

Abstract
Perinatal insults, including intrauterine growth restriction, preterm birth, maternal exposure to toxins, or dietary deficiencies produce deviations in the epigenome of lung cells. Occurrence of perinatal insults often coincides with the final stages of lung development. The result of epigenome disruptions in response to perinatal insults during lung development may be long-term structural and functional impairment of the lung and development of lung disease. Understanding the contribution of epigenetic mechanisms to life-long lung disease following perinatal insults is the focus of the developmental origins of adult lung disease field. DNA methylation, histone modifications, and microRNA changes are all observed in various forms of lung disease. However, the perinatal contribution to such epigenetic mechanisms is poorly understood. Here we discuss the developmental origins of adult lung disease, the interplay between perinatal events, lung development and disease, and the role that epigenetic mechanisms play in connecting these events.
Keywords: Lung development, epigenetic, developmental origins, programming
Introduction
Do the origins of adult lung disease lie in early life events? How is individual susceptibility to adult lung disease programmed by perinatal environmental conditions? Answers to these questions are at the center of the emerging ‘Developmental Origins of Adult Lung Disease’ field. Research is focused on understanding the contribution of perinatal events to adult lung disease, as well as uncovering the mechanisms connecting perinatal events to adult lung disease. A key mechanism under investigation involves plasticity of the lung epigenome.
Key Concepts in the Developmental Origins of Adult Lung Disease
The developmental origins of disease field evolved following David Barker’s observation that low birth weight (a surrogate for poor in utero conditions) predisposed to adult cardio-metabolic disease and early death (Barker and Osmond, 1986). More recently, the developmental origins of disease field has expanded to include lung disease (Joss-Moore et al., 2011, Harding and Maritz, 2012, Stocks et al., 2013).
Two main concepts are associated with the developmental origins of adult lung disease. The first relies on the idea that lung function tracks a predictable percentile over the life course (Figure 1). A consequence of percentile tracking of lung function is that failure to reach normal, maximal lung function by early adulthood results in a lower lung function at later age; deficits that can become significant when considered in combination with the reduction in lung function that accompanies normal aging. When perinatal events alter the growth and development of the lung, thus reducing lung function in the neonatal and childhood period, the result is reduced lung function throughout life. The second concept in the developmental origins of adult lung disease involves the idea that the lung has specialized cells that undergo programing and subsequent remodeling during normal lung development. When lung development is interrupted by noxious stimuli the result is divergence of cellular programming, producing dysfunctional remodeling of the lung, either during development or later in response to injury (Figure 2).
Figure 1. Variance of lung function with age.

Lung function, represented by FEV1 as a % of maximal value, varies with age and reaches a maximum in the early 20s. The solid line represents FEV1 variation with age under conditions of normal growth, and in the absence of disease or additional insults (e.g. smoking). The dashed line represents FEV1 in the case of reduced lung growth and/or development during early in life. Failure to achieve normal maximal lung function, even with normal age-related decline, produces respiratory symptoms (shaded area). The dotted line represents a more rapid decline in lung function as a result of additional insults (e.g. smoking), in which case respiratory symptoms are observed at earlier ages. Figure adapted from (Weiss, 2010, Stocks et al., 2013).
Figure 2. The impact of an injury stimulus to the immature lung.
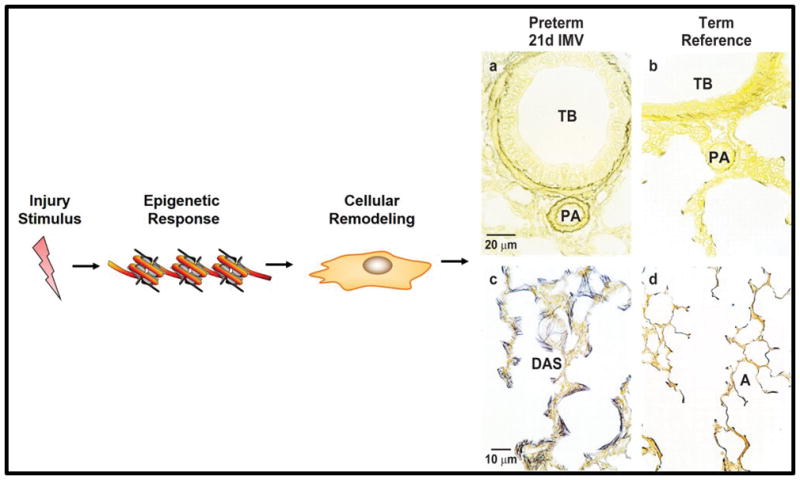
An injury stimulus to the immature lung may prompt an epigenetic response and subsequent cellular remodeling. The schematic shows cellular remodeling for the lamb lung mesenchyme in response to preterm birth with support by intermittent mandatory ventilation with oxygen-rich gas for 21days. Histopathological outcomes are shown in panels a–d. Panel a illustrates accumulation of smooth muscle cells surrounding a terminal bronchiole (TB) and its adjacent pulmonary arteriole (PA) compared to an age-matched term reference lamb (panel b). Panel c shows distended distal airspaces (DAS) with aberrant, excessive accumulation of mature cross-linked elastic fibers (black). This architecture is unlike the normal delicate, lacy features of an age-matched term reference lamb (panel d) with anatomic alveoli (A), thin walls and concentrated elastin at the tip of secondary septa.
Perinatal insults, including intrauterine growth restriction (IUGR), premature birth, maternal exposure to toxins, or dietary deficiency are linked to several lung disease outcomes and remodeling disorders (Stocks et al., 2013) (Table 1). The pathophysiology of lung disease with perinatal origins is likely a combination of incomplete lung growth and development, as well as reprogramming of specific cells within the lung. Lung growth and development, as well as reprogramming of specific lung cells, involves precise spatial and temporal activation of gene expression programs. Spatial and temporal gene expression programs are regulated by epigenetic mechanisms.
Table 1.
Perinatal insults and associated lung disease reported in human subjects.
| Perinatal Insult | Associated Lung Disease | Representative References |
|---|---|---|
| Intrauterine growth restriction with and without preterm birth | Bronchopulmonary Dysplasia Reduced Lung Function Chronic Obstructive Pulmonary Disease |
(Barker et al., 1991, Caudri et al., 2007, Hancox et al., 2009, Kindlund et al., 2010, Jobe, 2012) |
| Maternal Tobacco Smoke Exposure | Reduced Lung Function Asthma and Wheezing Airway Reactivity |
(Gilliland et al., 2003, Jaakkola and Gissler, 2004, Alati et al., 2006, Hylkema and Blacquiere, 2009) |
| Air Pollution | Asthma Allergic Airway Disease Chronic Obstructive Pulmonary Disease |
(Miller and Marty, 2010, Jedrychowski et al., 2013, Wright and Brunst, 2013, Gaffin et al., 2014, Urman et al., 2014) |
| Maternal Diet | Asthma Allergic Airway Disease |
(Erkkola et al., 2009, Checkley et al., 2010, Nwaru et al., 2010) |
The Epigenetic Contribution to the Developmental Origins of adult lung disease
The interaction between genetics, epigenetics, and the environment contribute to the developmental origins of adult lung disease. We focus on the epigenetic component because epigenetic mechanisms regulate the timing, magnitude, and cell-specificity of gene expression and resulting transcriptomes. Epigenetic regulation of gene expression is accomplished by directing the interactions among the transcription machinery, transcription factors, and specific regions of DNA. Excellent reviews of the general mechanisms of epigenetic regulation of gene expression are available (Klose and Bird, 2006, Zentner and Henikoff, 2013, Voss and Hager, 2014) so we will not review them.
Lung Developmental Timing
To consider the role of epigenetic mechanisms in the developmental origins of adult lung disease, an understanding of the timing of lung development is helpful. Human lung development passes through 5 distinct stages: embryonic (4–7 weeks gestation), pseudoglandular (5–17 weeks gestation), canalicular (16–27 weeks gestation), saccular (28–31 weeks gestation), and alveolar (32 weeks gestation though early postnatal years) (Albertine and Pysher, 2004, Burri, 2006). Because animal models are often used to study the molecular mechanisms of the developmental origins of adult lung disease, the timing of lung development in non-human mammalian species is also important. Lung development in non-human mammalian species follows the same distinct stages as human lung development. In the sheep, the timing is of lung development is similar to that of the human, with alveolar formation largely complete by, or shortly after, term birth. This is in contrast to the rat and mouse, in which the lung is developmentally more immature at term birth. The rat and mouse lung is in the saccular stage of development at term birth (Figure 3). Distinct from humans and sheep, rats and mice undergo alveolar formation during the second and third weeks of postnatal life after term birth. Normal newborn rats and mice pups do not need ventilation support or supplemental oxygen to live.
Figure 3. Timeline of rat lung development.
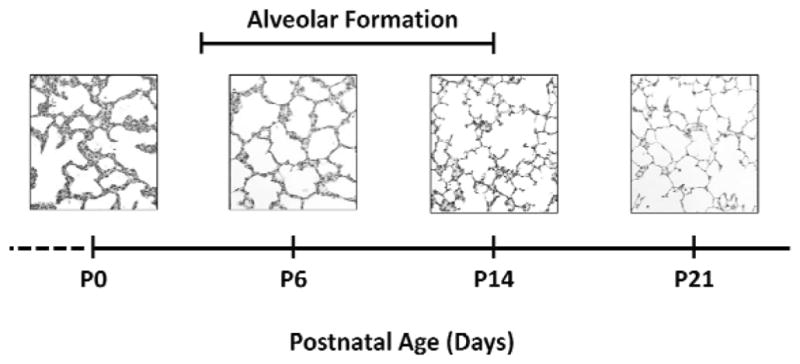
Representative images of female rat lungs at postnatal day 0 (P0), P6, P14 and P21. At P0, the rat lung is at the saccular stage of lung development. Alveolar formation takes place from approximately P4 to P14. At P21 alveoli have characteristic histological features of long, straight alveolar walls and numerous, long secondary septa. All panels original magnification 80 × (scale bar is 50 μm).
The lung is developmentally dynamic during times of common perinatal insults. Perinatal insults that produce long-term changes in the human lung tend to occur during the second half of gestation and the early postnatal period. Because of this timing, the saccular and alveolar stages of lung development are most relevant to our discussion. Large animal models, such as sheep, provide opportunities for physiologic and molecular studies in a similar developmental context to humans. Sheep models of preterm birth and respiratory support, as well as models of intrauterine growth restriction (IUGR), are used to study the developmental origins of adult lung disease (Albertine et al., 1999, Rozance et al., 2011, Null et al., 2014). Uniquely, the preterm lamb model recapitulates the human clinical occurrence of preterm birth that is frequently associated with respiratory distress and failure. Respiratory failure necessitates ventilator support with oxygen-rich gas to keep the preterm neonates alive.
Rat and mouse models are also valuable tools for understanding the developmental origins of adult lung disease, particularly for molecular manipulation. Immaturity of the lungs at birth in rat and mouse allows postnatal manipulation during the saccular and alveolar stages of lung development. Rat and mouse models used to study the effects of perinatal events on lung outcomes include models of IUGR induced by uteroplacental insufficiency, malnutrition, maternal toxins and tobacco smoke, as well as models of postnatal hypoxia and hyperoxia (Bassi et al., 1984, Bhaskaran et al., 2012, Hilgendorff et al., 2012, Olave et al., 2012, Rehan et al., 2012, Joss-Moore et al., 2013).
Epigenetic Effects in the Lung
Environmentally-induced disruptions to the normal epigenetic characteristics of DNA within distinct cells during development leads to alterations in gene expression and thus, changes in development. Three major consequences can arise from changes in lung cell epigenetics and transcriptomes that occur at developmentally sensitive time points. First, the lung can assume an alteration in structure and/or function secondary to changes in cell differentiation and cell-to-cell communications. Secondly, when the epigenome of a cell is altered at a developmentally sensitive time point, this “new” epigenetic platform becomes the basis upon which subsequent epigenetic changes are built. The potential result is an increasing deviation from “normal” during and after development. The final consequence of changing lung cell epigenetics during development is that the new epigenome is also primed to direct gene expression differently in response to future lung injury and repair.
Epigenetic mechanisms include DNA methylation, histone protein modifications, and non-coding RNA’s, including microRNA. Involvement of each of these epigenetic mechanisms in the developmental origins of adult lung disease will be considered below.
DNA methylation
DNA methylation occurs primarily as the addition of a methyl group to the 5′ position of a cytosine followed by a guanine (CpG). CpG density in the mammalian genome is relatively low, with CpGs clustered in dense regions known as CpG “islands” (Gardiner-Garden and Frommer, 1987). CpG islands are commonly found in promoter regions of mammalian genes, and are often unmethylated (Klose and Bird, 2006, Huh et al., 2013). Unlike promoter regions, coding regions and regions between genes contain low density CpGs, and are more frequently methylated (Illingworth et al., 2008, Liang et al., 2011, Maunakea et al., 2013). Low density CpGs located in the coding region tend to be associated with exons more often than with introns, and may have a role in alternative exon usage (Choi, 2010).
Changes in DNA methylation following preterm-birth, intrauterine growth restriction (IUGR), or maternal toxin exposure are identifiable in placenta and peripheral tissues such as blood leucocytes (Ferreira et al., 2011, Suter et al., 2011, Hogg et al., 2012, Jiang et al., 2012). However, to our knowledge, no studies document DNA methylation changes in human lung tissue associated with perinatal events.
DNA methylation changes are linked to immune responses associated with the development of adult lung disease. Asthma development is linked to maternal folate intake during pregnancy (Haberg et al., 2009, Whitrow et al., 2009). Dietary intake of folate leads to the production of S-adenosyl-L-methionine (SAM), a universal methyl donor and precursor for DNA methylation. In mice, a maternal diet high in methyl donors increases offspring airway inflammation, serum IgE levels, and airway hyperesponsiveness. Additionally, the phenotypic changes in mice offspring occur in conjunction with hypermethylation of the Run3x gene, a mediator of inflammation (Hollingsworth et al., 2008). Control of activated T-helper cells is also regulated by DNA methylation, again linking epigenetic mechanisms to the development of allergic airway disease (Kabesch et al., 2010, Yang and Schwartz, 2011).
Despite no human evidence of lung DNA methylation changes following perinatal insults, abundant evidence shows lung DNA methylation changes in the presence of adult lung disease. One large study of DNA methylation in peripheral blood of adults (smokers and non-smokers) with chronic obstructive pulmonary disease (COPD) identified numerous differentially methylated genes, compared to matched subjects without COPD (Qiu et al., 2012). Differentially methylated regions identified are generally hypomethylated and 70% of the sites are outside of CpG islands. Gene ontology (GO) analysis identified expected GO pathways associated with the differentially methylated genes, including, response to stress and external stimuli as well as wound healing cascades. Because the study identified differential methylation in non-lung tissue, difficulties arise in assessing whether the changes are causative, a marker of lung injury, or a marker of the initial COPD stimulus.
While not distinguishing between causative and marker status, one finding of the study does suggest specificity of peripheral blood differential methylation in COPD patients. Mutations of the SERPINA1 gene, which codes for α1- antitrypsinogen, are causally associated with COPD. Two differentially methylated CpG sites within the SERPINA1 gene were identified by the study. Methylation status of one of the SERPINA1 sites ranked highest in COPD cases, as well as highest for association with lung function (Qiu et al., 2012). These data raise interesting questions. Given that few of the genes identified as being differentially methylated could be predicted based on smoking intensity, what is the trigger for differential methylation and what is the timing of occurrence? A complete understanding of potential perinatal contributions to DNA methylation changes observed in adult lung disease is an important research directive.
Mechanistic insight into myofibroblast differentiation has provided insight into the role of DNA methylation in lung cellular reprogramming and remodeling. The myofibroblast has a pro-fibrotic phenotype characterized by expression of extracellular matrix (ECM) genes such as elastin, collagen, and α-smooth muscle actin (α-SMA). Myofibroblasts have features of both fibroblasts and smooth muscle cells, and appear to be the main source of excessive elastin production in lung diseases, such as bronchopulmonary dysplasia (BPD) and idiopathic pulmonary fibrosis (IPF). Activation of injury cascades result in proliferation of resident fibroblasts, as well as epithelial mesenchymal transitions. The resulting myofibroblast foci exhibit accumulation of ECM, dysregulated wound repair and lung remodeling (Bland et al., 2007, Yang and Schwartz, 2014).
The involvement of DNA methylation in myofibroblast differentiation is exemplified by Thy-1 promoter methylation (Hagood, 2014). Thy-1, also known as CD90, is a cell-surface antigen expressed on fibroblasts. Thy-1 positive fibroblasts have lipid inclusions and an anti-fibrotic phenotype, while Thy-1 negative fibroblasts differentiate into myofibroblasts (Zhou et al., 2004). Thy-1 also independently plays a role in alveolar formation (Nicola et al., 2009). One means by which Thy-1 maintains a lipogenic fibroblast phenotype is by stimulating peroxisome proliferator activated receptor gamma (PPARγ) signaling and PPARγ directed transcription of lipid enhancing genes (Varisco et al., 2012). Hypermethylation of the Thy-1 promoter silences Thy 1, producing myofibroblast differentiation. DNA hypermethylation of Thy 1 is observed in myofibroblasts located in fibroblastic foci in IPF, and in mice exposed to the fibrotic agent bleomycin. Hypermethylation of lung Thy-1 can also be induced by hypoxia (Sanders et al., 2008, Robinson et al., 2012). The perturbation of Thy-1 methylation, PPARγ activity, and subsequent myofibroblast differentiation is likely important in the lung response to perinatal insults.
Histone Modifications
While DNA methylation is one of the better studied and more easily assessed epigenetic modification, histone modifications provide greater variability in effects and often interact with DNA methylation. A core of 8 histone proteins, forming a unit called a nucleosome, is surrounded by two wraps of 147 base pairs of DNA (Luger et al., 1997). Adjacent nucleosomes are connected by short pieces of linker DNA. The nucleosome core is occupied by the globular portion of the histone proteins, while the unstructured, N-terminal “tails” of the histone proteins extend freely from the nucleosome (Luger et al., 1997). Histone tails provide the location for many (but not all) post-translational, covalent modifications (reviewed in (Zentner and Henikoff, 2013)). The modifications made to histone proteins include acetylation, mono, di and tri methylation, phosphorylation, and ubiquitylation.
The combinatorial complexity of histone modifications along a gene is high because of the potential number of modifications, modifiable amino acids, and the number of nucleosomes along the length of a gene. However, genome-wide mapping of global patterns of histone modifications using chromatin immunoprecipitation with parallel DNA sequencing (ChIP-seq), have revealed that patterns of histone modifications are associated with distinct elements within the DNA of a genome (Huff et al., 2010, Zentner and Henikoff, 2013). For example, promoters tend to have high levels of histone 3 (H3), lysine 4 (K4) trimethylation (me3), while putative enhancers are characterized by enriched H3K4me1 alone or with H3K27acetylation (ac) or H3K27me3 (Rada-Iglesias et al., 2011, Zentner et al., 2011). Gene bodies tend to be enriched with H3K36me2 (or me3) in association with transcriptional activation. H3K4me2 (or me3) and K36me2 (or me3) may contribute to regulating stability of the nucleosome during RNA polymerase II transit (Wagner and Carpenter, 2012, Zentner and Henikoff, 2013). Also enriched in the promoter and body of the gene is H4K20me1 (Smolle and Workman, 2013). H4K20me1 may function in transcriptional initiation and promoter clearance as well as nucleosome stability in the body of the gene (Karlic et al., 2010). H4K20me1 plays a critical role in target gene activation in wingless (Wnt) signaling (Li et al., 2011). Successful Wnt signaling requires H4K20me1 in enhancer regions of target genes. Because Wnt signaling is essential for both lung development and repair after lung injury, H4K20me1 and Wnt signaling is particularly interesting in the context of the developmental origins of adult lung disease (Dasgupta et al., 2009, Crosby and Waters, 2010, Villar et al., 2011).
Animal models demonstrate that perinatal events are associated with histone changes along genes within multiple tissues (Fu et al., 2004, Park et al., 2008, Zinkhan et al., 2012). For several reasons, our group has focused on histone changes to the lung PPARγ gene. Firstly, the nuclear receptor family transcription factor PPARγ is a key player in lung development and lung repair, and is involved in epithelial-mesenchymal transitions, lipid homeostasis, and inflammatory control (Lian et al., 2005, Simon et al., 2006, Cerny et al., 2008, Wang et al., 2009, Joss-Moore et al., 2010). Secondly, PPARγ activity is responsive to long chain fatty acids. As such, nutritional status may influence functional output of PPARγ and nutrition may provide a means for manipulating PPARγ activity. Lastly, PPARγ directly regulates the transcription of several chromatin modifying enzymes via PPAR response elements (PPRE) (Wakabayashi et al., 2009). One of these PPARγ responsive genes is the set domain containing histone methyltransferase, Setd8, which places the H4K20me1.
The expression and epigenetics of lung PPARγ and Setd8 are susceptible to perinatal insults, including IUGR (Joss-Moore et al., 2010). In the rat lung, IUGR decreases PPARγ and Setd8 expression, as well as genome-wide and gene-specific levels of H4K20me1 in both male and female rat lung (Joss-Moore et al., 2010). The PPARγ-Setd8-H4K20me1 axis can be enhanced with dietary long chain fatty acid activation of PPARγ. Supplementation of IUGR rats with the PPARγ agonist, docosahexanoic acid (DHA), restores PPARγ and Setd8 levels, as well as global and gene-specific H4K20me1 (Joss-Moore et al., 2010). Findings such as these suggest a role for nutritional approaches to treat perturbations in epigenetic regulation of gene expression following a perinatal insult.
IUGR in the rat also induces other histone modification changes along the PPARγ gene in the lung. Sex-specific alterations in H3 methylation along the PPARγ gene in IUGR include decreased H3K9me3 in male neonatal rats and increased H3K9me3 in female neonatal rats (Joss-Moore et al., 2011). Sex-divergent H3K9me3 changes occurring in response to IUGR are interesting because basal H3K9me3 is not different between control male and female rats. Sex-divergent changes in lung PPARγ epigenetics following IUGR demonstrates that males respond differently than females to the insult of IUGR.
IUGR also has sex-divergent effects on the methyl binding protein MeCp2, which bridges DNA methylation and histone modifications. MeCp2 is essential for myofibroblast differentiation and pulmonary fibrosis (Hu et al., 2011). One means by which MeCp2 affects myofibroblast differentiation is by binding to methylated DNA, influencing H3K9me3 placement, and promoting transcriptional repression of targets such as PPARγ (Fuks et al., 2003). In the developing rat lung, IUGR affects both MeCP2 expression and MeCP2 occupancy of the PPARγ promoters in a sex-divergent manner (Joss-Moore et al., 2011). In female rat lung, IUGR increases MeCP2 expression and MeCP2 occupancy of the PPARγ promoters in conjunction with decreased PPARγ expression. In contrast, in developing male rat lung, IUGR does not affect MeCP2 expression or MeCP2 occupancy of the PPARγ promoters, however PPAR expression is still decreased (Joss-Moore et al., 2011). These results suggest that the sex-divergent responses to IUGR in the lung originate with the regulation of gene expression.
Explanations for sex-divergent PPARγ regulation effects in response to perinatal insults are lacking. In disorders of placental insufficiency and preterm birth, potential explanations include sex-specific abnormalities in placental lipid transfer to the fetus, sex-specific inability to maintain lipid homeostasis ex-utero, and increased estrogen receptor inhibition of PPARγ signaling (Chu et al., 2014, Martin, 2014).
Multiple cues direct placement of histone modifications, including availability and activity of enzymes that place and remove marks. Fine regulation of gene expression relies on the dynamic nature of histone modifications, thus exemplifying the importance of chromatin modifying enzymes. Genes actively transcribed, or genes that are poised for transcription pending an activating signal (such as transcription factor binding to an enhancer), are characterized by rapid acetylation and deacetylation (Zhang and Nelson, 1988, Spencer and Davie, 2001, Barth and Imhof, 2010). Rapid local changes in histone acetylation occur on nucleosomes at or near promoters at the time of activation of transcription from poised genes (Waterborg, 2002). Acetyl groups are placed on histones by histone acetyltransferase (HAT) enzymes and removed by histone deacetylatase (HDAC) enzymes. Under basal conditions, chromatin that is being rapidly acetylated and deacetylated is enriched in HAT and HDAC activity. While mechanisms are still being elucidated, evidence suggests that rapid acetylation and deacetylation may facilitate nucleosome mobilization during polymerase transit, thus physically facilitating transcriptional activation and elongation (Walia et al., 1998, Reinke et al., 2001, Waterborg, 2002).
The HDAC family of enzymes has received attention in the context of the developmental origins of lung disease. Preterm lambs managed by invasive mechanical ventilation (MV) develop a BPD phenotype, with arrested alveolar development and excessive and disordered elasin deposition (Albertine et al., 1999, Bland et al., 2007). Lungs of preterm lambs managed by MV also have increased HDAC1 and genome-wide histone hypoacetylation. In contrast, lungs of preterm lambs managed by non-invasive high-frequency nasal ventilation do not develop BPD, have normal alveolar development and normal EMC deposition. Lungs of preterm lambs managed by high-frequency nasal ventilation have genome-wide histone hyperacetylation. Histone modifications H3K14ac, H3K18ac, and H3K27ac, are lower in lungs of preterm lambs managed by MV than high frequency nasal ventilation (Hamvas et al., 2013). Collectively, these data suggest that MV upsets the acetylation-deacetylation equilibrium in the lung by increasing deacetylation.
When preterm lambs are treated with the HDAC inhibitors, valproic acid or trichostatin A, all histone modifications increase in the MV group. Measures of alveolar formation are improved with increased histone acetylation that is triggered experimentally by inhibiting HDACs in preterm lambs managed by MV (Hamvas et al., 2013).
While descriptive studies have initiated the field of epigenetics in developmental origins of adult lung disease, studies assessing phenotypic changes that result from manipulation of epigenetic responses, such as those described above, are critical to moving the field forward.
Non-coding RNA
Non-coding RNA is functional RNA that is not translated into a protein product. Long non-coding RNA and short microRNAs (miRNA) effect the translation of target mRNA’s into protein. The involvement of long non-coding RNA in lung disease and development is limited, and what is known is largely in the context of cancerous lung cells (Booton and Lindsay, 2014). miRNA, on the other hand, is better understood in the context of lung disease and development.
miRNA are small non-coding RNAs that bind sequence-specifically to the 3′UTR of target mRNAs and prevent translation by accelerating mRNA degradation, or by blocking the passage of the ribosome (reviewed in (Dogini et al., 2014)). Pre-miRNAs may be transcribed from independent genes, or from within introns or exons of coding genes, either as a single miRNA or in clusters. Pre-miRNAs are processed by Drosha and Dicer to produce miRNAs of approximately 21 base-pairs. The transcription of miRNAs is controlled by the same epigenetic mechanisms as coding genes, including DNA methylation and histone modifications.
Given the number of known miRNA genes, and the ability of a single miRNA to have multiple targets, the involvement of miRNA’s in developmental and disease processes is not surprising. One particular aspect relevant to the developmental origins of disease is the role of miRNAs in the response to cellular stress. In response to cellular stressors, miRNAs interact with other cell-stress response pathways and direct translation to stress responses (Leung and Sharp, 2010, Emde and Hornstein, 2014). Interestingly, a subset of miRNAs also controls the expression of chromatin modifying enzymes, including DNA methyltransferases and histone deacetylases (Sato et al., 2011, Dakhlallah et al., 2013). Taken together, these data suggest an epigenetic-miRNA regulatory circuit that may be important in the transcriptional response to cellular stressors such as those induced by perinatal insults (Leung and Sharp, 2010, Sato et al., 2011, Emde and Hornstein, 2014).
Little is known about the effects of perinatal events on long-term expression and regulation of lung miRNAs. One study reported expression profiles of miRNA in peripheral blood of late-preterm infants with and without BPD (Wu et al., 2013). A 4-miRNA signature was identified as characterizing preterm infants with BPD. The miRNAs that differed in magnitude between preterm infants with BPD and those without were miR-152, miR-30a-3p, miR-133b, and miR-7 (Wu et al., 2013). The presence of a miRNA signature in peripheral blood that might serve as a biomarker is an exciting prospect. However, much more investigation is required to understand the local miRNA status in the human lung under developmental and disease conditions.
Steps toward understanding the effects of perinatal events on lung miRNA expression and regulation have been made in animal models. Differential miRNA expression has been reported in a number of rat and mouse models designed to mimic the histopathology of BPD. Neonatal rat and mouse lungs examined during normal development and in conjunction with injurious stimuli, such as hyperoxia, demonstrate that lung miRNA profiles are dynamic and vary with development and injury (Bhaskaran et al., 2012, Dong et al., 2012, Yang et al., 2012). Neonatal rat lungs exposed to hyperoxia are characterized by down-regulation of miR-342, miR-335, miR-150, miR-126, and miR-151, and up-regulation of miR-21 and miR-34a (Bhaskaran et al., 2012). Similarly, hyperoxia altered miRNA profiles, including increased miR-29. Gene ontology enrichment and pathway analysis demonstrated that genes involved in a variety of lung developmental processes are targets of altered miRNA (Dong et al., 2012).
One miRNA cluster, the miR17~92 cluster, is particularly interesting from a lung disease perspective. While not studied in the context of perinatal insults, the miR17~92 cluster has been studied in the context of lung repair and remodeling (Mendell, 2008, Jevnaker et al., 2011, Dakhlallah et al., 2013). The miR17~92 cluster codes for 6 miRNAs in a single open reading frame, miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92–1. The miR17~92 cluster is altered in human IPF and in mouse models of bleomycin-induced fibrosis, and is important in fibrotic gene expression. In an elegant study, lung tissue from IPF patients was compared to controls and showed significant down regulation of the miR17~92 cluster in IPF relative to control lung (Dakhlallah et al., 2013). Furthermore, the miRNA changes were inversely proportional to DNA methylation levels within the CpG-dense promoter of the cluster. Interestingly, the DNA methyltransferase DNMT1, which is a predicted target of the cluster, inversely correlated with expression of the cluster, suggesting that miR17~92 cluster may play a role in regulation of DMNT1 expression in the lung (Dakhlallah et al., 2013). Finally, DNA methylation was manipulated with the DNA demethylating compound 5′-aza-2′deoxyctidine and effects were reversed (Dakhlallah et al., 2013). Studies, such as this one, highlight the need for research that examine the presence and persistence of lung epigenetic-miRNA regulatory pathways in animal models of perinatal insults.
Conclusions and Research Priorities
The developmental origins of adult lung disease field is at a critical point in understanding the epigenetic mechanisms leading to adult lung disease after perinatal insult. More importantly, mechanistic understanding will lead to the development of targeted, specific, interventions that may alleviate, or even prevent, the development of adult lung disease in susceptible individuals.
Despite this positive outlook, the developmental origins of adult lung disease field faces many challenges. Human clinical research into the developmental origins of adult lung disease is needed. However, to design informative human studies, several concepts must be addressed. First, accessible tissue sources for study need to be validated. Secondly, potential epigenetic processes need to be identified as either biomarkers or causative agents.
Much of the mechanistic work needed to facilitate informative human studies in the developmental origins of adult lung disease field will fall to research using animal models. Animal models successful for understanding the developmental origins of adult lung disease require several characteristics. Perinatal insults should be administered to mimic the human scenario as closely as possible. In response to the perinatal insult, animal models should display a clinically relevant lung disease phenotype, with objective measures of disease severity, including structural, functional, and molecular characteristics. Objective measures of disease severity within animal models, will facilitate short and long-term studies examining the effects of perinatal insults, and potential interventions, on disease of the lung. Manipulation of epigenetic modifications performed in the perinatal period, may have positive effects on the lung in the short-term. However, manipulation of epigenetic modifications in the perinatal period may also have detrimental effects in the long-term. Long-term studies will clarify whether a particular intervention is effective in alleviating adult disease in the developmental origins of lung disease. An extension of these long-term studies involves addition of an adult lung injury or a “second hit” on the lung, after the perinatal insult.
Techniques utilized to understand the involvement of epigenetic modifications in the developmental origins of lung disease are varied and include genome wide and site-specific approaches. Choice of approach will be influenced by cost, resolution of data, and contribution to understanding the biological phenomena. Combined understanding of DNA methylation in the context of histone modifications will be important, as will the role of non-coding RNA in the context of specific targets. High resolution sequencing based approaches to understanding DNA methylation and histone modifications are promising and yield high quality information. However, sequencing based approaches to genome wide methylation and histone modifications are expensive and highly dependent upon sophisticated bioinformatics to ensure that accurate conclusions are derived from the data. Single molecule approaches, such as targeted bisulfite sequencing and traditional chromatin immunoprecipitation are useful in the context of understanding disruptions to select genes. An important caveat for single gene epigenetic analysis, however, is the need to consider the entire gene of interest and not just 5′ regulatory regions.
A significant challenge to the field lies in distinguishing epigenetic phenomena that are event biomarkers, from epigenetic phenomena with a causative role. Once causative epigenetic mechanisms are identified, selective focus on mechanisms that are amenable to manipulation will be important. Furthermore, potential interventions need to be considered in the context of non-lung organs. This concept is particularly important in the developmental origins of adult lung disease because the timing of perinatal insults coincides with not only lung development, but also development of brain, kidney, and other susceptible organs. One way to minimize “collateral damage” in treating the lung in the context of developmental origins of adult lung disease is to aim at normalizing conditions that are perturbed in sequence or in parallel by the perinatal event. For this reason, approaches involving nutritional manipulation may prove promising.
In summary, perinatal insults such as IUGR, preterm birth, maternal toxin exposure and dietary deficiency that occur during critical periods of lung development predispose to impaired lung function, structure, and molecular characteristics in later life. The development of adult lung disease following perinatal insults involves plasticity of the epigenome and subsequent alterations in lung cell transcriptomes. A comprehensive understanding of the epigenetic mechanisms driving the developmental origins of adult lung disease has the exciting potential to lead to treatments that reduce or eliminate lung disease in susceptible individuals.
Acknowledgments
This work was supported by National Institute of Heath grants HL062875 (KHA), HL110002 (KHA), HL07744 (KHA) and DK084036 (LJM) and the Department of Pediatrics at the University of Utah.
References
- Alati R, Al Mamun A, O’Callaghan M, Najman JM, Williams GM. In utero and postnatal maternal smoking and asthma in adolescence. Epidemiology. 2006;17(2):138–144. doi: 10.1097/01.ede.0000198148.02347.33. [DOI] [PubMed] [Google Scholar]
- Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, Carlton DP, Bland RD. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med. 1999;159(3):945–958. doi: 10.1164/ajrccm.159.3.9804027. [DOI] [PubMed] [Google Scholar]
- Albertine KH, Pysher TJ. Impaired lung growth after injury in premature lung. In: Polin FWRA, Abman SH, editors. Fetal and Neonatal Physiology. New York: Saunders; 2004. pp. 942–949. [Google Scholar]
- Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO. Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ. 1991;303(6804):671–675. doi: 10.1136/bmj.303.6804.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1(8489):1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci. 2010;35(11):618–626. doi: 10.1016/j.tibs.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Bassi JA, Rosso P, Moessinger AC, Blanc WA, James LS. Fetal growth retardation due to maternal tobacco smoke exposure in the rat. Pediatr Res. 1984;18(2):127–130. doi: 10.1203/00006450-198402000-00002. [DOI] [PubMed] [Google Scholar]
- Bhaskaran M, Xi D, Wang Y, Huang C, Narasaraju T, Shu W, Zhao C, Xiao X, More S, Breshears M, Liu L. Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiol Genomics. 2012;44(20):970–980. doi: 10.1152/physiolgenomics.00145.2011. doi:910.1152/physiolgenomics.00145.02011. Epub 02012 Aug 00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland RD, Xu L, Ertsey R, Rabinovitch M, Albertine KH, Wynn KA, Kumar VH, Ryan RM, Swartz DD, Csiszar K, Fong KS. Dysregulation of pulmonary elastin synthesis and assembly in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1370–1384. doi: 10.1152/ajplung.00367.2006. [DOI] [PubMed] [Google Scholar]
- Booton R, Lindsay MA. Emerging role of MicroRNAs and long noncoding RNAs in respiratory disease. Chest. 2014;146(1):193–204. doi: 10.1378/chest.13-2736. doi:110.1378/chest.1313-2736. [DOI] [PubMed] [Google Scholar]
- Burri PH. Structural aspects of postnatal lung development - alveolar formation and growth. Biol Neonate. 2006;89(4):313–322. doi: 10.1159/000092868. [DOI] [PubMed] [Google Scholar]
- Caudri D, Wijga A, Gehring U, Smit HA, Brunekreef B, Kerkhof M, Hoekstra M, Gerritsen J, de Jongste JC. Respiratory symptoms in the first 7 years of life and birth weight at term: the PIAMA Birth Cohort. Am J Respir Crit Care Med. 2007;175(10):1078–1085. doi: 10.1164/rccm.200610-1441OC. Epub 2007 Feb 1078. [DOI] [PubMed] [Google Scholar]
- Cerny L, Torday JS, Rehan VK. Prevention and treatment of bronchopulmonary dysplasia: contemporary status and future outlook. Lung. 2008;186(2):75–89. doi: 10.1007/s00408-007-9069-z. [DOI] [PubMed] [Google Scholar]
- Checkley W, West KP, Jr, Wise RA, Baldwin MR, Wu L, LeClerq SC, Christian P, Katz J, Tielsch JM, Khatry S, Sommer A. Maternal vitamin A supplementation and lung function in offspring. N Engl J Med. 2010;362(19):1784–1794. doi: 10.1056/NEJMoa0907441. doi:1710.1056/NEJMoa0907441. [DOI] [PubMed] [Google Scholar]
- Choi JK. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 2010;11(7):R70. doi: 10.1186/gb-2010-11-7-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu R, van Hasselt A, Vlantis AC, Ng EK, Liu SY, Fan MD, Ng SK, Chan AB, Liu Z, Li XY, Chen GG. The cross-talk between estrogen receptor and peroxisome proliferator-activated receptor gamma in thyroid cancer. Cancer. 2014;120(1):142–153. doi: 10.1002/cncr.28383. doi:110.1002/cncr.28383. Epub 22013 Oct 28382. [DOI] [PubMed] [Google Scholar]
- Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L715–731. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, Mikhail A, Hitchcock CL, Wright VP, Nana-Sinkam SP, Piper MG, Marsh CB. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187(4):397–405. doi: 10.1164/rccm.201205-0888OC. doi:310.1164/rccm.201205-200888OC. Epub 202013 Jan 201210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta C, Sakurai R, Wang Y, Guo P, Ambalavanan N, Torday JS, Rehan VK. Hyperoxia-induced neonatal rat lung injury involves activation of TGF-{beta} and Wnt signaling and is protected by rosiglitazone. Am J Physiol Lung Cell Mol Physiol. 2009;296(6):L1031–1041. doi: 10.1152/ajplung.90392.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogini DB, Pascoal VD, Avansini SH, Vieira AS, Pereira TC, Lopes-Cendes I. The new world of RNAs. Genet Mol Biol. 2014;37(1 Suppl):285–293. doi: 10.1590/s1415-47572014000200014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Carey WA, Abel S, Collura C, Jiang G, Tomaszek S, Sutor S, Roden AC, Asmann YW, Prakash YS, Wigle DA. MicroRNA-mRNA interactions in a murine model of hyperoxia-induced bronchopulmonary dysplasia. BMC Genomics. 2012;13:204. doi: 10.1186/1471-2164-1113-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emde A, Hornstein E. miRNAs at the interface of cellular stress and disease. Embo J. 2014:27. doi: 10.15252/embj.201488142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkkola M, Kaila M, Nwaru BI, Kronberg-Kippila C, Ahonen S, Nevalainen J, Veijola R, Pekkanen J, Ilonen J, Simell O, Knip M, Virtanen SM. Maternal vitamin D intake during pregnancy is inversely associated with asthma and allergic rhinitis in 5-year-old children. Clin Exp Allergy. 2009;39(6):875–882. doi: 10.1111/j.1365-2222.2009.03234.x. doi:810.1111/j.1365-2222.2009.03234.x. [DOI] [PubMed] [Google Scholar]
- Ferreira JC, Choufani S, Grafodatskaya D, Butcher DT, Zhao C, Chitayat D, Shuman C, Kingdom J, Keating S, Weksberg R. WNT2 promoter methylation in human placenta is associated with low birthweight percentile in the neonate. Epigenetics. 2011;6(4):440–449. doi: 10.4161/epi.6.4.14554. Epub 2011 Apr 2011. [DOI] [PubMed] [Google Scholar]
- Fu Q, McKnight RA, Yu X, Wang L, Callaway CW, Lane RH. Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiol Genomics. 2004;20(1):108–116. doi: 10.1152/physiolgenomics.00175.2004. [DOI] [PubMed] [Google Scholar]
- Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278(6):4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- Gaffin JM, Kanchongkittiphon W, Phipatanakul W. Perinatal and early childhood environmental factors influencing allergic asthma immunopathogenesis. Int Immunopharmacol. 2014;18(14):00219–00217. doi: 10.1016/j.intimp.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196(2):261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gilliland FD, Berhane K, Li YF, Rappaport EB, Peters JM. Effects of early onset asthma and in utero exposure to maternal smoking on childhood lung function. Am J Respir Crit Care Med. 2003;167(6):917–924. doi: 10.1164/rccm.200206-616OC. [DOI] [PubMed] [Google Scholar]
- Haberg SE, London SJ, Stigum H, Nafstad P, Nystad W. Folic acid supplements in pregnancy and early childhood respiratory health. Arch Dis Child. 2009;94(3):180–184. doi: 10.1136/adc.2008.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagood JS. Beyond the genome: epigenetic mechanisms in lung remodeling. Physiology (Bethesda) 2014;29(3):177–185. doi: 10.1152/physiol.00048.2013. doi:110.1152/physiol.00048.02013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamvas A, Deterding R, Balch WE, Schwartz DA, Albertine KH, Whitsett JA, Cardoso WV, Kotton DN, Kourembanas S, Hagood JS. Diffuse lung disease in children: Summary of a scientific conference. Pediatr Pulmonol. 2013 doi: 10.1002/ppul.22805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox RJ, Poulton R, Greene JM, McLachlan CR, Pearce MS, Sears MR. Associations between birth weight, early childhood weight gain and adult lung function. Thorax. 2009;64(3):228–232. doi: 10.1136/thx.2008.103978. doi:210.1136/thx.2008.103978. Epub 102008 Dec 103973. [DOI] [PubMed] [Google Scholar]
- Harding R, Maritz G. Maternal and fetal origins of lung disease in adulthood. Semin Fetal Neonatal Med. 2012;17(2):67–72. doi: 10.1016/j.siny.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Hilgendorff A, Parai K, Ertsey R, Juliana Rey-Parra G, Thebaud B, Tamosiuniene R, Jain N, Navarro EF, Starcher BC, Nicolls MR, Rabinovitch M, Bland RD. Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am J Physiol Lung Cell Mol Physiol. 2012;303(3):L215–227. doi: 10.1152/ajplung.00405.2011. doi:210.1152/ajplung.00405.02011. Epub 02012 Jun 00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg K, Price EM, Hanna CW, Robinson WP. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther. 2012;92(6):716–726. doi: 10.1038/clpt.2012.141. doi:710.1038/clpt.2012.1141. Epub 2012 Oct 1010. [DOI] [PubMed] [Google Scholar]
- Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, Bailey N, Potts EN, Whitehead G, Brass DM, Schwartz DA. In utero supplementation with methyl donors enhances allergic airway disease in mice. J Clin Invest. 2008;118(10):3462–3469. doi: 10.1172/JCI34378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Essential role of MeCP2 in the regulation of myofibroblast differentiation during pulmonary fibrosis. Am J Pathol. 2011;178(4):1500–1508. doi: 10.1016/j.ajpath.2011.01.002. doi:1510.1016/j.ajpath.2011.1501.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff JT, Plocik AM, Guthrie C, Yamamoto KR. Reciprocal intronic and exonic histone modification regions in humans. Nat Struct Mol Biol. 2010;17(12):1495–1499. doi: 10.1038/nsmb.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh I, Zeng J, Park T, Yi SV. DNA methylation and transcriptional noise. Epigenetics Chromatin. 2013;6(1):9. doi: 10.1186/1756-8935-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylkema MN, Blacquiere MJ. Intrauterine effects of maternal smoking on sensitization, asthma, and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6(8):660–662. doi: 10.1513/pats.200907-065DP. [DOI] [PubMed] [Google Scholar]
- Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, Jackson D, Clee C, Plumb R, Rogers J, Humphray S, Cox T, Langford C, Bird A. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6(1):e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola JJ, Gissler M. Maternal smoking in pregnancy, fetal development, and childhood asthma. Am J Public Health. 2004;94(1):136–140. doi: 10.2105/ajph.94.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrychowski WA, Perera FP, Spengler JD, Mroz E, Stigter L, Flak E, Majewska R, Klimaszewska-Rembiasz M, Jacek R. Intrauterine exposure to fine particulate matter as a risk factor for increased susceptibility to acute broncho-pulmonary infections in early childhood. Int J Hyg Environ Health. 2013;216(4):395–401. doi: 10.1016/j.ijheh.2012.12.014. doi:310.1016/j.ijheh.2012.1012.1014. Epub 2013 Jan 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevnaker AM, Khuu C, Kjole E, Bryne M, Osmundsen H. Expression of members of the miRNA17-92 cluster during development and in carcinogenesis. J Cell Physiol. 2011;226(9):2257–2266. doi: 10.1002/jcp.22562. doi:2210.1002/jcp.22562. [DOI] [PubMed] [Google Scholar]
- Jiang X, Yan J, West AA, Perry CA, Malysheva OV, Devapatla S, Pressman E, Vermeylen F, Caudill MA. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012;26(8):3563–3574. doi: 10.1096/fj.12-207894. doi:3510.1096/fj.3512-207894. Epub 202012 May 207891. [DOI] [PubMed] [Google Scholar]
- Jobe AH. What is BPD in 2012 and what will BPD become? Early Hum Dev. 2012;88(Suppl 2):S27–28. doi: 10.1016/S0378-3782(12)70009-9. [DOI] [PubMed] [Google Scholar]
- Joss-Moore L, Carroll T, Yang Y, Fitzhugh M, Metcalfe D, Oman J, Hale M, Dong L, Wang ZM, Yu X, Callaway CW, O’Brien E, McKnight RA, Lane RH, Albertine KH. Intrauterine growth restriction transiently delays alveolar formation and disrupts retinoic acid receptor expression in the lung of female rat pups. Pediatr Res. 2013;73(5):612–620. doi: 10.1038/pr.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joss-Moore LA, Albertine KH, Lane RH. Epigenetics and the developmental origins of lung disease. Mol Genet Metab. 2011;104(1–2):61–66. doi: 10.1016/j.ymgme.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joss-Moore LA, Wang Y, Baack ML, Yao J, Norris AW, Yu X, Callaway CW, McKnight RA, Albertine KH, Lane RH. IUGR decreases PPARgamma and SETD8 Expression in neonatal rat lung and these effects are ameliorated by maternal DHA supplementation. Early Hum Dev. 2010;86(12):785–791. doi: 10.1016/j.earlhumdev.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joss-Moore LA, Wang Y, Ogata EM, Sainz AJ, Yu X, Callaway CW, McKnight RA, Albertine KH, Lane RH. IUGR differentially alters MeCP2 expression and H3K9Me3 of the PPARgamma gene in male and female rat lungs during alveolarization. Birth Defects Res A Clin Mol Teratol. 2011;91(8):672–681. doi: 10.1002/bdra.20783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabesch M, Michel S, Tost J. Epigenetic mechanisms and the relationship to childhood asthma. Eur Respir J. 2010;36(4):950–961. doi: 10.1183/09031936.00019310. [DOI] [PubMed] [Google Scholar]
- Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107(7):2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindlund K, Thomsen SF, Stensballe LG, Skytthe A, Kyvik KO, Backer V, Bisgaard H. Birth weight and risk of asthma in 3–9-year-old twins: exploring the fetal origins hypothesis. Thorax. 2010;65(2):146–149. doi: 10.1136/thx.2009.117101. doi:110.1136/thx.2009.117101. Epub 112009 Dec 117108. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40(2):205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Nie F, Wang S, Li L. Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc Natl Acad Sci U S A. 2011;108(8):3116–3123. doi: 10.1073/pnas.1009353108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X, Yan C, Qin Y, Knox L, Li T, Du H. Neutral lipids and peroxisome proliferator-activated receptor-{gamma} control pulmonary gene expression and inflammation-triggered pathogenesis in lysosomal acid lipase knockout mice. Am J Pathol. 2005;167(3):813–821. doi: 10.1016/s0002-9440(10)62053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, Fujiwara K, Igarashi J, Nagase H, Held WA. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics. 2011;12(1):231. doi: 10.1186/1471-2164-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Martin CR. Fatty Acid Requirements in Preterm Infants and Their Role in Health and Disease. Clin Perinatol. 2014;41(2):363–382. doi: 10.1016/j.clp.2014.02.007. doi:310.1016/j.clp.2014.1002.1007. Epub 2014 Apr 1013. [DOI] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013 doi: 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133(2):217–222. doi: 10.1016/j.cell.2008.04.001. doi:210.1016/j.cell.2008.1004.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MD, Marty MA. Impact of environmental chemicals on lung development. Environ Health Perspect. 2010;118(8):1155–1164. doi: 10.1289/ehp.0901856. doi:1110.1289/ehp.0901856. Epub 0902010 May 0901855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola T, Hagood JS, James ML, Macewen MW, Williams TA, Hewitt MM, Schwiebert L, Bulger A, Oparil S, Chen YF, Ambalavanan N. Loss of Thy-1 inhibits alveolar development in the newborn mouse lung. Am J Physiol Lung Cell Mol Physiol. 2009;296(5):L738–750. doi: 10.1152/ajplung.90603.2008. doi:710.1152/ajplung.90603.92008. Epub 92009 Mar 90606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Null DM, Alvord J, Leavitt W, Wint A, Dahl MJ, Presson AP, Lane RH, DiGeronimo RJ, Yoder BA, Albertine KH. High-frequency nasal ventilation for 21 d maintains gas exchange with lower respiratory pressures and promotes alveolarization in preterm lambs. Pediatr Res. 2014;75(4):507–516. doi: 10.1038/pr.2013.254. doi:510.1038/pr.2013.1254. Epub 2013 Dec 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaru BI, Ahonen S, Kaila M, Erkkola M, Haapala AM, Kronberg-Kippila C, Veijola R, Ilonen J, Simell O, Knip M, Virtanen SM. Maternal diet during pregnancy and allergic sensitization in the offspring by 5 yrs of age: a prospective cohort study. Pediatr Allergy Immunol. 2010;21(1 Pt 1):29–37. doi: 10.1111/j.1399-3038.2009.00949.x. Epub 02009 Dec 00949. [DOI] [PubMed] [Google Scholar]
- Olave N, Nicola T, Zhang W, Bulger A, James M, Oparil S, Chen YF, Ambalavanan N. Transforming growth factor-beta regulates endothelin-1 signaling in the newborn mouse lung during hypoxia exposure. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L857–865. doi: 10.1152/ajplung.00258.2011. doi:810.1152/ajplung.00258.02011. Epub 02012 Jan 00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118(6):2316–2324. doi: 10.1172/JCI33655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, Rennard S, Agusti A, Anderson W, Lomas DA, DeMeo DL. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185(4):373–381. doi: 10.1164/rccm.201108-1382OC. doi:310.1164/rccm.201108-201382OC. Epub 202011 Dec 201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan VK, Sakurai R, Li Y, Karadag A, Corral J, Bellusci S, Xue YY, Belperio J, Torday JS. Effects of maternal food restriction on offspring lung extracellular matrix deposition and long term pulmonary function in an experimental rat model. Pediatr Pulmonol. 2012;47(2):162–171. doi: 10.1002/ppul.21532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke H, Gregory PD, Horz W. A transient histone hyperacetylation signal marks nucleosomes for remodeling at the PHO8 promoter in vivo. Mol Cell. 2001;7(3):529–538. doi: 10.1016/s1097-2765(01)00200-3. [DOI] [PubMed] [Google Scholar]
- Robinson CM, Neary R, Levendale A, Watson CJ, Baugh JA. Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir Res. 2012;13:74. doi: 10.1186/1465-9921-1113-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozance PJ, Seedorf GJ, Brown A, Roe G, O’Meara MC, Gien J, Tang JR, Abman SH. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2011;301(6):L860–871. doi: 10.1152/ajplung.00197.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, Hagood JS. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39(5):610–618. doi: 10.1165/rcmb.2007-0322OC. doi:610.1165/rcmb.2007-0322OC. Epub 2008 Jun 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. 2011;278(10):1598–1609. doi: 10.1111/j.1742-4658.2011.08089.x. [DOI] [PubMed] [Google Scholar]
- Simon DM, Arikan MC, Srisuma S, Bhattacharya S, Andalcio T, Shapiro SD, Mariani TJ. Epithelial cell PPARgamma is an endogenous regulator of normal lung maturation and maintenance. Proc Am Thorac Soc. 2006;3(6):510–511. doi: 10.1513/pats.200603-034MS. [DOI] [PubMed] [Google Scholar]
- Smolle M, Workman JL. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta. 2013;1829(1):84–97. doi: 10.1016/j.bbagrm.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer VA, Davie JR. Dynamically acetylated histone association with transcriptionally active and competent genes in the avian adult beta-globin gene domain. J Biol Chem. 2001;276(37):34810–34815. doi: 10.1074/jbc.M104886200. [DOI] [PubMed] [Google Scholar]
- Stocks J, Hislop A, Sonnappa S. Early lung development: lifelong effect on respiratory health and disease. Lancet Respir Med. 2013;1(9):728–742. doi: 10.1016/S2213-2600(13)70118-8. doi:710.1016/S2213-2600(1013)70118-70118. Epub 72013 Aug 70113. [DOI] [PubMed] [Google Scholar]
- Suter M, Ma J, Harris A, Patterson L, Brown KA, Shope C, Showalter L, Abramovici A, Aagaard-Tillery KM. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6(11):1284–1294. doi: 10.4161/epi.6.11.17819. doi:1210.4161/epi.1286.1211.17819. Epub 12011 Nov 17811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urman R, McConnell R, Islam T, Avol EL, Lurmann FW, Vora H, Linn WS, Rappaport EB, Gilliland FD, Gauderman WJ. Associations of children’s lung function with ambient air pollution: joint effects of regional and near-roadway pollutants. Thorax. 2014;69(6):540–547. doi: 10.1136/thoraxjnl-2012-203159. doi:510.1136/thoraxjnl-2012-203159. Epub 202013 Nov 203119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varisco BM, Ambalavanan N, Whitsett JA, Hagood JS. Thy-1 signals through PPARgamma to promote lipofibroblast differentiation in the developing lung. Am J Respir Cell Mol Biol. 2012;46(6):765–772. doi: 10.1165/rcmb.2011-0316OC. doi:710.1165/rcmb.2011-0316OC. Epub 2012 Jan 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar J, Cabrera NE, Valladares F, Casula M, Flores C, Blanch L, Quilez ME, Santana-Rodriguez N, Kacmarek RM, Slutsky AS. Activation of the Wnt/beta-catenin signaling pathway by mechanical ventilation is associated with ventilator-induced pulmonary fibrosis in healthy lungs. PLoS One. 2011;6(9):e23914. doi: 10.1371/journal.pone.0023914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet. 2014;15(2):69–81. doi: 10.1038/nrg3623. Epub 2013 Dec 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13(2):115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Okamura M, Tsutsumi S, Nishikawa NS, Tanaka T, Sakakibara I, Kitakami J, Ihara S, Hashimoto Y, Hamakubo T, Kodama T, Aburatani H, Sakai J. The peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha heterodimer targets the histone modification enzyme PR-Set7/Setd8 gene and regulates adipogenesis through a positive feedback loop. Mol Cell Biol. 2009;29(13):3544–3555. doi: 10.1128/MCB.01856-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia H, Chen HY, Sun JM, Holth LT, Davie JR. Histone acetylation is required to maintain the unfolded nucleosome structure associated with transcribing DNA. J Biol Chem. 1998;273(23):14516–14522. doi: 10.1074/jbc.273.23.14516. [DOI] [PubMed] [Google Scholar]
- Wang Y, Santos J, Sakurai R, Shin E, Cerny L, Torday JS, Rehan VK. Peroxisome proliferator-activated receptor gamma agonists enhance lung maturation in a neonatal rat model. Pediatr Res. 2009;65(2):150–155. doi: 10.1203/PDR.0b013e3181938c40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterborg JH. Dynamics of histone acetylation in vivo. A function for acetylation turnover? Biochem Cell Biol. 2002;80(3):363–378. doi: 10.1139/o02-080. [DOI] [PubMed] [Google Scholar]
- Weiss ST. Lung function and airway diseases. Nat Genet. 2010;42(1):14–16. doi: 10.1038/ng0110-1014. [DOI] [PubMed] [Google Scholar]
- Whitrow MJ, Moore VM, Rumbold AR, Davies MJ. Effect of supplemental folic acid in pregnancy on childhood asthma: a prospective birth cohort study. Am J Epidemiol. 2009;170(12):1486–1493. doi: 10.1093/aje/kwp315. [DOI] [PubMed] [Google Scholar]
- Wright RJ, Brunst KJ. Programming of respiratory health in childhood: influence of outdoor air pollution. Curr Opin Pediatr. 2013;25(2):232–239. doi: 10.1097/MOP.0b013e32835e78cc. doi:210.1097/MOP.1090b1013e32835e32878cc. [DOI] [PubMed] [Google Scholar]
- Wu YT, Chen WJ, Hsieh WS, Tsao PN, Yu SL, Lai CY, Lee WC, Jeng SF. MicroRNA expression aberration associated with bronchopulmonary dysplasia in preterm infants: a preliminary study. Respir Care. 2013;58(9):1527–1535. doi: 10.4187/respcare.02166. doi:1510.4187/respcare.02166. Epub 02013 Mar 02112. [DOI] [PubMed] [Google Scholar]
- Yang IV, Schwartz DA. Epigenetic control of gene expression in the lung. Am J Respir Crit Care Med. 2011;183(10):1295–1301. doi: 10.1164/rccm.201010-1579PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2014;31(14):00117–00110. doi: 10.1016/j.trsl.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kai G, Pu XD, Qing K, Guo XR, Zhou XY. Expression profile of microRNAs in fetal lung development of Sprague-Dawley rats. Int J Mol Med. 2012;29(3):393–402. doi: 10.3892/ijmm.2011.855. doi:310.3892/ijmm.2011.3855. Epub 2011 Dec 3896. [DOI] [PubMed] [Google Scholar]
- Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20(3):259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21(8):1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DE, Nelson DA. Histone acetylation in chicken erythrocytes. Rates of acetylation and evidence that histones in both active and potentially active chromatin are rapidly modified. Biochem J. 1988;250(1):233–240. doi: 10.1042/bj2500233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Hagood JS, Murphy-Ullrich JE. Thy-1 expression regulates the ability of rat lung fibroblasts to activate transforming growth factor-beta in response to fibrogenic stimuli. Am J Pathol. 2004;165(2):659–669. doi: 10.1016/s0002-9440(10)63330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkhan EK, Fu Q, Wang Y, Yu X, Callaway CW, Segar JL, Scholz TD, McKnight RA, Joss-Moore L, Lane RH. Maternal Hyperglycemia Disrupts Histone 3 Lysine 36 Trimethylation of the IGF-1 Gene. J Nutr Metab. 2012;2012:930364. doi: 10.1155/2012/930364. [DOI] [PMC free article] [PubMed] [Google Scholar]