV rat hippocampal neurons as in (A).
Scale bar represents 10µm (vertical) and 10 seconds (horizontal). Graphs depict means ± SEM; n ≥ 20 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (****p < 0.0001).
To determine if Lis1 is also required to mediate the effects of CDK5 on transport of somatodendritic cargo, we again used the Lis1 K147A mutant that is incapable of binding dynein. Expression of Lis1 K147A led to similar defects in sorting at the AIS as were observed upon expression of the Ndel1 5A mutant or upon CDK5 inhibition. TfR puncta were transported through the AIS and into the axon, rather than stopping and returning to the cell body (Figure 5A, right). As expected, the density of TfR puncta within the axon increased, while the density within the AIS decreased, without altering the total density of TfR to enter the AIS (Figure 5B–D). Taken together, these results indicate that CDK5-dependent phosphorylation of Ndel1, formation of an Ndel1/Lis1/dynein complex, and subsequent activation of dynein is required for proper transport of somatodendritic cargo out of the AIS and back into the cell body.
Inhibition of the CDK5-dependent regulation of the Ndel1/Lis1/dynein complex causes mislocalization of Golgi fragments to the axon
As TfR-positive vesicles were mis-sorted in response to CDK5 inhibition or inhibition of the Ndel1/Lis1/dynein complex, we hypothesized that this mechanism might be more broadly applicable to other somatodendritic cargos. In particular, we explored whether Golgi fragments might become mislocalized to the axon under these conditions, and whether Golgi mislocalization might be sufficient to explain the disruption of microtubule polarity observed under these conditions. Microtubules nucleate from their slow growing minus ends,50 which are typically embedded in γ-tubulin surrounding the centrosomal microtubule organizing center within the cell body prior to the development of neuronal polarity.51 However, microtubules have also been shown to nucleate from the Golgi in neurons.52 We thus hypothesized that mislocalization of Golgi fragments to the axon might explain the observed increases in mis-oriented microtubules.
To localize the Golgi, we expressed mCherry-tagged GM130, a cis-Golgi matrix marker,53 in 7 DIV hippocampal neurons. We found that under control conditions, Golgi bodies predominantly localized to the soma (Figure 6A), consistent with prior studies.54 Inhibition of CDK5 increased the Golgi signal in both the axon and dendrites of hippocampal neurons (Figure 6B). While CDK5 inhibition altered Golgi localization in dendrites, CDK5 hyper-activation via p25 expression did not, suggesting that proper Golgi localization requires a minimum level of CDK5 activity but is not affected by upregulation of CDK5 (Figure 6C; p<0.01).
Figure 6. Inhibition of CDK5 or phospho-deficient Ndel1 mutant causes mislocalization of Golgi bodies.

(A) Live-cell fluorescence imaging of 7 DIV rat hippocampal neurons expressing mCherry-tagged Golgi marker GM130. Region of the AIS designated by white arrows, as indicated by co-transfection with NaV II–III. Individual Golgi bodies outside of the soma indicated by yellow arrowheads.
(B) Live-cell fluorescence imaging of 7 DIV rat hippocampal neurons expressing mCherry-tagged Golgi marker GM130 and BFP-tagged dnCDK5. Region of the AIS designated by white arrows, as indicated by co-transfection with NaV II–III. Individual Golgi bodies outside of the soma indicated by yellow arrowheads.
(C) Quantification of density of Golgi bodies in axons vs. dendrites of 7 DIV rat hippocampal neurons transfected with different CDK5 constructs as in (A) and (B).
(D) Live-cell fluorescence imaging of 7 DIV rat hippocampal neurons expressing mCherry-GM130 and untagged Ndel1 5A mutant. Region of the AIS designated by white arrows, as indicated by co-transfection with NaV II–III. Individual Golgi bodies outside of the soma indicated by yellow arrowheads.
(E) Quantification of density of Golgi bodies in axons vs. dendrites of 7 DIV rat hippocampal neurons transfected with Ndel1 5A mutant as in (D).
(F) Quantification of percent total retrograde EB3 comets originating within 15µm of the nearest Golgi body for the indicated CDK5 activity and Ndel1 mutant in rat hippocampal axons.
(G) Histogram of distance between retrograde-directed EB3 comets and the nearest axonal Golgi fragment for the indicated CDK5 activity and Ndel1 mutant in 7 DIV rat hippocampal neurons.
(H) Kymographs of EB3 motility (top row) and Golgi location (middle row) in 7 DIV rat hippocampal neurons. Schematic (bottom row) depicts all visible EB3 anterograde-directed comets (black), all visible retrograde-directed EB3 comets (green) and the location of any Golgi bodies (red).
Scale bar represents 10µm in (A), (B), and (D), and 5µm (horizontal) and 1 minute (vertical) in (G). Graphs depict means ± SEM; n ≥ 18 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (**p<0.01, ***p<0.001, ****p < 0.0001).
To determine whether these CDK5-dependent changes were mediated by the Ndel1/Lis1/dynein complex, we tested the effect of expression the phosphorylation-deficient Ndel1 5A mutant on Golgi localization. Expression of this construct was sufficient to significantly increase the density of Golgi fragments in both axons and dendrites in comparison to control neurons (Figure 6D, E; p<0.01). Kymograph analysis indicates that some of the axonal Golgi fragments identified in either dnCDK5- or Ndel1 5A- expressing neurons displayed a low level of motility (Figure S5A). These findings suggest that restriction of Golgi fragments to the soma is CDK5-dependent, and raise the possibility that mis-localized Golgi fragments may serve as a locus from which mis-oriented microtubules could polymerize.
Altered dynein regulation increases mis-polarized microtubules associated with axonal Golgi fragments
The microtubule nucleation protein γ-tubulin is associated with the Golgi, and consequently the Golgi can serve as a secondary site for microtubule nucleation within the cell.55,56 Therefore, we hypothesized that the abnormally polarized plus-end-in microtubules we observed upon CDK5 inhibition or long-term dynein disruption via expression of mutant forms of Ndel1 or Lis1 might be nucleated from aberrantly localized Golgi fragments in the AIS and axon. To test this hypothesis, we transfected 7 DIV rat hippocampal neurons with tagged EB3 to identify microtubules, GM130 to identify Golgi, and an AIS marker to unambiguously identify the axon. We compared the localization of these markers in live cell assays in neurons expressing either dnCDK5 or the Ndel1 5A mutant.
As expected, both Golgi fragments and retrograde EB3 comets were rarely observed along the axons of control neurons. Hyper-activation of CDK5 did not affect either distribution (Figure 6F). However, when CDK5 was inhibited or Ndel1 phosphorylation by CDK5 was blocked, we did observe significant changes in both measures. Under these conditions, approximately 40% of total retrograde axonal EB3 comets were found in close proximity, within 15µm, of the nearest axonal Golgi body (Figure 6F; p<0.01). The majority of retrograde EB3 comets were found within 5µm of a Golgi fragment; this was especially pronounced for neurons expressing dnCDK5 or Ndel1 5A (Figure 6G). This close spatial proximity (Figure 6H) suggests that mis-localized axonal Golgi-associated γ-tubulin might be serving as a point of nucleation and stabilization for mis-oriented axonal microtubules that are observed when dynein activity is disrupted. Thus, CDK5 inhibition or alterations in the regulated formation of the Ndel1/Lis1/dynein complex disrupts not only the effective sorting of axonal and somatodendritic cargo at the AIS, but additionally alters the organization of the structure of the microtubule tracks along which the axonal cargos are transported.
Temporally regulated dynein activation in the AIS is required for proper sorting of somatodendritic cargo
Altered CDK5 activity, or disruption of the Ndel1/Lis1-dependent regulation of dynein, leads to both defects in the polarity of axonal microtubules (Figures 2 and 3) and defective sorting at the AIS (Figures 4 and 5). Potentially, these observations are directly linked, as the polarity defects might in turn cause defects in cargo sorting. Alternatively, the dysregulation of cargo sorting may occur on a different time scale than changes in microtubule polarity, leading to different short-term and long-term effects of dynein inhibition. To differentiate between these mechanisms, we compared the effects of acute dynein inhibition via the dynein inhibitor ciliobrevin D to the longer-term inhibition of dynein induced by expression of mutant forms of CDK5, Ndel1, or Lis1.
We found that short-term addition of ciliobrevin D57 to the culture media of 7 DIV rat hippocampal neurons was sufficient to cause a pronounced defect in the sorting of TfR puncta at the AIS (Figure 7A). This led to a significant increase in the density of TfR puncta aberrantly located in the axon (Figure 7B; p<0.0001). Under these same conditions, we did not observe significant alterations in the polarity of microtubules in the axon and AIS, as assessed by the directionality of EB3 comet tails (Figure 7C). Together, these observations indicate that dynein activity is actively required for sorting at the AIS, although over a longer time course mis-organization of axonal microtubules caused by mis-regulation of dynein may further contribute to sorting deficits.
Figure 7. CDK5 differentially disrupts somatodendritic cargo trafficking and microtubule polarity in a temporally specific manner, and regulates dynein activation and axonal microtubule polarity.

(A) Kymographs of TfR motion in 7 DIV rat hippocampal neurons in the presence of the dynein inhibitor ciliobrevin D. Individual runs highlighted to the right: runs shown in pink, axon in light green and AIS, as determined by co-expression of NaV II–III, in dark green. Yellow arrow marks the same run on both the kymograph and the cartoon.
(B) Quantification of the density of TfR puncta in the AIS or the axon with short-term dynein inhibition with ciliobrevin D in 7 DIV rat hippocampal neurons as in (A).
(C) Kymograph of EB3 motion in the axons of 7 DIV rat hippocampal neurons expressing mCherry-EB3 after 1.5–3 hours of dynein inhibition with ciliobrevin D. Schematic below highlights all anterograde-directed EB3 comets in axons (black), and any visible retrograde-directed EB3 comets (one, green).
(D) Quantification of the percent of TfR puncta rebounding from and passing through the AIS/axon boundary with expression and exposure to various CDK5 and dynein activators and inhibitors in 7 DIV rat hippocampal neurons (from Figures 4–5, 7).
(E) AnkG (green) and CDK5/p35 (orange) localize to the neuronal membrane in the AIS. Ndel1 (yellow) binds to AnkG at the membrane, until phosphorylation by CDK5 causes it to release. Phosphorylated Ndel1 binds to Lis1 (blue) in a 2:2 ratio, which then recruits dynein (red). The Ndel1/Lis1/dynein complex engages the microtubule (purple), initiating retrograde-directed axonal transport. This activation can also occur at the membrane, activating cortical dynein.
(F) Axonal microtubules are uniformly oriented with their plus-ends out, while microtubules in the soma are regulated differently. Minus-end-out microtubules that enter the axon from the soma are returned to the soma by the actions of dynein attached to correctly polarized microtubules. Plus-end-out microtubules that enter the axon are propelled forward and permitted to stay in the axon.
(G) Golgi bodies (burgundy) contain γ-tubulin (indigo) at the cis-Golgi membrane. The minus-end of microtubules are stabilized through interactions with the γ-tubulin at the axonal Golgi bodies.
Scale bar represents 10µm and 10 seconds (A) or 30 seconds (C). Graphs depict means ± SEM; n ≥ 20 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (****p < 0.0001).
Discussion
Neurons rely on precise sorting of organelles and proteins to maintain polarized axonal and dendritic compartments. The principal site for sorting has been identified as coincident with the AIS4,5 or the pre-AIS (PAEZ7), but there is little consensus with regard to the mechanisms responsible for this sorting. Here we identify a molecular mechanism regulated by CDK5 and driven by dynein in conjunction with its effectors Ndel1 and Lis1. Recent work demonstrated Ndel1 localization to the AIS17; here we show that p35, the upstream activator of CDK5-dependent Ndel1 phosphorylation, is also highly enriched in the AIS. Further, we demonstrate that inhibition of CDK5 or abrogation of the CDK5-dependent phosphorylation of Ndel1 significantly affects the fidelity of sorting of somatodendritic cargo at the AIS (Figure 7D). These observations, in conjunction with previous observations in C. elegans,24,26 establish CDK5 as an upstream regulator of the polarized trafficking of both axonal and somatodendritic cargos.
Ndel1 and Lis1 act together to bind dynein and induce the formation of a high-affinity microtubule binding state.27,28,39,41,58,59 In this high affinity state, dynein binds tightly to the microtubule and is not disassociated by millimolar concentrations of ATP. This Ndel1/Lis1/dynein complex is transiently immobilized, as the binding of Lis1 to dynein sterically blocks the swing of the linker domain that is integral to the mechanochemisty of the dynein ATPase cycle.28 Despite this apparent inhibitory effect, Lis1 is actually required for initiation of axonal transport of both larger60 and smaller organelles29 along the axon. The model which best reconciles these results predicts that the transient binding of Ndel1 and Lis1 to dynein forms a high-affinity structure that promotes the recruitment of the motor-cargo complex to the microtubule.27 Dissociation of Lis1 from its binding site on the dynein head domain relieves this inhibition and promotes active transport.61,62
Ndel1 is highly enriched in the AIS,17 as is the CDK5 activator p35, as shown here. Further, we can now show that Ndel1/Lis1 complex provides tightly controlled spatial and temporal regulation of dynein motor activity (Figure 7E). Consistent with this model, inhibition or depletion of CDK5, blocking the CDK5-dependent phosphorylation of Ndel1, blocking the binding or Lis1 to dynein, or acutely blocking dynein activity pharmacologically all result in the same effect: disruption of the integrity of axodendritic sorting at the AIS.
While acute inhibition of dynein is sufficient to disrupt sorting, we also note that more prolonged inhibition of this pathway has multiple downstream effects on neuronal cytoarchitecture. First, alterations in the activity of CDK5 dramatically change both the length and the internal organization of the AIS. Previous work in Drosophila had noted a link between CDK5 activity and AIS length, thus suggesting a conserved function for the kinase when combined with our observations. However, we also noted an unexpected effect on the organization of actin rings in the AIS. These rings are a relatively recent discovery stemming from superresolution studies,13,35 and have been suggested to provide stability to the axon. The disarray in the organization of the actin rings observed upon dysregulation of CDK5 activity suggests that this kinase may be important for the maintenance of the mechanical stability of the axon. Further studies will be required to fully assess the underlying mechanisms as well as the effects of CDK5-dependent alterations on axonal integrity in response to mechanical stresses.
In parallel to the disruption of the actin cytoskeleton, we noted that inhibition of CDK5 activity also leads to pronounced remodeling of axonal microtubules. Specifically, we observed a 20% increase in mis-oriented, minus-end-out microtubules, a striking finding given the almost completely unipolar array normally found in the axons of hippocampal neurons.15 Our work, in conjunction with previous Drosophila data implicating dynein in the maintenance of microtubule organization in axons,18 raises the fundamental but unresolved question of how dynein controls axonal microtubule polarity. One model postulates that dynein activity is required to translocate mis-polarized microtubules out of the AIS and into the cell body (Figure 7F). In this model, microtubules with incorrect orientation may enter the AIS from the soma at random, but are then efficiently returned to the soma through the activity of dynein affixed to properly oriented microtubules. This would ensure that only microtubules with their plus-ends out would be able to enter the AIS and axon. In this model, inhibition of dynein would result in a gradual spread of mis-oriented microtubules from the AIS and into the axon. We did not observe significant increases in mis-oriented microtubules with short-term dynein inhibition (Figure 7C), however it is possible that permissive movement of large microtubules from the cell body into the axon occurs slowly, and would not have been captured during our three hour dynein inhibition. In support of this model, recent work has demonstrated that after 24 and 48 hours of dynein inhibition with ciliobrevin D, microtubule polarity in rat neurons becomes progressively less organized.63 Further work with continued observation of microtubule growth and orientation over longer periods following dynein inhibition will be necessary to fully confirm the underlying mechanism.
In an alternative model, misplaced microtubules may arise in the axon from local nucleation. Dynein is known to maintain both Golgi localization and Golgi integrity, and inhibition of dynein function leads to Golgi fragmentation.64,65 Here, we show that CDK5 is an upstream regulator of dynein activity at the Golgi, as inhibition of the CDK5-dependent regulation of the Ndel1/Lis1/dynein complex leads to Golgi fragmentation. Strikingly, the resulting Golgi fragments do not remain sequestered in the soma, but instead are distributed through the axon and dendritic processes. We propose that the misorganization of the normally highly unipolar axonal microtubule cytoskeleton may be dependent on Golgi fragmentation. In support of this hypothesis, we noted a significant increase in actively growing mis-oriented microtubule plus-ends in close proximity to Golgi fragments along the axon. We propose that axonal Golgi and associated γ-tubulin act to stabilize the minus-end of mis-polarized microtubules, thereby supporting the aberrant growth of mis-oriented microtubules (Figure 7G), in addition to any pre-formed microtubule which may enter the axon as a result of dynein inhibition.
Identification of a key role for CDK5 as a regulator of dynein-mediated functions raises interesting implications for both neurodevelopment and neurodegeneration. Previously, CDK5 activity has been shown to affect the transport of dynein-dependent cargos in the mid-axon.25,27 Here, we define a new function for CDK5 in sorting at the AIS and maintenance of a highly polarized axonal microtubule cytoskeleton in a dynein-dependent fashion. Dynein is localized throughout the neuron during the initial states of neuronal development66 and as such can play diverse roles in neuronal differentiation, migration, polarity development, and pan-neuronal transport.67,68 In adult neurons, dynein’s role in sorting at the AIS may become particularly relevant. Failure of the filter function within the AIS due to changes in cytoskeletal structure have been linked to Alzheimer’s disease. Axonal pathology in the APP/PS1 mouse model of Alzheimer’s is due in part to a decrease in AnkG and thus impairment of the AIS, which can be rescued by forced expression of AnkG.69 Further, CDK5 activation is altered in a number of neurodegenerative diseases including Alzheimer’s70 and ALS71; dysregulated CDK5 may adversely impact polarized sorting in neurons as well as affecting transport along the axon, leading to further enhancement of cellular stress.
Overall, we identify CDK5 as a master regulator of dynein activity within the AIS. Though a mechanism involving phosphorylation of Ndel1, and formation of an Ndel1/Lis1/dynein complex, CDK5 helps to coordinate many dynein-dependent processes. Without CDK5-based activation of dynein, hippocampal neurons exhibit disruption of microtubule polarity, mistrafficking of somatodendritic cargo, disruption of the Golgi, and mislocalization of Golgi fragments throughout the neuron. Therefore, coherence of the axon initial segment, and thus regulated axonal sorting requires tightly controlled dynein activity, which is regulated by CDK5.
Materials and Methods
Constructs and Antibodies
Mammalian expression plasmids include Ndel1, Ndel1 5A, and Lis1 (D.S. Smith, University of South Carolina), p25-BFP and dnCDK5-BFP (described in Reference 27), YFP-NavII-III (Addgene), AnkG-GFP (V. Bennett, Duke University), mCherry- and EGFP-EB3 (Addgene), mCherry-GM130 (M. Marks, University of Pennsylvania), and Halo-STOP (Addgene). TfR-RFP (Addgene) was cloned to replace the RFP moiety with a Halo sequence, mCherry was inserted into the p35 plasmid to create mCherry-p35, and a GFP tag was cloned into the Ndel1 and Ndel1 5A plasmids. SNAPf was added to replace the mCherry tag in GM130 to permit visualization with far-red. Depletion of the rat mRNA transcript of CDK5 was achieved using the short (19 bp) siRNA sequence (forward strand): 5′-AAGCCGUACCCGAUGUAUC-3′, which has been extensively validated,45,72,73 referred to here as “siRNA KD”. A pool of four siRNA oligonucleotides (“siCtrl”, Dharmacon ON-TARGET siRNA Controls) was used as an siRNA control. Antibodies obtained from commercial sources include rabbit-anti-AnkG (Santa Cruz), goat-anti-BFP (Santa Cruz), mouse-anti-α-tubulin (Sigma-Aldrich), mouse-anti-HaloTag (Promega), Alexa Fluor 405 and 488 anti-goat, Alexa Fluor 350, 488, 555, and 594 anti-mouse, and Alexa Fluor 488 and 555 anti-rabbit (Invitrogen and Life Sciences). Other chemical reagents include roscovitine (Cell Signaling) and Alexa Fluor 488 and 594 Phalloidin (Invitrogen).
Primary Neuron Culture and Transfection
Primary hippocampal neurons from E18–20 Sprague Dawley rat embryos were dissociated at the Neuron Culture Service Center at the University of Pennsylvania, as described.74 Neurons were plated for 6–8 hours in minimal essential media (MEM, Gibco) supplemented with 10% heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, and 33 mM glucose. A full media change was then performed and neurons were subsequently maintained in neurobasal medium (Gibco) supplemented with 2% B-27 (Invitrogen), 2mM GlutaMAX (Gibco), 33 mM glucose, 100 U/ml penicillin, and 100 µg/ml streptomycin in a 37°C incubator with 5% CO2. Neurons were plated at a density of 180,000 – 230,000 cells/ml on poly-L-lysine (0.5 mg/ml; Sigma) coated glass bottom dishes (MatTek). At 3 DIV, 1 µM AraC was added and a half-volume media change was performed. 6–7 DIV hippocampal neurons were transfected with 1–3 constructs as necessary using Lipofectamine 2000 reagent (Invitrogen), and a second half-media change was performed. Neurons were further incubated for 16–36 hours, prior to imaging or fixation.
Live Cell Imaging
Neurons were maintained in Hibernate E (Brainbits) supplemented with 2% B-27 and 2mM GlutaMAX for the duration of imaging. Imaging of live cells was performed on an inverted epifluorescence microscope (DM1600B, Leica Camera AG) or Ultraview Vox spinning disc confocal system (PerkinElmer) in an environmental chamber at 37°C. Digital images were acquired with an ORCA-R2 (Hamamatsu) using LAS-AF software (Leica), or with an Ultraview Photokinesis (PerkinElmer) unit on an inverted Nikon Ti microscope with a C91005A0 EM-CCD (Hamamatsu) camera controlled by Volocity software (PerkinElmer). Images were acquired at 2 frames per second for 2 minutes for TfR, 1 frame per second for 3 minutes for EB3 and GM130, and 1 frame every 2 seconds for 2 minutes for p35.
STED Fixation and Imaging
Hippocampal neurons were isolated and cultured as above, but plated at a density of 250,000 cells/coverslip. Coverslips (Fisherbrand 12-544A #1.5) were conditioned prior to neuron plating with an overnight acid wash in 0.25% acetic acid, followed by three rinses in EtOH, and an additional overnight coating with poly-L-lysine at 37°C.
At 7–8 DIV, neurons were fixed in a 37°C mixture of 4% PFA and 4% sucrose for 8 minutes, washed three times with BPS, permeabilized for 5 minutes in 0.1% Triton X-100 in BPS, and blocked for 1 hour in 5% goat serum, 1% BSA, and 0.1% Triton X-100 in BPS. Coverslips were placed in a humidity chamber and incubated in primary antibody diluted in blocking buffer for 1–1.5 hours at room temperature, then washed three times for five minutes in BPS. The coverslips were then incubated in a light-protected humidity chamber in secondary antibody diluted in blocking buffer for 1 hour, followed by three five-minute washes in PBS. Coverslips were then rinsed once in Milli-Q water and mounted in ProLong Gold (Life Technologies).
STED microscopy was performed on a Leica DMI 6000 Inverted laser scanning confocal microscope equipped with 592nm and 660nm STED depletion laser.
Motility and Density Analysis
Kymographs were generated using MetaMorph or Fiji. Density of puncta was determined by line scan within the first 15 frames of the video on the kymograph and normalized by kymograph length.
Intensity and Distance Quantification
Intensity of p35 and Ndel1 fluorescence was analyzed using Fiji. ROI were taken around the AIS, dendrite (>20µm from the cell body), axon (>20µm from end of the AIS), and background near the AIS. Each ROI was ~10µm other than the background selection, which was larger. Average intensity was quantified for each region, after background subtraction.
The distance over which AnkG signal persisted was calculated from STED or epifluorescence microscopy images. In both cases, a segmented line was drawn along the AIS and axon, starting at the cell body. The segment line, width of 3–5px, was straightened in Fiji, and the intensity values along the line were exported to Excel. A rolling average was calculated for each value based on the 5 nearest values. The data was then normalized to the maximum (maximum set to 1), and the half-maximum value (approximately 0.72) was calculated using the starting intensity and the maximum for the individual neuron. Individual normalized straightened AIS intensity measurements were aligned by setting the half-maximum at 0 microns, and input as grouped data sets in Prism (GraphPad). The end of signal intensity over background was determined empirically, and set at 0.4 for fixed-cell and 0.5 for live-cell imaging. The length over which signal persisted was calculated by binning each experimental condition by biological replicate, and determining the background for each. The point at which the signal crossed to background was considered to be the length of AnkG signal. These average signal lengths were used to determine differences between conditions.
Halo and SNAP Ligand Treatment
Halo-tagged TfR was incubated with HaloTag TMR Ligand (Promega) or HaloTag Oregon Green Ligand (Promega). For single-color experiments, TMR was used. Each plate required 0.25µl TMR ligand diluted in 375µl culture media to create a 10× working stock. The 10× stock was further diluted into 1× stock using 1.8ml media and 200ml 10× stock per dish. Media on the plates was removed and replaced with 2ml of the media containing TMR ligand. The plates were returned to the incubator for 15 minutes, with occasional gentle rocking of the dish. Unbound ligand was then removed by rinsing twice with 2ml fresh warm media, and the plates were incubated in the third and final wash for 30 min at 37°C. Plates were then treated as normal for imaging. For dual-labeling, plates were first incubated with Oregon Green (non-cell permeant) for 15 minutes, rinsed once, and then incubated as above with cell-permeant TMR ligand.
SNAP staining was performed similarly to HaloTag labeling, but neurons were incubated for 25 rather than 15 minutes in the presence of the label. SNAP-Cell 647-SiR (NE Biolabs) was used to visualize SNAPf-tagged GM130 in the far-red channel.
Supplementary Material
Figure S1, Related to Figure 1: Inhibition of CDK5 disrupts periodic axonal actin ring structures.
(A) STED super-resolution images of the AIS from 8 DIV rat hippocampal neurons stained with phalloidin. Cartoons of visible actin rings depicted below each image. Scale bar represent 2µm.
(B) Quantification of separation between adjacent actin rings from STED super-resolution images of 8 DIV rat hippocampal neurons. Each value represents the average separation of rings within one axon; only regions with consecutive visible rings were analyzed (minimum of 4 rings), largest region with consecutive rings was chosen for analysis per axon. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (***p < 0.001).
Figure S2, Related to Figure 2: CDK5 activity has no effect on axonal microtubule bundling or microtubule directionality in dendrites.
(A) STED super-resolution images of the AIS from 9 DIV rat hippocampal neurons stained for α-tubulin. Scale bar represents 2µm.
(B) Quantification of the number of microtubule bundles from STED super-resolution images of 9 DIV rat hippocampal neurons. Graph depicts means ± SEM; n ≥ 12 neurons from at least three biological replicates. No significant differences noted between conditions (one-way ANOVA with Tukey’s post-hoc test).
(C) Kymographs of EB3 motion in live-cell fluorescence microscopy in dendrites of 7 DIV rat hippocampal neurons expressing mCherry-EB3. Scale bar represents 10µm (horizontal) and 30 seconds (vertical).
Figure S3, Related to Figure 3: Dynein cofactor Ndel1 localizes to the AIS.
(A) Live-cell fluorescence imaging of 7 DIV rat hippocampal neurons expressing Halo-tagged TfR (soma and dendritic marker) and GFP-tagged Ndel1 (left) or Ndel1 5A mutant (right).
(B) Quantification of Ndel1 fluorescence intensity from the AIS, axon, and dendrite of 7 DIV hippocampal neurons expressing wild type or mutant Ndel1 as in (A). Graphs depict means ± SEM; n ≥ 21 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (*p < 0.05, ****p < 0.0001).
Figure S4, Related to Figure 4: CDK5 inhibition disrupts proper motility of somatodendritic TfR puncta.
(A) Kymographs of TfR motion in 7 DIV rat hippocampal neurons expressing indicated CDK5 constructs. Individual runs highlighted to the right: runs shown in pink, axon in light green and AIS, as determined by co-expression of NaV II–III, in dark green. Yellow arrow marks the same run on both the kymograph and the cartoon. Videos recorded at 100× with a confocal microscope for higher magnification than Figure 4C.
Figure S5, Related to Figure 6: Golgi fragments are present and motile in the axon when CDK5 is inhibited or unable to phosphorylate Ndel1.
(A) Kymographs of mCherry-tagged GM130 in 7 DIV rat hippocampal neurons co-expressing the indicated construct.
Synopsis.
Klinman et al. demonstrate evidence of localized dynein activation at the AIS, and show that upstream CDK5 activation is required to maintain the normal organization of the AIS and its role as a cargo filter. They determine that disruption of the CDK5-Ndel1-Lis1-dynein mechanism results in aberrant permeability of the AIS to dendritic cargo and changes in axonal microtubule polarity, in a time-dependent fashion. These findings indicate an essential homeostatic function for dynein and CDK5 in controlling AIS integrity.
Acknowledgments
This work was supported by NIH pre-doctoral training grant 5T32AG000255 to E.K. and NIH grants R37NS060698 and R01GM48661 to E.L.F.H. We thank Andrea Stout, Andrea Stavoe, Amy Ghiretti, and Jerome Molleston for their invaluable assistance.
Footnotes
The authors have no conflict of interest to declare.
References
- 1.Lasiecka ZM, Winckler B. Mechanisms of polarized membrane trafficking in neurons -- focusing in on endosomes. Mol Cell Neurosci. 2011;48(4):278–87. doi: 10.1016/j.mcn.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dotti CG, Sullivan Ca, Banker Ga. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8(4):1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maeder CI, Shen K, Hoogenraad CC. Axon and dendritic trafficking. Curr Opin Neurobiol. 2014;27:165–70. doi: 10.1016/j.conb.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 4.Nakada C, Ritchie K, Oba Y, et al. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol. 2003;5(7):626–633. doi: 10.1038/ncb1009. [DOI] [PubMed] [Google Scholar]
- 5.Song A-H, Wang D, Chen G, et al. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136(6):1148–60. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999 Feb;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- 7.Farías GG, Guardia CM, Britt DJ, Guo X, Bonifacino JS. Sorting of Dendritic and Axonal Vesicles at the Pre-axonal Exclusion Zone. Cell Rep. 2015;13(6):1221–32. doi: 10.1016/j.celrep.2015.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183(4):635–40. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11(8):552–562. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- 10.Jones SL, Korobova F, Svitkina T. Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J Cell Biol. 2014;205(1):67–81. doi: 10.1083/jcb.201401045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155(5):739–46. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi T, Storrie B, Simons K, Dotti CG. A functional barrier to movement of lipids in polarized neurons. Nature. 1992;359:647–650. doi: 10.1038/359647a0. [DOI] [PubMed] [Google Scholar]
- 13.D’Este E, Kamin D, Göttfert F, El-Hady A, Hell SW. STED nanoscopy reveals the ubiquity of subcortical cytoskeleton periodicity in living neurons. Cell Rep. 2015;10(8):1246–51. doi: 10.1016/j.celrep.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe K, Al-Bassam S, Miyazaki Y, Wandless TJ, Webster P, Arnold DB. Networks of polarized actin filaments in the axon initial segment provide a mechanism for sorting axonal and dendritic proteins. Cell Rep. 2012;2(6):1546–53. doi: 10.1016/j.celrep.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baas PW, Deitch JS, Black MM, Banker GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci. 1988;85(21):8335–9. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stepanova T, Slemmer J, Hoogenraad CC, et al. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23(7):2655–64. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuijpers M, van de Willige D, Freal A, et al. Dynein Regulator NDEL1 Controls Polarized Cargo Transport at the Axon Initial Segment. Neuron. 2016;89(3):461–71. doi: 10.1016/j.neuron.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y, Wildonger J, Ye B, et al. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol. 2008;10(10):1172–80. doi: 10.1038/ncb1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur ELF. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84(2):292–309. doi: 10.1016/j.neuron.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumazawa A, Mita N, Hirasawa M, et al. Cyclin-dependent kinase 5 is required for normal cerebellar development. Mol Cell Neurosci. 2013;52:97–105. doi: 10.1016/j.mcn.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohshima T, Ward J, Huh C, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci. 1996;93(20):11173–8. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10(7):816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 23.Morfini G, Szebenyi G, Brown H, et al. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO. 2004;23(11):2235–45. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ou C-Y, Poon VY, Maeder CI, et al. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell. 2010;141(5):846–58. doi: 10.1016/j.cell.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pandey JP, Smith DS. A Cdk5-dependent switch regulates Lis1/Ndel1/dynein-driven organelle transport in adult axons. J Neurosci. 2011;31(47):17207–19. doi: 10.1523/JNEUROSCI.4108-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodwin PR, Sasaki JM, Juo P. Cyclin-dependent kinase 5 regulates the polarized trafficking of neuropeptide-containing dense-core vesicles in Caenorhabditis elegans motor neurons. J Neurosci. 2012;32(24):8158–72. doi: 10.1523/JNEUROSCI.0251-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klinman E, Holzbaur ELF. Stress-Induced CDK5 Activation Disrupts Axonal Transport via Lis1/Ndel1/Dynein. Cell Rep. 2015;12(3):462–73. doi: 10.1016/j.celrep.2015.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toropova K, Zou S, Roberts AJ, et al. Lis1 regulates dynein by sterically blocking its mechanochemical cycle. Elife. 2014;3:1–25. doi: 10.7554/eLife.03372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moughamian AJ, Osborn GE, Lazarus JE, Maday S, Holzbaur ELF. Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J Neurosci. 2013;33(32):13190–203. doi: 10.1523/JNEUROSCI.0935-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagmann H, Taniguchi Y, Pippin JW, et al. Cyclin I and p35 determine the subcellular distribution of Cdk5. Am J Physiol Cell Physiol. 2015;308(4):C339–47. doi: 10.1152/ajpcell.00168.2014. [DOI] [PubMed] [Google Scholar]
- 31.Dhariwala Fa, Rajadhyaksha MS. An unusual member of the Cdk family: Cdk5. Cell Mol Neurobiol. 2008;28(3):351–69. doi: 10.1007/s10571-007-9242-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buffington SA, Rasband MN. The axon initial segment in nervous system disease and injury. Eur J Neurosci. 2011;34(10):1609–19. doi: 10.1111/j.1460-9568.2011.07875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grubb MS, Burrone J. Building and maintaining the axon initial segment. Curr Opin Neurobiol. 2010;20(4):481–488. doi: 10.1016/j.conb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trunova S, Baek B, Giniger E. Cdk5 Regulates the Size of an Axon Initial Segment-Like Compartment in Mushroom Body Neurons of the Drosophila Central Brain. J Neurosci. 2011;31(29):10451–62. doi: 10.1523/JNEUROSCI.0117-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339(6118):452–6. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kevenaar JT, Hoogenraad CC. The axonal cytoskeleton: from organization to function. Front Mol Neurosci. 2015;8:44. doi: 10.3389/fnmol.2015.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McIntosh BB, Holzbaur ELF, Ostap EM. Control of the initiation and termination of kinesin-1-driven transport by myosin-Ic and nonmuscle tropomyosin. Curr Biol. 2015;25(4):523–9. doi: 10.1016/j.cub.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Beuningen SFB, Will L, Harterink M, et al. TRIM46 Controls Neuronal Polarity and Axon Specification by Driving the Formation of Parallel Microtubule Arrays. Neuron. 2015:1208–1226. doi: 10.1016/j.neuron.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 39.Niethammer M, Smith DS, Ayala R, et al. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28(3):697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 40.Hebbar S, Mesngon MT, Guillotte AM, Desai B, Ayala R, Smith DS. Lis1 and Ndel1 influence the timing of nuclear envelope breakdown in neural stem cells. J Cell Biol. 2008;182(6):1063–71. doi: 10.1083/jcb.200803071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zyłkiewicz E, Kijańska M, Choi W-C, Derewenda U, Derewenda ZS, Stukenberg PT. The N-terminal coiled-coil of Ndel1 is a regulated scaffold that recruits LIS1 to dynein. J Cell Biol. 2011;192(3):433–45. doi: 10.1083/jcb.201011142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasaki S, Shionoya A, Ishida M, et al. A LIS1/NUDEL/Cytoplasmic Dynein Heavy Chain Complex in the Developing and Adult Nervous System. Neuron. 2000;28(3):681–696. doi: 10.1016/S0896-6273(00)00146-X. [DOI] [PubMed] [Google Scholar]
- 43.Arthur AL, Yang SZ, Abellaneda AM, Wildonger J. Dendrite arborization requires the dynein cofactor NudE. J Cell Sci. 2015;128(11):2191–201. doi: 10.1242/jcs.170316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petersen JD, Kaech S, Banker G. Selective Microtubule-Based Transport of Dendritic Membrane Proteins Arises in Concert with Axon Specification. J Neurosci. 2014;34(12):4135–4147. doi: 10.1523/JNEUROSCI.3779-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng Y-L, Li B-S, Kanungo J, et al. Cdk5 Modulation of mitogen-activated protein kinase signaling regulates neuronal survival. Mol Biol Cell. 2007;18(2):404–13. doi: 10.1091/mbc.E06-09-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dixit R, Ross JL, Goldman YE, Holzbaur ELF. Differential Regulation of Dynein and Kinesin Motor Proteins by Tau. Science (80-) 2008;319:8–11. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo X, Farías GG, Mattera R, Bonifacino JS. Rab5 and its effector FHF contribute to neuronal polarity through dynein-dependent retrieval of somatodendritic proteins from the axon. Proc Natl Acad Sci. 2016;113:E5318–E5327. doi: 10.1073/pnas.1601844113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonanomi D, Benfenati F, Valtorta F. Protein sorting in the synaptic vesicle life cycle. Prog Neurobiol. 2006;80(4):177–217. doi: 10.1016/j.pneurobio.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 50.Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol. 2015;16(12):711–26. doi: 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- 51.Alieva IB, Berezinskaya T, Borisy GG, Vorobjev IA. Centrosome nucleates numerous ephemeral microtubules and only few of them participate in the radial array. Cell Biol Int. 2015;39(11):1203–16. doi: 10.1002/cbin.10492. [DOI] [PubMed] [Google Scholar]
- 52.Ori-McKenney KM, Jan LY, Jan Y-N. Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron. 2012;76(5):921–30. doi: 10.1016/j.neuron.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q, Wang F, Cao J, et al. Nudel promotes axonal lysosome clearance and endo-lysosome formation via dynein-mediated transport. Traffic. 2009;10(9):1337–1349. doi: 10.1111/j.1600-0854.2009.00945.x. [DOI] [PubMed] [Google Scholar]
- 54.Zhou W, Chang J, Wang X, et al. GM130 is required for compartmental organization of dendritic golgi outposts. Curr Biol. 2014;24(11):1227–33. doi: 10.1016/j.cub.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chabin-brion K, Perez F, Drechou A, Pou C. The Golgi Complex Is a Microtubule-organizing Organelle. Mol Biol Cell. 2001 Jul;12:2047–2060. doi: 10.1091/mbc.12.7.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanders AAWM, Kaverina I. Nucleation and Dynamics of Golgi-derived Microtubules. Front Neurosci. 2015;9:431. doi: 10.3389/fnins.2015.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sainath R, Gallo G. The dynein inhibitor Ciliobrevin D inhibits the bidirectional transport of organelles along sensory axons and impairs NGF-mediated regulation of growth cones and axon branches. Dev Neurobiol. 2015;75(7):757–77. doi: 10.1002/dneu.22246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang J, Roberts AJ, Leschziner AE, Reck-Peterson SL. Lis1 acts as a “clutch” between the ATPase and microtubule-binding domains of the dynein motor. Cell. 2012;150(5):975–86. doi: 10.1016/j.cell.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKenney RJ, Vershinin M, Kunwar A, Vallee RB, Gross SP. LIS1 and NudE induce a persistent dynein force-producing state. Cell. 2010;141(2):304–14. doi: 10.1016/j.cell.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yi JY, Ori-McKenney KM, McKenney RJ, Vershinin M, Gross SP, Vallee RB. High-resolution imaging reveals indirect coordination of opposite motors and a role for LIS1 in high-load axonal transport. J Cell Biol. 2011;195(2):193–201. doi: 10.1083/jcb.201104076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lenz JH, Schuchardt I, Straube A, Steinberg G. A dynein loading zone for retrograde endosome motility at microtubule plus-ends. EMBO J. 2006;25(11):2275–86. doi: 10.1038/sj.emboj.7601119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Egan MJ, Tan K, Reck-Peterson SL. Lis1 is an initiation factor for dynein-driven organelle transport. J Cell Biol. 2012;197(7):971–82. doi: 10.1083/jcb.201112101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rao AN, Patil A, Black MM, et al. Cytoplasmic Dynein Transports Axonal Microtubules in a Polarity-Sorting Manner. Cell Rep. 2017;19(11):2210–2219. doi: 10.1016/j.celrep.2017.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palmer KJ, Hughes H, Stephens DJ. Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. Mol Biol Cell. 2009;20(12):2885–99. doi: 10.1091/mbc.E08-12-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. 1998;141(1):51–9. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Twelvetrees AE, Pernigo S, Sanger A, et al. The Dynamic Localization of Cytoplasmic Dynein in Neurons Is Driven by Kinesin-1. Neuron. 2016;90(5):1000–15. doi: 10.1016/j.neuron.2016.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lipka J, Kuijpers M, Jaworski J, Hoogenraad CC. Mutations in cytoplasmic dynein and its regulators cause malformations of cortical development and neurodegenerative diseases. Biochem Soc Trans. 2013;41(6):1605–12. doi: 10.1042/BST20130188. [DOI] [PubMed] [Google Scholar]
- 68.Wynshaw-Boris A. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin Genet. 2007;72(4):296–304. doi: 10.1111/j.1399-0004.2007.00888.x. [DOI] [PubMed] [Google Scholar]
- 69.Sun X, Wu Y, Gu M, et al. Selective filtering defect at the axon initial segment in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 2014;111(39):14271–6. doi: 10.1073/pnas.1411837111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Su SC, Tsai L-H. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–91. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- 71.Nguyen MD, Larivière RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30(1):135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- 72.Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolaños JP, Almeida A. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 2008;27(20):2736–45. doi: 10.1038/emboj.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Veas-Pérez de Tudela M, Maestre C, Delgado-Esteban M, Bolaños JP, Almeida A. Cdk5-mediated inhibition of APC/C-Cdh1 switches on the cyclin D1-Cdk4-pRb pathway causing aberrant S-phase entry of postmitotic neurons. Sci Rep. 2015;5:18180. doi: 10.1038/srep18180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilcox KS, Buchhalter J, Dichter MA. Properties of inhibitory and excitatory synapses between hippocampal neurons in very low density cultures. Synapse. 1994;18(2):128–51. doi: 10.1002/syn.890180206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, Related to Figure 1: Inhibition of CDK5 disrupts periodic axonal actin ring structures.
(A) STED super-resolution images of the AIS from 8 DIV rat hippocampal neurons stained with phalloidin. Cartoons of visible actin rings depicted below each image. Scale bar represent 2µm.
(B) Quantification of separation between adjacent actin rings from STED super-resolution images of 8 DIV rat hippocampal neurons. Each value represents the average separation of rings within one axon; only regions with consecutive visible rings were analyzed (minimum of 4 rings), largest region with consecutive rings was chosen for analysis per axon. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (***p < 0.001).
Figure S2, Related to Figure 2: CDK5 activity has no effect on axonal microtubule bundling or microtubule directionality in dendrites.
(A) STED super-resolution images of the AIS from 9 DIV rat hippocampal neurons stained for α-tubulin. Scale bar represents 2µm.
(B) Quantification of the number of microtubule bundles from STED super-resolution images of 9 DIV rat hippocampal neurons. Graph depicts means ± SEM; n ≥ 12 neurons from at least three biological replicates. No significant differences noted between conditions (one-way ANOVA with Tukey’s post-hoc test).
(C) Kymographs of EB3 motion in live-cell fluorescence microscopy in dendrites of 7 DIV rat hippocampal neurons expressing mCherry-EB3. Scale bar represents 10µm (horizontal) and 30 seconds (vertical).
Figure S3, Related to Figure 3: Dynein cofactor Ndel1 localizes to the AIS.
(A) Live-cell fluorescence imaging of 7 DIV rat hippocampal neurons expressing Halo-tagged TfR (soma and dendritic marker) and GFP-tagged Ndel1 (left) or Ndel1 5A mutant (right).
(B) Quantification of Ndel1 fluorescence intensity from the AIS, axon, and dendrite of 7 DIV hippocampal neurons expressing wild type or mutant Ndel1 as in (A). Graphs depict means ± SEM; n ≥ 21 neurons from at least three biological replicates. Values that differ significantly (one-way ANOVA with Tukey’s post-hoc test) are noted on graphs (*p < 0.05, ****p < 0.0001).
Figure S4, Related to Figure 4: CDK5 inhibition disrupts proper motility of somatodendritic TfR puncta.
(A) Kymographs of TfR motion in 7 DIV rat hippocampal neurons expressing indicated CDK5 constructs. Individual runs highlighted to the right: runs shown in pink, axon in light green and AIS, as determined by co-expression of NaV II–III, in dark green. Yellow arrow marks the same run on both the kymograph and the cartoon. Videos recorded at 100× with a confocal microscope for higher magnification than Figure 4C.
Figure S5, Related to Figure 6: Golgi fragments are present and motile in the axon when CDK5 is inhibited or unable to phosphorylate Ndel1.
(A) Kymographs of mCherry-tagged GM130 in 7 DIV rat hippocampal neurons co-expressing the indicated construct.