

Abstract
Osteogenesis imperfecta (OI) is a hereditary disease characterized by bone fragility caused by mutations in the proteins that support the formation of the extracellular matrix in the bone. The diagnosis of OI begins with clinical suspicion, from phenotypic findings at birth, low-impact fractures during childhood or family history that may lead to it. However, the variability in the semiology of the disease does not allow establishing an early diagnosis in all cases, and unfortunately, specific clinical data provided by the literature only report 28 patients with OI type XI. This information is limited and heterogeneous, and therefore, detailed information on the natural history of this disease is not yet available. This paper reports the case of a male patient who, despite undergoing multidisciplinary management, did not have a diagnosis for a long period of time, and could only be given one with the use of whole-exome sequencing. The use of the next-generation sequencing in patients with ultrarare genetic diseases, including skeletal dysplasias, should be justified when clear clinical criteria and an improvement in the quality of life of the patients and their families are intended while reducing economic and time costs. Thus, this case report corresponds to the 29th patient affected with OI type XI, and the 18th mutation in FKBP10, causative of this pathology.
Keywords: osteogenesis imperfecta type XI, FKBP10, diagnostic odyssey
Introduction
Osteogenesis imperfecta (OI) is a hereditary disease characterized by bone fragility due to mutations in proteins that help support the formation of the extracellular matrix in the bone.10 The severity of the disease varies depending on the gene involved, and the disease may be lethal during the first year of life or the patient may achieve a longer survival rate. The first classification of this disease33 described four types of OI; in 2009, the Nosology and Constitutional Disorders ICHG of the Skeleton (INCDS) proposed a new classification, creating five groups and considering the clinical manifestations, the radiological findings, and the molecular alteration.3,31
The molecular findings of OI were first described in 1983, when a deletion in the COL1A1 gene was demonstrated in a patient with OI type II.8 In 2006, a mutation of the CRTAP gene was found in a patient with OI type II, being the first OI gene discovered with an autosomal recessive inheritance pattern.2 Currently, 18 genes involved in the different types of OI have been recognized,22,37 of which 90% correspond to heterozygous mutations of the COL1A1 and COL1A2 genes.25
The diagnosis of OI begins with clinical suspicion, from phenotypic findings at birth, low-impact fractures during childhood or family history that may lead to it.12 Imaging studies allow evidencing the extended bone involvement, although, in some circumstances, molecular tools are necessary.9 However, the variability in the semiology of the disease does not allow establishing an early diagnosis in all cases.
This paper reports the case of a male patient who, despite undergoing multidisciplinary management, did not have a diagnosis for a long period of time, and could only be given one with the use of new health technologies like whole-exome sequencing (WES). The etiology of his condition could be established to support treatment, follow-up, and prognosis for decision-making, thus improving the quality of life of the patient and his family.11
Clinical case
The clinical case, accompanied by the clinical history, photos, and laboratory results presented in this report, has the approval to be published by the parents of the patient and witnesses with the endorsement of the ethics committee of the Military Hospital of Colombia.
The medical genetics service of Hospital Militar received a 40-month-old patient in 2011, referred by the endocrinology service due to low height, delayed psychomotor development, and suspicion of mucopolysaccharidosis (MPS). The patient is the first-born child of a 25-year-old mother and a 21-year-old father, who are first-degree relatives. The mother had antenatal controls (+), eutocic delivery at 39 weeks, 2700 g at birth, 49 cm, and no NICU admission. Pathologies include left hip dysplasia and scoliosis; his first fracture occurred at 6 months in the right radius; parents deny prior hospitalizations, as well as surgical and allergic history.
Regarding development milestones, he held his head up at 9 months, sat up at 12 months, and began to produce disyllables at 18 months. He presented unstable asymmetric gait at age 2, stopped walking at 3 due to bilateral hip pain and muscle weakness, and started producing words at 2, with a high-pitched voice. Figure 1 shows the family tree of the patient.
Figure 1.

Family tree of the patient with OI type XI.
Abbreviation: OI, osteogenesis imperfecta.
The initial clinical examination showed a height of 76.5 cm (−5.50 SD), weight of 7.4 kg (−7.07 SD), cephalic perimeter of 49 cm (−0.38 SD), and BMI of 12.6 (−3.98 SD). Figure 2A and B shows his anthropometric evolution until the age of 7. The patient exhibited usual non-dysmorphic facies, without blue sclera or tooth involvement, asymmetric thorax with severe rotoscoliosis, and 60° kyphosis (Figure 3A). In addition, asymmetric limbs caused by the shortening of left lower limb, genu valgus, and hypotrophy were found, while no retractions, arthrogryposis, or pterygia (Figure 3B) was observed. No neurological alterations were identified either.
Figure 2.

Anthropometric evolution of a patient with OI type III: (A) weight vs. age and (B) height vs. age. Growth charts were developed by Frank Rauch for patients with OI type III.
Abbreviation: OI, osteogenesis imperfecta.
Figure 3.
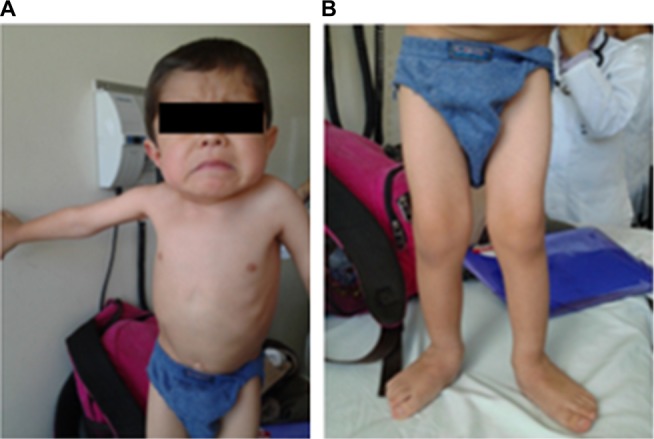
Physical examination of a patient with OI type III: (A) thorax and (B) lower limbs with hypotrophy and genu valgus.
Abbreviation: OI, osteogenesis imperfecta.
The first diagnostic suspicion focused on MPS type IVA, which was subsequently ruled out due to normal GALNS enzyme activity. Paraclinical tests of bone metabolism were requested, initially finding hyperphosphatemia with hypercalciuria, and parathyroid hormone, Vitamin D 25, and serum calcium values within normal ranges. The patient was referred to nephrology service, where little mobilization of the patient was considered as the cause. The paraclinical follow-up is shown in Figure 4A–E.
Figure 4.

Paraclinical follow-up of a patient with OI type III: (A) values of 25-hydroxyvitamin D, (B) serum levels of parathyroid hormone, (C) levels of alkaline phosphatase, (D) levels of serum calcium, and (E) level of inorganic phosphorus in serum.
Abbreviations: OI, osteogenesis imperfecta; PTH, parathyroid hormone.
At the age of 5 years and 5 months, the patient suffered a second fracture in the proximal left femur, a third fracture at 6 years in the proximal left femur, and a fourth fracture at 6 years and 6 months in the left proximal humerus. Given this history, OI was considered, and at the age of 6 years and 6 months, a WES was requested since the symptoms of the patient were not clinically or radiologically compatible with OI type I or III.
No radiological alterations in the metaphyses and epiphyses of the long bones were seen; the callus of the fractures did not show hyper-growth, and some reduction in the bone mineral mass was observed. Iliac bones showed an abnormality in their morphology with hypoplasia in the lowest portion and left coxa valga. The spine showed severe thoracolumbar rotoscoliosis, and bone densitometry in L1–L4 showed a Z score of −6.6.
Finally, at the age of 7 years and 7 months, the result of the WES was obtained, showing a homozygous mutation, not reported in the literature, in the gene FKBP10, with a change in the nucleotide NM_021939.3 c.612C>G Chr17:41,818,412 C>G (GRCh38/hg18), p. Tyr204X, which generated a stop codon in exon 4, as seen in Figures S1 and S2. The patient had a fifth and last fracture in the left proximal humerus at the age of 6 years and 8 months.
Discussion
One of the first manifestations of OI is the onset of fractures at early ages;14 nevertheless, there are other etiologies that should be suspected. First, it is necessary to establish whether the fracture was traumatic or not,4 and once a traumatic event is ruled out – including child abuse – different diagnostic possibilities, such as inborn errors of metabolism, antenatal viral infection, insensitivity to pain, and skeletal neoplasms or dysplasias, should be considered.6
When approaching these diseases, Bronicki et al established four groups of pathologies based on radiographic findings: those with decreased bone density, increased bone density, presence of thin bones, and without radiographic manifestations,6 finding in the first-group syndromes such as Cole–Carpenter syndrome, Hajdu–Cheney syndrome, hypophosphatemia, mucolipidosis II, Stuve–Wiedemann syndrome, or Bruck syndrome,1 the latter being a differential diagnosis and with allelic heterogeneity with OI type XI.
The clinical history, the biochemical paraclinical tests, and the radiology of the patient described in this study did not provide enough information for a definitive clinical approach, and only after new genomic health technologies were available in the country, establishing a definitive diagnosis was possible. This report exposes the 29th case of a patient affected with OI type XI and the 18th mutation in the FKBP10 gene that causes this pathology. This diagnosis was given when the patient was already 7 years old, considering that his first fracture occurred at the age of 6 months. Only with the use of robust molecular tools such as the exome, this new mutation could be identified.
The OI type XI is due to the mutation in the FKBPP10 gene that encodes for the FKBP65 protein.30 This protein has a molecular weight of 65 kDa, has the function of a chaperone, and is located in the lumen of the endoplasmic reticulum23 in colocalization with tropoelastin, helping in the folding of the three helices of the type 1 procollagen.23 Its chaperone activity is compared with the protein disulfide isomerase, and it has another function of peptidyl prolyl isomerase type, which assists in the folding of tropoelastin 1.
In 1979, Van Dijk and Sillence28,32 classified OI into four types: type I with mild involvement, caused by a quantitative deficiency of collagen; type II with lethal involvement; and types III and IV with severe and moderate involvement, respectively.23 However, the classification has constantly changed due to the broad phenotypic spectrum, the heterogeneity in the mechanisms of inheritance, and the allelic variation (involvement of new genes).16
In 2004, Rauch and Glorieux27 expanded the classification by adding types V, VI, and VII, and describing the recessive inheritance for the types VI and VII as a new finding.13 In 2009, the INCDS proposed a new classification, creating five groups based on clinical manifestations, radiological findings, and molecular alteration17 as follows: 1) OI type 1 or nondeforming OI with blue sclera; 2) OI type 2 or perinatal lethal OI in which involvement is much more severe; 3) progressively deforming OI or OI type 3, with multiple neonatal or infant fractures that lead to bone deformities; 4) the common variable OI type 4, with recurrent fractures, deformities in long bones and vertebrae, normal sclera, and mild hearing impairment;19 and 5) OI type 5, or OI with calcifications in interosseous membranes, which also shows greater susceptibility to the formation of hyperplastic bone callus after a fracture or a surgical intervention.17 Our patient belongs to type 3, according to this classification.
The diagnosis of OI is variable and depends mostly on the clinical manifestations presented by the patient. Being diagnosed helps patients and relatives to understand better the prognosis and development of the disease, and also allows health care personnel to have more clarity in their interventions.7 Therefore, insisting on adequate endophenotypic descriptions of these cases is critical; in addition, using health technologies can lead to the etiologic finding in an adequate and timely manner.15
Unfortunately, the specific clinical data provided by the literature on the 28 patients with OI type XI are limited and heterogeneous, and therefore, detailed information on the natural history of this disease is not available yet. For example, regarding the age of onset of the first fracture, 13 of the 28 individuals exhibited an average at 6 months (the same as the Colombian patient); the number of fractures reported ranged from one to 100 in seven of the 28 patients (Colombian patient, five fractures); in 24 of the 28 patients, the first bone affected was long; of the 28 patients reported, 13 used wheelchairs, and three did not; the patients alive so far (22 of 28) are within the age range from 5 months to 46 years, and the average age of molecular diagnosis was 16.2 years (±12.6), with the age of diagnosis of our patient being the third earliest reported in the literature (Table 1).
Table 1.
Compendium of patients with OI with mutations in the gene FKBP10
| Gender | Age at 1st fracture (months) | Number of fractures at 10 years | Group of the 1st affected bone | Age at clinical diagnosis | Age at molecular diagnosis (years) | Use of wheelchair | Age at the time of the study (years) | Type of mutation | Consanguinity | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | NA | NA | Long | NA | NA | NA | NA | Homozygous 33-base-pair deletion (c.321_353del), deletion of 11 aa (Gly107 Leu117del) | Yes | Alanay et al1 |
| 2 | M | NA | NA | Long | NA | NA | NA | NA | Homozygous 33-base-pair deletion (c.321_353del), deletion of 11 aa (Gly107 Leu117del) | Yes | Alanay et al1 |
| 3 | F | NA | NA | Long | NA | NA | NA | NA | Homozygous 33-base pair deletion (c.321_353del), deletion of 11 aa (Gly107 Leu117del) | Yes | Alanay et al1 |
| 4 | F | NA | NA | Long | NA | NA | NA | NA | Homozygous 33-base-pair deletion (c.321_353del), deletion of 11 aa (Gly107 Leu117del) | Yes | Alanay et al1 |
| 5 | F | NA | NA | Long | NA | 12 | No | 12 | Homozygous c.831_832insC (p.Gly278ArgfsX295) stop codon 17 aa downstream | Yes | Alanay et al1 |
| 6 | M | NA | NA | Long | NA | 16 | Yes | 16 | Homozygous c.831_832insC (p.Gly278ArgfsX295) stop codon 17 aa downstream | Yes | Alanay et al1 |
| 7 | M | NA | NA | Long | NA | 18 | Yes | 18 | Homozygous c.831_832insC (p.Gly278ArgfsX295) stop codon 17 aa downstream | Yes | Alanay et al1 |
| 8 | M | 9 | 70–100 in lifetime | Long | 9 years | 46 | Yes | 46 | Homozygous into a stop codon (c.1207C>T, FKBP65-p.Arg403) | Yes | Steinlein et al34 |
| 9 | M | 1 | 70–100 in lifetime | Long | 1 year | 44 | Yes | 44 | Homozygous into a stop codon (c.1207C>T, FKBP65-p.Arg403) | Yes | Steinlein et al34 |
| 10 | M | 1 | 70–100 in lifetime | Long | 1 year | 43 | Yes | 43 | Homozygous into a stop codon (c.1207C>T, FKBP65-p.Arg403) | Yes | Steinlein et al34 |
| 11 | M | 1 | NA | NA | Birth | 7 | Yes | 7 | Homozygous mutation c.976delA (p.Met326Trpfs∗39) in exon 6 of the FKBP10 gene | Yes | Seyedhassani et al35 |
| 12 | F | 1 | 5–6 | Long | 2 years | 6 | Yes | 11 | c.743dupC | Yes | Shaheen et al36 |
| 13 | F | 6 | 2–3 | NA | 5 years | 14 | No | 14 | c.743dupC | Yes | Shaheen et al36 |
| 14 | M | 1 | 3 | Long | 8 years | 17 | Yes | 17 | c.743dupC | Yes | Shaheen et al36 |
| 15 | M | 48 | 1–2 | NA | 48 years | NA | Yes | 12 | c.743dupC | Yes | Shaheen et al36 |
| 16 | M | 1 | NA | NA | 10 months | 1 | NA | 0.8 | c.831dupC | Yes | Shaheen et al36 |
| 17 | F | 1 | ND | Long | 1 year | 12 | No | 12 | Homozygous FKBP10 c.1271_1272delCCinsA mutation | Yes | Barnes et al3 |
| 18 | M | NA | NA | Long | NA | 17 | Yes | 17 | Homozygous substitution of G with A at nucleotide position 1490 (c.1490G4A; p.Trp497) | Yes | Umair et al32 |
| 19 | M | NA | NA | Long | NA | 10 | Yes | 10 | Homozygous substitution of G with A at nucleotide position 1490 (c.1490G4A; p.Trp497) | Yes | Umair et al32 |
| 20 | F | NA | NA | Long | NA | NA | NA | NA | Homozygous substitution of G with A at nucleotide position 1490 (c.1490G4A; p.Trp497) | Yes | Umair et al32 |
| 21 | F | NA | NA | Long | NA | 15 | NA | 15 | Homozygous substitution of nucleotide G with A at position 344 (c.344G4A; p.R115Q) | Yes | Umair et al32 |
| 22 | F | NA | NA | Long | NA | NA | NA | NA | Homozygous substitution of nucleotide G with A at position 344 (c.344G4A; p.R115Q) | Yes | Umair et al32 |
| 23 | F | NA | NA | Long | NA | 11 | NA | 11 | Homozygous substitution of nucleotide G with A at position 344 (c.344G4A; p.R115Q) | Yes | Umair et al32 |
| 24 | M | NA | NA | Long | NA | 12 | NA | 14 | Homozygous duplication of a nucleotide C at position 831 (c.831dupC; p.Gly278ArgfsX295) | Yes | Umair et al32 |
| 25 | M | NA | NA | Long | NA | 17 | NA | 17 | Homozygous duplication of a nucleotide C at position 831 (c.831dupC; p.Gly278ArgfsX295) | Yes | Umair et al32 |
| 26 | F | NA | NA | Long | NA | 18 | Yes | 18 | Homozygous duplication of a nucleotide C at position 831 (c.831dupC; p.Gly278ArgfsX295) | Yes | Umair et al32 |
| 27 | F | 1 | 3–5 years | Long | 3 years | 3 | Yes | 3 | c.21dupC p.(Ser8Glnfs 67) | Yes | Caparrós et al37 |
| 28 | F | 1 | NA | Long | 5 months | 1 | No | 0.4 | c.689T>C p.(Ile230Thr) | Yes | Caparrós et al37 |
| 29 | M | 6 | 1 | Long | 7 years | 7 | No | 7 | Homozygous MN-021939.3:C612>G Chr 17 (GRCh 38): 41818412C>G | Yes | Case report of this article |
Note: This table was created by the authors of this article based on the results reported in the articles referenced in the last column of the table.
Abbreviations: OI, osteogenesis imperfecta; NA, not available.
With the increased use of molecular tools, multiple genes involved in OI have been found. Genes COL1A1, COL1A2, and others are classified in seven groups depending on the molecular pathway affected in the following: synthesis of collagen; defects in the structure or in the amount of collagen available; defects in the “prolyl-3-hydroxylase complex”, in the lysyl hydroxylase telopeptide, and in the chaperone proteins; and regulation of the bone metabolism with involvement of genes with unknown functions.20,21 Currently, proper genetic counseling to establish the risk of recurrence or to project the behavior of some types of OI is possible if the mutated gene is identified, which allows predicting whether bone deformities will be found or not, as in types 1 and 4, respectively, and hence the importance of the molecular result.1,26
Molecular aids to diagnose OI consist in, besides the Sanger sequencing, next-generation sequencing (NGS) tools such as multigenic panels and exome sequencing, covering between 4,600 and 21,000 genes. The advantages of NGS over Sanger include a higher diagnostic performance, and a higher performance in relation to costs and time of analysis.1 In the particular case of this patient, who has a wide clinical heterogeneity (fractures, low stature, high-pitched voice, severe scoliosis, and absence of severe bone deformities), when using exome sequencing, the target of genes to be analyzed is greater, increasing the probability of finding the affected gene.
In the case of this patient, the use of multigenic panel was not considered because of the cost, which is higher than for WES; moreover, OI was not initially suspected and – as mentioned above – there was high clinical heterogeneity. According to the guidelines of the Canadian College of Medical Genetics, in such a case, using WES is advisable.5 Some studies have analyzed the costs generated by the diagnostic odyssey in specific diseases, such as epilepsy19 or chronic neurodegenerative diseases5 among others; however, so far, reports of the costs generated for patients with skeletal dysplasia are not found. Costs, during the period of time in which a definitive diagnosis is not given, include the time lost by the patient and his family, the decreased quality of life, and the economic losses.19
Soden et al analyzed the repercussions of the diagnostic odyssey in 119 patients with neurodegenerative diseases, describing an average time of 8 years for obtaining a final diagnosis, which is similar to that reported in our patient (7 years), with an economic cost of US$19,000 per family (US$3,248–55,321) spent in tests performed before the WES/whole genomic sequencing.29
Information on quality of life was more difficult to assess due to the absence of a quantitative scale; however, Joshi et al38 mentioned that when the diagnosis is established in patients with epileptic encephalopathies, general recognition by each family is possible because they know the origin and prognosis of the pathology in each patient. With this in mind, advantages in both economy and quality of life can be evidenced in the cases of rare diseases with difficult diagnoses, since the use of health technologies such as NGS is cost-effective even in cases of bone dysplasias such as OI.
Conclusion
This case report describes the second Latin American case with a molecularly confirmed diagnosis of OI type XI – the first case was reported by Alanay in 20101 – with a new mutation in the gene FKBP10, consanguineous parents, and a moderate clinical evolution, in relation to what was reported for the other 28 patients studied worldwide. Only with the use of advanced molecular aids, such as the WES, was it possible to find the etiological element and to finish the diagnostic odyssey of nearly 7 years, which will have an impact, in the short term, on the integral management of the patient and his family. The use of the NGS in patients with ultrarare genetic diseases, including skeletal dysplasias, should be justified when clear clinical criteria and an improvement in the quality of life of the patients and their families are intended while reducing economic and time costs.
Supplementary materials
Image of Chr17 variant (GRCh38):g.41818412C>G seen by NGS.
Abbreviation: NGS, next-generation sequencing.
Image of the electropherogram of the confirmation of the pathogenic variant Chr17 (GRCh38):g.41818412C>G p.Tyr204*.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Alanay Y, Avaygan H, Camacho N, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86(4):551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes AM, Chang W, Morello R, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. 2006;355(26):2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes AM, Chang W, Morello R, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. 2006;355(26):2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boycott K, Hartley T, Adam S, et al. Canadian College of Medical Geneticists The clinical application of genome-wide sequencing for monogenic diseases in Canada: Position Statement of the Canadian College of Medical Geneticists. J Med Genet. 2015;52(7):431–437. doi: 10.1136/jmedgenet-2015-103144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bregou Bourgeois A, Aubry-Rozier B, Bonafé L, Laurent-Applegate L, Pioletti DP, Zambelli PY. Osteogenesis imperfecta: from diagnosis and multidisciplinary treatment to future perspectives. Swiss Med Wkly. 2016;146:w14322. doi: 10.4414/smw.2016.14322. [DOI] [PubMed] [Google Scholar]
- 6.Bronicki LM, Stevenson RE, Spranger JW. Beyond osteogenesis imperfecta: causes of fractures during infancy and childhood. Am J Med Genet C Semin Med Genet. 2015;169(4):314–327. doi: 10.1002/ajmg.c.31466. [DOI] [PubMed] [Google Scholar]
- 7.Byers PH, Pyott SM. Recessively inherited forms of osteogenesis imperfecta. Annu Rev Genet. 2012;46:475–497. doi: 10.1146/annurev-genet-110711-155608. [DOI] [PubMed] [Google Scholar]
- 8.Carmichael N, Tsipis J, Windmueller G, Mandel L, Estrella E. “Is it going to hurt?”: the impact of the diagnostic odyssey on children and their families. J Genet Couns. 2015;24(2):325–335. doi: 10.1007/s10897-014-9773-9. [DOI] [PubMed] [Google Scholar]
- 9.Chu ML, Williams CJ, Pepe G, Hirsch JL, Prockop DJ, Ramirez F. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nature. 1983;304(5921):78–80. doi: 10.1038/304078a0. [DOI] [PubMed] [Google Scholar]
- 10.Cozzolino M, Perelli F, Maggio L, et al. Management of osteogenesis imperfecta type I in pregnancy; a review of literature applied to clinical practice. Arch Gynecol Obstet. 2016;293(6):1153–1159. doi: 10.1007/s00404-016-4012-2. [DOI] [PubMed] [Google Scholar]
- 11.Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2014;(7):CD005088. doi: 10.1002/14651858.CD005088.pub3. [DOI] [PubMed] [Google Scholar]
- 12.Emery SC, Karpinski NC, Hansen L, Masliah E. Abnormalities in central nervous system development in osteogenesis imperfecta type II. Pediatr Dev Pathol. 1999;2(2):124–130. doi: 10.1007/s100249900100. [DOI] [PubMed] [Google Scholar]
- 13.Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339(14):947–952. doi: 10.1056/NEJM199810013391402. [DOI] [PubMed] [Google Scholar]
- 14.Glorieux FH, Rauch F, Plotkin H, et al. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000;15(9):1650–1658. doi: 10.1359/jbmr.2000.15.9.1650. [DOI] [PubMed] [Google Scholar]
- 15.Hennedige AA, Jayasinghe J, Khajeh J, Macfarlane TV. Systematic review on the incidence of bisphosphonate related osteonecrosis of the jaw in children diagnosed with osteogenesis imperfecta. J Oral Maxillofac Res. 2014;4(4):e1. doi: 10.5037/jomr.2013.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoyer-Kuhn H, Netzer C, Semler O. Osteogenesis imperfecta: pathophysiology and treatment. Wien Med Wochenschr. 2015;165(13–14):278–284. doi: 10.1007/s10354-015-0361-x. [DOI] [PubMed] [Google Scholar]
- 17.Hoyer-Kuhn H, Semler O, Schoenau E. Effect of denosumab on the growing skeleton in osteogenesis imperfecta. J Clin Endocrinol Metab. 2014;99(11):3954–3955. doi: 10.1210/jc.2014-3072. [DOI] [PubMed] [Google Scholar]
- 18.Kamoun-Goldrat AS, Le Merrer MF. Osteogenesis imperfecta and dentinogenesis imperfecta: diagnostic frontiers and importance in dentofacial orthopedics. Orthod Fr. 2007;78(2):89–99. doi: 10.1051/orthodfr:2007010. French [with English abstract] [DOI] [PubMed] [Google Scholar]
- 19.Kuurila K, Kaitila I, Johansson R, Grénman R. Hearing loss in Finnish adults with osteogenesis imperfecta: a nationwide survey. Ann Otol Rhinol Laryngol. 2002;111(10):939–946. doi: 10.1177/000348940211101014. [DOI] [PubMed] [Google Scholar]
- 20.Lindert U, Cabral WA, Ausavarat S, et al. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun. 2016;7:11920. doi: 10.1038/ncomms11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28(3):209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morello R, Bertin TK, Chen Y, et al. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127(2):291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 23.Paterson CR, McAllion S, Miller R. Heterogeneity of osteogenesis imperfecta type I. J Med Genet. 1983;20(3):203–205. doi: 10.1136/jmg.20.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363(9418):1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 25.Semler O, Netzer C, Hoyer-Kuhn H, Becker J, Eysel P, Schoenau E. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. 2012;12(3):183–188. [PubMed] [Google Scholar]
- 26.Shi CG, Zhang Y, Yuan W. Efficacy of bisphosphonates on bone mineral density and fracture rate in patients with osteogenesis imperfecta: a systematic review and meta-analysis. Am J Ther. 2016;23(3):e894–e904. doi: 10.1097/MJT.0000000000000236. [DOI] [PubMed] [Google Scholar]
- 27.Sillence DO. Craniocervical abnormalities in osteogenesis imperfecta: genetic and molecular correlation. Pediatr Radiol. 1994;24(6):427–430. doi: 10.1007/BF02011910. [DOI] [PubMed] [Google Scholar]
- 28.Sillence DO, Barlow KK, Cole WG, Dietrich S, Garber AP, Rimoin DL. Osteogenesis imperfecta type III. Delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet. 1986;23(3):821–832. doi: 10.1002/ajmg.1320230309. [DOI] [PubMed] [Google Scholar]
- 29.Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Umair M, Hassan A, Jan A, et al. Homozygous sequence variants in the FKBP10 gene underlie osteogenesis imperfecta in consanguineous families. J Hum Genet. 2016;61(3):207–213. doi: 10.1038/jhg.2015.129. [DOI] [PubMed] [Google Scholar]
- 31.Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B. What is new in genetics and osteogenesis imperfecta classification? J Pediatr (Rio J) 2014;90(6):536–541. doi: 10.1016/j.jped.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Umair M, Hassan A, Jan A, et al. Homozygous sequence variants in the FKBP10 gene underlie osteogenesis imperfecta in consanguineous families. J Hum Genet. 2016;61(3):207–13. doi: 10.1038/jhg.2015.129. [DOI] [PubMed] [Google Scholar]
- 33.Warman ML, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A(5):943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinlein OK, Aichinger E, Trucks H, Sander T. Mutations in FKBP10 can cause a severe form of isolated Osteogenesis imperfecta. BMC Med Genet. 2011;12:152. doi: 10.1186/1471-2350-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seyedhassani SM, Hashemi-Gorji F, Yavari M, Harazi F, Yassaee VR. Novel FKBP10 Mutation in a Patient with Osteogenesis Imperfecta Type XI. Fetal Pediatr Pathol. 2016;35(5):353–358. doi: 10.1080/15513815.2016.1191567. [DOI] [PubMed] [Google Scholar]
- 36.Shaheen R, Al-Owain M, Faqeih E, Al-Hashmi N, Awaji A, Al-Zayed Z, et al. Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am J Med Genet A. 2011;155A(6):1448–52. doi: 10.1002/ajmg.a.34025. [DOI] [PubMed] [Google Scholar]
- 37.Caparros-Martin JA, Valencia M, Pulido V, et al. Clinical and Molecular Analysis in Families With Autosomal Recessive Osteogenesis Imperfecta Identifies Mutations in Five Genes and Suggests Genotype-Phenotype Correlations. American Journal of Medical Genetics Part A. 2013;161(6):1354–1369. doi: 10.1002/ajmg.a.35938. [DOI] [PubMed] [Google Scholar]
- 38.Joshi C, Kolbe DL, Mansilla MA, Mason SO, Smith RJ, Campbell CA. Reducing the Cost of the Diagnostic Odyssey in Early Onset Epileptic Encephalopathies. Biomed Res Int. 2016;2016:6421039. doi: 10.1155/2016/6421039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Image of Chr17 variant (GRCh38):g.41818412C>G seen by NGS.
Abbreviation: NGS, next-generation sequencing.
Image of the electropherogram of the confirmation of the pathogenic variant Chr17 (GRCh38):g.41818412C>G p.Tyr204*.