

MrpL35 and Mrp7, components of the mitoribosome, play a role in regulating the COX assembly process and influence the molecular environments of the Mss51, Coa3, and Cox14 proteins, chaperones involved in posttranslational assembly events for the COX complex.
Abstract
Mitoribosomes perform the synthesis of the core components of the oxidative phosphorylation (OXPHOS) system encoded by the mitochondrial genome. We provide evidence that MrpL35 (mL38), a mitospecific component of the yeast mitoribosomal central protuberance, assembles into a subcomplex with MrpL7 (uL5), Mrp7 (bL27), and MrpL36 (bL31) and mitospecific proteins MrpL17 (mL46) and MrpL28 (mL40). We isolated respiratory defective mrpL35 mutant yeast strains, which do not display an overall inhibition in mitochondrial protein synthesis but rather have a problem in cytochrome c oxidase complex (COX) assembly. Our findings indicate that MrpL35, with its partner Mrp7, play a key role in coordinating the synthesis of the Cox1 subunit with its assembly into the COX enzyme and in a manner that involves the Cox14 and Coa3 proteins. We propose that MrpL35 and Mrp7 are regulatory subunits of the mitoribosome acting to coordinate protein synthesis and OXPHOS assembly events and thus the bioenergetic capacity of the mitochondria.
INTRODUCTION
Mitochondrial ribosomes (mitoribosomes) perform the synthesis of a small, but important, subset of the mitochondrial proteome, which is encoded by the mitochondrial genome and represents key components of the oxidative phosphorylation (OXPHOS) system (Borst and Grivell, 1978; O’Brien, 2003; Smits et al., 2007; Kehrein et al., 2015; Greber and Ban, 2016; Ott et al., 2016; Mai et al., 2017). Thus the regulation and activity of mitoribosomes have a major potential impact on an organism’s capacity to generate its energy through aerobic respiration. Due to the endosymbiotic origin of mitochondria, the mitoribosomes are evolutionarily related to bacterial ribosomes and thus share a number of overlapping features, such as antibiotic sensitivities and similarities in protein subunit composition (O’Brien, 2002, 2003; Gruschke et al., 2010; De Silva et al., 2015; Greber and Ban, 2016; Ott et al., 2016). Despite these similarities, mitoribosomes display important compositional and functional differences from their prokaryotic ancestors. Mitoribosomes contain a higher protein:rRNA ratio than bacterial ribosomes, and this extra protein content manifests itself in two ways. First, several of the evolutionarily conserved mitoribosomal proteins are larger than their bacterial counterparts, having acquired additional, extension sequences (often C-terminal) and being termed mitochondrial-specific or “mitospecific” sequences. Second, mitoribosomes contain a number of additional novel proteins, that is, mitospecific ribosomal proteins, which are often conserved throughout eukaryotes but do not have counterparts in the prokaryotic ribosomes (Graack et al., 1988; Koc et al., 2000; O’Brien, 2002; van der Sluis et al., 2015). The significant advances in the structural analysis of mitoribosomes, including from the yeast Saccharomyces cerevisiae, have greatly enhanced our knowledge of the location of these mitospecific elements within the mitoribosome structures (Amunts et al., 2014; Brown et al., 2014; Greber et al., 2014a, b, 2015; Kaushal et al., 2014; Amunts et al., 2015; Kaushal et al., 2015; Desai et al., 2017). However, little is known about the function of these mitospecific protein features and how they may contribute to, or regulate, the translational process of the mitoribosome.
Compositional differences between the mitochondrial and bacterial ribosomes are thought to reflect a divergence in function of the mitochondrial translational apparatus from the bacterial. The mitochondrial proteome, in contrast to the bacterial, is mosaic in origin. The proteins synthesized by mitoribosomes are destined to become components of oligomeric complexes coassembled with these imported nuclear encoded partners, a process facilitated by OXPHOS complex-specific chaperone proteins. Thus there is a need for a close coordination between the activity of the mitochondrial translational apparatus and the components involved in the import and assembly of their cytosol-synthesized partners (Gruschke and Ott, 2010; De Silva et al., 2015; Kehrein et al., 2015; Ott et al., 2016).
The cytochrome c oxidase complex (COX), the terminal electron acceptor in the mitochondrial OXPHOS machinery, is an oligomeric enzyme complex composed of mitochondrial translation products (Cox1, Cox2, and Cox3) partnered with multiple nuclearly encoded ones, imported from the cytosol. The assembly of this complex is a multi-step, chaperone-facilitated process that involves the uniting of protein subunits from two origins and in a strict stoichiometric manner. Cox1, with its metal centers (two hemes [a+a3] and one copper, CuB), forms a central aspect of the catalytic core of the COX complex. The synthesis and assembly of Cox1 into the COX complex is tightly regulated, with the proposed purpose of minimizing accumulation of potentially reactive Cox1-assembly subcomplexes (Khalimonchuk et al., 2010; Mick et al., 2011; McStay et al., 2013; Dennerlein and Rehling, 2015; Mayorga et al., 2016; Richter-Dennerlein et al., 2016; Soto and Barrientos, 2016). In yeast, Cox1 synthesis by the mitoribosomes is under the strict feedback regulation from the downstream COX assembly process and directly involves the Mss51 protein (Figure 1). Mss51, a heme sensing protein, promotes the translation of Cox1 by binding to the 5′-UTR of the COX1 mRNA (Perez-Martinez et al., 2003; Barrientos et al., 2004; Pierrel et al., 2007; Perez-Martinez et al., 2009; Fontanesi et al., 2010; Mick et al., 2010; Fontanesi et al., 2011; Soto et al., 2012; Mayorga et al., 2016). The Mss51 protein can also interact with the newly synthesized Cox1 protein (via the C-terminal end of Cox1), as it is cotranslationally inserted into the membrane, where it forms an Mss51-containing Cox1-assembly intermediate (COA) with the Cox14 and Coa3(Cox25) proteins (Mick et al., 2010; Fontanesi et al., 2011; Clemente et al., 2013; McStay et al., 2013; Garcia-Villegas et al., 2017). Limiting in levels, the Mss51 remains bound to the Cox1 protein-Coa3 intermediate and thus unavailable to bind the COX1 mRNA to promote further rounds of Cox1 synthesis, until the assembly of Cox1 together with the nuclear encoded COX subunits proceeds. Further progression of Cox1 in its assembly pathway causes the release and recycling of Mss51, making it available to initiate the next round of Cox1 protein synthesis. The presence of Cox14 and Coa3 are required to trap Mss51 in the Cox1-assembly intermediate and they thus act as negative Cox1-translational regulators (Mick et al., 2010 , 2011; Fontanesi et al., 2011). It is currently unclear what molecular mechanism drives the subsequent recycling of Mss51 from this Cox1-intermediate and whether, and how, the Cox14 or Coa3 proteins may be directly involved.
FIGURE 1:

Overview of the Cox1 synthesis and assembly pathway. Cox1 is synthesized on mitoribosomes tethered to the inner membrane at the site of the cotranslation insertion machinery containing the Oxa1 protein. Synthesis of Cox1 is promoted on activation by the translational activators Mss51 (and Pet309, not depicted) by their binding to the 5′-UTR of the COX1 mRNA. Following insertion into the membrane, the newly synthesized Cox1 protein forms an assembly intermediate with COX-specific assembly factors, including Cox14 and Coa3, which support the recruitment of Mss51 and its binding to the C-terminal end of the Cox1 protein. Further maturation of Cox1 (e.g., incorporation of heme) and assembly with nuclear encoded COX partner subunits, facilitated by specific chaperones, leads to the assembly of Cox1 into the active COX enzyme and promotes the release of Mss51, which in turn activates the next round of Cox1 synthesis. Abbreviations: IM, inner mitochondrial membrane; IMS, intermembrane space; Matrix, mitochondrial matrix space.
In this present study, we have used the yeast model S. cerevisiae to investigate the functional relevance of the mitospecific ribosomal protein, MrpL35 (also known as mL38 using the standardized mitoribosomal protein nomenclature). Conserved throughout eukaryotes (where in higher eukaryotes is referred to as MrpL38), MrpL35 (mL38) is a component of the central protuberance region of the large (54S) mitoribosomal subunit, where its functional relevance is unknown. Our evidence indicates that MrpL35 (mL38) protein is coassembled into a subcomplex with two other mitochondrial-specific ribosomal proteins, MrpL17 (mL46) and MrpL28 (mL40), and together with proteins that are homologues of bacterial ribosomal proteins, MrpL7 (uL5), Mrp7 (bL27), and MrpL36 (bL31), respectively. MrpL35/mL38 proteins contain a C-terminal domain that displays homology to members of the conserved phosphatidylethanolamine-binding protein domain (PEBP) family. The PEBP family comprises a diverse group of proteins present in many organisms and whose functions have been linked to the regulation of key cell signaling pathways (Banfield et al., 1998; Bruun et al., 1998; Serre et al., 1998; Yousuf et al., 2014). Through mutational analysis of the PEBP-like domain, we demonstrate here that MrpL35 plays a key role in coordinating the synthesis and assembly of the COX complex. Our findings demonstrate a connection among MrpL35, Cox1 synthesis, and the COX assembly components, in particular Cox14 and Coa3, and thus illustrate a link between the mitochondrial translation apparatus and the downstream OXPHOS assembly chaperones.
RESULTS
Mutation of residue Y275 of MrpL35 results in a temperature-sensitive phenotype
The S. cerevisiae yeast mrpL35 null mutant strain (ΔmrpL35) expressing the wild-type MrpL35 protein (from a plasmid-borne gene) exhibited the ability to grow on the nonfermentable carbon (glycerol) source, whereas the strain lacking MrpL35 (i.e., ΔmrpL35+empty plasmid) was completely unable to do so (Figure 2A). This result illustrates the essential nature of MrpL35 for respiratory/OXPHOS-based growth. Through mL38 protein sequence alignments, we sought to identify highly conserved residues present in the PEBP-like homology domain, located at the C-terminal end of the MrpL35 (mL38) proteins. One of these residues was a conserved Tyr(Y) residue, corresponding to Tyr(Y)275 of the MrpL35 protein from S. cerevisiae, present in all mL38 proteins analyzed, and located on the periphery of the PEBP-like domain of MrpL35 and in a region of the protein that is adjacent to its neighboring protein Mrp7 (bL27) (Figure 2B). We targeted the Y275 residue (indicated in yellow in Figure 2B) for site-directed mutagenesis, initially changing it either to Ala(A) or Asp(D). Expression of the resulting derivatives, mrpL35Y275A and mrpL35Y275D, respectively, supported respiratory-based growth of the ΔmrpL35 mutant at 30°C but not at the elevated temperature of 37°C (Figure 2A). In contrast, the relatively conservative exchange of Y275 for another aromatic residue Phe(F) did not appear to impact MrpL35’s ability to support aerobic growth at both temperatures tested (Figure 2A). Thus we conclude that the highly conserved Y275 residue must play a critical structural/functional role in the MrpL35 protein that is compromised by mutation to a smaller, or negatively charged, residue.
FIGURE 2:
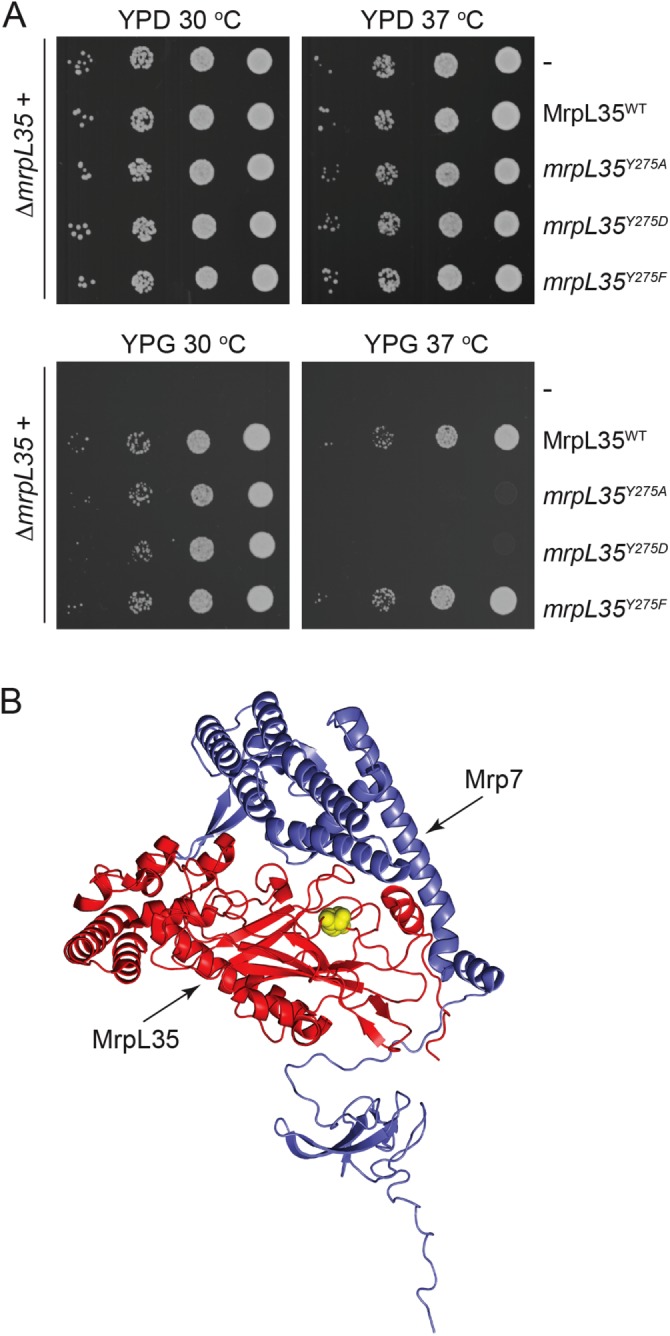
Mutation of residue Y275 of MrpL35 can result in a respiratory deficient phenotype. (A) Serial 10-fold dilutions of ΔmrpL35 strain harboring a pRS413 plasmid containing gene inserts encoding either the wild-type MrpL35 protein (MrpL35WT), no insert (-) or genes encoding the mutated mrpL35 derivatives (mrpL35Y275A, mrpL35Y275D, or mrpL35Y275F), as indicated, were spotted on YP plates containing glucose (YPD) (2 d) or glycerol (YPG) (3 d) and grown at 30° and 37°C. (B) The images of MrpL35 (in red) and Mrp7 (in slate blue) structures were generated using PDB ID:5MRC. The position of the Y275 residue in MrpL35 targeted for mutagenesis is indicated in yellow.
Mutation of Y275 residue does not prevent mitoribosome translation
Using an in vivo radiolabeling approach in the presence of cycloheximide (to block cytosolic protein synthesis), we initially investigated if the translational capacity of the mitoribosome was inhibited in mrpL35Y275A and mrpL35Y275D mutants grown either at the permissive and nonpermissive temperature. In S. cerevisiae, the mitoribosomes synthesize eight proteins, Var1 (a small ribosomal subunit component) and OXPHOS subunits, Cox1, Cox2, and Cox3 of the COX (core subunits of the COX enzyme), and cytochrome b (of the cytochrome bc1 enzyme) and F1Fo-ATP synthase subunits, Atp6, Atp8, and Atp9. Both mutant strains displayed normal capacity for mitochondrial protein synthesis at the permissive temperature, and they retained the ability to synthesize also at 37°C (Figure 3A). Thus we conclude that the lack of OXPHOS-based growth of the mrpL35 mutants at 37°C is not due to an absence of mitoribosomal translation at the elevated temperature. An overall reduction in total translation levels, in particular for the mrpL35Y275D mutant, was observed at 37°C, however, and may indicate a role for MrpL35 in mediating efficient mitochondrial translation or simply that the mitochondria of these cells were bioenergetically compromised after the prolonged growth period at the nonpermissive temperature, thus indirectly affecting their capacity for the energy-driven process of translation. When analyzed in isolated mitochondria, where exogenously added energy (ATP, GTP, and OXPHOS substrate NADH) were supplemented, the mrpL35Y275D mitochondria displayed normal level of protein synthesis (Figure 4A).
FIGURE 3:
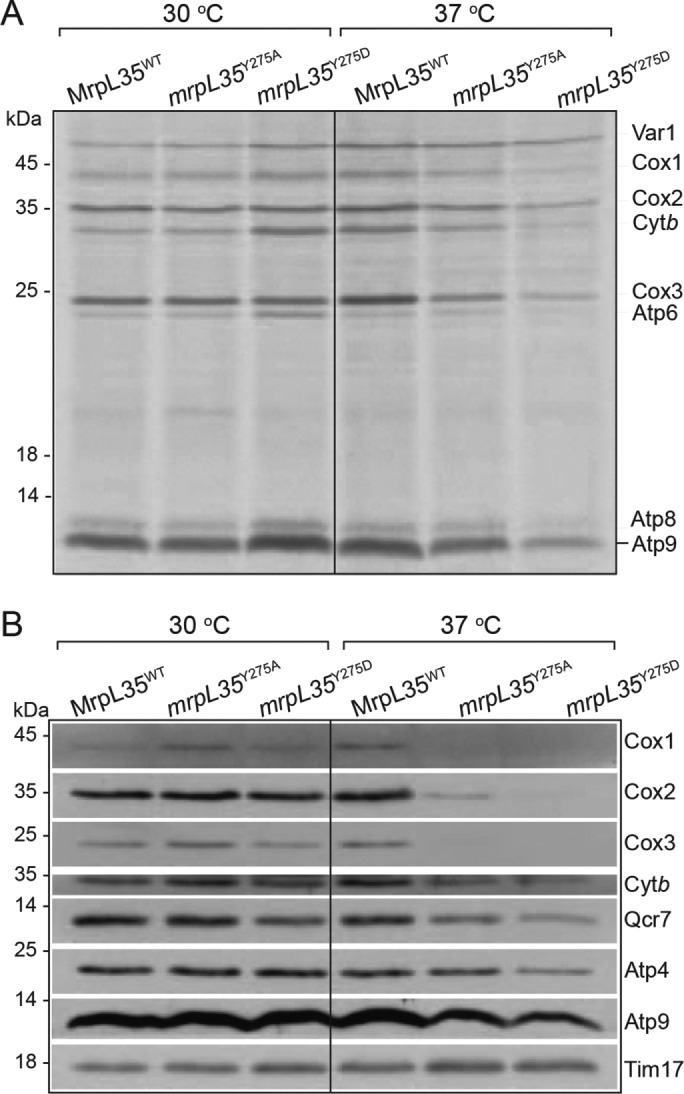
The mrpL35 mutants maintain the ability to translate even at nonpermissive temperature. (A) MrpL35 wild-type mrpL35Y275A and mrpL35Y275D mutant strains were grown on minimal media supplemented with galactose grown at either 30°C or 37°C for 24 h, as indicated. Mitochondrial translation capacity was monitored in vivo for 10 min with [35S]methionine and in the presence of cycloheximide (200 ug/ml). Equivalent amounts of cells were reisolated and proteins extracted and analyzed by SDS–PAGE, Western blotting, and autoradiography. (B) The indicated strains were grown as described in A, total cellular proteins were extracted, and steady-state levels of indicated mitochondrial proteins were measured following SDS–PAGE, Western blotting, and immunedecorated with antibodies indicated. Tim17 served as loading control.
FIGURE 4:

The mrpL35 mutants are COX assembly defective and display an altered Cox1 synthesis profile. (A) Mitochondria were isolated from indicated strains, which had been grown at 30°C. Following a preincubation for 10 min at 37°C, in organello labeling in the presence of [35S]methionine was performed for 20 min and 30 min, as indicated, followed by a chase for 5 min in the presence of puromycin and cold methionine. Mitochondria were reisolated and samples analyzed by SDS–PAGE, Western blotting, and autoradiography (left panel). The amounts of newly synthesized Cox1 were quantified by subsequent phosphorimaging and expressed as a ratio of the levels of Cox2 and Cox3 synthesized in the same time period (30 min) (right panels). (B) Steady-state levels of the OXPHOS subunits in mitochondria (25, 50, 100 µg) isolated from indicated strains detailed in A were determined following SDS–PAGE, Western blotting, and immunedecoration. Tim44 was used as a loading control (upper panel). The amounts of Cox1, Qcr7, and Atp4 were quantified, normalized to Tim44 levels, and expressed as a percentage of the wild-type control (lower panels). (C) Mitochondria isolated from indicated strains were solubilized in DDM (0.6%) and subjected to BN–PAGE analysis, Western blotting, and immunedecoration with antibodies against the cytochrome bc1 (bc1) subunit, cytochrome c1 (α−Cytc1), Cox3 (COX), and F1Fo-ATP synthase (F1Fo) subunits (α−F1α/β) (upper panels). The amounts of these complexes were quantified in the mutant mitochondria and expressed as a percentage of the control wild type (lower panels). (D) The specific activities of the cytochrome bc1 (top panel) and COX (bottom panel) enzymes were measured in the mrpL35 mutant and wild-type MrpL35WT control (WT) mitochondria, as described under Materials and Methods and expressed as a percentage of the wild-type control. Measurements were performed in triplicate and the statistical analysis and values were analyzed for statistical significance by Student’s t test. Data are presented as means ± SD of percentage of control. (E) In organello translation in the presence of [35S]methionine was performed for 30 min in the indicated mitochondria (upper panel) and further analyzed as described in A. The steady-state levels of indicated OXPHOS subunits were analyzed (lower panel), as described in B. Tim17 is used as a loading control.
Western blotting of the protein extracts from the cells grown at the different temperatures indicated that the COX subunits, Cox1, Cox2, and Cox3, were strongly reduced in the mrpL35Y275A and more so in the mrpL35Y275D mutant strains grown at the nonpermissive temperature of 37°C (Figure 3B). Although the mrpL35 mutants display the capacity to synthesize these COX subunits at 37°C, their strongly reduced steady-state protein amounts are indicative of a possible COX assembly defect at elevated temperatures, which would promote turnover of the newly synthesized Cox1, Cox2, and Cox3 proteins. Control experiments demonstrated that the preexisting COX complex assembled in the mutant at the permissive temperature did not display an enhanced proteolytic instability (i.e. relative to the wild-type control assayed in parallel) when shifted to 37°C (Supplemental Figure S1). These results indicate that if assembled at the permissive temperature, the COX complex in the mutant cells displays a similar resilience as that of the wild-type control when shifted to 37°C. We conclude therefore that the reduced levels of COX complex in the mrpL35 mutants when grown at 37°C is indicative of a compromised ability to initially assemble the COX enzyme when the mitoribosomal protein synthesis and assembly processes occur at the nonpermissive temperature.
Although somewhat reduced, the levels of cytochrome b and Atp9 (and of Qcr7 and Atp4, two proteins dependent on the presence of cytochrome b- and Atp9-containing complexes, respectively, for their stability), were clearly less affected in the mrpL35 mutants than the COX subunits were (Figure 3B).
The mrpL35 Y275 mutants display a cytochrome c oxidase defect
To analyze the OXPHOS defect in the mrpL35Y275A and mrpL35Y275D mutant strains in more depth, mitochondria were isolated from cultures grown in galactose/lactate media at the permissive temperature of 30°C. First, in organello translation in the presence of [35S]methionine was performed, where we observed that the overall translational capacity of the mitochondrial ribosomes was not adversely affected in the mrpL35 mutants (Figure 4A). The mrpL35 Y275D mutant mitochondria, however, displayed an altered translation profile with the levels of Cox1, cytochrome b, and Atp6 elevated, relative to the wild-type control, and the levels of Cox2 were somewhat reduced (Figure 4A, left panel). The enhanced levels of newly synthesized Cox1 protein in the mrpL35 mutant mitochondria, in particular in the mrpL35Y275D mutant, indicated that the tightly regulated Cox1 synthesis pathway may be misregulated (Figure 4A, right panels).
Western blotting of the isolated mrpL35 mutant mitochondrial preparations showed that the steady-state amounts of COX subunits Cox1, Cox2, and Cox3 were reduced, in particular in the mrpL35Y275D mitochondria, indicating that although synthesized at relatively normal (Cox2 and Cox3) or elevated (Cox1) levels in organello, the COX subunits fail to efficiently assemble in the mrpL35 mutant (Figure 4B). Blue native gel electrophoresis (BN–PAGE) analysis of the individual OXPHOS complexes confirmed that the levels of assembled COX complex were strongly reduced in the mrpL35 mutant mitochondria relative to the other OXPHOS complexes analyzed (Figure 4C, upper and lower panels). A parallel reduction in the level of COX enzyme activity was also measured in the mrpL35 mutants (Figure 4D). In contrast, the levels of the cytochrome bc1 and F1Fo-ATP synthase complexes, as indicated by steady-state analysis of individual subunits, BN–PAGE analysis of the complexes and their levels and assembly states, and the measurement of the cytochrome bc1 enzyme activity were not affected to the same extent as the COX complex was in the mrpL35Y275A and mrpL35Y275D mitochondria (Figure 4, B–D).
The ability of the mrpL35Y275 mutants to continue to synthesize Cox1, and moreover at elevated levels relative to the wild-type MrpL35 control, was not anticipated given the COX complex assembly defect observed in these mitochondria. As outlined earlier, synthesis of Cox1 by the mitoribosomes is under strict feedback regulation with its assembly into the COX complex (Figure 1). Hence, as previously reported, Cox1 synthesis is characteristically reduced in COX assembly mutants, for example, Δcox10 and Δcox18 mitochondria, where the steady-state levels of COX subunits are strongly reduced (Figure 4E). In contrast, the mrpL35Y275 mutant, despite having strongly reduced COX subunit levels, not only retains the ability but also displays enhanced capacity to synthesize the Cox1 protein (Figure 4E).
Taken together, these data demonstrate that mutation of the MrpL35 protein at residue Y275 does not cause an overall inhibition in mitochondrial protein translation but rather results in an altered translational profile and a notable misregulated Cox1 synthesis behavior and a COX assembly defect.
Mutation in the Y275 residue of MrpL35 reveals a mitoribosomal connection to the COX assembly chaperones, Coa3 and Cox14
The observed elevated levels of Cox1 synthesis in the absence of assembly of the COX complex suggests that the Mss51 feedback regulatory system may be perturbed in the mrpL35 mutant mitochondria. Analysis of the steady-state levels of Mss51, Cox14, Shy1, and Coa3 assembly factors indicated them to be present in the mrpL35 mutant mitochondria at amounts similar to the wild-type MrpL35 control (Figure 5A). Thus the aberrant Cox1 synthesis and/or lack of efficient COX enzyme assembly observed in the mrpL35 mutant mitochondria was not due to altered levels of these key COX regulatory/assembly factors.
FIGURE 5:

mrpL35 mutant exhibits alterations in Cox14 and Coa3 behavior. (A) Steady-state levels of proteins in isolated mrpL35 mutant mitochondria were determined as described in Figure 4B. (B) In organello translation in the presence [35S]methionine was performed for 30 min in isolated MrpL35WT or mrpL35Y275D mitochondria harboring Coa3His, or not, as indicated. Following translation, mitochondria were solubilized with 0.6% DDM and affinity purification of Coa3His on Ni-NTA beads was performed, followed by SDS–PAGE, Western blotting, and autoradiography (upper panel). The recovery of Coa3His on the Ni-NTA beads was demonstrated by immunedecoration of the resulting blot with anti-His antibody (lower panel). Total, 5% of solubilized material; Bound, 100% of affinity purified material recovered from the Ni-NTA beads. (C) Same as B, except the in organello step was omitted, mitochondria were solubilized in 1% digitonin, and immunedecoration with indicated antibodies was performed following Western blotting. The Cox14 and novel, slower migrating Cox14* species enriched with the affinity purified Coa3His complex is indicated. (D) BN–PAGE analysis of Coa3-containing complexes following solubilization of indicated isolated mitochondria with 1% digitonin. Western blotting and immunedecoration with Coa3 antisera was performed and protein complex that nonspecifically reacts with the Coa3 antiserum is indicated by **. (E) Chemical cross-linking using DSG (+) or the mock treatment of solvent DMSO alone (-) was performed on mitochondria isolated from the indicated yeast strains. Following SDS–PAGE and Western blotting, decoration with Coa3- antisera was performed. Note the area encompassing the 14.4- to 25-kDa range of the gel is only shown (upper panel), as no Coa3-specific cross-link adducts were observed in the higher molecular mass range of the gel (not shown). The minus cross-linking (DMSO) control is only shown for the MrpL35WT sample, as this reflected the results for all other mitochondrial types analyzed, that is, the complete absence of any Coa3-immunereactive material in this molecular weight range. The positions of the Coa3 monomer (lower panel and lighter exposure) and the four DSG-Coa3 adducts observed (upper panel) are indicated. A protein (14 kDa) that nonspecifically reacts with the Coa3 antiserum and observed in the lower panel, is indicated by **.
Following insertion into the membrane, newly synthesized Cox1 forms a COA assembly intermediate with Coa3 and other factors (including Cox14, Mss51, Coa1, and Coa2), which facilitates its subsequent assembly into the COX enzyme. We therefore tested whether radiolabeled Cox1 in the mrpL35 mutants could be recovered in a complex with Coa3, and to do so we employed MrpL35 wild-type and mrpL35Y275D mutant mitochondria harboring a His-tagged Coa3 derivative (Coa3His). Following in organello synthesis, radiolabeled Cox1 protein was recovered in a specific manner with the affinity-purified Coa3His species from both the control and mrpL35 mutant mitochondria (Figure 5B). The observed association of Cox1 with Coa3His in the mrpL35Y275D mitochondria suggests that the COX assembly defect in this mutant is downstream of the Cox1 synthesis and Coa3-Cox1 assembly intermediate formation events.
Affinity-purified Coa3His complexes were also analyzed to ascertain whether the association of Coa3 with Mss51, Shy1, and Cox14 was perturbed in the mrpL35 mutant (Figure 5C). Both Shy1 and Mss51 copurified with Coa3His; however, the levels of both proteins associated with the Coa3His complex from the mrpL35Y275D mitochondria were reduced, relative to those from the parallel wild-type control. Cox14 also detected in the affinity purified Coa3His complex from the mrpL35Y275D mitochondria; however, a significant fraction of the protein recovered was present as a novel, slower migrating species, termed here Cox14* (Figure 5C). A smaller proportion of this novel Cox14* species (relative to mature Cox14 protein) was also observed in the Coa3His complex purified from wild-type MrpL35 control mitochondria. The enrichment of this novel Cox14* species with the Coa3-complex from the mrpL35Y275D mitochondria would indicate that the behavior of the Cox14 protein is altered when the function of the MrpL35 protein is compromised.
BN–PAGE analysis indicated that the assembly of Coa3-containing complexes was significantly affected in the mrpL35Y275D mutant mitochondria (Figure 5D). In the wild-type MrpL35 control mitochondria, Coa3 was recovered in COA complexes of ∼240–480 kDa in mass, which have been proposed to also include the Cox1–Coa3 assembly intermediates of the COX complex (McStay et al., 2013; Mayorga et al., 2016). The level of these Coa3–COA complexes was strongly reduced in the mrpL35Y275D mutant mitochondria, indicating that despite the fact that newly synthesized Cox1 could associate with Coa3 (Figure 5B), the assembly state of the predominant Coa3–COA-containing complexes was strongly perturbed in this mutant. As the steady-state levels of the Coa3 protein were not reduced in the mrpL35Y275D mitochondria (Figure 5A), we conclude that the majority of the Coa3 protein in the mutant must be present in complexes smaller than 60 kDa under these conditions and thus not retained in the BN–PAGE gel. In contrast to the mrpL35Y275D mutant, the assembly state of the Coa3 complex did not appear to be affected in the mrpL35Y275A mutant mitochondria. As these mitochondria had both been isolated from cells grown at the permissive temperature, it appears that the phenotype of the mrpL35Y275D mutant is more severe and penetrated into cells grown at 30°C, in contrast to the mrpL35Y275A mutant. Consistently reduced COX levels were observed in these isolated mrpL35Y275D mitochondria and to a lesser extent in the mrpL35Y275A mitochondria (Figure 4, B–D). The aberrant assembly state of Coa3 observed in the mrpL35Y275D mutant was not simply due to the absence of an assembled COX enzyme, as Coa3-containing COA complexes were detected in mitochondria from the Δcox10 and Δcox18 COX assembly mutants, analyzed in parallel. The perturbed assembly of the Coa3–COA complexes in the mrpL35Y275D mutant mitochondria mirrored, however, that observed in the Δmss51 and wild-type/ρ0 mitochondria (Figure 5D). In contrast to these mitochondria where Cox1 synthesis is absent (Barrientos et al., 2004), the mrpL35Y275D mitochondria retain the capacity to synthesize Cox1 and form Cox1–Coa3 intermediates. We conclude therefore that in the ribosomal mutant mrpL35Y275D, the further (or stable) assembly of Coa3–Cox1 into larger Coa3–COA complexes was perturbed.
Finally, the molecular environment of Coa3 in intact mrpL35 mutant mitochondria was independently probed with the chemical cross-linking agent disuccinimidyl glutarate (DSG) (Figure 5E). In the wild-type MrpL35 control mitochondria, endogenous Coa3 (8 kDa) was observed to form three small adducts in the presence of DSG, which corresponded to Coa3 cross-linked to partners of ∼14, 12, and 8 kDa, respectively. A reduction in the formation of the smaller two of the Coa3-adducts, and a parallel acquisition of a novel fourth Coa3-adduct, corresponding to Coa3 cross-linked to an 11-kDa protein, was observed instead in the mrpL35Y275D mitochondria. The Coa3-adduct profile in the mrpL35Y275D mitochondria bore similarities to that obtained in the Δmss51 and Δcox14 mutant mitochondria, analyzed in parallel, where loss of the Coa3+12 kDa and Coa3+8 kDa adducts, and the gain of the novel Coa3+11 kDa adduct, was observed (Figure 5E). The altered Coa3-adduct profile in these mitochondria may be indicative of their COX-assembly defective phenotype, as it was also observed in Δcox18 mitochondria and also in the ribosomal mutant mrp20ΔC mitochondria, which are defective in mitochondrial translation and, consequently, COX assembly. Although the identities of the Coa3 cross-linked partners in these adducts are unknown at this time, the observed altered Coa3 cross-linking pattern in the mrpL35Y275D mitochondria is further evidence of an aberrant Coa3 condition in this mitoribosomal mutant.
In summary, the molecular environment of Coa3, and the nature of the Cox14 species associated with it, is altered in the mrpL35 mutant. These perturbations may impact the productive nature of the Cox1-assembly intermediate formed with the Coa3 and thus the further assembly of Cox1 into the COX enzyme.
Mutation of Y275 residue perturbs the close relationship of MrpL35 with Mrp7 protein
The cryo-EM structures of yeast mitoribosomes indicate MrpL35 (mL38) to be a peripheral component of the central protuberance where it is embraced by mitospecific elements of the Mrp7 (bL27) protein. To gain more insight into MrpL35 and the possible effect of the Y275 mutation on its interactions with partner proteins, we affinity purified His-tagged wild-type and mutant MrpL35 derivatives, which had been expressed in the ribosome assembly yeast mutant, mrp20ΔC (Kaur and Stuart, 2011). Stable assembly of the 54S particles has been reported to be defective in this mrp20ΔC mutant, and instead ribosomal proteins can be purified in subassemblies or subcomplexes, enabling their compositional analysis (Kaur and Stuart, 2011). The affinity purified wild-type MrpL35His was recovered in a complex with other proteins, which were shown either through specific antibody decorations or mass spectrometry analysis (Supplemental Table S1) to include Mrp7 (bL27), MrpL36 (bL31), and MrpL7 (uL5) and two mitospecific proteins, MrpL17 (mL46) and MrpL28 (mL40) (Figure 6A). Western blotting with available antibodies against Mrp7 (bL27) and MrpL36 (bL31) confirmed their specificity of their association with MrpL35His, as they were not detected in the Mrp20 ribosomal exit pore subcomplex, purified parallel via MrpL25His (mL59), a known component of the Mrp20 exit pore subcomplex (Kaur and Stuart, 2011) (Figure 6, B and C). Reciprocal affinity purifications of MrpL7His, MrpL17His, and Mrp7His from the mrp20ΔC mutant strain also confirmed their specific association with Mrp7, MrpL36, and MrpL35 proteins (Figure 6B). The ability of Mrp7 to be copurified with the His-tagged derivatives of mrpL35Y275A and mrpL35Y275D proteins was strongly reduced relative to the wild-type MrpL35His control (Figure 6C). The recovery of MrpL36 (bL31) in the mutant mrpL35 subcomplexes was also reduced, albeit not to the same extent as the Mrp7 protein. These results indicate that the interaction of MrpL35 with Mrp7 (and possibly other components of the central protuberance subcomplex) is perturbed through the mutation of the Y275 residue, such that it does not remain associated with the Y275 mutated mrpL35 protein under these detergent solubilization and purification conditions.
FIGURE 6:
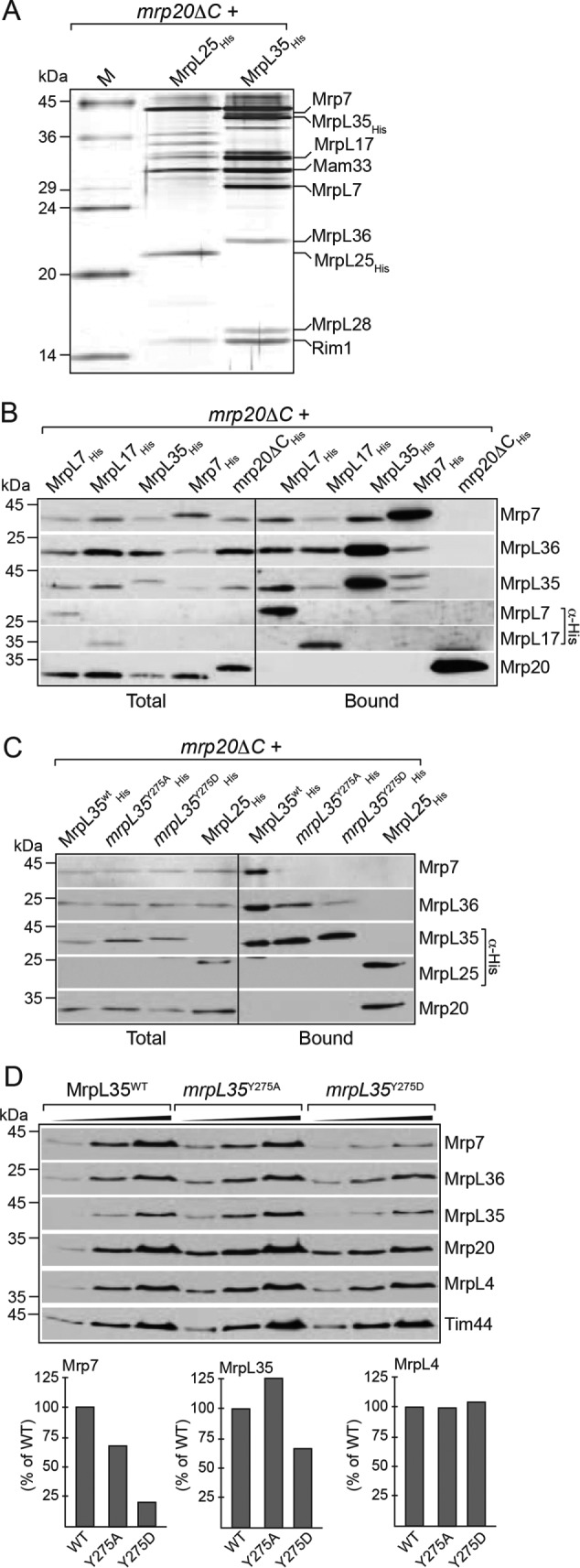
Mutation of MrpL35 perturbs the association with and stability of Mrp7. (A) Isolated mrp20ΔC mitochondria harboring His-tagged derivatives of wild-type MrpL35 or MrpL25, as indicated, were solubilized with 1.0% Triton X-100 and subjected to affinity purification protocol with Ni-NTA beads. The Ni-NTA purified material was further analyzed by SDS–PAGE and silver staining. The positions of proteins identified to copurify with the MrpL35His and MrpL25His proteins are indicated. Note Rim1 and Mam33 were found to copurify with both the MrpL35His and MrpL25His subcomplexes, which was confirmed by Western blotting (Rim1) and reciprocal Mam33His purification studies (not shown). M, molecular mass marker proteins (kDa) used for SDS–PAGE. (B) Affinity purification of His-tagged MrpL7, MrpL17, MrpL35, Mrp7, and mrp20ΔC derivatives from mrp20ΔC mitochondria was performed as described in A with the exception that following SDS–PAGE, Western blotting and immunedecoration with indicated specific antibodies was performed. Total, 5% of solubilized material; Bound, 100% of affinity purified material recovered from the Ni-NTA beads. (C) Affinity purification of His-tagged MrpL35WT, mrpL35Y275A, or mrpL35Y275D derivatives or MrpL25His from mrp20ΔC mitochondria was performed and analyzed as described in B. (D). The steady-state amounts of mitoribosomal proteins were determined by analyzing indicated mitochondria, as described in Figure 4B (upper panel). The amounts of Mrp7, MrpL35, and MrpL4 were quantified, normalized to Tim44, and expressed as a percentage of the wild-type control (lower panel).
Steady-state analysis of protein levels indicated that a reduction in Mrp7 levels was observed in the mrpL35Y275D mutant (Figure 6D, upper and lower panels). Mutation of the Y275 to Ala(A) residue did not greatly compromise the stability of the MrpL35 protein, though a decrease in MrpL35 steady-state levels was seen with the mrpL35Y275D mitochondria. In contrast to Mrp7, the levels of MrpL36, another central protuberance constituent, or other 54S particle proteins, for example, Mrp20 and MrpL4, were relatively unaffected in the mrpL35 mitochondria (Figure 6D).
Taken together, we conclude that the MrpL35 can assemble into a central protuberance subcomplex with the MrpL36, Mrp7, MrpL7, MrpL17, and MrpL28 proteins and that the association with, and possibly stability of, Mrp7 is perturbed through the mutation of the Y275 residue in MrpL35.
DISCUSSION
Mitochondrial translation and the assembly of the OXPHOS complexes are processes of critical importance to aerobic metabolism and cell viability. Mutation and impairment of mitoribosome capacity has been shown to be directly associated with a range of pathophysiological neuromuscular conditions (Lightowlers et al., 2014; De Silva et al., 2015). Investigating the molecular details of mitoribosome activity, regulation, and coupling of translation to the downstream OXPHOS assembly events is therefore critical for our understanding of normal and dysfunctional cellular metabolism. MrpL35 (mL38) is a mitospecific component of the yeast mitoribosomal 54S particle. Located in the central protuberance region, aspects of MrpL35 and the mitospecific domain of its partner protein Mrp7 (bL27) form an externally exposed element of this region of the mitoribosome (Amunts et al., 2014; Desai et al., 2017). Mutation of residue Tyr(Y)275 of the MrpL35 protein was found to compromise its ability to support aerobic growth and OXPHOS assembly, in particular at elevated temperatures, with the exchange for the negatively charged Asp(D) residue being more deleterious than Ala(A). Surprisingly, the molecular basis for the defective OXPHOS function in the mrpL35 mutants was not due to a primary inhibition in mitochondrial protein translation, but rather our OXPHOS complex analysis supports a pronounced COX assembly defect in these mrpL35 mutants. The consequences of the mrpL35 mutation for the assembly of the cytochrome bc1 or F1Fo-ATP synthase complexes, which both also contain mitoribosome synthesized subunits, were not as pronounced as they were for the COX complex assembly. As the mrpL35 mutants retained the capacity to synthesize the mitochondrially encoded COX subunits, the strong reduction in COX complex steady-state levels indicates that a defect may lie in a posttranslational step required for the assembly of the COX complex. Furthermore, the elevated levels of Cox1 synthesized in the mutants, despite their gross COX assembly defect, points to a breakdown in the Mss51/Cox14/Coa3-dependent feedback loop used to negatively regulate Cox1 synthesis under conditions when further assembly of Cox1 into a COX complex is perturbed.
Our data indicate that newly synthesized Cox1 can form the Cox1–Coa3-containing assembly intermediate in the mrpL35 mutant, but the further progression of Cox1 assembly from this intermediate into the Coa3–COA intermediate complex, and thus subsequently to the fully assembled COX enzyme, are defective. Both Cox14 and Coa3 act as negative regulators of the Cox1 synthesis regulatory pathway (Mick et al., 2010; Fontanesi et al., 2011). Thus the elevated levels of Cox1 synthesized in the mrpL35 may also be indicative of a breakdown in the function of the Cox14 and/or Coa3 proteins. We observed the presence of a novel form of Cox14, the Cox14* species enriched with the purified Coa3 complex and observed both in the wild type, and at greatly increased proportions in Coa3 complexes from the mrpL35 mutant. The Cox14* species has not been reported in the literature to date and may represent a novel, post–translationally modified form of the Cox14 protein. The abundance of Cox14* with Coa3 complex in the mrpL35 mutant would indicate its recycling to its regular Cox14 status is perturbed in this mitoribosomal mutant, which in turn may stall the progression of Cox1–Coa3 assembly intermediate. Both BN–PAGE and chemical cross-linking data further illustrate that the stable assembly state of the Coa3–COA complexes is defective in the mrpL35Y275D mutant. Although the nature of this modification is currently unknown, we speculate here that the formation of Cox14*, and its possible recycling to Cox14, may influence the productive nature of the Cox1–Coa3 complex as an assembly intermediate and/or its ability to recruit or retain Mss51 and thus Cox14’s ability to negatively control the Cox1 synthesis levels.
MrpL35, like the other members of the mL38 family, contains a PEBP-like domain in its C-terminal region. PEBP proteins are a highly diverse group of proteins (i.e., not exclusively mitoribosomal proteins) involved in signaling pathways. PEBP-domains form anion ligand-binding pockets, which can interfere with kinase-based signaling pathways when they form complexes with anionic ligands (Banfield et al., 1998; Banfield and Brady, 2000; Yousuf et al., 2014). The cryoEM structural analyses of mitoribosomes indicates that the PEBP-region of MrpL35/mL38 proteins is exposed to an exterior surface of the CP (Amunts et al., 2014, 2015; Greber et al., 2015; Desai et al., 2017). This aspect of the MrpL35 may represent a binding site for an external regulatory ligand or feature of a nonribosomal protein (e.g., a phosphorylated residue) in the central protuberance of the mitoribosome. Residue Y275 is conserved in all MrpL35 (mL38) proteins and is located in the PEBP-like domain of MrpL35 (mL38). It is unclear whether the PEBP-like domain of MrpL35 binds anionic ligands, but the conserved nature of this domain, and the detrimental effect of the Y275 mutation, may suggest a role for MrpL35 in a signaling pathway involving the mitoribosome. It is important to note that the region of MrpL35 where residue Y275 is located is directly adjacent to the fungal-specific, mitospecific domain of Mrp7 (bL27), which embraces the PEBP-like domain of MrpL35 (Amunts et al., 2014; Desai et al., 2017). We observed that mutation of residue Y275 destabilized the association of MrpL35 with its partner protein Mrp7 and led to a reduction in Mrp7 levels in the mutant mitochondria. Some of the reported phenotypes in the mrpL35 mutants could therefore be attributed to a disturbance in the relationship of MrpL35 with Mrp7 protein rather than (or in addition to) a defect in MrpL35.
How does mutation of MrpL35, a mitoribosomal protein of the central protuberance, affect the COX assembly pathway and at the level of the Coa3/Cox14 proteins? One possibility is a physical connection between elements of the central protuberance, involving the PEBP-domain of MrpL35 and/or Mrp7, and the COX assembly chaperones, such as Cox14 and Coa3 proteins in the membrane, which is important for the correct operation of the COX assembly chaperones. The mitoribosomes are physically tethered to the inner membrane; however, to date this association has been shown to involve aspects of the 54S particle in the vicinity of the polypeptide exit channel and thus distanced from the central protuberance region (Jia et al., 2009; Gruschke et al., 2010; Haque et al., 2010; Pfeffer et al., 2015). It is therefore unlikely that MrpL35 in context of the translating ribosome can have a direct physical connection to the Cox14/Coa3 proteins; however, we cannot rule out that MrpL35 (and/or Mrp7) independent of the assembled ribosome (e.g., alone or in context of a central protuberance subcomplex) could do so. Furthermore, it is also possible that MrpL35 and/or components of the central protuberance may serve to recruit Mss51 to the ribosome and thus modulate its capacity to act as a Cox1 translational activator; however, we have not obtained data to support a Mss51-MrpL35 (or Mrp7) interaction to date. Another possibility is that MrpL35, through its PEBP-like domain, serves to affect COX assembly by recruiting a common anionic factor important for signaling between the ribosome and the Coa3-Cox14-Mss51 proteins and thereby to balance the Cox1 synthesis rate with the downstream chaperoned assembly events. Mutation of the Y275 residue may impair this ligand binding and thus promote more Cox1 synthesis in a manner that is uncoupled from the downstream assembly events. Finally, the mutation of the MrpL35 protein could also cause alterations in the ribosomal structure/organization that affects the accessibility of the newly synthesized Cox1 protein to the Mss51 protein following its insertion into the membrane. Mss51 binds to the extreme C-terminal tail of the newly synthesized Cox1 protein, a step required to recruit the chaperone into the Cox1–Coa3 assembly intermediate (Shingu-Vazquez et al., 2010; Garcia-Villegas et al., 2017). Factors that hinder the access to the C-terminal region of the newly synthesized Cox1 may influence the recruitment of Mss51 to the Cox1–Coa3 complex. Delayed recycling of the ribosome at the termination of translation and/or impaired disengaging of the ribosome from Oxa1, the membrane site of cotranslational insertion of proteins, may both prevent Mss51’s timely access to the C-tail of Cox1. Interestingly, yeast mutants deficient in the ribosome-recycling factor (Rrf1), which promotes ribosomal subunit dissociation and recycling, display an OXPHOS assembly defect that was not caused by an inhibition in mitoribosomal translation per se (Ostojic et al., 2016). Rather, similarly to the mrpL35 mutants, the rrf1 mutant mitochondria displayed an altered profile of synthesis products, including increased levels of newly synthesized Cox1, and reduction in COX complex assembly levels. However, in contrast to the mrpL35 mutants, the yeast rrf1 mutants also exhibit a strong impairment in the assembly of the F1Fo–ATP synthesis complex, in addition to the observed COX assembly defect (Ostojic et al., 2016). The molecular defect underpinning the COX assembly deficiency in the mrpL35 Y275 mutant would thus appear to be different from that rrf1 mutant and ribosome recycling where a more pleiotropic OXPHOS assembly defect is described. Taking this together with other mrpL35 results reported here, namely i) the maintained ability to form the Cox1-Coa3 intermediate, ii) the enrichment of Cox14* with Coa3, and iii) the altered assembly state of the Coa3 complex (as evidenced by chemical cross-linking and BN–PAGE approaches), we favor that the mrpL35Y275 mutation does not cause a general ribosome translation inhibition defect but rather has direct consequences for functional integrity of the Mss51, Coa3, or Cox14 proteins.
In summary, our findings here unexpectedly highlight that components of the mitoribosome, MrpL35, and possibly its closely associated Mrp7 protein, play a role in regulating the COX assembly process, in particular by influencing the molecular environments of the Mss51, Coa3, and Cox14 proteins. Furthermore, this study also represents the first report of a novel Cox14 species, Cox14* associated with the Cox1–Coa3 complex, suggesting that Cox14 may be regulated by a posttranslational event. Future experimentation will ascertain the nature of the Cox14 modification and the role it plays in Cox14’s ability to support Coa3, Mss51, and the productive Cox1-assembly pathway and how this may involve the mitoribosomes and MrpL35.
MATERIALS AND METHODS
Yeast strains and growth conditions
All S. cerevisiae strains used in this study are in the haploid W303-1A genetic background (W303-1A, mat a leu2, trp1, ura3, his3, ade2). They include the ΔmrpL35 (MRPL35::KAN) mutant and those harboring plasmid borne copies of the wild-type MRPL35 gene or mrpL35 mutant derivatives, created through a plasmid shuffling approach, as described below. The W303-1A-based strain mrp20ΔC + GAL10-mrp20ΔC (termed here mrp20ΔC) was also employed (Kaur and Stuart, 2011). Yeast strains were cultured using standard protocols on either minimal medium supplemented as appropriate with uracil, tryptophan, leucine, adenine, histidine, or on full YP media, as indicated. Mitochondria were isolated from cultures grown at 30°C in minimal medium and containing 0.5% lactate and 2% galactose as carbon sources.
Generation of mrpL35 mutants
The yeast chromosomal MRPL35 open reading frame (ORF) was replaced with the KAN cassette in a haploid W303-1A yeast strain harboring a wild-type MRPL35 (MRPL35WT) genomic insert (corresponding to the MRPL35 ORF plus 650 base pairs 5′ and 300 base pairs 3′) cloned into a pRS316 URA3-based plasmid. The resulting strain then transformed with the centromeric plasmid pRS413 (HIS3) harboring either a similar genomic fragment containing the wild-type or mutant mrpL35 derivative or no insert (i.e., the empty plasmid). Following plasmid shuffling process, involving growth on 5′FOA to promote the loss of the pRS316(URA3)-MRPL35WT plasmid, ΔmrpL35 transformants harboring only the pRS413(HIS3) plasmids were selected. The resulting strains (ΔmrpL35+MRPL35WT or ΔmrpL35+mrpL35Y275 derivatives) are referred to here as the ΔmrpL35 null or MrpL35WT (wild-type) and the mrpL35Y275A, mrpL35Y275D, or mrpL35Y275F mutant strains. Mutations in the Y275 residue of MRPL35 gene in the pRS413(HIS3)-MRPL35 plasmid were generated using a PCR site-directed mutagenesis strategy.
Generation of His-tagged MrpL35 and Coa3 derivatives
The Yip351-LEU2 vector containing the open reading frame encoding either the wild-type MrpL35 or mrpL35Y275D derivatives or the Coa3 protein as C-terminal His12-tagged proteins and downstream of the galactose-inducible GAL10 promoter were created. The resulting plasmids were linearized with BstEII and integrated into the leu2 loci of indicated yeast strains and LEU+ transformants were selected. The presence of His-tagged derivative proteins in isolated mitochondria was confirmed using Western blot approaches with a His-specific antibody.
Affinity purification of His-tagged proteins
Isolated mitochondria (200 μg protein) harboring the His-tagged protein derivatives were solubilized in lysis buffer (160 mM KCl, 20 mM HEPES-KOH, 10 mM MgCl2, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], pH 7.4), with 1.0% Triton X-100, 0.6% n-dodecyl β-d-maltoside (DDM), or 1% digitonin, as indicated, for 30 min on ice. Following a clarifying spin, Ni-NTA purification of His-tagged proteins was performed as previously described (Jia et al., 2007).
BN–PAGE analysis
BN–PAGE analysis of digitonin (1%) or DDM (0.6%) solubilized mitochondrial extracts (30 μg protein) was performed using Invitrogen Nu–PAGE gradient (3–12%) gels according to the manufacturer’s protocol and followed by Western blotting and antibody decoration, as indicated.
Miscellaneous
All experiments presented were performed at least three times; results were consistently reproducible, and representative examples of data are given in the figures. Spectral measurements of specific enzyme activities of the cytochrome bc1 and COX complexes were performed as previously described (Tzagoloff et al., 1975). In organello translation with [35S]methionine and chemical cross-linking with DSG (7.7 Å, homobifunctional, amine-specific), were performed as previously described (Hell et al., 2001). Mitochondrial isolation, protein determination, and SDS–PAGE were performed as previously described (Dienhart and Stuart, 2008). The Cox1 and Cox3 antibodies used in this study were commercially obtained (Cox1: Molecular Probes, anti-yeast Cox1, mouse monoclonal 11D8-B7, lot# 6251-1; Cox3: Invitrogen/Novex Anti-Cox3 monoclonal, 459300, lot#H3578). All other antibodies used were rabbit polyclonal against the respective purified yeast proteins and generated either in the Stuart lab or received as gifts (see Acknowledgments). Antigen–antibody complexes were detected by horseradish peroxidase–coupled secondary antibodies and chemiluminescence detection on x-ray films. Resulting signals were quantified following scanning using Image Studio software.
Supplementary Material
Acknowledgments
We thank Sneha Potdar and Hannah Govendik for their preliminary work on the MrpL35 mutational analysis. We acknowledge and thank Vera Strogolova and Jessica Anderson for their helpful discussions. We are grateful to T. Mason and M. Ott for the valuable gifts of the ribosomal antisera and to A. Barrientos and F. Fontanesi for the kind gifts of the Coa3 and Mss51 null mutants and antisera. The research was supported by National Science Foundation (NSF) grants MCB 0744067 and MCB 1157722 to R.A.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSF.
Abbreviations used:
- BN–PAGE
blue native–polyacrylamide electrophoresis
- COX
cytochrome c oxidase
- PEBP
phosphatidylethanolamine-binding protein domain
- OXPHOS
oxidative phosphorylation.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-04-0239) on September 20, 2017.
REFERENCES
- Amunts A, Brown A, Bai XC, Llacer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343:1485–1489. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V. Ribosome. The structure of the human mitochondrial ribosome. Science. 2015;348:95–98. doi: 10.1126/science.aaa1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield MJ, Barker JJ, Perry AC, Brady RL. Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Structure. 1998;6:1245–1254. doi: 10.1016/s0969-2126(98)00125-7. [DOI] [PubMed] [Google Scholar]
- Banfield MJ, Brady RL. The structure of Antirrhinum centroradialis protein (CEN) suggests a role as a kinase regulator. J Mol Biol. 2000;297:1159–1170. doi: 10.1006/jmbi.2000.3619. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Zambrano A, Tzagoloff A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 2004;23:3472–3482. doi: 10.1038/sj.emboj.7600358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst P, Grivell LA. The mitochondrial genome of yeast. Cell. 1978;15:705–723. doi: 10.1016/0092-8674(78)90257-x. [DOI] [PubMed] [Google Scholar]
- Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science. 2014;346:718–722. doi: 10.1126/science.1258026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruun AW, Svendsen I, Sorensen SO, Kielland-Brandt MC, Winther JR. A high-affinity inhibitor of yeast carboxypeptidase Y is encoded by TFS1 and shows homology to a family of lipid binding proteins. Biochemistry. 1998;37:3351–3357. doi: 10.1021/bi971286w. [DOI] [PubMed] [Google Scholar]
- Clemente P, Peralta S, Cruz-Bermudez A, Echevarria L, Fontanesi F, Barrientos A, Fernandez-Moreno MA, Garesse R. hCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria. J Biol Chem. 2013;288:8321–8331. doi: 10.1074/jbc.M112.422220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva D, Tu YT, Amunts A, Fontanesi F, Barrientos A. Mitochondrial ribosome assembly in health and disease. Cell Cycle. 2015;14:2226–2250. doi: 10.1080/15384101.2015.1053672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennerlein S, Rehling P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J Cell Sci. 2015;128:833–837. doi: 10.1242/jcs.161729. [DOI] [PubMed] [Google Scholar]
- Desai N, Brown A, Amunts A, Ramakrishnan V. The structure of the yeast mitochondrial ribosome. Science. 2017;355:528–531. doi: 10.1126/science.aal2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienhart MK, Stuart RA. The yeast Aac2 protein exists in physical association with the cytochrome bc1-COX supercomplex and the TIM23 machinery. Mol Biol Cell. 2008;19:3934–3943. doi: 10.1091/mbc.E08-04-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanesi F, Clemente P, Barrientos A. Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J Biol Chem. 2011;286:555–566. doi: 10.1074/jbc.M110.188805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanesi F, Soto IC, Horn D, Barrientos A. Mss51 and Ssc1 facilitate translational regulation of cytochrome c oxidase biogenesis. Mol Cell Biol. 2010;30:245–259. doi: 10.1128/MCB.00983-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Villegas R, Camacho-Villasana Y, Shingu-Vazquez MA, Cabrera-Orefice A, Uribe-Carvajal S, Fox TD, Perez-Martinez X. The Cox1 C-terminal domain is a central regulator of cytochrome c oxidase biogenesis in yeast mitochondria. J Biol Chem. 2017;292:10912–10925. doi: 10.1074/jbc.M116.773077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graack HR, Grohmann L, Choli T. Mitochondrial ribosomes of yeast: isolation of individual proteins and N-terminal sequencing. FEBS Lett. 1988;242:4–8. doi: 10.1016/0014-5793(88)80975-x. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Ban N. Structure and function of the mitochondrial ribosome. Annu Rev Biochem. 2016;85:103–132. doi: 10.1146/annurev-biochem-060815-014343. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science. 2015;348:303–308. doi: 10.1126/science.aaa3872. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R, Ban N. The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014a;515:283–286. doi: 10.1038/nature13895. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014b;505:515–519. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- Gruschke S, Grone K, Heublein M, Holz S, Israel L, Imhof A, Herrmann JM, Ott M. Proteins at the polypeptide tunnel exit of the yeast mitochondrial ribosome. J Biol Chem. 2010;285:19022–19028. doi: 10.1074/jbc.M110.113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruschke S, Ott M. The polypeptide tunnel exit of the mitochondrial ribosome is tailored to meet the specific requirements of the organelle. BioEssays. 2010;32:1050–1057. doi: 10.1002/bies.201000081. [DOI] [PubMed] [Google Scholar]
- Haque ME, Spremulli LL, Fecko CJ. Identification of protein-protein and protein-ribosome interacting regions of the C-terminal tail of human mitochondrial inner membrane protein Oxa1L. J Biol Chem. 2010;285:34991–34998. doi: 10.1074/jbc.M110.163808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell K, Neupert W, Stuart RA. Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. EMBO J. 2001;20:1281–1288. doi: 10.1093/emboj/20.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Dienhart MK, Stuart RA. Oxa1 directly interacts with Atp9 and mediates its assembly into the mitochondrial F1Fo-ATP synthase complex. Mol Biol Cell. 2007;18:1897–1908. doi: 10.1091/mbc.E06-10-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Kaur J, Stuart RA. Mapping of the Saccharomyces cerevisiae Oxa1-mitochondrial ribosome interface and identification of MrpL40, a ribosomal protein in close proximity to Oxa1 and critical for oxidative phosphorylation complex assembly. Eukaryot Cell. 2009;8:1792–1802. doi: 10.1128/EC.00219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J, Stuart RA. Truncation of the Mrp20 protein reveals new ribosome-assembly subcomplex in mitochondria. EMBO Rep. 2011;12:950–955. doi: 10.1038/embor.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal PS, Sharma MR, Agrawal RK. The 55S mammalian mitochondrial ribosome and its tRNA-exit region. Biochimie. 2015;114:119–126. doi: 10.1016/j.biochi.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal PS, Sharma MR, Booth TM, Haque EM, Tung CS, Sanbonmatsu KY, Spremulli LL, Agrawal RK. Cryo-EM structure of the small subunit of the mammalian mitochondrial ribosome. Proc Natl Acad Sci USA. 2014;111:7284–7289. doi: 10.1073/pnas.1401657111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrein K, Schilling R, Moller-Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T, Ott M. Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep. 2015;10:843–853. doi: 10.1016/j.celrep.2015.01.012. [DOI] [PubMed] [Google Scholar]
- Khalimonchuk O, Bestwick M, Meunier B, Watts TC, Winge DR. Formation of the redox cofactor centers during Cox1 maturation in yeast cytochrome oxidase. Mol Cell Biol. 2010;30:1004–1017. doi: 10.1128/MCB.00640-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc EC, Burkhart W, Blackburn K, Moseley A, Koc H, Spremulli LL. A proteomics approach to the identification of mammalian mitochondrial small subunit ribosomal proteins. J Biol Chem. 2000;275:32585–32591. doi: 10.1074/jbc.M003596200. [DOI] [PubMed] [Google Scholar]
- Lightowlers RN, Rozanska A, Chrzanowska-Lightowlers ZM. Mitochondrial protein synthesis: figuring the fundamentals, complexities and complications, of mammalian mitochondrial translation. FEBS Lett. 2014;588:2496–2503. doi: 10.1016/j.febslet.2014.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai N, Chrzanowska-Lightowlers ZM, Lightowlers RN. The process of mammalian mitochondrial protein synthesis. Cell Tissue Res. 2017;367:5–20. doi: 10.1007/s00441-016-2456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayorga JP, Camacho-Villasana Y, Shingu-Vazquez M, Garcia-Villegas R, Zamudio-Ochoa A, Garcia-Guerrero AE, Hernandez G, Perez-Martinez X. A novel function of Pet54 in regulation of Cox1 synthesis in Saccharomyces cerevisiae mitochondria. J Biol Chem. 2016;291:9343–9355. doi: 10.1074/jbc.M116.721985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay GP, Su CH, Tzagoloff A. Stabilization of Cox1p intermediates by the Cox14p-Coa3p complex. FEBS Lett. 2013;587:943–949. doi: 10.1016/j.febslet.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick DU, Fox TD, Rehling P. Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat Rev Mol Cell Biol. 2011;12:14–20. doi: 10.1038/nrm3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick DU, Vukotic M, Piechura H, Meyer HE, Warscheid B, Deckers M, Rehling P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J Cell Biol. 2010;191:141–154. doi: 10.1083/jcb.201007026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien TW. Evolution of a protein-rich mitochondrial ribosome: implications for human genetic disease. Gene. 2002;286:73–79. doi: 10.1016/s0378-1119(01)00808-3. [DOI] [PubMed] [Google Scholar]
- O’Brien TW. Properties of human mitochondrial ribosomes. IUBMB Life. 2003;55:505–513. doi: 10.1080/15216540310001626610. [DOI] [PubMed] [Google Scholar]
- Ostojic J, Panozzo C, Bourand-Plantefol A, Herbert CJ, Dujardin G, Bonnefoy N. Ribosome recycling defects modify the balance between the synthesis and assembly of specific subunits of the oxidative phosphorylation complexes in yeast mitochondria. Nucleic Acids Res. 2016;44:5785–5797. doi: 10.1093/nar/gkw490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Amunts A, Brown A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu Rev Biochem. 2016;85:77–101. doi: 10.1146/annurev-biochem-060815-014334. [DOI] [PubMed] [Google Scholar]
- Perez-Martinez X, Broadley SA, Fox TD. Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J. 2003;22:5951–5961. doi: 10.1093/emboj/cdg566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Martinez X, Butler CA, Shingu-Vazquez M, Fox TD. Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol Biol Cell. 2009;20:4371–4380. doi: 10.1091/mbc.E09-06-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Woellhaf MW, Herrmann JM, Forster F. Organization of the mitochondrial translation machinery studied in situ by cryoelectron tomography. Nat Commun. 2015;6:6019. doi: 10.1038/ncomms7019. [DOI] [PubMed] [Google Scholar]
- Pierrel F, Bestwick ML, Cobine PA, Khalimonchuk O, Cricco JA, Winge DR. Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion in cytochrome c oxidase assembly. EMBO J. 2007;26:4335–4346. doi: 10.1038/sj.emboj.7601861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Dennerlein R, Oeljeklaus S, Lorenzi I, Ronsor C, Bareth B, Schendzielorz AB, Wang C, Warscheid B, Rehling P, Dennerlein S. Mitochondrial protein synthesis adapts to influx of nuclear-encoded protein. Cell. 2016;167:471–483.e410. doi: 10.1016/j.cell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre L, Vallee B, Bureaud N, Schoentgen F, Zelwer C. Crystal structure of the phosphatidylethanolamine-binding protein from bovine brain: a novel structural class of phospholipid-binding proteins. Structure. 1998;6:1255–1265. doi: 10.1016/s0969-2126(98)00126-9. [DOI] [PubMed] [Google Scholar]
- Shingu-Vazquez M, Camacho-Villasana Y, Sandoval-Romero L, Butler CA, Fox TD, Perez-Martinez X. The carboxyl-terminal end of Cox1 is required for feedback assembly regulation of Cox1 synthesis in Saccharomyces cerevisiae mitochondria. J Biol Chem. 2010;285:34382–34389. doi: 10.1074/jbc.M110.161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, Ettema TJ. Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res. 2007;35:4686–4703. doi: 10.1093/nar/gkm441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto IC, Barrientos A. Mitochondrial cytochrome c oxidase biogenesis is regulated by the redox state of a heme-binding translational activator. Antioxid Redox Signal. 2016;24:281–298. doi: 10.1089/ars.2015.6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto IC, Fontanesi F, Myers RS, Hamel P, Barrientos A. A heme-sensing mechanism in the translational regulation of mitochondrial cytochrome c oxidase biogenesis. Cell Metab. 2012;16:801–813. doi: 10.1016/j.cmet.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzagoloff A, Akai A, Needleman RB. Assembly of the mitochondrial membrane system. Characterization of nuclear mutants of Saccharomyces cerevisiae with defects in mitochondrial ATPase and respiratory enzymes. J Biol Chem. 1975;250:8228–8235. [PubMed] [Google Scholar]
- van der Sluis EO, Bauerschmitt H, Becker T, Mielke T, Frauenfeld J, Berninghausen O, Neupert W, Herrmann JM, Beckmann R. Parallel structural evolution of mitochondrial ribosomes and OXPHOS complexes. Genome Biol Evol. 2015;7:1235–1251. doi: 10.1093/gbe/evv061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousuf S, Duan M, Moen EL, Cross-Knorr S, Brilliant K, Bonavida B, LaValle T, Yeung KC, Al-Mulla F, Chin E, Chatterjee D. Raf kinase inhibitor protein (RKIP) blocks signal transducer and activator of transcription 3 (STAT3) activation in breast and prostate cancer. PLoS One. 2014;9:e92478. doi: 10.1371/journal.pone.0092478. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.