

Nck promotes breast carcinoma progression and metastasis by directing the polarized interaction of carcinoma cells with collagen fibrils, decreasing actin turnover, and enhancing the localization and activity of MMP14 at the cell surface through modulation of the spatiotemporal activation of Cdc42 and RhoA.
Abstract
Although it is known that noncatalytic region of tyrosine kinase (Nck) regulates cell adhesion and migration by bridging tyrosine phosphorylation with cytoskeletal remodeling, the role of Nck in tumorigenesis and metastasis has remained undetermined. Here we report that Nck is required for the growth and vascularization of primary tumors and lung metastases in a breast cancer xenograft model as well as extravasation following injection of carcinoma cells into the tail vein. We provide evidence that Nck directs the polarization of cell–matrix interactions for efficient migration in three-dimensional microenvironments. We show that Nck advances breast carcinoma cell invasion by regulating actin dynamics at invadopodia and enhancing focalized extracellular matrix proteolysis by directing the delivery and accumulation of MMP14 at the cell surface. We find that Nck-dependent cytoskeletal changes are mechanistically linked to enhanced RhoA but restricted spatiotemporal activation of Cdc42. Using a combination of protein silencing and forced expression of wild-type/constitutively active variants, we provide evidence that Nck is an upstream regulator of RhoA-dependent, MMP14-mediated breast carcinoma cell invasion. By identifying Nck as an important driver of breast carcinoma progression and metastasis, these results lay the groundwork for future studies assessing the therapeutic potential of targeting Nck in aggressive cancers.
INTRODUCTION
Metastasis, the outgrowth of secondary tumors following the successful colonization of distant organs by malignant cells, is the major cause of cancer death. Metastasis is regarded as a stepwise progression undertaken by transformed cells (Nguyen et al., 2009) that includes complex morphological and functional changes, collectively termed epithelial–mesenchymal transition (Ye and Weinberg, 2015), as well as the breaching of physical barriers represented by basement membranes and connective tissues (Sabeh et al., 2009; Kelley et al., 2014), the escape from immune surveillance mechanisms (Kitamura et al., 2015), and the homing and colonization of target organs (Nguyen et al., 2009). To a large extent, cytoskeletal remodeling is involved in the orchestration of cellular processes enabling the progression of metastasis. Thus, the identification of molecular mechanisms underlying cytoskeletal changes that promote invasion and metastasis holds great promise for the development of new targeted therapies in aggressive cancers.
Based on in vitro models, the past decade has witnessed significant advances in the understanding of structural and functional properties of invadosomes (Linder, 2009), including podosomes and invadopodia. These actin-based membrane protrusions facilitate, under specific physiological (podosomes) or pathological (invadopodia) conditions, cell invasion through tissues (Linder, 2009; Murphy and Courtneidge, 2011; Beaty and Condeelis, 2014). Although increasing evidence links the presence of such structures with progression of developmental (Santiago-Medina et al., 2015; Lohmer et al., 2016) and pathological processes (Gligorijevic et al., 2012; Leong et al., 2014; Seano et al., 2014), there remain significant challenges in determining the molecular underpinnings of invadosome biogenesis and their functional significance. Noticeably, the formation and function of invadopodia in cancer cells appears highly dependent on the activities of core molecular components including the Rho GTPases (Nakahara et al., 2003; Yamaguchi et al., 2005; Bravo-Cordero et al., 2011; Juin et al., 2014; Razidlo et al., 2014), the nucleation promoting factor neuronal Wiskott-Aldrich syndrome protein (N-WASp) (Yamaguchi et al., 2005; Gligorijevic et al., 2012), the scaffolding protein Tks5 (Seals et al., 2005; Stylli et al., 2009; Sharma et al., 2013; Leong et al., 2014), and the membrane-type matrix metalloproteinase MMP14 (Artym et al., 2006; Steffen et al., 2008). Although core invadopodial components are recruited in response to the engagement of cell surface receptors by growth factors (Yamaguchi et al., 2005; Rajadurai et al., 2012) and extracellular matrix fibrils (Mueller et al., 1999; Enderling et al., 2008; Artym et al., 2015), how extracellular cues are integrated to provide spatiotemporal coordination of the invadopodial molecular machinery remains poorly understood.
The family of noncatalytic region of tyrosine kinase (Nck) adaptors (Chen et al., 1998), comprising the paralogues Nck1/α and Nck2/β (herein Nck), is involved in the propagation of extracellular signals that induce tyrosine phosphorylation (Li and She, 2000). By virtue of its modular architecture, consisting of three N-terminal Src homology (SH) 3 domains and one C-terminal SH2 domain, Nck contributes to the organization of molecular ensembles that modulate the actin cytoskeleton (Li et al., 2001; Buday et al., 2002). Initially linked to pathogen-induced actin polymerization (Frischknecht et al., 1999; Gruenheid et al., 2001; Campellone et al., 2002), the role of Nck in regulation of actin dynamics in various cellular processes, including cell locomotion (Rivera et al., 2006; Chaki et al., 2013), was subsequently established. We, among others, demonstrated that Nck promotes localized actin polymerization through robust activation of N-WASp (Rivera et al., 2004, 2009; Ditlev et al., 2012). Although previous studies implicated Nck in actin polymerization during invadopodia formation and extracellular matrix (ECM) degradation (Stylli et al., 2009; Oser et al., 2010), the functional significance of Nck in tumor progression and metastasis and the underlying molecular mechanisms have not been elucidated. Here we show that Nck promotes primary tumor growth and progression to metastatic disease by directing the polarized interaction of breast carcinoma cells with collagen fibrils, thus enabling efficient three-dimensional (3D) migration. In addition, our results indicate that Nck regulates actin dynamics and enhances the localization and activity of MMP14 at the cell surface through a mechanism that involves modulation of the spatiotemporal activation of cell division control protein homologue 42 (Cdc42) and transforming protein RhoA (RhoA).
RESULTS
Nck silencing reduces growth and invasiveness of breast carcinoma cells in vitro
We used the triple-negative human breast carcinoma cell line MDA-MB-231 that is known to express both Nck1 and Nck2 (Oser et al., 2010; Labelle-Cote et al., 2011) (Supplemental Figure 1A). To begin to test the role of Nck in mammary carcinoma growth and invasion, cellular levels of Nck 1 and Nck2 were reduced by transfection of specific siRNA oligonucleotides. Because MMP14 is required for cancer cell invasion (Sabeh et al., 2009), we used MMP14 silencing as an additional control. The efficacy of small interference RNA (siRNA) and short hairpin RNA (shRNA) reagents in the silencing of target proteins was confirmed by Western blotting (Figure 1A and Supplemental Figure 1). The cellular levels of target proteins were reduced by 70–90% following silencing with either siRNAs or shRNAs (Supplemental Figure 1, B and C). Importantly, Nck silencing did not change the total cellular levels of MMP14 and, conversely, MMP14 silencing did not alter the total cellular levels of Nck. We performed Western blotting using an anti–pan-Nck antibody to confirm the specificity of siRNA reagents in cells expressing EYFP-Nck1 or mCherry-Nck2 fusion proteins (Supplemental Figure 1, D and E). In addition, functional experiments including reexpression of siRNA-resistant Nck in Nck-silenced cells (rescue approach) argue for the specificity of siRNA reagents (Supplemental Figure 2). The specificity of shRNA reagents was previously validated in our laboratory (Chaki et al., 2013).
FIGURE 1:

Nck silencing decreases mammary carcinoma cell invasion. Small interfering (si) RNA or short hairpin (sh) RNA was used, respectively, for transient or stable target protein silencing in MDA-MB-231 breast carcinoma cells. (A) Representative Western blots showing efficacy of target protein silencing. GAPDH levels were used as loading control. (B) Individual cell invasion expressed as percentage invasion (invaded cells/migrated cells × 100). Cells were subjected to migration or invasion assays using BD BioCoat Invasion Chambers in the presence of 10 ng/ml EGF used as a chemoattractant (n = 3 independent experiments). To assess migration, cells were seeded on uncoated control inserts and allowed to migrate for 5 h. To assess invasion, cells were seeded on growth factor–reduced coated inserts and allowed to invade for 18 h. (C) Representative images of spheroids at day 0 (left insets) and day 3 in a laminin-rich matrix. (D) Representative images of spheroids at day 0 (left insets) and day 1 of invasion in fibrillar collagen I matrices. Boxed areas were magnified to show morphology of invading cells (right insets). In C and D, scale bar equals 500 μm. (E) Box-and-whisker plots showing invasion distance (day 3) in a laminin-rich matrix (siScr, n = 21; siMMP14, n = 20; siNck, n = 18). (F) Box-and-whisker plots showing invasion distance (day 1) in fibrillar collagen I (shScr, n = 9; shMMP14, n = 7; shNck, n = 9). To determine invasion distance, the extreme diameter of each spheroid was measured using FIJI at four different angles and the average diameter calculated. The average diameter for time zero was the subtracted from each time point to determine the average invasion distance. Panels B, E, and F summarize data from at least three independent experiments. (G) Spheroid growth represented as total spheroid area during days 0–5 of spheroid formation (n = 2, three spheroids/condition/experiment). *p < 0.05.
Although a role for Nck1 in matrix proteolysis and serum-stimulated invasion of breast carcinoma cells was previously reported (Oser et al., 2010), the potential contribution of Nck2 was not investigated. Of note, MDA-MB-231 cells display abundant expression of Nck2 protein (Oser et al., 2010; Labelle-Cote et al., 2011) (Supplemental Figure 1A). We chose to determine the effects of the combined depletion of Nck1 and Nck2, rather than their independent contribution, because Nck1 and Nck2 share ∼68% amino acid sequence identity, and their functions are regarded as highly overlapping. Indeed, whereas simultaneous inactivation of Nck (both Nck1 and Nck2) leads to embryonic lethality, mice with inactivation of single Nck genes (either Nck1 or Nck2) are viable and do not exhibit an overt phenotype (Bladt et al., 2003; Jones et al., 2006; Clouthier et al., 2015). In addition, the binding specificities of Nck1 versus Nck2 SH2 domains were found to be essentially indistinguishable (Frese et al., 2006), a finding that further supports the notion of functional overlap between Nck1 and Nck2. To test the role of Nck (both Nck1 and Nck2) in epidermal growth factor (EGF)-stimulated proteolytic invasion, we determined invasion potential taking into account changes in cell migration. The invasiveness of individual cells in a laminin-rich matrix was assessed using BD BioCoat Invasion Chambers. Invasion was significantly decreased (p < 0.05) when Nck or MMP14-silenced cells were compared with scramble (Src) controls (Figure 1B). We then tested the invasive potential of multicellular tumor spheroids (MTS) embedded in a 3D laminin-rich matrix. Transiently silencing Nck, but not MMP14, resulted in significantly reduced (p < 0.05) invasion as early as 1 day after the MTS were embedded in the matrix and that difference persisted throughout the experimental period (Figure 1, C and E, and Supplemental Figure 3). We also tested MTS invasion in type I fibrillar collagen, a major extracellular matrix constituent (Maller et al., 2010; Mouw et al., 2014) known to play an important role in mammary gland development and tumor progression (Schedin and Keely, 2011). We found that silencing Nck or MMP14 resulted in significantly reduced (p < 0.05) invasion in fibrillar collagen I (Figure 1, D and F). In addition to invasiveness, we also determined the role of Nck in the growth of MTS by analyzing the change in spheroid area. Nck silencing resulted in significantly smaller (p < 0.05) spheroids when compared with shScr and untreated parental MDA-MB-231 cells (Figure 1G, and Supplemental Figure 4). Collectively, these results suggest that Nck adaptors are required for invasion in three-dimensional laminin-rich and collagen I matrices that are typically enriched in basement membranes and connective tissue, respectively.
Coordinated tumor cell–matrix interactions are disrupted by Nck silencing
We speculated that the reduced MTS invasion resulting from Nck silencing (Figure 1, C–F) was due, at least in part, to suboptimal interactions of breast carcinoma cells with the matrix in 3D microenvironments. Using high-resolution two-photon excited fluorescence (TPEF) and second harmonic generation (SHG) microscopy (Bai et al., 2014a, b) to image F-actin and collagen, respectively, we were able to qualitatively and quantitatively examine the role of Nck in coordinating cell–matrix interactions. To this end, we imaged collagen I gel-embedded MTS of MDA-MB-231 cells that were allowed to invade for 3 d. Consistent with results of invasion distance in collagen determined for populations of MTS (Figure 1, D and F), analysis of images collected at various distances from the spheroid showed that cells expressing shNck did not invade as far into the collagen matrix as cells expressing shScr (Figure 2A). We also observed that colocalized F-actin and collagen signal, indicating areas of cell-matrix interaction, had a distribution that reflected the direction of cell migration in control (shScr) but not shNck-expressing (shNck) cells. Indeed, the colocalized signal distributed predominantly at the “front” of the migrating control cells (Figure 2B). This coordinated distribution, however, appeared disrupted in shNck cells. A difference in the distribution was readily apparent in xy as well as xz and yz optical sections. To quantify such a difference, we thresholded the F-actin and collagen images and took their intersection to create a binary mask where F-actin and collagen were colocalized. We then measured the angular distribution of this interaction mask with θ = 0 taken to be the direction of the long axis of the cell pointed away from the spheroid. The results, presented in both polar and rectangular plots in Figure 2C, show that the interaction is heavily weighted toward the front in the shScr control but not in shNck cells.
FIGURE 2:

Nck depletion disrupts directional cell–matrix interactions. Spheroids of MDA-MB-231 cells expressing short hairpin (sh) RNAs encoding nontargeting sequences (shScr) or sequences targeting Nck (shNck) embedded in collagen I matrices were imaged using high-resolution two-photon excited fluorescence (TPEF) and second harmonic generation (SHG) microscopy. (A) Representative images of MDA-MB-231 cells fixed and stained for F-actin with fluorescent phalloidin (TPEF, red) during invasion in collagen I (SHG, green). Scale bar represents 50 μm. (B) Representative optical sections of cell–matrix interactions, represented by the colocalization between F-actin and collagen I, showing overlapping signals (yellow, arrowhead) predominantly in the leading edge of the cells expressing shScr but not in cells expressing shNck. Colocalized signal was converted into a binary interaction mask for further processing with theta (θ) equal to zero, corresponding to the direction of the long axis of the cell pointing away from the spheroid. Scale bar represents 10 μm. (C) Rectangular and polar plots of the angular distribution of the interaction masks showing polarization of cell–matrix interactions in cells expressing shScr. The polarized interaction is disrupted in cells expressing shNck, suggesting Nck helps coordinate the polarity of cell–matrix interactions required for efficient invasion. (D) The polarity of cell–matrix interactions was measured by an independent method comparing the displacement between the nucleus center and center of mass of the interaction mask treating each pixel in the mask as a unit mass. The center-of-mass displacement (CoM) refers to the displacement of the center of mass of the interaction mask (assuming a unit mass per pixel) from the centroid of an ideal mask with no angular inhomogeneity. In the case that the interaction mask was completely radially symmetric, the CoM displacement would be zero, while in the case that the interaction mask was radially asymmetric, the CoM displacement would be nonzero. This analysis also shows that cell–matrix interactions are more directed in cells expressing shScr compared with shNck. ***p < 0.001. (E, F) Scatter plots of CoM displacement vs. orientation angle, where an angle of zero corresponds to the direction of the normal vector of the spheroid, and aspect ratio (length/width) showing that cells expressing shNck are similarly aligned and stretched to cells expressing shScr but yet not properly polarized. Legends display interaction masks from data points replaced by an asterisk. Results shown summarize data from four assays in two independent experiments.
We also analyzed the degree of polarization of cell–matrix interactions using a complementary approach, that is, determination of the center-of-mass (CoM) displacement. This measurement refers to the displacement of the center of mass of the interaction mask (assuming a unit mass per pixel) from the centroid of an ideal mask with no angular inhomogeneity. If the interaction mask was completely radially symmetric, then the CoM displacement would be zero, while if the interaction mask was radially asymmetric, then the CoM displacement would be nonzero. In agreement with results from angular distribution, the data show that the CoM displacement was significantly greater (p < 0.001) for the shScr cells compared with shNck cells in the spheroid assays (Figure 2D).
To gain additional insights into the relationship between the degree of polarization of cell–matrix interactions and cell orientation or cell morphology, we analyzed the distribution of the center-of-mass displacement as a function of the migration angle (measured relative to a normal vector pointing out from the spheroid) or the aspect ratio of the cells. These measures were not significantly different between shScr and shNck cultures, as reflected by the scatter plots of the CoM displacement versus migration angle (Figure 2E) or aspect ratio (Figure 2F). Collectively, these findings suggest that, although seemingly dispensable for cell orientation and elongation, Nck is required for the proper polarization of cell–matrix interactions for efficient migration in 3D microenvironments.
Ablation of Nck reduces primary tumor growth, extravasation, and metastasis
The above results showing that abrogation of Nck signaling decreases invasiveness of breast carcinoma cells in vitro led us to test the role of Nck in tumorigenesis and metastasis. Breast carcinoma cells stably expressing nontargeting or Nck-targeting shRNAs were injected bilaterally into the flanks of nude mice and tumor formation was monitored over 9 wk. No differences were observed in primary tumor initiation. All mice developed primary tumors and macroscopically detectable tumors formed in 13/14 shScr and 12/14 shNck injection sites. The size of visible tumors (Figure 3A), measured with calipers at weekly intervals, was reduced as a result of Nck silencing. Indeed, tumors derived from Nck silenced (shNck) versus control (shScr) cells displayed slower growth rates (p < 0.05) from the sixth week postinjection until the termination of the experiment (Figure 3B). In addition, the difference in tumor mass (Figure 3C), determined following dissection at killing (week 9), approached statistical significance (shScr vs. shNck, p = 0.071). Histological examination of H&E preparations revealed that shScr tumors infiltrated the surrounding tissues to a greater extent and appear more vascularized, as evidenced by the readily distinguishable capillaries filled with red blood cells, when compared with shNck tumors (unpublished data). To confirm this notion, we performed immunohistochemistry with an antibody against Factor VIII–related antigen (von Willebrand factor), a useful marker for endothelial cells. Histological examination and quantitative image analysis showed significantly increased (p < 0.05) angiogenesis in primary tumors derived from shScr control versus Nck-silenced cells (Figure 3, D and E).
FIGURE 3:

Silencing of Nck reduces growth and vascularization of primary tumors and lung metastasis. Six- to 9-wk-old nude mice (NU/J) received subcutaneous, bilateral flank injections of MDA-MB-231 cells expressing short hairpin (sh) RNAs encoding nontargeting sequences (shScr) or sequences targeting Nck (shNck). (A) Representative images of mice bearing subcutaneous tumors induced by shScr and shNck cells at 8 wk postinjection. Notice the more vascularized aspect of shScr tumors. (B) Tumor volume (mean ± SEM) determined at weekly intervals. *p < 0.05. (C) Tumor mass (mean ± SEM) determined after dissection at killing 9 wk postinjection. (D) Representative images of tumor sections subjected to immunohistochemistry with an anti–Factor VIII antibody (highlighted by circles in shScr tissue) to detect neovascularization. (E) Quantification of Factor VIII–positive areas (n = 10 fields/condition) was performed using FIJI. (F) Representative images of H&E (top) and vimentin immunohistochemistry (bottom) of lung sections showing the presence of micrometastases in animals injected with shScr but not shNck cells. (G) Proportion of animals with lung metastases as determined by qPCR amplification of human GAPDH. Positive samples were determined based on a greater than sixfold increase in human GAPDH expression level in the lungs. (H) Examples of lymph node (H&E staining) and lung (vimentin immunohistochemistry) micrometastases present in animals injected with shScr carcinoma cells. In D, F, and H scale bar represents 100 μm.
Lung samples were processed and subjected to real-time PCR to detect expression of human GAPDH, which was used as an indicator of the presence of human cells in the mouse tissue (Yin et al., 2009). We confirmed the specificity of the probes used in this study by testing them against cDNAs from human breast cancer cells and mouse tissue. A conservative threshold of sixfold was selected to minimize false-positive detection when using a single reference gene (Kozera and Rapacz, 2013). Quantitation of human GAPDH fold change approached significance (p = 0.0552, unpublished data), an indicator strongly suggesting decreased lung metastases in the Nck silenced (shNck) compared with the scrambled (shScr) population. More importantly, four of seven and zero of seven mice injected with shScr and shNck cells, respectively, had a greater than sixfold increase in expression of human GAPDH in the lung tissue (Figure 3G). Histological examination of H&E stained sections revealed micrometastases in lungs from animals injected with shScr but not shNck cells (Figure 3F, top panel). We confirmed these observations by immunostaining lung tissues with an antibody recognizing vimentin (Figure 3, F and H, bottom panels), an intermediate filament highly expressed in various cancers that correlates with poor prognosis (Satelli and Li, 2011). Interestingly, occasional metastases were also observed in local lymph nodes of animals bearing shScr but not shNck xenografts (Figure 4H, top panel).
FIGURE 4:

Nck is required for breast carcinoma cell extravasation. Eight-week-old nude mice (n = 5/treatment) were injected, via the tail vein, with fluorescently labeled control (shScr) or Nck-silenced (shNck) breast carcinoma cells (5 × 105 cells/animal). Mice were killed 36 h after injections and lungs harvested for histological processing. (A) Representative confocal images of lung sections stained with FITC-Isolectin-B4 (endothelial cell marker) to visualize the vasculature (green, arrowhead). The presence of fluorescently labeled carcinoma cells (magenta) in the extravascular compartment is indicated (arrow). Boxed regions in top panels are shown at higher magnification in bottom panels. (B) Quantification (mean ± SD) of breast carcinoma cells or clusters (foci) of cells in the extravascular compartment. Totals of 82 and 70 fields from control shScr and shNck lungs (n = 5/group) were evaluated by confocal microscopy, *p < 0.01. (C) Proportion of lungs and optical fields with presence of fluorescently labeled breast carcinoma cells. *p < 0.0001.
We considered the possibility that decreased metastasis resulting from Nck silencing in the xenograft model could be related to the smaller size and/or decreased angiogenesis of the corresponding primary tumors. To rule out such possibility, we performed an extravasation assay by directly injecting fluorescently labeled control (shScr) or Nck-silenced (shNck) human breast carcinoma cells into the tail vein of female nude mice. Animals were killed 36 h after injections and lungs harvested for histological processing and microscopic assessment. Consistent with the notion that Nck is required for breast carcinoma cell invasion in vivo, the results show increased extravasation potential of shScr control versus shNck cells (Figure 4, A and B). Indeed, lungs harvested from animals injected with shScr control cells displayed increased an (p < 0.05) number of cells/foci per field than those from animals injected with shNck cells. In addition, extravasation was detected in a higher proportion of lungs and microscopic fields in mice injected with shScr versus shNck cells (Figure 4C). Taken together, these results demonstrate that Nck expression promotes metastasis of breast carcinoma cells.
Loss of Nck reduces matrix degradation and alters invadopodia dynamics
In light of the above results, we hypothesized that Nck depletion reduces mammary carcinoma cell invasiveness by altering the activity of matrix metalloproteinases (MMPs) at the membrane. To test this hypothesis, we employed an assay that enables the quantitative analysis of functional and structural features of invadopodia (Figure 5A), actin-based organelles formed by tumor cells that are involved in localized remodeling of the extracellular matrix (Artym et al., 2006, 2009). Colocalization of F-actin and cortactin has been used previously to identify bona fide invadopodia (Artym et al., 2006). Therefore, our analysis identifies invadopodia as colocalization of F-actin and cortactin, whereas actin puncta are defined as focalized F-actin accumulation without cortactin (Figure 5B).
FIGURE 5:

Nck regulates key cellular mechanisms during invasion of breast carcinoma. (A) Cartoon representing the working hypothesis that Nck-dependent actin remodeling mediates the targeted delivery of matrix metalloproteinases (MMPs) to enable focalized matrix degradation. (B) Distinction between actin puncta and invadopodia. A region of interest at the ventral surface of a cell containing both structures is shown. Invadopodia were defined as actin puncta that colocalized with cortactin (CTTN). Scale bar equals 1 µm. (C) Representative confocal images of MDA-MB-231 cells transfected with small interference (si) RNA oligonucleotides encoding nontargeting sequences (siScr) or sequences targeting MMP14 (siMMP14) or Nck (siNck). Cells were plated on fluorescent gelatin-coated coverslips and incubated for 6 h before fixation and labeling for F-actin (Actin) and the invadopodia marker CTTN. Boxed areas were magnified and single channels/merge images in the XY planes are displayed. Scale bar 10 μm. (D) Percentage (mean ± SD) of cells actively degrading gelatin. A minimum of 99 cells/condition were analyzed. (E) Degradation index calculated as degradation area per active cell/cell area (siScr-n = 56, siMMP14-n = 13, siNck-n = 36). (F) Area occupied by invadopodia. (G) Distribution of cells in the population as a function of invadopodia number/cell. (H) Number of actin puncta/cell. (I) Distribution of cells in the population as a function of number of puncta/cell. Panels F–I summarize data extracted from a minimum of 30 cells/condition and a range of 313–715 invadopodia/actin puncta. *p < 0.05, ***p < 0.001.
Since gelatin degradation requires the focalized activity of MMPs at invadopodia, we first determined whether Nck modulates the gelatin degrading capacity of MDA-MB-231. We found that Nck as well as MMP14 silencing significantly decreased (p < 0.05) the percentage of actively degrading cells when compared with siScr (Figure 5, C and D). In addition, the degradation index differed significantly (p < 0.05) among subsets of cells actively degrading the matrix; siScr, siNck, and siMMP14 resulted in high, moderate, and low levels of fluorescent gelatin degradation, respectively (Figure 5E). These findings suggest that reduced matrix remodeling resulting from Nck silencing is due, at least in part, to decreased accumulation/activity of MMPs at discrete sites on the cell surface.
Features of actin-based structures at the ventral surface were also compared among cell populations. The results show that silencing of Nck or MMP14 significantly decreased (p < 0.05) the size and number of invadopodia (Figure 5, F and G) and number of actin puncta per cell (Figure 5, H and I). In both Nck- and MMP14-silenced populations, invadopodia were ∼20–25% smaller than in the scrambled control population. In addition, the distribution of cells according to the number of actin puncta or invadopodia identified per cell was altered in Nck- and MMP14-silenced populations when compared with the scrambled control. Remarkably, only 10–20% of the Nck- or MMP14-silenced but 40–70% of siScr population had greater than 20 invadopodia/cell (Figure 5G) or greater than two actin puncta/cell (Figure 5I). Superresolution imaging showed the presence of actin cometlike structures, particularly prominent in control cells, associated with invadopodia (Supplemental Movie 1). Whereas cortactin was enriched at the tips of these structures in both control and Nck-silenced cells, the accumulation of F-actin was decreased in Nck-silenced versus control cells. In sum, Nck silencing-induced reduction in invasive capacity of breast carcinoma cells is associated with impaired invadopodia dynamics and matrix degradation, presumably, through a decrease in the focalized delivery/activation of MMPs.
Nck regulates the focalized, ventral localization but not bulk membrane levels of MMP14
We hypothesized that Nck regulates the partitioning of MMP14 between surface exposed and internal pools. To test this prediction, cells were plated on poly-l-lysine-coated glass and subsequently fixed, but not permeabilized, and subjected to immunostaining using an antibody that recognizes the extracellular, catalytic domain of MMP14. This approach enabled the detection of the endogenous, surface exposed MMP14. Compared to control cells transfected with nontargeting siRNA (siScr), silencing of MMP14 significantly decreased (p < 0.05) the levels of surface exposed MMP14. However, Nck silencing did not affect the bulk levels of MMP14 at the cell surface (Supplemental Figure 5). Similar findings were obtained when the plasma membrane levels of MMP14 were analyzed by flow cytometry (unpublished data). Therefore, results from these experiments suggest that, at least under steady conditions, the pool of MMP14 exposed to the cell surface is not dependent on Nck signaling. To determine whether loss of Nck altered the localization of MMP14 specifically at the ventral cell surface, we performed time-lapse total internal reflection fluorescence (TIRF) imaging of MDA-MB-231 breast carcinoma cells coexpressing Lifeact EYFP, an F-actin probe (Riedl et al., 2008), and mCherry-tagged MMP14 (Steffen et al., 2008). Inspection of time-lapse series suggested that silencing of Nck decreased the number of MMP14-containing vesicles being delivered to the ventral surface (Figure 6A and Supplemental Movie 2). To quantify the fraction of mCherry-MMP14 exposed to the ventral surface over time, we calculated a displacement index (Figure 5B) following methods previously described (Castro-Castro et al., 2012). We determined that silencing of Nck resulted in a greater than 60% decrease (p < 0.01) in MMP14 mobility at the ventral cell surface (Figure 6B). To rule out potential artifacts resulting from ectopic expression of MMP14, we also performed immunostaining of endogenous MMP14 in cells plated on fluorescent gelatin (Figure 6C). Analysis of confocal sections of the ventral cell surface revealed a significant reduction (p < 0.05) in MMP14 signal in the most ventral focal planes encompassing the cell membrane and membrane proximal areas (Figure 6D) although the total cellular levels of MMP14 remained unchanged (Figure 6E). These observations suggested that interference with Nck signaling reduces the accumulation of MMP14 at the ventral cell surface; however, the thin layer of gelatin used in the assay limits the extension of invadopodia. Thus, to gain additional insights into the modulation of MMP14 targeting directly to invadopodia by Nck, we used a previously described chemoinvasion assay (Schoumacher et al., 2010) that relies on filters containing 11-µm-long × 1-µm-diameter vertical channels that allow invadopodia elongation (Figure 6F). MDA-MB-231 breast carcinoma cells stably expressing mCherry-MMP14 were seeded on matrix-coated filters, allowed to form invadopodia overnight, fixed, and subjected to confocal imaging. Remarkably, the accumulation of mCherry-MMP14 at the various confocal planes of z-stacks encompassing the long invadopodia was significantly decreased (p < 0.05) in Nck-silenced versus control scrambled cells, and, as expected, the mCherry-MMP14 signal was minimal in MMP14-silenced cells (Figure 6, F and G). Collectively, these results support the notion that Nck regulates the polarized, ventral cell surface accumulation of MMP14 and, distinctively, its targeting to invadopodia.
FIGURE 6:

Nck depletion decreases the ventral surface mobility and accumulation of MMP14 in breast carcinoma cells. (A) Representative TIRF images taken from time-lapse series of MDA-MB-231 cells expressing mCherry-MMP14. Images are presented using an intensity modulated display to associate intensity (surface localization) with color and hue (red = high activity; blue = low activity). (B) Displacement index calculation and bar graph depicting MMP14 displacement index (mean ± SEM). The displacement index, an estimate of MMP14 mobility (Castro-Castro et al., 2012), was calculated as the total area of MMP14 over the entire time-lapse (Σ A1-101) divided by the area of MMP14 in the initial frame (A1) of the series. A total of 20 cells (time-lapse series) from three independent experiments were analyzed. (C) Cartoon representation of the confocal Z-plane imaging scheme (left) and representative images of MDA-MB-231cells (XY projections) and focal planes of the ventral surface (XZ projections). Cells were transiently transfected with small interference (si) RNA oligonucleotides encoding nontargeting sequences (siScr) or sequences targeting MMP14 (siMMP14) or Nck1 and Nck2 (siNck) were seeded on coverslips coated with fluorescent gelatin and immunolabeled with anti-MMP14 (MMP14) and fluorescent phalloidin (F-actin). (D) Bar graph depicting relative intensities (mean ± SEM, arbitrary units) of endogenous MMP14 at the various focal planes of the ventral surface (siScr-n = 81, siMMP14-n = 28, siNck-n = 52). (E) Bar graph showing relative intensities (mean ± SEM, arbitrary units) of total cellular MMP14 (sum of focal planes encompassing whole cell body). Results shown in A–E summarize data from three independent experiments. *p < 0.05, ***p < 0.001. (F) Cartoon representation of the confocal Z-plane plane imaging scheme of invadopodia in chemoinvasion assay and representative Z-projections of elongated invadopodia. (G) Bar graph depicting relative intensities (mean ± SEM, arbitrary units) of mCherry-MMP14 at the various focal planes of elongated invadopodia. The data summarizes at least 70 invadopodia from images of 20 cells/condition obtained in two independent experiments. **p < 0.001 siScr vs. siMMp14 or siNck, *p < 0.01 siMMP14 vs. siNck. In A, B, C, and F, scale bar represents 10 µm.
Nck regulates actin dynamics but not cortactin accumulation at invadopodia
To determine the ratio of actin to cortactin accumulation at invadopodia, we obtained z-stacks of confocal images from cells seeded on fluorescent gelatin and subjected to immunostaining of endogenous cortactin and F-actin labeling. We found a significant decrease (∼25%) in the ratio of F-actin to cortactin when Nck- and MMP14-silenced cells were compared with scrambled controls (Figure 7, A and B). The implications of these findings are twofold: the recruitment of cortactin, an F-actin binding and invadopodial core protein, is likely to be Nck-independent and, on the other hand, actin dynamics at invadopodia is likely to be dependent on Nck signaling. These findings prompted us to perform analysis of actin dynamics at invadopodia using fluorescence recovery after photobleaching (FRAP) following protocols previously established in our group (Barhoumi et al., 1993). FRAP allows the determination of the rate constant for actin diffusion into the local area of actin based structures (Figure 7C). Using MDA-MB-231 cells expressing actin-EYFP, we determined that the rate of actin recovery at puncta was significantly decreased (p < 0.05) in Nck-silenced cells relative to the scrambled controls (Figure 7, D and E). In sum, these results suggest that abrogation of Nck signaling in breast carcinoma cells leads to accumulation of core invadopodial components at a suboptimal ratio and decreased invadopodial actin turnover.
FIGURE 7:

Nck regulates cortactin accumulation and actin dynamics at invadopodia. (A) Representative Z-projections of confocal stacks from the ventral surface of MDA-MB-231 cells seeded on fluorescent gelatin and stained for endogenous cortactin (CTTN) and F-actin. Invadopodia were defined as colocalization of F-actin clusters with cortactin. (B) Ratio of F-actin to cortactin area (mean ± SEM) at invadopodia. A minimum of 30 cells/condition and a range of 313–667 invadopodia were analyzed. (C) Representative confocal images of a cell expressing actin-EYFP (left image) and magnification of a region of interest (ROI, boxed area shown at higher magnification to the right) before and after photobleaching. Scale bar, 2 µm. The plot shows normalized prebleaching intensity and postbleaching recovery for ROIs from single cells. (D) Representative best fit curves showing normalized fluorescence recovery after photobleaching. Results shown are from a single experiment representative of data from three independent experiments. (E) Rate (mean ± SEM) of fluorescence recovery after photobleaching of actin-EYFP in regions of interest containing invadopodia. Data collected from a minimum of 30 images (3–15 puncta per image) from two culture dishes per experimental group. In panels B and E, results summarize data from three independent experiments. ***p < 0.001.
Nck modulates the balance of Cdc42/RhoA activation and acts as an upstream regulator of RhoA-dependent, MMP14-mediated breast carcinoma cell invasion
A number of studies have documented a critical role for Cdc42 signaling in tumorigenesis and metastasis (Arias-Romero and Chernoff, 2013). Although Cdc42 has been involved in regulation of invadopodia in cancer cells (Nakahara et al., 2003; Yamaguchi et al., 2005), how Nck modulates Cdc42 activity remains undetermined. We hypothesized that Nck promotes breast carcinoma cell motility and invasion by enhancing the activation of Cdc42. To test this hypothesis, we first determined spatiotemporal patterns of Cdc42 activation by time-lapse confocal imaging of the ventral surface of MDA-MB-231 cells expressing the Raichu Cdc42-FRET biosensor (Aoki and Matsuda, 2009). Nck-silenced versus control cells showed, surprisingly, a significant increase (p < 0.05) in the overall levels of active Cdc42 at the ventral cell surface (Figure 8A). Interestingly, Nck-silenced cells appeared to undergo futile, fast cycles of membrane protrusion/retraction in multiple directions that coincided with increased Cdc42 activation (Supplemental Movie 3). In these cells, clusters of active Cdc42 puncta in the center of the ventral cell surface were also readily apparent. In contrast, control cells displayed slightly more persistent and oriented patterns of membrane protrusion/Cdc42 activation and decreased clustering of active Cdc42 puncta in the center of the ventral cell surface. Consistent with these results, we observed that the overall levels of endogenous Cdc42 activation, determined by affinity pull-down assays with immobilized Pak1 rac/Cdc42 (p21) binding domain (PBD) (Benard and Bokoch, 2002), were slightly greater than twofold in lysates from Nck-silenced versus controlled cells (Figure 8B).
FIGURE 8:

Nck modulates the balance of Cdc42/RhoA activation and acts as an upstream regulator of RhoA-dependent, MMP14-mediated breast carcinoma cell invasion. (A) MDA-MB-231 cells expressing the Raichu Cdc42 FRET biosensor and either short hairpin (sh) RNAs encoding nontargeting sequences (shScr) or sequences targeting Nck (shNck) were plated on fibronectin in complete medium. Confocal time-lapse images of the ventral surface of the cells were acquired every minute for 30–60 min. The left panel shows representative FRET/CFP ratio images taken from time-lapse series. Images are presented using an intensity modulated display to associate intensity (activity) with color and hue (red = high activity; blue = low activity). Scale bar, 10 µm. The right panel shows quantitative analysis of FRET activity (mean ± SEM) in whole cells. FRET ratios were calculated for each cell in two to four fields (two to four cells per field) and every time interval (n = 642). Results shown summarize data from three independent experiments. ***p < 0.001. (B) Representative Western blots (left panel) showing GTP-loaded (active) Cdc42, total Cdc42, Nck, and GAPDH (loading control) levels in lysates from MDA-MB-231 stably expressing shRNAs encoding nontargeting (shScr) and Nck-targeting (shNck) sequences. Quantification of active Cdc42 is shown in the right panel. The intensity of bands corresponding to active Cdc42 (Cdc42GTP) was normalized to the intensity of the bands corresponding to total Cdc42 (Cdc42) and expressed as fold change relative to control shScr levels. Results summarize two independent experiments. *p < 0.05. (C) Cells expressing the Raichu RhoA FRET probe were cultured in complete medium and imaged every minute for at least 30 min. The left panel shows representative FRET/CFP ratio images taken from time-lapse series. Scale bar, 10 µm. The right panel shows quantitative analysis of RhoA FRET activity (mean ± SEM) in whole cells. FRET ratios were collected for each cell and every time interval (n = 947). Results shown summarize data from two experiments. ***p < 0.001. (D) Representative Western blots (left panel) showing RhoA and GAPDH levels in samples from control (shScr) and Nck-silenced (shNck) cells left untreated or stimulated with EGF. The right panel shows quantification of active RhoA. Levels of active RhoA (RhoAGTP) in extracts from starved cells left untreated (0 min) or stimulated with EGF (5 and 10 min) were determined by ELISA and values normalized to levels of total RhoA quantified by Western blotting. Results summarize two independent experiments. *p < 0.05. (E–G) MDA-MB-231 cells were transduced with viruses harboring vectors encoding (E) EGFP, (F) constitutively active EGFP-tagged RhoAQ63A (RhoA-CA-EGFP), or (G) wild-type EGFP-tagged human Nck2 (hNck2-EGFP). The newly created stable cell lines were subsequently transfected with control, nontargeting siRNA oligonucleotides (siScr), or siRNA oligonucleotides targeting RhoA (siRhoA), MMP14 (siMMP14), or Nck (siNck) as indicated. Cells were subjected to overnight starvation before seeding onto 8-µm-pore filters precoated with a laminin-rich matrix (Matrigel). Cells were allowed to invade for 10 h toward bottom chambers of transwell invasion plates loaded with complete medium. Panels E–G display representative Western blots showing cellular levels of target proteins or GAPDH (loading control), representative wild field images of migrated cells and quantitative analysis of relative migration. Bar graphs represent mean ± SEM (n = 3). *p < 0.05, **p < 0.01.
Because RhoA overexpression has been involved in neoplastic transformation of mammary epithelial cells (Zhao et al., 2009) and is critical in breast cancer progression and invasion (Wu et al., 2009; Chan et al., 2010; Gilkes et al., 2014; Schlienger et al., 2014), we examined spatiotemporal patterns of RhoA activation using a previously described FRET biosensor (Nakamura et al., 2006; Aoki and Matsuda, 2009). Time-lapse imaging of MDA-MD-231 cells expressing the Raichu RhoA FRET probe showed decreased overall RhoA activation associated with unproductive, multidirectional protrusive activity, and loss of front-back cell polarity and directional motility in Nck-silenced cells (Figure 8C and Supplemental Movie 4). In contrast, control cells displayed robust activation of RhoA, particularly at the leading edge, and in dorsal ruffle- and invadosome-like structures. In addition, control cells were more motile and exhibited polarized organization and directional displacement (Supplemental Movie 4). Importantly, basal and EGF-stimulated levels of active RhoA (RhoAGTP) in cell extracts were significantly reduced (p < 0.05) in Nck-depleted versus control cells (Figure 8D). Thus, ablation of Nck leads to a shift in the balance of Rho GTPase activity distinguished by impaired RhoA and increased, spatially unrestricted Cdc42 activation.
Invasiveness of breast carcinoma cells is dependent on the activity of MMP14 at the cell surface (Munoz-Najar et al., 2006; Bravo-Cordero et al., 2007; Yu et al., 2012; Rosse et al., 2014; Marchesin et al., 2015). Thus, to further define the molecular mechanism linking Nck, RhoA activation, and MMP14 surface activity, we combined analysis of breast carcinoma cell invasion with signaling manipulation by protein silencing and expression of wild-type/mutant variants (Figure 8, E–G). Silencing of Nck, RhoA, or MMP14 reduced invasiveness of MDA-MB-231 cells (enhanced green fluorescent protein [EGFP], control) to a similar extent (Figure 8E), a finding suggesting the involvement of these proteins in a common pathway. To test the dependence of breast carcinoma cell invasion on Rho-A activation, we generated a cell line expressing moderate levels of an EGFP-tagged, constitutively active (CA, RhoAQ63L) RhoA (Figure 8F). Silencing of MMP14, but not Nck, reduced significantly (p < 0.05) the invasive capacity of MDA-MB-231 cells expressing RhoA-CA. These findings suggested that MMP14 is, indeed, a key downstream effector of RhoA-driven carcinoma cell invasion. On the basis of these results, we speculated that Nck may act as an upstream regulator of RhoA-dependent, MMP14-mediated carcinoma cell invasion. To test this prediction, cells stably expressing moderate levels of a EGFP-tagged, wild-type human Nck2 (hNck2-EGFP) were transfected with nontargeting siRNA oligonucleotides (siScr), or siRNA oligonucleotides targeting RhoA or MMP14. The results showed that Nck-driven carcinoma cell invasion is abrogated by depletion of RhoA or MMP14 (Figure 8G). Taken together, these results suggest that Nck modulates the balance of Rho GTPase activation and supports an important role for Nck as an upstream regulator of RhoA-dependent, MMP14-mediated breast carcinoma cell invasion.
DISCUSSION
This study uncovers an important role for Nck in breast carcinoma progression, extravasation, and metastasis and highlights a mechanism whereby Nck links actin dynamics and enhanced cell surface localization/activity of MMP14 by regulating the balance between RhoA and Cdc42 activation (Figure 9). It is known that tumor cells rely on distinct signaling modules that specify adhesive, proteolytic, and protrusive functions to breach physical barriers during tissue invasion and metastasis (Friedl and Alexander, 2011; Revach and Geiger, 2014). However, the identity of critical molecular players involved in the articulation of such programs has remained elusive. Here we provide evidence that Nck potentiates tumor cell invasion and metastasis by directing the polarized interaction of carcinoma cells with collagen fibrils, promoting actin turnover at invadopodia, and enhancing focalized proteolysis of the extracellular matrix. We propose that, by bridging core functions enabling tumor cell invasion and formation of metastases, Nck constitutes a potentially valuable therapeutic target in aggressive cancers.
FIGURE 9:
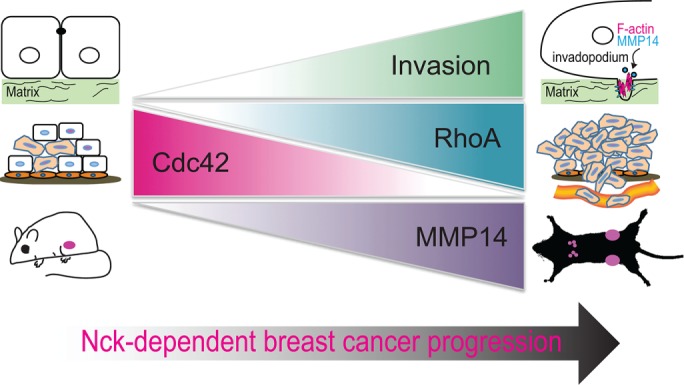
Role of Nck in malignant progression of breast cancer: a working model. Present findings and those from the available literature support an important role for Nck in the malignant progression of breast cancer by fostering primary tumor growth, extracellular matrix remodeling and invasion of carcinoma cells through a mechanism that involves an imbalance in Cdc42/RhoA activation and increased MMP14 activity. Nck also contributes to formation of metastases by enabling tumor cell extravasation and, presumably, promoting tumor cell homing and colonization.
Unexpectedly, tumors derived from Nck silenced versus control cells displayed slower growth rates and reduced mass. Abrogation of Nck also reduced the growth of multicellular tumor spheroids in 3D laminin-rich matrices (Figure 1G) and impaired the growth curve of MDA-MB-231 cells in long-term two-dimensional cultures (unpublished data). We speculate that Nck may contribute to regulation of tumor cell proliferation through modulation of Cdc42/aPKC activation (Chaki et al., 2015). Of note, the polarity protein Par6, overexpressed in breast cancer cell lines and primary breast tumors, was shown to induce breast cancer cell proliferation through a mechanism involving Cdc42/aPKC (Nolan et al., 2008). Our results also suggest impaired vascularization as a factor contributing to the slower growth rates of Nck-silenced versus control primary tumors. It is possible that Nck signaling in tumor cells contributes to the establishment of a reactive stroma through paracrine mediators. Thus, a potential role of Nck in expression of tumor cell-derived factors that modify the tumor microenvironment, including vascularization, warrants further investigation.
The present results suggest that the polarized interaction between tumor cells and extracellular matrix fibrils constitutes an important mechanism by which Nck regulates tissue invasion. These findings are consistent with previous observations of asymmetry in cell–matrix interactions and cytoskeletal organization consisting in bundles of aligned collagen fibers, prominent F-actin accumulation, and redistribution of β1 integrin toward the leading edge of migrating tumor cells (Hegerfeldt et al., 2002). Collagen remodeling by mammary carcinoma cells leads to the thickening of fibrils and their alignment perpendicular to the tumor boundary (Zhang et al., 2013). Such collagen arrangement enhances the migration efficiency of tumor cells by restricting protrusions along the aligned fibers and increasing directional persistence (Riching et al., 2014). Thus, it is possible that a similar mechanism underlies the more efficient migration of control versus Nck-silenced cells observed in the present study.
Recent findings highlight the significance of invadosomes in developmental (Santiago-Medina et al., 2015; Lohmer et al., 2016) and pathological (Gligorijevic et al., 2012; Leong et al., 2014; Seano et al., 2014) processes. Our results showing that Nck promotes tumor cell extravasation and metastasis are consistent with previous studies that identified invadopodia as key subcellular structures involved in intravasation (Gligorijevic et al., 2012) and extravasation (Leong et al., 2014). We show here that Nck acts in the coordination of proteolytic and protrusive functions of invadopodia. Our FRAP data showing decreased actin turnover in Nck-silenced versus control cells suggest that invadopodia-associated Nck modulates the reciprocal regulation between N-WASp turnover and actin dynamics (Delatour et al., 2008; Weisswange et al., 2009). In addition to a dendritic F-actin array polymerized by the Arp2/3 complex, invadopodia display bundles of linear actin filaments polymerized by the formin family of actin nucleators (Lizarraga et al., 2009; Schoumacher et al., 2010). Because Nck integrates dendritic and linear actin nucleation (Borinskaya et al., 2016), we propose that Nck coordinates the molecular machineries directing the assembly of the complex actin network enabling the protrusive function of invadopodia.
It is well established that Rho GTPases play an important role in tumor progression and metastasis (Vega and Ridley, 2008). Rho GTPases are regulated by ∼85 guanine exchange factors (GEFs) (Goicoechea et al., 2014) and a slightly smaller number GTPase-activating proteins (GAPs) (Huang et al., 2017) whose aberrant expression is frequently linked to cancer (Vigil et al., 2010). Of significance, we found that Nck contributes to the reciprocal regulation of Cdc42 and RhoA activation in breast carcinoma cells. Changes in the balance of Cdc42/RhoA activation have been shown to modulate invasiveness and the metastatic potential of cancer cells (Bendris et al., 2016; Kedziora et al., 2016). Surprisingly, Nck depletion led to increased, spatiotemporally dysregulated Cdc42 activation and, conversely, decreased activation of RhoA. Our data suggest that Nck modulates the balance of Cdc42/RhoA activation by fine-tuning the rate of GTP loading/hydrolysis through the localized enrichment of regulatory factors (Figure 9). For example, it is possible that Nck facilitates Cdc42-driven tumor cell invasion by damping its activity through the localized recruitment of a Cdc42-directed GTPase activating protein (GAP). Consistent with this notion, multiple activation/inactivation cycles of Rac at the cell front are necessary for the spatiotemporal regulation of protrusions and efficient directional migration (Parrini et al., 2011). Similarly, Nck may modulate RhoA activation by directing, in space and time, the activity of a RhoA-directed GEF or, alternatively, stabilize RhoA in its active state by inhibiting/sequestering a RhoA-directed GAP. Thus, findings from this work provide the groundwork for the formulation of testable hypotheses and underscore the need for future studies to identify critical factors linking Nck signaling with Rho GTPase activation.
The intracellular trafficking of MMP14 is known to regulate invadopodia-associated matrix proteolysis (Poincloux et al., 2009; Frittoli et al., 2011); however, the molecular mechanisms underlying the surface targeting and delivery of MMP14 remain under active investigation (Linder, 2015; Castro-Castro et al., 2016). Here we provide evidence that Nck regulates the polarized delivery of MMP14 to the ventral cell surface and its accumulation at invadopodia through fine-tuning the balance between RhoA and Cdc42 activation. Interestingly, invadosome remodeling appears to be dictated by the balance between Cdc42 and RhoA activity (Kedziora et al., 2016). Key roles for Cdc42 and RhoA (Sakurai-Yageta et al., 2008) as well as the downstream effector aPKC (Rosse et al., 2014) in the trafficking of MMP14 to the cell surface and breast cancer progression have been identified. Importantly, our findings support a role for Nck as an upstream regulator of RhoA-dependent, MMP14-mediated breast carcinoma cell invasion. Thus, our results suggest that through the fine-tuning of Cdc42/RhoA activation, Nck contributes to invadopodia biogenesis (focalized actin assembly) and maturation (targeted delivery of MMP14). These results also stimulate new hypotheses and are likely to prompt new investigations on the role of Nck in direct regulation of the subcellular distribution/activity of GEF and/or GAPs controlling the activation of Rho GTPases. It should be noted, however, that type and dimensionality of the extracellular matrix could potentially influence the modulatory role of Nck in the activation of Cdc42/RhoA. Although evaluation of such factors through specific comparisons was beyond the scope of this work, context-dependent regulation of tumor cell invasion by Nck warrants further investigation.
In summary, we have identified an important role for Nck in breast carcinoma progression, extravasation, and metastasis (summarized in Figure 9). Differential modulation of RhoA and Cdc42 activation appears as an important mechanism by which Nck directs cytoskeletal changes that enable polarized interaction of tumor cells with collagen fibrils and focalized extracellular proteolysis by increased cell surface accumulation of MMP14. Emerging technologies are enabling the targeting of protein–protein interactions (Makley and Gestwicki, 2013), a largely untapped resource with vast potential for therapeutic intervention in cancer. Results from this study pave the way for the development of pharmacological agents that, by blocking Nck signaling, may prove effective in the treatment of aggressive cancers.
MATERIALS AND METHODS
Cell culture, reagents, and standard assays
MDA-MB-231 (CRM-HTB-26) and 293T (CRL-3216) were purchased from the American Type Culture Collection (Manassas, VA). pSuper.puro/hygro carrying nontargeting (shScr) or targeting sequences (human Nck1 or Nck2; shNck) were previously described (Chaki et al., 2013). RhoA(Q63L)-EGFP in MSCV vector has been previously described (Sreenivasappa et al., 2014). Small interference RNA SmartPool siRNA reagents were purchased from Dharmacon (Lafayette, CO). We used previously described Raichu Cdc42 and RhoA Förster resonance energy transfer (FRET) probes (Aoki and Matsuda, 2009) and pcDNA3.1 MT1-MMP mCherry (Sakurai-Yageta et al., 2008). Recombinant hEGF and Matrigel were from BD Biosciences (San Jose, CA). Antibodies used include anti-Nck1/2 (Millipore, Billerica, MA; 06-288), anti-Nck2 (3313, kindly provided by Louise Larose, Montreal Diabetes Research Center) (Labelle-Cote et al., 2011; Latreille et al., 2011), anti-MMP14 (Millipore; MAB3328), anti-cortactin (Millipore #05-180), anti-GAPDH (Invitrogen, Carlsbad, CA; 43700), goat anti-mouse immunoglobulin-G–horseradish peroxidase (IgG-HRP; Santa Cruz Biotechnology, Dallas, TX; sc-2055), goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, sc-2030), Alexa Fluor 647 goat anti-mouse IgG (H + L; Life Technologies; A-21235). Other reagents included NucBlue Live ReadyProbes (Life Technologies, R37605) and Texas Red-X Phalloidin (Life Technologies; T7471).
Xenograft studies
MDA-MB-231 cells (2 × 106) were injected subcutaneously into the flanks of 6- to 9-wk-old female nude mice in 75% growth factor reduced matrigel (9.9 mg/ml). Tumors were measured weekly using calipers and volumes calculated as previously described (Blouw et al., 2015). Xenograft tumors and mouse lungs were harvested, weighed, and snap-frozen in liquid nitrogen then stored at –80°C for DNA/RNA purification or fixed in 4% paraformaldehyde and paraffin embedded for immunohistochemistry. Animal procedures were approved by the Texas A&M Animal Care and Use Committee (AUP 2014-0194).
Extravasation assay
Control shRNA or Nck-depleted (shNck) MDA-MB-231 breast carcinoma cells were fluorescently labeled with CellTracker Orange CMRA dye (Molecular Probes, Eugene, OR) before injection (5 × 105 cells) into the tail vein of 8-wk-old female nude mice (n = 5/treatment). Animals were killed 36 h after receiving cell injections, and lungs were collected for histological processing. Paraffin embedded sections (4 µm thick) spaced every 50 µm of lung tissue were subjected to staining with fluorescein isothiocyanate (FITC)-Isolectin-B4 (Vector Laboratories, Burlingame, CA), en endothelial cell marker for visualization of the lung vasculature. At least five tissue sections/lung were examined using a Zeiss LSM780 confocal microscope equipped with a Plan-Apo 20X/0.8NA objective.
Three-dimensional assays
Three-dimensional growth assays were performed using the manufacturer’s protocol (Trevigen, Gaithersburg, MD). Three-dimensional Spheroid invasion assay in collagen I was adapted from manufacturer’s literature (Trevigen/Corning) and previously described protocols (Artym and Matsumoto, 2010; Guzman et al., 2014). Briefly, MDA-MB-231 cells were subjected to spheroid formation in 3D culture qualified 96-well spheroid formation plate (Trevigen) in the presence of 10× spheroid formation matrix diluted to 1× in complete media. Formed spheroids were then embedded in 2 mg/ml acid-extracted type I rat-tail collagen polymerized at 37°C and allowed to invade in the presence of EGF (5 ng/ml) used as a chemoattractant in serum-reduced media (2% fetal bovine serum [FBS]).
Transwell invasion
Invasion was measured using control and Corning BioCoat (Corning, NY) inserts according to the manufacturer’s protocol. MDA-MB-231 cells were starved overnight prior to seeding 50,000 cells in starvation media (0.2% FBS) in the upper chamber and allowed to migrate (5 h) or invade (18 h), toward starvation media with 10 ng/ml EGF in the lower chamber. Percentage invasion was calculated as described (Albini and Benelli, 2007).
Imaging
Spheroid invasion in type I collagen (rat-tail tendon 354236; BD Biosciences, San Jose, CA) gels were fixed, stained with Alexa Fluor 488-conjugated phalloidin (Thermo Fisher Scientific, Grand Island, NY), and imaged with a custom-built nonlinear optical microscope previously described (Larson and Yeh, 2006). All images were processed and analyzed with custom Matlab (Mathworks, Natick, MA) routines. Maximum intensity projection images were generated for qualitative analysis. Two-dimensional images of raw photon counts from individual optical sections were used for quantitative analysis. Migrating cells that were not in close contact with other cells were analyzed. The center of each cell nucleus (or the centroid of the entire cell if the nucleus location was ambiguous) was marked from an overlay of the F-actin and collagen images. F-actin (TPEF) and collagen (SHG) images were then manually thresholded to create binary masks of each signal. The intersection of these masks represented areas of cell–matrix interaction and the angular distribution of the cell–matrix interaction was calculated by creating a grid of theta values centered on the nucleus, multiplying the grid with the interaction mask, and counting the number of pixels with each theta value. We also compared the location of the center of each cell nucleus to the calculated center of mass of the interaction mask (assuming a unit mass for each pixel). The distance between these two points (that would overlap perfectly if the cell–matrix interaction was distributed perfectly symmetrically around the cell nucleus) was called the CoM displacement. Aspect ratio (ratio of the long and short axis of the cell) and migration angle (relative to a unit vector normal to the spheroid outer surface) of cells were calculated from user-specified landmarks selected from the F-actin images.
FRAP was performed as previously described (Barhoumi et al., 1993; Li et al., 2010). Time-lapse series of MDA-MB-231 cells expressing the Raichu/Cdc42 or Raichu/RhoA single chain FRET probes were acquired every minute for 60 min using a Zeiss LSM780 confocal microscope equipped with a Plan-Apo 40X/1.40NA oil objective. For TIRF imaging, MDA-MB-231 cells transiently expressing pcDNA3.1 MT1-mCh were imaged on a Zeiss Axio Observer Z1 TIRF 3 microscope, equipped with a Plan-Apo 100X/1.46NA oil objective and Roper S/W PVCAM. Superresolution Structured Illumination Microscopy was performed using a Zeiss ELYRA S.1 superresolution microscope equipped with a Plan-Apo 63×/1.4oil objective.
Fluorescent gelatin degradation
We used a previously described protocol for the fluorescent gelatin degradation assay (Artym et al., 2009). Briefly, acid-washed glass coverslips were coated with 0.2% gelatin labeled with Alexa Fluor 488 (Invitrogen, Eugene, OR). MDA-MB-231 cells (7.5 × 104 in I ml of media) were seeded on gelatin-coated coverslips and incubated at 37° for 6 h. Cells were subsequently fixed, permeabilized and subjected to immunostaining with appropriate antibodies. Image acquisition was performed on an Olympus IX70 inverted microscope equipped with a 60× 1.4 NA objective and a Spot RT3 camera or, alternatively, a Zeiss Laser Scanning Microscope 780 equipped with a Plan-Apo 40× 1.4 NA oil objective. Images were processed using FIJI.
RNA isolation, reverse transcription, and quantitative PCR
RNA isolation from tissues was performed using Direct-zol RNA MiniPrep with a TRI-Reagent kit (Zymo Research, Irvine, CA; R2052). From the isolated total RNA 2 µg was subjected to reverse transcription into cDNA using the manufacturer’s protocol with the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA; 170-8990). Quantitative PCR (qPCR) was performed as previously described (Scribner et al., 2013) using primers for human glyceraldehyde 3-phosphate dehydrogenase (GAPDH; forward primer 5′-CCA GGT GGT CTC CTC TGA CTT C-3′; reverse primer 5′-CCA GGT GGT GAG GGC AAT G-3′. Mouse β-actin (forward primer 5′-GTT TGA GAC CTT CAA CAC CCC-3′; reverse primer 5′-GTG GCC ATC TCT TGC TCG AAG TC-3′) was used as a normalizing gene and data was analyzed by the ΔΔCT method (Hettinger et al., 2001).
Active Cdc42 and RhoA
GTP-loaded Cdc42 from cell extracts was enriched by pull down with immobilized PBD domains from Pak1 (Benard and Bokoch, 2002). GTP-loaded RhoA from cell extracts was determined by enzyme-linked immunosorbent assay (ELISA) (G-LISA; Cytoskeleton) following the vendor’s protocol.
Statistical methods
When comparing multiple groups with normal distributions ANOVA with a Tukey post-hoc was used. If data did not approximate a normal distribution, then a Mann-Whitney nonparametric analysis was performed or when applicable data was log transformed. Growth curves were analyzed using a general linear model to determine interaction between time and treatment, followed by a Tukey post-hoc analysis. For data depicted as a box-and-whisker plot the bottom and top of the boxes are the first and third quartile, respectively, whereas the band inside is the middle quartile (median). The ends of the whiskers extend to the lowest and highest values excluding outliers. The Grubbs outlier test was used to identify and remove outliers. Significant values where a specific p-value is not listed indicate p < 0.05.
Supplementary Material
Acknowledgments
We thank Michiyuki Matsuda (Kyoto University) for the kind gift of the FRET Raichu-Cdc42 and Raichu-RhoA probes and Philippe Chavrier (Institut Curie) for the kind gift of the mCherry-MT1-MMP plasmid. We thank Ivan Ivanov (TAMU) for advice on statistical analysis, Andy Ambrus (TAMU) for tissue processing for immunohistochemistry, and George Stoica and Brian Porter (TAMU) for help with the histopatological analysis. We are indebted to Robert Burghardt (Image Analysis Laboratory, TAMU) for facilitating access to the imaging instrumentation. D.C.M. was supported by the Air Force Institute of Technology, Civilian Institution Program and the US Air Force Academy Department of Biology. This investigation was partly supported by TAMU/AgriLife funds. The Image Analysis Laboratory is partly supported by the National Institutes of Health–National Center for Research Resources (1 S10 RR22532-01) and the Center for Translational Environmental Health Research (NIEHS P30ES023512).
Abbreviations used:
- Cdc42
cell division control protein homologue 42
- FRET
Förster resonance energy transfer
- Nck
noncatalytic region of tyrosine kinase
- N-WASp
neuronal Wiskott-Aldrich syndrome protein
- RhoA
transforming protein RhoA.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-02-0106) on September 27, 2017.
REFERENCES
- Albini A, Benelli R. The chemoinvasion assay: a method to assess tumor and endothelial cell invasion and its modulation. Nat Protoc. 2007;2:504–511. doi: 10.1038/nprot.2006.466. [DOI] [PubMed] [Google Scholar]
- Aoki K, Matsuda M. Visualization of small GTPase activity with fluorescence resonance energy transfer-based biosensors. Nat Protoc. 2009;4:1623–1631. doi: 10.1038/nprot.2009.175. [DOI] [PubMed] [Google Scholar]
- Arias-Romero LE, Chernoff J. Targeting Cdc42 in cancer. Expert Opin Ther Targets. 2013;17:1263–1273. doi: 10.1517/14728222.2013.828037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artym VV, Matsumoto K. Imaging cells in three-dimensional collagen matrix. Curr Protoc Cell Biol Chapter. 2010;10:Unit 10 18 11–20. doi: 10.1002/0471143030.cb1018s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artym VV, Swatkoski S, Matsumoto K, Campbell CB, Petrie RJ, Dimitriadis EK, Li X, Mueller SC, Bugge TH, Gucek M, Yamada KM. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J Cell Biol. 2015;208:331–350. doi: 10.1083/jcb.201405099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artym VV, Yamada KM, Mueller SC. ECM degradation assays for analyzing local cell invasion. Methods Mol Biol. 2009;522:211–219. doi: 10.1007/978-1-59745-413-1_15. [DOI] [PubMed] [Google Scholar]
- Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–3043. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- Bai Y, Lee PF, Gibbs HC, Bayless KJ, Yeh AT. Dynamic multicomponent engineered tissue reorganization and matrix deposition measured with an integrated nonlinear optical microscopy-optical coherence microscopy system. J Biomed Opt. 2014a;19:36014. doi: 10.1117/1.JBO.19.3.036014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Lee PF, Humphrey JD, Yeh AT. Sequential multimodal microscopic imaging and biaxial mechanical testing of living multicomponent tissue constructs. Ann Biomed Eng. 2014b;42:1791–1805. doi: 10.1007/s10439-014-1019-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoumi R, Bowen JA, Stein LS, Echols J, Burghardt RC. Concurrent analysis of intracellular glutathione content and gap junctional intercellular communication. Cytometry. 1993;14:747–756. doi: 10.1002/cyto.990140707. [DOI] [PubMed] [Google Scholar]
- Beaty BT, Condeelis J. Digging a little deeper: the stages of invadopodium formation and maturation. Eur J Cell Biol. 2014;93:438–444. doi: 10.1016/j.ejcb.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard V, Bokoch GM. Assay of Cdc42, Rac, and Rho GTPase activation by affinity methods. Methods Enzymol. 2002;345:349–359. doi: 10.1016/s0076-6879(02)45028-8. [DOI] [PubMed] [Google Scholar]
- Bendris N, Williams KC, Reis CR, Welf ES, Chen PH, Lemmers B, Hahne M, Leong HS, Schmid SL. SNX9 promotes metastasis by enhancing cancer cell invasion via differential regulation of RhoGTPases. Mol Biol Cell. 2016;27:1409–1419. doi: 10.1091/mbc.E16-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladt F, Aippersbach E, Gelkop S, Strasser GA, Nash P, Tafuri A, Gertler FB, Pawson T. The murine Nck SH2/SH3 adaptors are important for the development of mesoderm-derived embryonic structures and for regulating the cellular actin network. Mol Cell Biol. 2003;23:4586–4597. doi: 10.1128/MCB.23.13.4586-4597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouw B, Patel M, Iizuka S, Abdullah C, You WK, Huang X, Li JL, Diaz B, Stallcup WB, Courtneidge SA. The invadopodia scaffold protein Tks5 is required for the growth of human breast cancer cells in vitro and in vivo. PLoS One. 2015;10:e0121003. doi: 10.1371/journal.pone.0121003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borinskaya S, Velle KB, Campellone KG, Talman A, Alvarez D, Agaisse H, Wu YI, Loew LM, Mayer BJ. Integration of linear and dendritic actin nucleation in Nck-induced actin comets. Mol Biol Cell. 2016;27:247–259. doi: 10.1091/mbc.E14-11-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Marrero-Diaz R, Megias D, Genis L, Garcia-Grande A, Garcia MA, Arroyo AG, Montoya MC. MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J. 2007;26:1499–1510. doi: 10.1038/sj.emboj.7601606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Oser M, Chen X, Eddy R, Hodgson L, Condeelis J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr Biol. 2011;21:635–644. doi: 10.1016/j.cub.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buday L, Wunderlich L, Tamas P. The Nck family of adapter proteins: regulators of actin cytoskeleton. Cell Signal. 2002;14:723–731. doi: 10.1016/s0898-6568(02)00027-x. [DOI] [PubMed] [Google Scholar]
- Campellone KG, Giese A, Tipper DJ, Leong JM. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol Microbiol. 2002;43:1227–1241. doi: 10.1046/j.1365-2958.2002.02817.x. [DOI] [PubMed] [Google Scholar]
- Castro-Castro A, Janke C, Montagnac G, Paul-Gilloteaux P, Chavrier P. ATAT1/MEC-17 acetyltransferase and HDAC6 deacetylase control a balance of acetylation of alpha-tubulin and cortactin and regulate MT1-MMP trafficking and breast tumor cell invasion. Eur J Cell Biol. 2012;91:950–960. doi: 10.1016/j.ejcb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rosse C, Chavrier P. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu Rev Cell Dev Biol. 2016;32:555–576. doi: 10.1146/annurev-cellbio-111315-125227. [DOI] [PubMed] [Google Scholar]
- Chaki SP, Barhoumi R, Berginski ME, Sreenivasappa H, Trache A, Gomez SM, Rivera GM. Nck enables directional cell migration through the coordination of polarized membrane protrusion with adhesion dynamics. J Cell Sci. 2013;126:1637–1649. doi: 10.1242/jcs.119610. [DOI] [PubMed] [Google Scholar]
- Chaki SP, Barhoumi R, Rivera GM. Actin remodeling by Nck regulates endothelial lumen formation. Mol Biol Cell. 2015;26:3047–3060. doi: 10.1091/mbc.E15-06-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY, Wu J, Nakayama KI, Kang HY, Huang HY, et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol. 2010;12:457–467. doi: 10.1038/ncb2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, She H, Davis EM, Spicer CM, Kim L, Ren R, Le Beau MM, Li W. Identification of Nck family genes, chromosomal localization, expression, and signaling specificity. J Biol Chem. 1998;273:25171–25178. doi: 10.1074/jbc.273.39.25171. [DOI] [PubMed] [Google Scholar]
- Clouthier DL, Harris CN, Harris RA, Martin CE, Puri MC, Jones N. Requisite role for Nck adaptors in cardiovascular development, endothelial-to-mesenchymal transition, and directed cell migration. Mol Cell Biol. 2015;35:1573–1587. doi: 10.1128/MCB.00072-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatour V, Helfer E, Didry D, Le KH, Gaucher JF, Carlier MF, Romet-Lemonne G. Arp2/3 controls the motile behavior of N-WASP-functionalized GUVs and modulates N-WASP surface distribution by mediating transient links with actin filaments. Biophys J. 2008;94:4890–4905. doi: 10.1529/biophysj.107.118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditlev JA, Michalski PJ, Huber G, Rivera GM, Mohler WA, Loew LM, Mayer BJ. Stoichiometry of Nck-dependent actin polymerization in living cells. J Cell Biol. 2012;197:643–658. doi: 10.1083/jcb.201111113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enderling H, Alexander NR, Clark ES, Branch KM, Estrada L, Crooke C, Jourquin J, Lobdell N, Zaman MH, Guelcher SA, et al. Dependence of invadopodia function on collagen fiber spacing and cross-linking: computational modeling and experimental evidence. Biophys J. 2008;95:2203–2218. doi: 10.1529/biophysj.108.133199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese S, Schubert WD, Findeis AC, Marquardt T, Roske YS, Stradal TE, Heinz DW. The phosphotyrosine peptide binding specificity of Nck1 and Nck2 Src homology 2 domains. J Biol Chem. 2006;281:18236–18245. doi: 10.1074/jbc.M512917200. [DOI] [PubMed] [Google Scholar]
- Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Frischknecht F, Moreau V, Rottger S, Gonfloni S, Reckmann I, Superti-Furga G, Way M. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature. 1999;401:926–929. doi: 10.1038/44860. [DOI] [PubMed] [Google Scholar]
- Frittoli E, Palamidessi A, Disanza A, Scita G. Secretory and endo/exocytic trafficking in invadopodia formation: the MT1-MMP paradigm. Eur J Cell Biol. 2011;90:108–114. doi: 10.1016/j.ejcb.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Gilkes DM, Xiang L, Lee SJ, Chaturvedi P, Hubbi ME, Wirtz D, Semenza GL. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc. Natl. Acad. Sci. USA. 2014;111:E384–E393. doi: 10.1073/pnas.1321510111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125:724–734. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goicoechea SM, Awadia S, Garcia-Mata R. I’m coming to GEF you: Regulation of RhoGEFs during cell migration. Cell Adh Migr. 2014;8:535–549. doi: 10.4161/cam.28721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, Pawson T, Finlay BB. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat Cell Biol. 2001;3:856–859. doi: 10.1038/ncb0901-856. [DOI] [PubMed] [Google Scholar]
- Guzman A, Ziperstein MJ, Kaufman LJ. The effect of fibrillar matrix architecture on tumor cell invasion of physically challenging environments. Biomaterials. 2014;35:6954–6963. doi: 10.1016/j.biomaterials.2014.04.086. [DOI] [PubMed] [Google Scholar]
- Hegerfeldt Y, Tusch M, Brocker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002;62:2125–2130. [PubMed] [Google Scholar]
- Hettinger AM, Allen MR, Zhang BR, Goad DW, Malayer JR, Geisert RD. Presence of the acute phase protein, bikunin, in the endometrium of gilts during estrous cycle and early pregnancy. Biol Reprod. 2001;65:507–513. doi: 10.1095/biolreprod65.2.507. [DOI] [PubMed] [Google Scholar]
- Huang GH, Sun ZL, Li HJ, Feng DF. Rho GTPase-activating proteins: regulators of Rho GTPase activity in neuronal development and CNS diseases. Mol Cell Neurosci. 2017;80:18–31. doi: 10.1016/j.mcn.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, Huang H, Larose L, Li SS, Takano T, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- Juin A, Di Martino J, Leitinger B, Henriet E, Gary AS, Paysan L, Bomo J, Baffet G, Gauthier-Rouviere C, Rosenbaum J, et al. Discoidin domain receptor 1 controls linear invadosome formation via a Cdc42-Tuba pathway. J Cell Biol. 2014;207:517–533. doi: 10.1083/jcb.201404079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedziora KM, Leyton-Puig D, Argenzio E, Boumeester AJ, van Butselaar B, Yin T, Wu YI, van Leeuwen FN, Innocenti M, Jalink K, Moolenaar WH. Rapid remodeling of invadosomes by gi-coupled receptors: dissecting of the role of Rho GTPases. J Biol Chem. 2016;291:4323–4333. doi: 10.1074/jbc.M115.695940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LC, Lohmer LL, Hagedorn EJ, Sherwood DR. Traversing the basement membrane in vivo: a diversity of strategies. J Cell Biol. 2014;204:291–302. doi: 10.1083/jcb.201311112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozera B, Rapacz M. Reference genes in real-time PCR. J Appl Genet. 2013;54:391–406. doi: 10.1007/s13353-013-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labelle-Cote M, Dusseault J, Ismail S, Picard-Cloutier A, Siegel PM, Larose L. Nck2 promotes human melanoma cell proliferation, migration and invasion in vitro and primary melanoma-derived tumor growth in vivo. BMC Cancer. 2011;11:443. doi: 10.1186/1471-2407-11-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AM, Yeh AT. Ex vivo characterization of sub-10-fs pulses. Opt Lett. 2006;31:1681–1683. doi: 10.1364/ol.31.001681. [DOI] [PubMed] [Google Scholar]
- Latreille M, Laberge MK, Bourret G, Yamani L, Larose L. Deletion of Nck1 attenuates hepatic ER stress signaling and improves glucose tolerance and insulin signaling in liver of obese mice. Am J Physiol Endocrinol Metab. 2011;300:E423–E434. doi: 10.1152/ajpendo.00088.2010. [DOI] [PubMed] [Google Scholar]
- Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K, McPherson VA, et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014;8:1558–1570. doi: 10.1016/j.celrep.2014.07.050. [DOI] [PubMed] [Google Scholar]
- Li A, Dawson JC, Forero-Vargas M, Spence HJ, Yu X, Konig I, Anderson K, Machesky LM. The actin-bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr Biol. 2010;20:339–345. doi: 10.1016/j.cub.2009.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Fan J, Woodley DT. Nck/Dock: an adapter between cell surface receptors and the actin cytoskeleton. Oncogene. 2001;20:6403–6417. doi: 10.1038/sj.onc.1204782. [DOI] [PubMed] [Google Scholar]
- Li W, She H. The SH2 and SH3 adapter Nck: a two-gene family and a linker between tyrosine kinases and multiple signaling networks. Histol Histopathol. 2000;15:947–955. doi: 10.14670/HH-15.947. [DOI] [PubMed] [Google Scholar]
- Linder S. Invadosomes at a glance. J Cell Sci. 2009;122:3009–3013. doi: 10.1242/jcs.032631. [DOI] [PubMed] [Google Scholar]
- Linder S. MT1-MMP: endosomal delivery drives breast cancer metastasis. J Cell Biol. 2015;211:215–217. doi: 10.1083/jcb.201510009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizarraga F, Poincloux R, Romao M, Montagnac G, Le Dez G, Bonne I, Rigaill G, Raposo G, Chavrier P. Diaphanous-related formins are required for invadopodia formation and invasion of breast tumor cells. Cancer Res. 2009;69:2792–2800. doi: 10.1158/0008-5472.CAN-08-3709. [DOI] [PubMed] [Google Scholar]
- Lohmer LL, Clay MR, Naegeli KM, Chi Q, Ziel JW, Hagedorn EJ, Park JE, Jayadev R, Sherwood DR. A sensitized screen for genes promoting invadopodia function in vivo: CDC-42 and Rab GDI-1 direct distinct aspects of invadopodia formation. PLoS Genet. 2016;12:e1005786. doi: 10.1371/journal.pgen.1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makley LN, Gestwicki JE. Expanding the number of 'druggable’ targets: non-enzymes and protein-protein interactions. Chem Biol Drug Des. 2013;81:22–32. doi: 10.1111/cbdd.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maller O, Martinson H, Schedin P. Extracellular matrix composition reveals complex and dynamic stromal-epithelial interactions in the mammary gland. J Mammary Gland Biol Neoplasia. 2010;15:301–318. doi: 10.1007/s10911-010-9189-6. [DOI] [PubMed] [Google Scholar]
- Marchesin V, Castro-Castro A, Lodillinsky C, Castagnino A, Cyrta J, Bonsang-Kitzis H, Fuhrmann L, Irondelle M, Infante E, Montagnac G, et al. ARF6-JIP3/4 regulate endosomal tubules for MT1-MMP exocytosis in cancer invasion. J Cell Biol. 2015;211:339–358. doi: 10.1083/jcb.201506002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. 2014;15:771–785. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SC, Ghersi G, Akiyama SK, Sang QX, Howard L, Pineiro-Sanchez M, Nakahara H, Yeh Y, Chen WT. A novel protease-docking function of integrin at invadopodia. J Biol Chem. 1999;274:24947–24952. doi: 10.1074/jbc.274.35.24947. [DOI] [PubMed] [Google Scholar]
- Munoz-Najar UM, Neurath KM, Vumbaca F, Claffey KP. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene. 2006;25:2379–2392. doi: 10.1038/sj.onc.1209273. [DOI] [PubMed] [Google Scholar]
- Murphy DA, Courtneidge SA. The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara H, Otani T, Sasaki T, Miura Y, Takai Y, Kogo M. Involvement of Cdc42 and Rac small G proteins in invadopodia formation of RPMI7951 cells. Genes Cells. 2003;8:1019–1027. doi: 10.1111/j.1365-2443.2003.00695.x. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Kurokawa K, Kiyokawa E, Matsuda M. Analysis of the spatiotemporal activation of rho GTPases using Raichu probes. Methods Enzymol. 2006;406:315–332. doi: 10.1016/S0076-6879(06)06023-X. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Nolan ME, Aranda V, Lee S, Lakshmi B, Basu S, Allred DC, Muthuswamy SK. The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 2008;68:8201–8209. doi: 10.1158/0008-5472.CAN-07-6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser M, Mader CC, Gil-Henn H, Magalhaes M, Bravo-Cordero JJ, Koleske AJ, Condeelis J. Specific tyrosine phosphorylation sites on cortactin regulate Nck1-dependent actin polymerization in invadopodia. J Cell Sci. 2010;123:3662–3673. doi: 10.1242/jcs.068163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrini MC, Sadou-Dubourgnoux A, Aoki K, Kunida K, Biondini M, Hatzoglou A, Poullet P, Formstecher E, Yeaman C, Matsuda M, et al. SH3BP1, an exocyst-associated RhoGAP, inactivates Rac1 at the front to drive cell motility. Mol Cell. 2011;42:650–661. doi: 10.1016/j.molcel.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. 2009;122:3015–3024. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- Rajadurai CV, Havrylov S, Zaoui K, Vaillancourt R, Stuible M, Naujokas M, Zuo D, Tremblay ML, Park M. Met receptor tyrosine kinase signals through a cortactin-Gab1 scaffold complex, to mediate invadopodia. J Cell Sci. 2012;125:2940–2953. doi: 10.1242/jcs.100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razidlo GL, Schroeder B, Chen J, Billadeau DD, McNiven MA. Vav1 as a central regulator of invadopodia assembly. Curr Biol. 2014;24:86–93. doi: 10.1016/j.cub.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revach OY, Geiger B. The interplay between the proteolytic, invasive, and adhesive domains of invadopodia and their roles in cancer invasion. Cell Adh Migr. 2014;8:215–225. doi: 10.4161/cam.27842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, Ponik SM, Bass BR, Crone WC, Jiang Y, Weaver AM, et al. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys J. 2014;107:2546–2558. doi: 10.1016/j.bpj.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera GM, Antoku S, Gelkop S, Shin NY, Hanks SK, Pawson T, Mayer BJ. Requirement of Nck adaptors for actin dynamics and cell migration stimulated by platelet-derived growth factor B. Proc Natl Acad Sci USA. 2006;103:9536–9541. doi: 10.1073/pnas.0603786103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera GM, Briceno CA, Takeshima F, Snapper SB, Mayer BJ. Inducible clustering of membrane-targeted SH3 domains of the adaptor protein Nck triggers localized actin polymerization. Curr Biol. 2004;14:11–22. doi: 10.1016/j.cub.2003.12.033. [DOI] [PubMed] [Google Scholar]
- Rivera GM, Vasilescu D, Papayannopoulos V, Lim WA, Mayer BJ. A reciprocal interdependence between Nck and PI(4,5)P(2) promotes localized N-WASp-mediated actin polymerization in living cells. Mol Cell. 2009;36:525–535. doi: 10.1016/j.molcel.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse C, Lodillinsky C, Fuhrmann L, Nourieh M, Monteiro P, Irondelle M, Lagoutte E, Vacher S, Waharte F, Paul-Gilloteaux P, et al. Control of MT1-MMP transport by atypical PKC during breast-cancer progression. Proc Natl Acad Sci USA. 2014;111:E1872–E1879. doi: 10.1073/pnas.1400749111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol. 2009;185:11–19. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai-Yageta M, Recchi C, Le Dez G, Sibarita JB, Daviet L, Camonis J, D’Souza-Schorey C, Chavrier P. The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J Cell Biol. 2008;181:985–998. doi: 10.1083/jcb.200709076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Medina M, Gregus KA, Nichol RH, O’Toole SM, Gomez TM. Regulation of ECM degradation and axon guidance by growth cone invadosomes. Development. 2015;142:486–496. doi: 10.1242/dev.108266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol. 2011;3:a003228. doi: 10.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlienger S, Campbell S, Claing A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol Biol Cell. 2014;25:17–29. doi: 10.1091/mbc.E13-06-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoumacher M, Goldman RD, Louvard D, Vignjevic DM. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J Cell Biol. 2010;189:541–556. doi: 10.1083/jcb.200909113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scribner KC, Behbod F, Porter WW. Regulation of DCIS to invasive breast cancer progression by Singleminded-2s (SIM2s) Oncogene. 2013;32:2631–2639. doi: 10.1038/onc.2012.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DF, Azucena EF, Jr, Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, Courtneidge SA. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–165. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Seano G, Chiaverina G, Gagliardi PA, di Blasio L, Puliafito A, Bouvard C, Sessa R, Tarone G, Sorokin L, Helley D, et al. Endothelial podosome rosettes regulate vascular branching in tumour angiogenesis. Nat Cell Biol. 2014;16:931, 931–941. doi: 10.1038/ncb3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VP, Eddy R, Entenberg D, Kai M, Gertler FB, Condeelis J. Tks5 and SHIP2 regulate invadopodium maturation, but not initiation, in breast carcinoma cells. Curr Biol. 2013;23:2079–2089. doi: 10.1016/j.cub.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasappa H, Chaki SP, Lim SM, Trzeciakowski JP, Davidson MW, Rivera GM, Trache A. Selective regulation of cytoskeletal tension and cell-matrix adhesion by RhoA and Src. Integr Biol (Camb) 2014;6:743–754. doi: 10.1039/c4ib00019f. [DOI] [PubMed] [Google Scholar]
- Steffen A, Le Dez G, Poincloux R, Recchi C, Nassoy P, Rottner K, Galli T, Chavrier P. MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr Biol. 2008;18:926–931. doi: 10.1016/j.cub.2008.05.044. [DOI] [PubMed] [Google Scholar]
- Stylli SS, Stacey TT, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J Cell Sci. 2009;122:2727–2740. doi: 10.1242/jcs.046680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy. Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisswange I, Newsome TP, Schleich S, Way M. The rate of N-WASP exchange limits the extent of ARP2/3-complex-dependent actin-based motility. Nature. 2009;458:87–91. doi: 10.1038/nature07773. [DOI] [PubMed] [Google Scholar]
- Wu D, Asiedu M, Wei Q. Myosin-interacting guanine exchange factor (MyoGEF) regulates the invasion activity of MDA-MB-231 breast cancer cells through activation of RhoA and RhoC. Oncogene. 2009;28:2219–2230. doi: 10.1038/onc.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin LL, Chung CM, Chen J, Fok KL, Ng CP, Jia RR, Ren X, Zhou J, Zhang T, Zhao XH, et al. A suppressor of multiple extracellular matrix-degrading proteases and cancer metastasis. J Cell Mol Med. 2009;13:4034–4041. doi: 10.1111/j.1582-4934.2008.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Zech T, McDonald L, Gonzalez EG, Li A, Macpherson I, Schwarz JP, Spence H, Futo K, Timpson P, et al. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J Cell Biol. 2012;199:527–544. doi: 10.1083/jcb.201203025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, Keely PJ, Longmore GD. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15:677–687. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Lu L, Pokhriyal N, Ma H, Duan L, Lin S, Jafari N, Band H, Band V. Overexpression of RhoA induces preneoplastic transformation of primary mammary epithelial cells. Cancer Res. 2009;69:483–491. doi: 10.1158/0008-5472.CAN-08-2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.