

Abstract
Staphylococcus epidermidis is an opportunistic pathogen that can form robust biofilms that render the bacteria resistant to antibiotic action and immune responses. Intercellular adhesion in S. epidermidis biofilms is mediated by the cell wall-associated Accumulation-Associated Protein (Aap), via zinc-mediated self-assembly of its B-repeat region. This region contains up to 17 nearly-identical sequence repeats, with each repeat assumed to be functionally equivalent. However, Aap B-repeats exist as two subtypes, defined by a cluster of consensus or variant amino acids. These variable residues are positioned near the zinc-binding (and dimerization) site and the stability determinant for the B-repeat fold. We have characterized four B-repeat constructs to assess the functional relevance of the two Aap B-repeat subtypes. Analytical ultracentrifugation experiments demonstrated that constructs with the variant sequence show reduced or absent Zn2+-induced dimerization. Likewise, circular dichroism thermal denaturation experiments showed that the variant sequence could significantly stabilize the fold, depending on its location within the construct. Crystal structures of three of the constructs revealed that the side chains from the variant sequence form an extensive bonding network that can stabilize the fold. Furthermore, altered distribution of charged residues between consensus and variant sequences changes the electrostatic potential in the vicinity of the Zn2+ binding site, providing a mechanistic explanation for the loss of zinc-induced dimerization in the variant constructs. These data suggest an assembly code that defines preferred oligomerization modes of the B-repeat region of Aap and a slip-grip model for initial contact and firm intercellular adhesion during biofilm formation.
Keywords: Biofilm, intercellular adhesion, Aap, stability, dimerization, Staphylococcus
INTRODUCTION
Staphylococcus epidermidis is a commensal gram-positive bacterium of the skin and is the staphylococcal species that most frequently colonizes humans [1]. Furthermore, its frequent occurrence in the skin microbiome makes it a prime candidate for hospital-acquired infection. Coagulase-negative Staphylococci (CoNS), of which S. epidermidis is the predominant species, are the most common cause of bloodstream and device-related infections [2,3]. The primary virulence factor for S. epidermidis is its ability to form robust biofilms [4]. Biofilms are specialized colonies of cells typically adherent on a surface (either biologic or abiotic, such as an implanted device) that are highly adhesive and cohesive. Formation of a biofilm renders S. epidermidis infections nearly impossible to eliminate without removing the device [5]. This is due to the fact that S. epidermidis cells in a biofilm evade phagocytic killing [6] and are in a quiescent mode of growth that is less sensitive to antibiotics [7,8].
During the formation of a biofilm, individual S. epidermidis cells first adhere to the surface; S. epidermidis can attach directly to plastic or glass or indirectly via host matrix proteins [9]. The Staphylococci then associate with one another, accumulating to form a tightly woven community of cells [8]. The biofilm bacteria can also secrete an extracellular matrix primarily comprised of the linear polysaccharide poly-N-acetyl-glucosamine (PNAG) and modified derivatives as well as extracellular DNA [9]. In addition to extracellular matrix, cell wall-attached proteins of S. epidermidis such as the accumulation-associated protein (Aap) are critically important in mediating intercellular adhesion within a biofilm [10]. Highlighting the importance of protein-mediated biofilm formation, it was recently demonstrated that S. epidermidis infection in a rat catheter model was dependent on the expression of Aap, but independent of PNAG [11].
Aap was initially identified as a protein essential for the accumulation of bacterial cells into a biofilm on a glass surface [12]. It is a long, thread-like protein covalently attached to the cell wall at its C-terminus by sortase A, via the threonine residue in the LPXTG motif of Aap [13,14]. Aap is comprised of multiple distinct functional regions. The N-terminal A-domain, containing unstructured repeats followed by a lectin domain, plays an important role in the initial attachment of cells to the surface [11,15,16]. However, the primary functional domain for the accumulation of cells into a biofilm is the B-repeat domain [17], comprised of 12-17 nearly identical, 128-amino acid repeats [18]. Each repeat is further divided into two subdomains: a 78-amino acid G5 subdomain and a 50-amino acid subdomain interspersed between each G5 that we term the spacer domain (also called the E domain [19]). We previously characterized a protein construct (Brpt1.5) comprised of the C-terminal intact B-repeat and capping G5 domain ‘half repeat’ [20]. Biophysical characterization revealed that Brpt1.5 is an elongated protein capable of self-assembly in the presence of Zn2+. Further data from biophysical experiments and biofilm growth assays indicated that longer B-repeat constructs assembled at progressively lower zinc concentrations, and that the full-length B-repeat region from Aap can assemble at physiological (low micromolar) levels of Zn2+ [20]. Taken together, these experiments led to a model in which tandem B-repeats mediate intercellular adhesion via modular assembly of individual domains along the length of the B-repeat region of Aap. Due to the high sequence identity between Aap B-repeats, it was initially assumed that each repeat was equally capable of assembly.
Subsequently, the crystal structure of Brpt1.5 was solved in the presence of Zn2+ in four different crystal forms revealing an unusual, elongated fold and illustrating the configuration of the anti-parallel dimer and zinc-coordinating residues of this construct [21]. Each G5 domain in these structures is comprised of two successive three-stranded β-sheets connected by triple-helix-like regions; the spacer domain has a similar, but abbreviated, fold [21]. The β-sheets are essentially free-standing and are stabilized by the formation of an unusual “hydrophobic stack” formed by the interdigitation of side chains contributed by upstream and downstream loops at interdomain interfaces. The stack is made up of central phenylalanine and proline side chains surrounded by three uncharged polar residues. Mutation of the lynchpin phenylalanine residue to alanine caused a significant reduction in protein stability, equivalent to loss of the entire downstream G5 domain [21].
As predicted by the biophysical studies, dimeric Brpt1.5 crystallized in the presence of Zn2+ with two anti-parallel protein chains coordinating the Zn2+ in trans and forming dimer interfaces via surrounding residues. The Zn2+-binding sites on each Brpt1.5 protomer are comprised of similar regions within the N-terminal and C-terminal G5 subdomains. In both cases, the coordination site is on the face of the second β-sheet of each G5. In the case of the N-terminal G5 site, this is at the interface between the G5 and spacer subdomains. In the case of the C-terminal G5, there is no downstream spacer subdomain, so the equivalent second β-sheet occurs near the terminus of the protein. In each of the four structures, zinc is tetrahedrally coordinated by three residues, with the fourth position coordinated by water (or a crystallization solute). In two of the structures, the three residues involved were D21 and H75 in the N-terminal G5 and E203 in the C-terminal G5. In the third structure, zinc was coordinated by E19 and H75 in the N-terminal G5 and D149 in the C-terminal G5. In the fourth structure, we observed two different zinc coordination schemes in two crystallographically distinct dimers. In one dimer interface, zinc was coordinated by E75 from one protomer coupled with D149 and E203 from the second protomer. The second dimer interface resembled the original coordination scheme with D21, H75, and E203 serving as ligands.
A study by Corrigan et al. showed that cell-surface expression of five or more intact B-repeats from the S. aureus ortholog SasG (S. aureus surface protein G) supported biofilm formation, whereas expression of one, two, or four B-repeats was insufficient [22]. Using our Brpt1.5 dimer structure as a template, we modeled a zinc-induced dimer of five intact B-repeats with a C-terminal cap (i.e., Brpt5.5). The model revealed a rope-like, anti-parallel twisting of the two Aap protomers that would originate from adjacent cells [21]. The model indicated that inclusion of five intact B-repeats was the minimum length to allow a full 360° twist of the two protein strands, which may explain the length dependence for biofilm formation. This model was based on the assumption that each B-repeat was equally capable of zinc-induced self-assembly given the high sequence identity between repeats, thus forming a continuous, adhesive filament.
Comparison of the amino acid sequence of all twelve B-repeats and the capping G5 domain half-repeat from Aap (based on the RP62A strain) reveals that each shows 89-100% sequence identity. However, in five of the twelve repeats (2, 3, 6, 7 & 8 – hereafter referred to as “variant repeats”) there is a set of eight residues in the G5 domain that replaces the consensus sequence (Figure 1A-B). Interestingly, several of the variant amino acids were non-conservative substitutions replacing small non-polar residues with large charged amino acids (e.g., V→K, A→K) (Figure 1B). These substitutions occur together, such that each B-repeat contains one of two different sequence ‘cassettes’ that define it as a consensus or variant repeat. Although some of these variant amino acids are relatively distant from one another in primary sequence, they all are closely apposed in the three-dimensional structure of Brpt1.5, near the zinc binding site, the dimer interface, and the hydrophobic stack (Figure 1C). We have directly assessed the effect of the variant cassette on B-repeat structure, stability, and zinc-induced assembly using biophysical and structural approaches. We find that the conserved substitution of these residues in the B-repeat fold impacts both zinc-induced self-assembly and the intrinsic stability of the G5 fold, both of which have important functional implications for Aap in biofilm formation.
Figure 1. Domain architecture of the Aap B-repeat region and sequence alignment of constructs used in this study.
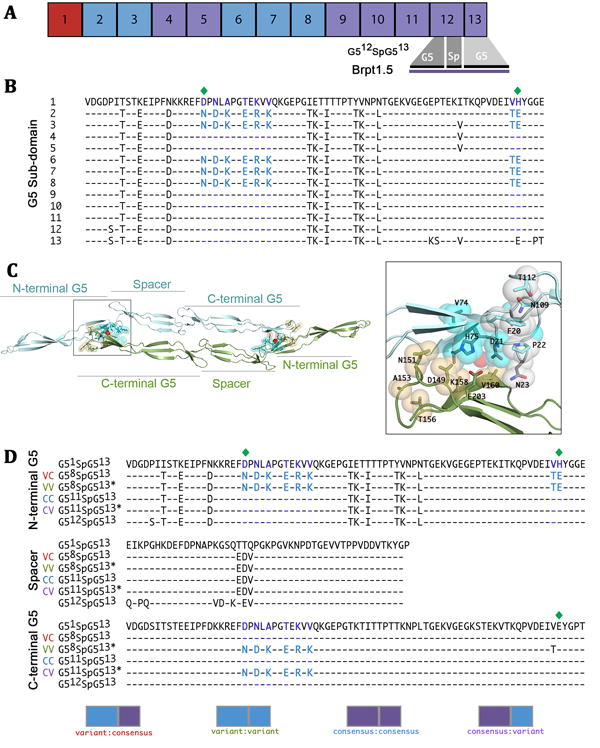
A) Distribution of variant (blue) and consensus (purple) repeats from the B-repeat region of Aap from S. epidermidis RP62A. The Brpt1.5 construct is comprised of a complete repeat (G5 and spacer subdomains) and a capping half-repeat. B) Sequence alignment of Aap B-repeats from S. epidermidis RP62A. Alternative amino acids found in variant repeats are highlighted in purple for consensus amino acids and blue for variant amino acids. Zinc-coordinating residues are indicated by a green diamond. Note that residues E99, D100 and V101 are present in some of the repeats but not others; however, these residues do not follow the same pattern of repetition as the variant cassette so they were not included in this study. C) The variant cassette residues encompass the dimerization interface and zinc-binding site and are immediately adjacent to the hydrophobic stack. The positions of variant residues are shown in cyan spheres in the N-terminal G5 and in orange spheres in the C-terminal G5 of the G512SpG513 dimer (PDB 4FUN). Zoom-in view of the zinc-binding site also shows the adjacent hydrophobic stack residues in white spheres. The residue at position 23 is part of the hydrophobic stack and is also one of the variant residues (consensus = N23, variant = D23). D) All sequences for constructs used in this study are compared to the first repeat (most divergent, G51) and last full repeat (used in previous studies, G512SpG513, CC). The consensus construct used in this study (G511SpG513, CC) contains the consensus sequence in both the N- and C-terminal G5 subdomains. The variant sequence is present in the N-terminal G5 of both G58SpG513 (VC) and G58SpG513* (VV), but only the latter contains the variant sequence in the C-terminal G5. The final construct (G511SpG513*, CV) contains the variant sequence only in the C-terminal G5. Note that G58SpG513* contains a Thr in position 202, whereas G511SpG513* contains Val at this position. Although the difference at this position was not intentional, subsequent data shows no functional effect of this substitution. Each construct is represented graphically where blue indicates a variant G5 and purple indicates a consensus G5.
MATERIALS AND METHODS
Alignment of B-repeats from strains of Staphylococcus epidermidis
Protein sequences for Aap were retrieved from the National Center for Biotechnology Information for the following Staphylococcus epidermidis strains: National Collection of Type Cultures (NCTC) 11047 (Accession no. HM587132), RP62A (Accession no. AJ249487), 1457 (Accession no. KJ920749), 5179 (Accession no. AY359815), and ATCC 12228 (Accession no. NP_763730). The individual B-repeats from each sequence were aligned to each other using T-Coffee server [23] to determine the number and location of variant and consensus repeats (Supplementary Figure S4).
Protein Expression and Purification
Four constructs were designed with conserved (C) or variant (V) B-repeat sequences in either N- or C-terminal positions or both, as described in more detail in Results. Note that G58SpG513* contains a Thr in position 202, whereas G511SpG513* contains Val at this position (see Figure 1D). Although the difference at this position was not intentional, subsequent data shows no functional effect of this substitution. Each construct was synthesized as a G-block by Integrated DNA Technologies, Inc. Single G-blocks were cloned into LifeTechnologies pENTR D-TOPO entry vector and then recombined into a pMAL-derived destination vector containing both His6 and maltose binding protein fusion tags. BLR(DE3) competent cells (LifeTechnologies) were transformed with this vector and grown at 37° C in 1L LB broth to an absorbance of 1.0-1.2 at 600 nm. Cultures were induced overnight with 250 μM IPTG at 20° C. Cells were harvested by centrifugation and resuspended in 20 mM Tris pH 7.4, 300 mM NaCl. Resuspended pellets were frozen and stored at -80° C. Thawed pellets were lysed by sonication in an ice bath and cell debris from the lysate was pelleted by centrifugation. Clarified lysate was filtered (0.2 micron) prior to loading onto a Ni-NTA column. Protein was eluted with 20 mM Tris pH 7.4, 300 mM NaCl, 1 M imidazole. His6 and MBP fusion tags were removed by tobacco etch virus protease cleavage. Cleaved protein was filtered (0.2 micron) and cleaved fusion tags were captured on a Ni-NTA column. Superdex75HiLoad 26/600 (GE Healthcare) was used as a final purification step.
Analytical Ultracentrifugation
Analytical ultracentrifugation (AUC) experiments were performed using a Beckman-Coulter ProteomeLab XL-I Protein Characterization System with an An-60 Ti four-hole rotor or an An-50 Ti six-hole rotor. For analysis of low-concentration assembly in the presence of zinc, protein samples were dialyzed for 24 hours in 20 mM Tris (pH 7.4) and 150 mM NaCl containing 0 or 5 mM ZnCl2 and concentrations were adjusted to 8 μM using dialysis buffer; absorbance data was collected at 230 nm. For analysis of high-concentration assembly in the presence of zinc, protein samples were dialyzed for 24 hours in 20 mM Tris pH 7.4, 150 mM NaCl, 5 mM ZnCl2 and adjusted to 80-85 μM using dialysis buffer; absorbance data was collected at 280 nm. All sedimentation velocity experiments were run at 20° C and 48,000 rpm. Sedimentation velocity data were analyzed using Sedfit [24]. Equilibrium experiments were performed at 20,000 rpm, 24,000 rpm and 32,000 rpm. Protein samples were dialyzed for 24 hours in 20 mM Tris pH 7.4, 150 mM NaCl, 5 mM ZnCl2 and concentrations were adjusted using dialysis buffer. Absorbance data was collected at 235 nm, 280 nm and 290 nm with protein at loading concentrations ranging from 7 to 103 μM. Raw sedimentation equilibrium data were trimmed using WinReEdit and globally fitted to a monomer-dimer equilibrium model using WinNonlin (www.rasmb.org/software), as previously described [20,21,25]. Buffer density and viscosity and protein partial specific volumes were calculated using the program SEDNTERP (www.rasmb.org/software).
Circular Dichroism
Circular dichroism (CD) wavelength and thermal denaturation data were collected on an AVIV model 215 spectrophotometer. For comparative stability analysis, protein samples were dialyzed into 20 mM Tris pH 7.4, 150 mM NaF for 24 hours and were adjusted to 45 μM using dialysis buffer. Thermal denaturation measurements were taken at 210 nm using a 0.05 cm pathlength cuvette. Data collection was at temperatures between 20-80 °C with 1.0 °C steps and 2 minutes between steps for equilibration. Fits for thermal denaturation data were determined using SigmaPlot according to the method previously described [21,26]. Each of three experiments was fitted independently. Each reported Tm is an average of the three individual fits with standard deviation. For each construct, a single representative fitted dataset has been plotted.
X-ray Crystallography
G58SpG513* construct
Protein was crystallized at 18 mg/mL by vapor diffusion hanging drop in 100 mM sodium acetate (pH 5), 175 mM CaCl2, and 22% polyethylene glycol (PEG) 6000. Crystal was transferred to a stabilizing solution of 100 mM sodium acetate pH 5.0, 200 mM CaCl2, 25% PEG 6000 for 30 minutes. It was then transferred to a solution of 100 mM sodium acetate pH 5.0, 250 mM CaCl2, 25% PEG 6000, 5% PEG 400 for two minutes. Finally, prior to freezing, it was soaked for 70 minutes in 100 mM sodium acetate pH 5, 200 mM CaCl2, 25% PEG 6000, 15% PEG 400. Data were collected to 2.3 Å at Advanced Photon Source (APS) Northeast Collaborative Access Team (NE-CAT) 24-ID-C on a Pilatus 6M detector. Data were processed using XDS [27]. The structure was solved by molecular replacement with an N-terminal truncated monomer from the previously solved Brpt1.5 structure 4FUP [21] using Phaser [28]. The structure was built in Coot [29] and refined using BUSTER [30]. BUSTER autoNCS was used while building the core of each monomer in the asymmetric unit but was not used when building the termini [31]. The final G58SpG513* model includes chain A residues 14-35, 71-143 and 163-199; chain B includes residues 16-34, 72-147, and 159-202.
G58SpG513 construct
Protein was crystallized at 7 mg/mL by vapor diffusion hanging drop in 100 mM sodium acetate (pH 5), 100 mM CaCl2, 20% PEG 6000. The crystal was transferred to a stabilizing solution of 100 mM sodium acetate pH 5.0, 200 mM CaCl2, 25% PEG 6000 and was cryoprotected in 100 mM sodium acetate pH 5.0, 200 mM CaCl2, 30% PEG 6000, 5% ethylene glycol. Data were collected remotely on the SSRL beamline 12-2 to 1.9 Å as part of the 2014 RapiData X-ray crystallography course at Brookhaven National Laboratory. Data were collected on a Pilatus 6M detector. The structure was solved by molecular replacement with a refined model of Brpt1.5 G58SpG513* using Phaser and was built and refined as described above with the addition of several rounds of refinement using PHENIX [32]. The final G58SpG513 model includes chain A residues 11-40, 65-141, and 165-196; chain B includes residues 17-33, 72-146, and 159-203.
G511SpG513 construct
Protein was crystallized at 20 mg/mL by vapor diffusion hanging drop in 100 mM sodium acetate(pH 5), 250 mM CaCl2, 24% PEG 6000. Crystal was soaked for one minute in 100 mM sodium acetate pH 5, 200 mM CaCl2, 30% PEG 6000, followed by a two minute soak in 100 mM sodium acetate pH 5, 200 mM CaCl2, 30% PEG 6000, 15% PEG 400 for two minutes. Finally, the crystal was soaked for five minutes in 100 mM sodium acetate pH 5, 200 mM CaCl2, 30% PEG 6000, 20% PEG 400. Data were collected on APS GM/CA beamline 23ID-B to 3.3 Å as part of the 2015 CCP4 X-ray crystallography workshop at APS. Structure was solved by molecular replacement with a refined model of Brpt1.5 G58SpG513* using Phaser and was built and refined as described above with the addition of several rounds of refinement using PHENIX [32]. The final G511SpG513 model includes chain A residues 14-33, 70-146, and 165-199 as well as chain B residues 17-27, 73-145, and 160-202. Crystal structure images were displayed using MacPyMOL: PyMOL Enhanced for Mac OS X (Copyright © Schrödinger, LLC) and Chimera [33].
Electrostatic Calculations
For electrostatic calculations the full-length, original consensus structure G512SpG513 (4FUN) was used as a model for each of the constructs described in this work (G511SpG513, G58SpG513, G511SpG513*, G58SpG513*) in order to investigate the entire fold, including the termini. Amino acid side chains in 4FUN were mutated to match the sequence of these constructs and, whenever possible, rotamer orientations were determined using the crystal structures described in this work. The biological unit in 4FUN is an anti-parallel dimer with a zinc ion at each of the two dimer interfaces. Zinc was removed from the pdb file prior to electrostatic calculations so that the isopotential maps generated represent the electrostatic character as it is experienced by the divalent cation. Electrostatic isopotential maps were calculated using the program DelPhi [34] with a grid size of 450, interior dielectric of 4, exterior dielectric of 80 and salt concentration of 150 mM. Surface charges from the 4FUN models and the calculated isopotential maps were displayed using Chimera with surface electrostatics displayed at −10/+10 and isopotential maps displayed in volume viewer at -2/+2.
RESULTS
Constructs Designed to Assess Functional Consequences of Repeat Variation
The Brpt1.5 construct from strain RP62A used in previous studies contained the 12th full repeat and the 13th G5 subdomain, which comprises the capping half-repeat (Figure 1A). Both of the G5 domains included in this original construct contain consensus residues. This construct has been fully characterized by X-ray crystallography and biophysical studies [20,21]. Crystallographic data confirmed an antiparallel, Zn2+-induced dimer and clearly identified the residues responsible for coordinating zinc at the dimer interfaces. The constructs in the current study were designed to assess the effect of incorporating the conserved variant residues in either or both of the N-terminal and C-terminal G5 subdomains of a Brpt1.5-length construct (Figure 1D). All constructs tested here follow the pattern of G5 domain-spacer-G5 domain (G5SpG5), with G5 combinations including consensus-consensus (CC), consensus-variant (CV), variant-consensus (VC), and variant-variant (VV). For the consensus (C) B-repeat sequence, we used repeat 11, which better represents the general consensus (the 12th repeat contains several divergent residues compared to the other consensus repeats). The consensus C-terminal cap uses the native 13th G5 domain (half-repeat) from the RP62A strain, which contains a few divergent residues that may be important for its role as a C-terminal capping domain. Thus, the control construct used in this study is G511SpG513 (CC), which is nearly identical to the original construct used for previous biophysical and crystallization studies (G512SpG513). For the variant (V) B-repeat sequence, we used repeat 8, which is the variant B-repeat closest to the C-terminal cap. This results in a variant-consensus (VC) construct designated G58SpG513. To test the effect of the variant sequence in the C-terminal position, the variant residues were simply swapped into the capping 13th G5 domain, designated G513*. The resulting final constructs analyzed were G511SpG513* (CV) and G58SpG513* (VV). These four constructs allowed us to test the impact of the variant versus consensus sequences on zinc-mediated assembly and intrinsic stability as well as positional effects of the N-terminal versus C-terminal position of the G5 domains.
The Variant G5 Sequence Negatively Impacts Zinc-Mediated Assembly of Brpt1.5
Sedimentation velocity AUC experiments confirmed that each construct sedimented as a single species with a sedimentation coefficient essentially identical to that of the previously characterized construct (G512SpG513) (Table S1). Each of the Brpt1.5 constructs was monomeric in the absence of zinc at a loading concentration of 8 μM, with a sedimentation coefficient of 1.53-1.57 S (Figure S1A). When analyzed in the presence of 5 mM ZnCl2, the consensus construct (G511SpG13, CC) assembled into a dimer as expected when tested at 8 μM loading concentration. However, under the same conditions the constructs with the variant cassette in either the N-terminal G5 (G58SpG513, VC), the C-terminal (G511SpG513*, CV) or in both G5 positions (G58SpG513*, VV) remained monomeric (Figure 2A). Thus, while the consensus construct (G511SpG13, CC) showed zinc-induced assembly at a protein concentration of 8 μM, each construct with a variant repeat was incompetent to assemble under these conditions.
Figure 2. Sedimentation velocity and sedimentation equilibrium AUC analysis of B-repeat constructs.
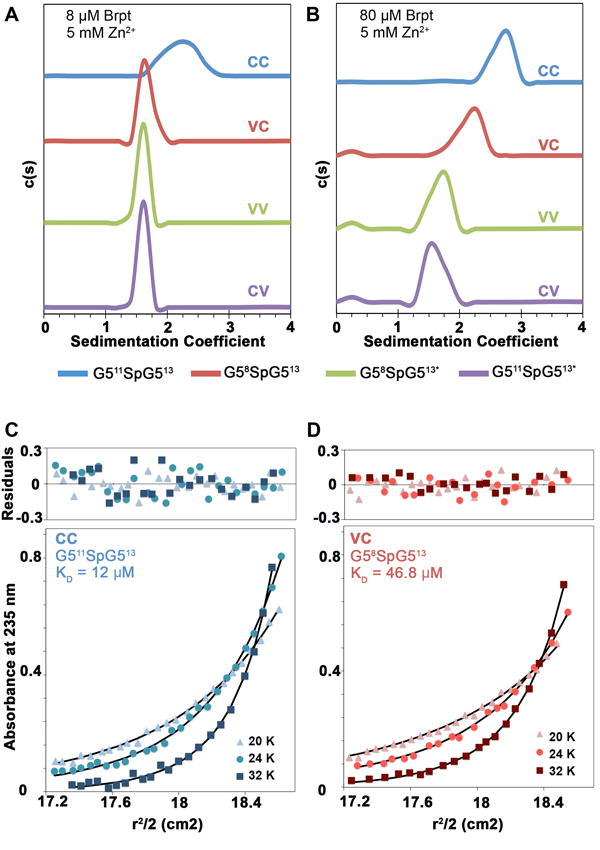
Four new Brpt1.5 constructs were analyzed by sedimentation velocity AUC to determine if they were assembly-competent in the presence of zinc, as observed for the initial Brpt1.5 construct, G512SpG513 (CC) [21]. In the presence of 5 mM Zn2+, only G511SpG513 (CC) assembled at 8 μM protein (A). However, at 80 μM protein, both G511SpG513 (CC) and G58SpG513 (VC) were assembly-competent in the presence of 5 mM Zn2+ (B). G58SpG513* (VV) and G511SpG513* (CV) did not assemble in the presence of Zn2+ even at 80 μM (B). Absorbance scans for sedimentation velocity data were collected at 230 nm for low concentration samples (8 μM) and at 280 nm for high concentration samples (80 μM). All experiments were conducted at 48,000 rpm, 20 °C and analyzed using Sedfit. To compare the zinc-mediated assembly of G511SpG513 (CC) and G58SpG513 (VC), sedimentation equilibrium experiments were done on protein concentrations ranging from 7 to 103 μM. The globally fitted monomer-dimer KD for G511SpG513 (CC) was 12.3 μM (C) and the calculated KD for G58SpG513 (VC) was 46.8 μM (D). The nearly four-fold difference in KD is consistent with the velocity experiments showing that G58SpG513 (VC) is not assembly-competent at 8 μM but self-assembles at 80 μM. Absorbance data were collected at 235 nm (shown), 280 nm and 290 nm at speeds of 20,000 rpm, 24,000 rpm and 32,000 rpm. Raw data were trimmed using WinReEdit and globally fitted to a monomer-dimer equilibrium model using WinNonlin.
We repeated the same experiments at ~80 μM protein to determine whether these variant constructs were in fact assembly-incompetent or merely had a reduced propensity for zinc-mediated dimerization. As expected, each construct at 80 μM was monomeric in the absence of Zn2+ (Figure S1B). In the presence of 5 mM ZnCl2, both G58SpG513* (VV) and G511SpG513* (CV) constructs remained monomeric while the consensus construct (G511SpG513, CC) formed a dimer. The sedimentation coefficient distribution for the N-terminal variant cassette construct (G58SpG513, VC) showed a significant shift toward a faster-sedimenting species, indicating a reaction boundary representing a monomer-dimer equilibrium (Figure 2B). However, the sedimentation coefficient for this VC construct (2.15 S) in the presence of Zn2+ was significantly lower than for the CC construct (2.68 S), indicating only partial assembly or possibly formation of a dimer with a less compact conformation than the CC construct.
To better understand the self-assembly of the G58SpG513 (VC) construct and to quantitatively compare it to G511SpG513 (CC), sedimentation equilibrium AUC experiments were conducted. Analysis of the CC and VC constructs at loading concentrations ranging from 7 μM to 103 μM in the presence of 5 mM ZnCl2 confirmed that both constructs were in a monomer-dimer equilibrium with dissociation constants of 12.3 μM for the consensus construct (G511SpG513, CC) (Figure 2C) and 46.8 μM for the N-terminal variant repeat construct (G58SpG513, VC) (Figure 2D). The equilibrium constants indicate that for G58SpG513 (VC) at 80 μM, the weight fraction of dimer would be 59% (compared to 73% for G511SpG513), so the difference in sedimentation coefficients observed for VC compared to CC in Figure 2B can be explained by reaction boundaries exhibiting differing extents of dimerization rather than a different conformation of the dimer.
Characterization of these constructs by analytical ultracentrifugation uncovered several important differences in their competence for zinc-mediated self-assembly. Incorporation of the variant amino acids in the C-terminal G5 domain renders Brpt1.5-length constructs assembly-incompetent. On the other hand, as long as the C-terminal G5 has the consensus residues Brpt1.5 is assembly-competent, but the dissociation constant is nearly 4-fold weaker with an N-terminal variant G5. These data highlight the importance of the C-terminal capping G5 subdomain for the function of the B-repeat region of Aap.
Variant G5 Sequence Confers Position-Dependent Stabilization of the Protein Fold
The finding that the C-terminal variant repeat renders Brpt1.5 assembly-incompetent led us to question whether the variant sequence altered secondary structure or influenced thermal stability of the variant constructs. Far-UV CD spectra demonstrated that all four constructs have similar secondary structure content consistent with the original G512SpG513 construct, with 41-45% β-sheet and 48-51% coil reported after deconvolution by the KD2 algorithm [35] on the DichroWeb server [36,37]. Thus, these data indicate that there were no overall changes in the G5 domain secondary structure conferred by the variant repeat, whether in the N-terminal or C-terminal position (Figure S2).
Circular dichroism thermal denaturation experiments demonstrated that the presence or absence of the variant residues in the N-terminal G5 does not affect the stability of the protein (Figure 3). Proteins were analyzed at 210 nm at temperatures ranging from 25-70°C and the data were fitted to a two-state unfolding model and plotted as fraction folded as a function of temperature. The Tm for G511SpG513, CC (43.7 ± 1.0 °C) and G58SpG513, VC (43.8 ± 0.5 °C) were comparable both to one another and to the previously published Tm for G512SpG513 (45.3 °C) [21]. Quite unexpectedly, introduction of the variant residues into the C-terminal G5 had a stabilizing effect, increasing the Tm by 5-7 degrees Celsius in both cases (for G511SpG513*, CV: Tm = 50.0 ± 0.8 °C; for G58SpG513*, VV: Tm = 49.3 ± 0.5 °C). This effect was independent of whether the variant amino acids were present in the N-terminal G5, showing a striking position-dependent effect of the variant residues.
Figure 3. CD thermal denaturation analysis of each B-repeat construct.
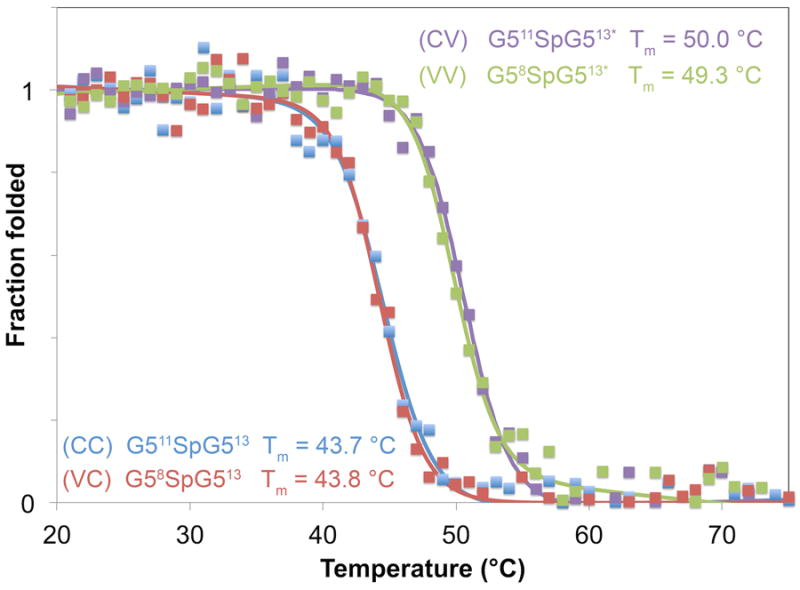
Stability of the overall protein fold for each construct was determined using CD thermal denaturation. Midpoints of the denaturation curves (Tm) were similar for G511SpG513 (CC) (blue Tm = 43.7 ± 1.0 °C) and G58SpG513 (VC) (red Tm = 43.8 ± 0.5 °C), both of which were comparable to the previously published Tm of Brpt1.5 G512SpG513 (CC). Interestingly, the Tm for constructs with a C-terminal variant G5 was elevated by 5-6 °C for G511SpG513* (CV) (purple Tm = 50.0 ± 0.8 °C) and G58SpG513* (VV) (green Tm = 49.3 ± 0.5 °C). Unfolding curves were collected at 210 nm between 20-80 °C and fits for two-state unfolding were calculated using SigmaPlot.
X-ray Crystallography of Monomeric Brpt1.5 Constructs
In order to understand the structural basis for position-specific effects of the variant sequence substitutions within the G5 domains, X-ray crystal structures were solved for three of the four constructs described in this study. Previously published structures of Aap B-repeat constructs were all crystallized in the presence of ZnCl2 to capture the dimeric assembly. In contrast, the current structures were crystallized in the absence of zinc to observe the monomeric structures, since the constructs with variant repeats have diminished or complete loss of zinc-induced dimerization. The two structures containing variant repeats in either the N-terminal G5 (G58SpG513, VC) or both N-and C-terminal G5s (G58SpG513*, VV) were crystallized and diffraction data were collected to 1.9 Å and 2.33 Å, respectively. The consensus construct (G511SpG513, CC) was also crystallized and was solved to 3.34 Å. In each case, the asymmetric unit contained two parallel, offset copies of the B-repeat protein, but none of these formed the anti-parallel dimer observed previously in the presence of Zn2+.
The central region of each molecule, encompassing the spacer subdomain and its adjacent interfaces with the G5 domains, exhibited well-resolved electron density with B-factors for most residues ranging between 20-50 Å2. However, due to the inherent flexibility in the B-repeat fold and the lack of stabilization from zinc-induced dimerization, the electron density became progressively less well-defined toward both the N- and C-termini of the constructs. Accordingly, the B-factors increased in the regions of poorly-resolved density, often exceeding 100 Å2. This disorder was most extensive in the N-terminal G5 domain; the first β-sheet in each N-terminal G5 domain could not be resolved in any of the constructs. The C-terminal G5 was also disordered but to a lesser degree. Multiple attempts to improve the quality of the electron density were unsuccessful, so models were built and refined using the regions of well-defined density (Figure 4A). Importantly, despite the missing regions of each model, the conserved variable residues in the N-terminal G5 domain were located in high-quality electron density, allowing analysis of the structural implications of these residue substitutions (Figure 4B-C).
Figure 4. Crystal structures of G511SpG513 (CC), G58SpG513 (VC) , and G58SpG513* (VV).
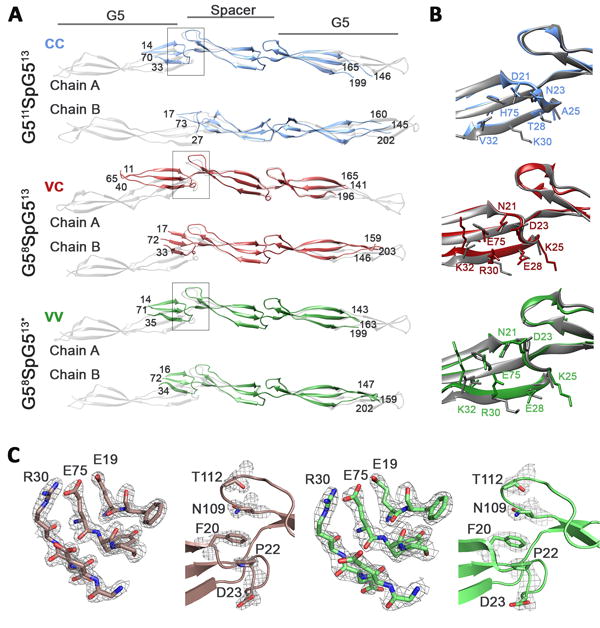
A) Regions built into well-resolved density for G511SpG513 (CC) G58SpG513 (VC) and G58SpG513* (VV) superimposed onto monomers of G512SpG513 (CC) (PDB 4FUN; gray). B) G511SpG513 (CC, blue), G58SpG513 (VC, red), and G58SpG513* (VV, green) superposed on the N-terminal consensus G5 from G512SpG513 (CC; PDB 4FUN). Consensus side chains from 4FUN are shown in gray . C) 2Fo-Fc electron density for residues at the dimer interface and hydrophobic stack in G58SpG513 (VC) and G58SpG513* (VV) is displayed in gray mesh contoured at 1.3 sigma.
Conserved Variant Residues
Each chain from the current study was superposed onto the single chain from the asymmetric unit of the G512SpG513 structure (PDB 4FUN [21]). The local alignment and side chain orientations within the Zn2+ binding site were similar overall (Figure 4B). However, the two structures with variant N-terminal G5 subdomains have differences in both the identity and orientation of the side chains (Figure 4B). The overarching difference observed when the variant sequence is present is a redistribution of charged and polar residues and reduced clustering of negatively-charged side chains near the zinc binding site. Notably, there is a swap of side chains: consensus residues D21 and N23 are instead N21 and D23 in the variant sequence. This switch allows hydrogen bonding to occur between N21 and E75 in the variant G5 (Figure 5A, B). In the variant G5, A25 is exchanged for a lysine that introduces a positively charged residue in the place of a non-polar one within a highly electronegative region. The introduction of a charged residue in the variant sequence also occurs with T28E. In general, the zinc-binding site in the consensus repeat is made up of a dense cluster of acidic residues, but in the variant repeat the density of clustered acidic residues is effectively decreased by interspersed positive charges. Further, at position 30, the lysine in the consensus G5 is exchanged for an arginine in the variant G5. Whereas the consensus lysine extends outwards from the sheet and into the solvent, the arginine is oriented in the opposite direction, either perpendicular to the β-sheet (in the case of G58SpG513), or even further inward towards the other strands in the sheet (in the case of G58SpG513*) (Figure 5B). The inward-facing orientation of the arginine brings it into salt bridge distance with E75. Because E75 in the center strand is also hydrogen-bonded to N21 on the upper strand, in the variant G5 a bonding network is formed by N21-E75-R30 across the entire face of the β-sheet (Figure 5B, D). An additional change from a non-polar to a charged amino acid occurs at position 32 where V32 is replaced by K32 in the variant G5. Because K32 is oriented toward the upper strand of the β-sheet, it is within salt bridge distance of E19 on the upper strand. The final two differences between consensus and variant G5 subdomains occur at positions 74 and 75 in the central β-strand. Position 74 is on the back side of the β-sheet, facing away from the zinc-binding site. In the consensus G5, this is a threonine and in the variant G5 it is a valine. Finally, H75 in the consensus repeat is replaced with E75 in the variant repeat. This is an important zinc-ligating residue in the dimeric Brpt1.5 structures; we showed previously that an H75E point mutant in the G512SpG513 construct was fully competent to dimerize in the presence of Zn2+ [21]. The sequence changes described here for the N-terminal consensus and variant G5 domains (Figure 5A, B) are recapitulated in the C-terminal domains (Figure 5C, D). The only exception is a difference between the N- and C-terminal consensus G5 domains, where position 75 is a histidine in the N-terminus and a glutamate in the C-terminus. This means there is one fewer hydrogen bond in the C-terminal arrangement (Figure 5C). Comparison of the bonding networks in two consensus structures and two variant structures are fully detailed in Figure S3.
Figure 5. Differences in side chain bonding networks in the consensus and variant G5 sub-domains.
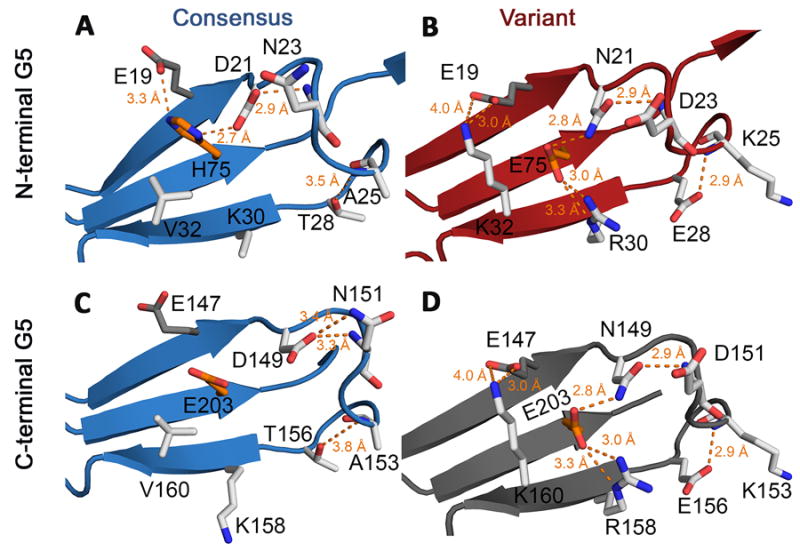
Residues shown as white sticks are those that are in the variant cassette. Residues shown in orange are key zinc-binding residues that are also part of the variant cassette. They indicate the general position where zinc would bind. Residue E19 is colored in dark gray; it is not part of the variant cassette but is an important side chain that interacts with variant residues. A) Environment around the variant residues in the N-terminal G5 domain from the consensus structure G512SpG513 (PDB 4FUO). Backbone is shown in blue cartoon representation. Two bonding interactions (hydrogen bonds or salt bridges) are possible with H75 in addition to the two side chain-backbone bonds between T28-A25 and D21-N23. B) The same site in the N-terminal variant from G58SpG513 (VC), with backbone residues shown in red cartoon representation. Compared to the maximum of four bonding interactions between side chains in the consensus G5, the variant G5 can form up to seven side chain bonding interactions. C) The C-terminal G5 domain from consensus structure 4FUO has one fewer side chain bond than the N-terminal domain. D) A model of the predicted variant C-terminal G5, with backbone in gray cartoon representation and side chain positions modeled as sticks based on the G58SpG513 N-terminal G5.
Variant G5 Amino Acids Alter Protein Surface Electrostatics
As described above, the variant G5 contains several charged residues that replace non-polar amino acids present in the consensus G5, including positive residues incorporated into the electronegative region surrounding the zinc binding site. To understand how these changes might affect the electrostatic potential in the region of the Zn2+ binding site and the hydrophobic stack, we used the program DelPhi to calculate electrostatic isopotential maps [34]. Since some regions from the monomeric structures presented here were not well resolved in electron density, we built models of each intact construct using G512SpG513 (PDB 4FUN) as a template, mutated to mimic the four constructs in this study, guided by the structural data on the well-resolved regions of the monomeric constructs. Each construct was then superimposed onto the Zn2+-induced G512SpG513 dimer to analyze the electrostatic potential in the dimer interface. The isopotential map for each protomer was superimposed on a surface representation of the structure (Figure 6). The dimer interface of each protomer is shown in a ‘flip-book’ view, with each protomer rotated 180° to illustrate the contacting surfaces (Figure 6A). The zinc-binding site on each protomer is indicated by a green circle.
Figure 6. Predicted surface electrostatics for variant and consensus residues at the Zn2+-binding site.
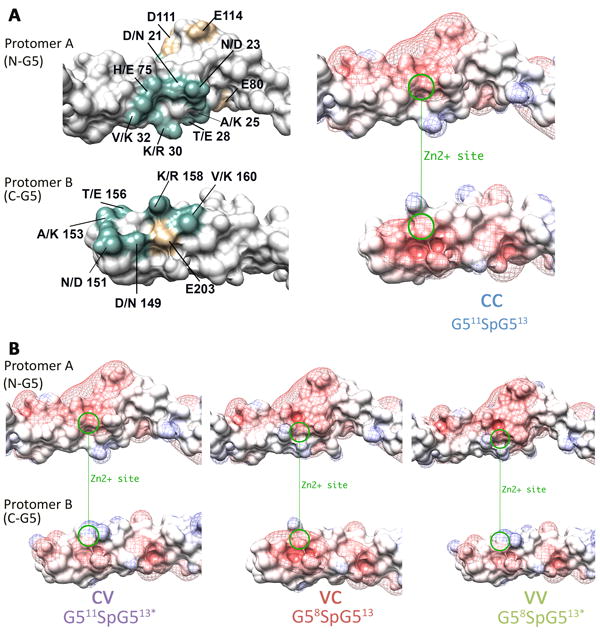
The dimerization interface is displayed in “flip book” orientation with both protomers rotated 180° to illustrate the contact surface. A) The presence of variant residues at the dimer interface are shown in teal next to an electrostatic representation of the G511SpG513 (CC) dimer interface. Additional residues that may contribute to local electrostatics are shown in tan. The Zn2+ binding site is illustrated with a green circle. Electrostatic changes that arise at the dimer interface when the variant residues are present are illustrated in B) G511SpG513* (CV), G58SpG513 (VC), and G58SpG513* (VV). All panels were generated in Chimera using surface representation colored by charge (negative=red, blue=positive), with surface electrostatics ranging from -10 to +10. The isopotential maps are displayed in mesh representation, colored by charge (red-blue) from -2 to +2.
For the consensus construct (G511SpG513), the zinc-binding site exhibits strong electronegative potential in both the N-terminal G5 (protomer A) and in the C-terminal G5 (protomer B) (Figure 6A). For the G511SpG513* (CV) construct the N-terminal G5 has the consensus sequence, but the C-terminal G5 has the variant amino acids. The introduction of V160K coupled with the inward-facing orientation of R158 results in the introduction of basic residues in this region of protomer B, diminishing the electronegative potential (Figure 6B). The reverse is true for G58SpG513 (VC) where the acidic patch surrounding the zinc-binding site is retained in the C-terminal G5 of protomer B, but the electrostatic characteristics of the N-terminal G5 of protomer A are altered (Figure 6B). Finally, when both N- and C-terminal G5 subdomains have the variant residues, as is the case for G58SpG513*, both zinc-binding sites have encroaching basic patches adjacent to one another, resulting in diminished electronegative potential on both sides of the prospective zinc-binding site (Figure 6B).
DISCUSSION
Position-specific effects of the variant cassette
Our studies indicate that the presence of the variant sequence in the C-terminal G5 has a marked effect on the biophysical characteristics of the construct. Using sedimentation velocity AUC we demonstrated that G511SpG513 (CC) and G58SpG513 (VC) are competent for zinc-mediated assembly. However, both G511SpG513* (CV) and G58SpG513* (VV), the constructs with the C-terminal G5 variant sequence, were incompetent for zinc-mediated assembly at the concentrations tested. Further, we measured circular dichroism thermal denaturation to demonstrate that these same two constructs were stabilized compared to G511SpG513 (CC) and G58SpG513 (VC). From these findings it is apparent that the variant sequence has a more profound effect when located in the C-terminal G5 than it does in the N-terminal G5.
Structural basis for position-specific Zn2+ dimerization effects
Analytical ultracentrifugation sedimentation equilibrium experiments demonstrated a four-fold difference between the dimerization constants of G511SpG513 (CC) and G58SpG513 (VC). This difference in affinity indicates that while the N-terminal G5 variant sequence does affect zinc-mediated dimerization, it does not render the construct assembly-incompetent as the C-terminal variant sequence does. Our previous work demonstrated the importance of electrostatics in the Zn2+-binding site: lowering the pH from 7.4 to 6.0 leads to the protonation of 3 ionizable residues and loss of dimerization [20]. Furthermore, the crystal structure of Brpt1.5 (G512SpG513) showed a dense cluster of acidic residues in the region of the Zn2+ ligation site. Alanine substitution of E19, D21, D149, E203, or D21/D149 together reduced dimerization in each case, confirming the importance of this cluster of acidic residues [21].
By calculating isopotential maps using DelPhi, we are able to visualize why the variant sequence is more detrimental to zinc-mediated assembly when positioned in the C-terminal G5. When the consensus sequence is present in either the N- or C-terminal G5, the large acidic patch is present near the zinc-binding site, as observed for the G512SpG513 construct. When the variant sequence is present in the N-terminal G5, several electropositive charges are introduced. However, the large acidic patch is not completely disrupted by the introduction of these charges. Part of the reason for this is because the electronegative charges making up the acidic patch are not limited to those in the consensus/variant sequence. The interface between the N-terminal G5 and the spacer is created by two loops. One of these loops is donated from the N-terminal G5; this is the loop that contains the predominant number of either the variant or consensus residues. The second loop is donated from the downstream spacer encompassing residues 106-116. Within this loop there are two acidic residues D111 and E114. In addition to the two loops, the sequence connecting the G5 domain to the subsequent spacer domain (residues 76-80) contains two acidic residues (E79 and E80). The presence of these acidic residues in the connecting segment and in the loop donated from the spacer help retain the acidic nature of the zinc-binding site. This is not the case when the variant sequence is present in the C-terminal G5. Without the contributing acidic residues from the spacer domain, the variant sequence disrupts the acidic patch in the C-terminal G5.
Structural basis for position-specific stability increase by variant residues
Just as the lack of a downstream spacer affects Zn2+-induced dimerization observed when the variant sequence is in the C-terminal G5 domain, its absence also plays a role in the increased relative stability seen with a C-terminal variant G5. We previously showed the major source of stability for Brpt1.5 is the hydrophobic stack at the interface between each G5:spacer and spacer:G5 junction and that this stack is formed from residues contributed by a loop from the G5 as well as a loop from the spacer [21]. In the N-terminal G5 of a Brpt1.5-length construct, the variant sequence is immediately adjacent to the hydrophobic stack, and one of the variant residues (D23N) is at the base of the hydrophobic stack. However, in the C-terminal G5 there is no hydrophobic stack because there is no spacer subdomain to contribute the second loop. This point is of utmost importance because it means that from a folding/stability perspective, the N- and C-terminal variant sequences are in quite different environments. The region of interest in the N-terminus is, due to its proximity to the hydrophobic stack, in one of the most stable segments of the protein. In contrast, at the C-terminus, the region of interest is located in a segment of the protein that is metastable.
Using the N-terminal variant sequence from both the G58SpG513 (VC) and G58SpG513* (VV) structures as a model, it is possible to infer how the variant amino acids stabilize the fold in the context of the C-terminal G5. While there are few opportunities for cross-strand side chain interactions with the consensus sequence, the variant sequence optimizes the number of stabilizing interactions among the amino acids in the final β-sheet of the C-terminal G5. Two of these interactions center on the zinc-coordinating residue E203. In the variant sequence, E203 is within hydrogen-bonding distance of N149 and can also form a double salt-bridge with R158. This forms a bonding network across all three strands of the β-sheet. This network is absent in the consensus sequence because there is an aspartate rather than an asparagine at position 149, and because K158 points away from E203 (unlike R158 which points toward E203). In the variant sequence there is also a double salt-bridge between E147 and K160, which spans the β-sheet. These stabilizing forces are present in the N-terminal G5 as well as the C-terminal G5. However, when the variant sequence is present in the N-terminal G5, the additional interactions do not contribute significantly, since that region of the protein is already nearly optimized for stability. In the C-terminal G5, the local environment lacks an intact hydrophobic stack and is metastable, so the additional bonding network formed by the variant residues measurably increases stability. The formation of bonding interactions across strands in the final β-sheet of the C-terminal G5 domain would act in parallel with the ‘mechanical clamps’ described in the related protein, SasG [38]. These clamps consist of long stretches of hydrogen bonds between strands in the G5 domain β-sheets identified in molecular dynamics simulations to provide resistance to tensile force. Our work presented here shows that in the case of Aap, additional hydrogen bonds and salt bridges from the variant residue side chains can further add to stability by forming inter-strand bonding networks.
The assembly code for Aap self-assembly at the cell surface
The present work demonstrates that two consensus sequence G5 domains can assemble in trans around Zn2+ with the highest affinity, with approximately 4-fold higher affinity than heterodimerization by consensus:variant G5 domains. In contrast, variant:variant homodimers do not form, even at high concentration. Thus, there will be preferred points of assembly along the Aap B-repeat region, located where consensus G5 domains occur (Figure 7). In the case of Aap from S. epidermidis RP62A,there are several potential assembly modes with up to four strong (consensus:consensus) G5:G5 interfaces complemented by up to eight weak (variant:consensus) interfaces. We propose a slip-grip model for intercellular assembly in a S. epidermidis biofilm in which initial capture events involve lower-affinity assembly with fewer G5:G5 interfaces (slip mode) when cells are further apart and there is less overlap between the Aap B-repeat regions. These weaker linkages can then reorder to form higher-affinity stable adhesion events when the cells are in closer proximity and have greater overlap between Aap molecules and a greater number of G5:G5 interfaces (Figure 7). The presence of two consensus G5 repeats in the middle of the B-repeat region (repeats 4 and 5) surrounded by variant repeats limits the number of slip mode assembly configurations that can form, helping to reduce the dimensionality of the intercellular assembly process.
Figure 7. Schematic of the assembly code for the B-repeat region of Aap.
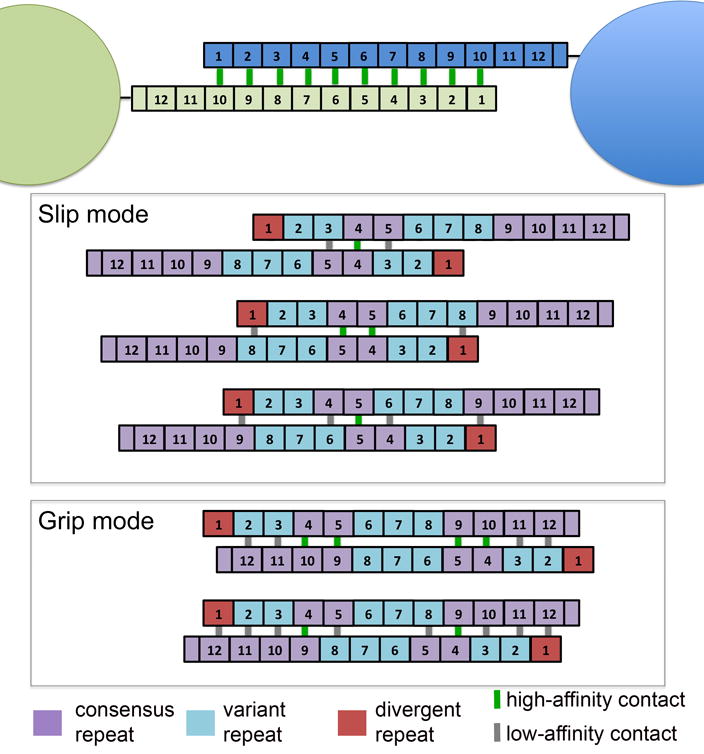
Updated model of zinc-mediated assembly of Aap B-repeat regions on adjacent cells accounting for the different assembly competencies of variant and consensus repeats. Top panel shows the original model with identical, high-affinity dimerization interfaces along the length of the B-repeat region of Aap. Gray boxes illustrate potential assembly modes that involve at least one high-affinity (consensus:consensus) G5:G5: interface. The upper box illustrates proposed ‘slip mode’ interactions with a limited number of G5:G5 interfaces; the lowest gray box illustrates proposed ‘grip mode’ interactions with substantial overlap of the B-repeat regions between the assembling Aap molecules and a higher number of G5:G5 interfaces. In each case, high-affinity (consensus:consensus) G5:G5 interfaces are portrayed by a green line between repeats; low-affinity (variant:consensus) interfaces are indicated by a gray line. In these illustrations, the 1st repeat is colored differently due to its divergent sequence. Preliminary data indicates that G51 shows low-affinity dimerization, similar to the variant repeats (data not shown).
Alignments of B-repeat sequences from several strains of S. epidermidis demonstrated that both variant and consensus repeats are present in each of the strains analyzed (Figure S4). The arrangement of variant and consensus repeats is very similar between strains NCTC 11047 and RP62A. Each of these two strains have sequences for Aap with 12.5 repeats with the variant repeats at positions 2, 3, 6, 7, and 8. However, the C-terminal capping half-repeat is a consensus subtype in RP62A but a variant in NCTC 11047. The slip-grip model applies equally well to these two strains. In the case of strain 1457, 6.5 repeats are expressed, all of which are consensus except for the C-terminal capping half-repeat. Because there are no variant repeats in segments that would overlap from adjacent cells of strain 1457, it is expected that this strain would have no low affinity contacts and would be in constant grip-mode. In strain 5179, of the 4.5 repeats only repeats 1 and 4 are consensus and the remaining repeats are variant. Strain ATCC 12228 contains variant repeats at positions 2 and 4 and at the capping half-repeat. Both of these strains would fit with the slip-grip model, but to a lesser degree than that of the longer Aap from RP62A and NCTC 11047. Several other gram-positive bacteria including Streptococcus gordonii and St. infantis, Corynebacterium urealyticum, and Finegoldia magna all express cell surface proteins with multiple tandem G5 repeats, although these lack spacer domains and instead feature short interdomain linkers. It is an intriguing possibility that similar slip-grip models of intercellular adhesion might apply to these proteins as well, although no functional data have been reported yet.
From a translational perspective, these findings suggest a preferred target region on Aap for antibody or small-molecule therapeutics aiming to prevent S. epidermidis biofilms. We have shown that the consensus repeats are required for Aap assembly because at the zinc-binding site they have a 3D surface with unique structural and electrostatic characteristics. Specifically, blocking this site in the consensus repeats would prevent both high- (consensus:consensus) and low- (variant:consensus) affinity contacts. Only the variant repeat binding sites would remain exposed, which are incompetent to form homomeric interactions. In summary, the data presented here show that subtle amino acid substitutions within nearly identical B-repeats of Aap tune the assembly properties of the G5 domain and could help to facilitate ordered intercellular assembly of S. epidermidis during biofilm growth.
Supplementary Material
Acknowledgments
Assistance with data collection was provided by beamline scientists at RapiData 2014 (Brookhaven National Laboratory/Stanford Synchrotron Radiation Lightsource) and the 2015 CCP4 workshop (Argonne National Laboratory). The authors also wish to thank Drs. Tom Thompson and Rhett Kovall for helpful discussions, suggestions, and assistance with data collection.
FUNDING INFORMATION
This work was supported by the National Institute of General Medical Sciences [grant number R01 GM094363 (to A.B.H.)]; the American Heart Association [predoctoral fellowship 13PRE16380011 (to C.L.S.)]; and the State of Ohio Eminent Scholar Program (to A.B.H.).
Abbreviations list
- Aap
accumulation-associated protein
- APS
Advanced Photon Source
- ATCC
American Type Culture Collection
- AUC
analytical ultracentrifugation
- CC
conserved-conserved
- CCP4
Collaborative Computational Project Number 4
- CD
circular dichroism
- CoNS
coagulase-negative Staphylococci
- CV
conserved-variant
- DNA
deoxyribonucleic acid
- GM/CA
General Medical Sciences and Cancer Institutes Structural Biology Facility
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- LB
Luria-Bertani
- MBP
maltose-binding protein
- NCBI
National Center for Biotechnology Information
- NCS
non-crystallographic symmetry
- NCTC
National Collection of Type Cultures
- NE-CAT
Northeast Collaborative Access Team
- NTA
nitrilotriacetic acid
- PEG
polyethylene glycol
- PNAG
poly-N-acetyl glucosamine
- Rpm
revolutions per minute
- SasG
S. aureus surface protein G
- SSRL
Stanford Synchrotron Radiation Lightsource
- TEV
tobacco etch virus
- VC
variant-conserved
- VV
variant-variant
Footnotes
AUTHOR CONTRIBUTION
Catherine Shelton, Deborah Conrady, and Andrew Herr designed the experiments. Catherine Shelton performed the experiments. Catherine Shelton, Deborah Conrady, and Andrew Herr analyzed the data, and Catherine Shelton and Andrew Herr wrote the paper.
ACCESSION NUMBERS
The coordinates and structure factor data for G511SpG513, G58SpG513*, and G58SpG513 have been deposited to the Protein Data Bank under accession codes 5UT7, 5UT8, and 5UT9, respectively.
References
- 1.Becker K, Heilmann C, Peters G. Coagulase-Negative Staphylococci. Clin Microbiol Rev. 2014;27:870–926. doi: 10.1128/CMR.00109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larru B, Gong W, Vendetti N, Sullivan KV, Localio R, Zaoutis TE, Gerber JS. Bloodstream Infections in Hospitalized Children: Epidemiology and Antimicrobial Susceptibilities. Pediatr Infect Dis J. 2016;35:507–510. doi: 10.1097/INF.0000000000001057. [DOI] [PubMed] [Google Scholar]
- 3.Rogers KL, Fey PD, Rupp ME. Coagulase-negative staphylococcal infections. Infect Dis Clin North Am. 2009;23:73–98. doi: 10.1016/j.idc.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Fey PD, Olson ME. Current concepts in biofilm formation of Staphylcoccus epidermidis. Future Microbiol. 2010;5:917–933. doi: 10.2217/fmb.10.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCann M, Gilmore B, Gorman S. Staphylococcus epidermidis device-related infections: pathogenesis and clinical management. J Pharm Pharmacol. 2008;60:1551–1571. doi: 10.1211/jpp/60.12.0001. [DOI] [PubMed] [Google Scholar]
- 6.Shommer NN, Christner M, Hentschke M, Ruckdeschel K, Aepfelbacher M, Rohde H. Staphylococcus epidermidis uses distinct mechanisms of biofilm formation to interfere with phagocytosis and activation of mouse macrophage-like cells 774A.1. Infect and Immun. 2011;79:2267–2276. doi: 10.1128/IAI.01142-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao Y, Surdevant D, Otto M. Genomewide analysis of gene expression in Staphylococcus epidermidis biofilms: insights into the pathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms. J Infect Dis. 2005;191:289–298. doi: 10.1086/426945. [DOI] [PubMed] [Google Scholar]
- 8.Otto M. Staphylococcus epidermidis the ‘accidental’ pathogen. Nat Rev Microbiol. 2009;7:555–567. doi: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otto M. Staphylococcal Biofilms. Curr Top Microbiol Immunol. 2008;322:207–208. doi: 10.1007/978-3-540-75418-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Speziale P, Pietrocola G, Foster TJ, Geoghegan JA. Protein-based biofilm matrices in Staphylococci. Front Cell Infect Microbiol. 2014;4:1–10. doi: 10.3389/fcimb.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaeffer CR, Woods KR, Longo GM, Kiedrowski MR, Paharik AE, Buttner H, Christner M, Boissy RJ, Horswill AR, Rohde H, Fey PD. Accumulation-associated protein enhances Staphylococcus epidermidis biofilm formation under dynamic conditions and Is required for infection in a rat catheter mode. Infect and Immun. 2015;83:214–225. doi: 10.1128/IAI.02177-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain M, Herrmann M, von Eiff C, Perdreau-Reminton F, Peters G. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect and Immun. 1997;65:519–524. doi: 10.1128/iai.65.2.519-524.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ton-That H, Liu G, Mazmanian SK, Faull KF, Schneewind O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc Natl Acad Sci U S A. 1999;96:12424–12429. doi: 10.1073/pnas.96.22.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowden MG, Chen W, Singvall J, Xu Y, Peacock SJ, Valtulina V, Speziale P, Höök M. Identification and preliminary characterization of cell-wall-anchored proteins of Staphylococcus epidermidis. Microbiology. 2005;151:1453–1464. doi: 10.1099/mic.0.27534-0. [DOI] [PubMed] [Google Scholar]
- 15.Macintosh RL, Brittan JL, Bhattacharya R, Jenkinson HF, Derrick J, Upton M, Handley PS. The Terminal A Domain of the Fibrillar Accumulation-Associated Protein (Aap) of Staphylococcus epidermidis. Mediates Adhesion to Human Corneocytes J Bacteriol. 2009;191:7007–7016. doi: 10.1128/JB.00764-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conlon BP, Geoghegan JA, Waters EM, McCarthy H, Rowe SE, Davies JR, Schaeffer CR, Foster TJ, Fey PD, O’Gara JP. Role for the A Domain of Unprocessed Accumulation-Associated Protein (Aap) in the Attachment Phase of Staphylococcus epidermidis Biofilm Phenotype. J Bacteriol. 2014;196:4268–4275. doi: 10.1128/JB.01946-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohde H, Burandt EC, Siemssen N, Frommelt L, Burdelski C, Wurster S, Scherpe S, Davies AP, Harris LG, Horstkotte MA, Knoblock JK, Ragunath C, Kaplan JB, Mack D. Polysaccharide intercellular adhesion or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials. 2007;28:1711–1720. doi: 10.1016/j.biomaterials.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 18.Rhode H, Burdelski C, Bartscht K, Hussain M, Buck F, Horstkotte MA, Knoblock JK, Hellmann C, Herrmann M, Mack D. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol Microbiol. 2005;55:1883–1895. doi: 10.1111/j.1365-2958.2005.04515.x. [DOI] [PubMed] [Google Scholar]
- 19.Gruszka DT, Wojdyla JA, Bingham RJ, Turkenburg JP, Manfield IW, Steward A, Leech AP, Geoghegan JA, Foster TJ, Clarke J, Potts JR. Staphylococcal biofilm-forming protein has a contiguous rod-like structure. Proc Natl Acad Sci U S A. 2012;109:E1011–E1018. doi: 10.1073/pnas.1119456109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conrady DG, Brescia CC, Katsunori H, Weiss AA, Hassett DJ, Herr AB. A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A. 2008;105:19456–19461. doi: 10.1073/pnas.0807717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conrady DG, Wilson JJ, Herr AB. Structural basis for Zn2+-dependent intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A. 2013;110:E202–E211. doi: 10.1073/pnas.1208134110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corrigan RM, Rigby D, Handley P, Foster TJ. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology. 2007;153:2435–2446. doi: 10.1099/mic.0.2007/006676-0. [DOI] [PubMed] [Google Scholar]
- 23.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 24.Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herr AB, White CL, Milburn C, Wu C, Bjorkman PJ. Bivalent Binding of IgA1 to FcαRI Suggests a Mechanism for Cytokine Activation of IgA Phagocytosis. J Mol Biol. 2003;327:645–657. doi: 10.1016/s0022-2836(03)00149-9. [DOI] [PubMed] [Google Scholar]
- 26.Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc. 2006;1:2327–2535. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy A, Grosse-Kunstleve RWG, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER version 2.10.1. Cambridge, United Kingdom: Global Phasing Ltd; 2016. [Google Scholar]
- 31.Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrheein C, Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera – A Visualization system for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 34.Li L, Li C, Sarkar S, Zhang J, Witham S, Zhang Z, Wang L, Smith N, Petukh M, Alexov E. DelPhi: a comprehensive suite for DelPhi software and associated resources. BMC Biophys. 2012;5:9. doi: 10.1186/2046-1682-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrae MA, Chacon P, Merelo JJ, Morán F. Evaluation of Secondary Structure of Proteins from UV Circular Dichroism Using an Unsupervised. Learning Neural Network Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 36.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 38.Gruszka DT, Whelan F, Farrance OE, Fung HK, Paci E, Jeffries CM, Svergun DI, Baldock C, Baumann CG, Brockwell DJ, Potts JR, Clarke J. Cooperative folding of intrinsically disordered domains drives assembly of a strong elongated protein. Nat Commun. 2015;6:7271. doi: 10.1038/ncomms8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.