

Abstract
As non-crystallizable polymers, microtubules have been the target of cryo-electron microscopy (cryo-EM) studies since the technique was first established. Over the years, image processing strategies have been developed that take care of the unique, pseudo-helical symmetry of the microtubule. With recent progress in data quality and data processing, cryo-EM reconstructions are now reaching resolutions that allow the generation of atomic models of microtubules and the factors that bind them. These include cellular partners that contribute to microtubule cellular functions, or small ligands that interfere with those functions in the treatment of cancer. The stage is set to generate a family portrait for all identified microtubule interacting proteins and to use cryo-EM as a drug development tool in the targeting of tubulin.
Tubulin organization within the Microtubule: symmetry or no symmetry?
The microtubule (MT) is a highly conserved and essential cytoskeletal polymer built of αβ-tubulin that undergoes remarkable dynamics powered by the energy of GTP hydrolysis. In the MT, tubulin dimers add head-to-tail making linear protofilaments (PFs), which themselves interact in parallel, with a certain lateral curvature and stagger, giving rise to the cylindrical shape of the MT. The contacts between PFs involve homotypic interactions (α-tubulin interacting with α-tubulin and β-tubulin with β-tubulin), except at a special site, referred to as the seam, in which heterotypic contacts are required to close the lattice of the most common MT assembly, containing 13 PFs (Fig. 1). This seam breaks true helical symmetry, although the MT can still be considered a helix of tubulin monomers if the differences between α- and β-tubulin are disregarded. Indeed, given the high similarity in the structure of the two tubulin subunits [1], MTs appear helical at low resolution, with an axial repeat of ~40 Å corresponding to the size of each tubulin protein (Fig. 1). This apparent symmetry is immediately broken when a MT-binding protein that recognizes specific sites on the αβ-tubulin dimer is bound to the MT surface (a couple of rare exceptions will be mentioned later). These geometrical considerations are important in structural studies of MTs and their interaction with binding partners. In this review, we briefly summarize the technical advances that have led to the progressive improvement in the resolution of cryo-EM structures of MTs that now allow the generation of atomic models, and then concentrate on the biological insights provided by recent MT structures.
Fig. 1. – Cryo-EM visualization of drug and protein binding on the microtubule surface.
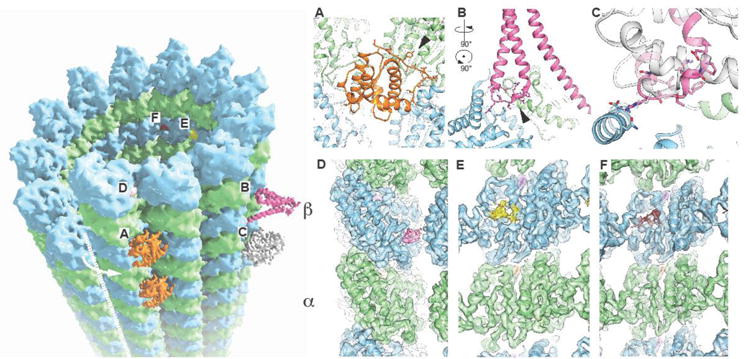
The MT lattice is shown (left) along with the location of associated MAPS: EB3 (A), PRC1 (B), kinesin (C), and small molecules: peloruside (D), Taxol (E), and zampanolide (F). The seam is indicated with a white dashed line and the arrow indicates the heterotypic (β/blue–α/green) interactions across the seam. Insets displayed on the right correspond to experimental cryo-EM maps at the site of interaction of each factor with tubulin.
Brief historical overview and how we have coped with the MT seam
As a polymer, the MT is not amenable to crystallization for X-ray studies and thus its structure has been pursued over the years using electron microscopy, both in vivo and in vitro. With the advent of cryo-EM to visualize frozen-hydrated samples, well preserved MTs became an obvious sample for structural characterization by this methodology. Many of the early cryo-EM MT studies involved the interaction of the motor protein kinesin with MTs. As helical Fourier methods were an obvious choice at the time, and given the problem caused by the seam mentioned above, many of such studies relied on the presence of a small percentage of MTs that in vitro are composed of 15 PFs organized in a 4-start helix [2–4], instead of the most common 3-start helix, 13-PF or 14-PF lattices. Such MTs do not have a seam and thus are true helical arrays of αβ-tubulin dimers. Given their scarcity, a typical reconstruction of these helical MTs used only one or a few MT images and was limited to a resolution of 20–25 Å. Downing and Li, with the use of a high-end (400 keV) electron microscope, the average of many more images, and ignoring the seam in the study of unbound MTs (and thus averaging α- and β-tubulin), dramatically improved the resolution to better than 10Å [5]. After that sharp resolution jump, the field stagnated for over a decade in terms of resolution improvement, with structures limited to 8 Å in the best of cases, although not in terms of new biological insights. The sub-nanometer resolution proved very useful in hybrid methodologies that involved docking crystal structures in a significant number of studies concerning the interaction of different cellular factors bound to MTs [6–8]. Through the last decade, image analysis strategies shifted from Fourier Bessel reconstruction to real space-based approaches, most significantly the Iterative Helical Real Space Reconstruction (IHRSR) method developed by Ed Egelman [9,10].
Our lab (i.e. Gregory Alushin and Gabriel Lander) implemented a pipeline for MT cryo-EM reconstruction that built on the IHRSR and in the use of non-helical averaging strategies as originally developed by Sindelar and Downing [11], to finally break the previous resolution barrier for the MT, obtaining structures at about 5 Å [12]. To reach that resolution required that α- and β-tubulin be distinguished and the seam located in order to align small, overlapping fragments of MTs (that ultimately corresponded to a total MT length of ~ 6 mm!). To be able to accomplish this in the context of the noisy images obtained by cryo-EM, and given the similarity between α- and β-tubulin, we used a kinesin motor domain as a fiducial for alignment of the tubulin dimer and the seam. The resulting ~5 Å structures were interpreted at the atomic level through the use of Rosetta modeling by using the fit to the cryo-EM map as an additional term in the energy function for minimization [13,14]. With the advent of direct electron detectors for data collection, it suddenly became possible to obtain higher resolution with significantly less data. Together with improvements to the computational scheme, including a method for accurately determining the position of the seam in our MT images (by Rui Zhang) [15], we have now reported a number of structures in the 4 to 3.5 Å resolution regime that allowed the generation of atomic models from the cryo-EM density maps. These studies have included MTs that were bound by different MT-associated proteins (MAPs), thus eliminating the need of kinesin as a fiducial. Although smaller than the kinesin head, these MAPs still provide sufficient signal for alignment of the seam, thus avoiding mixing of α- and β-tubulin. Our studies have most recently included the description of the effect of MT stabilizing drugs on MT structure, contributing to our understanding of how they exert their function. The resolution of our drug-bound MTs allowed the direct visualization of the binding site and thus supports the promise of cryo-EM for use in drug discovery/optimization.
Recent cryo-EM structures of MTs bound to associated factors
Within the last two years a number of cryo-EM structures of MTs bound to an associated protein factor have been reported at resolutions that allow atomic modeling (for an overview of recent structures over the last four years please see our previous review [16]). In almost all cases, the crystallographic structure of the MAP had been obtained in isolation, and the cryo-EM analysis enabled very accurate determination of the MT-MAP interaction surfaces using the crystal structure as a starting point for refinement into the density. An emergent theme is that some regions that interact with tubulin that may have been disordered in the crystal structure, or in a conformation dictated by crystal contacts, need to be (re)built as they become newly structured on the MT surface. An example is that of the +Tip protein EB3. Its calponin homology domain interacts with four adjacent tubulin dimers across both longitudinal and lateral interactions within the MT [8,17]. The interface in EB3 includes the extended N terminus (residues 1–16), which had to be rebuilt as it contacts helices H4 and H5 in α-tubulin and helix H12 in the adjacent β-tubulin (Fig. 1A) [17]. Another example is the MT crosslinker/organizer PRC1 [18]. The major MT-interaction region in this protein corresponds to a small spectrin domain of three alpha helices. One of the loops, connecting helices H7 and H8 (nomenclature from crystal structure PDB:3NRX [19]) that was disordered in the crystal, corresponds to the major contact point with the MT (Fig. 1B). Our atomic model of this loop, as built on our cryo-EM map, identified a key arginine residue (R381) that is conserved in kinesin (R278 in kinesin 1) and in both cases interacts with β-tubulin’s D427 on the MT surface (Fig. 1C). This unexpected conservation points to a convergence in the use of this tubulin region for interaction by different protein factors via very distinct structural modules. Folding or restructuring on the MT surface may be, therefore, a widespread feature of MAPs, and will likely be even more salient in intrinsically disordered proteins such as the classical MAPs tau and MAP2.
It is also interesting to point out that, based on the yet limited number of MAP structures bound to MTs, the same protein fold can bind very different surfaces on the MT. For example, the calponin homology (CH) domain of the Ndc80 protein within the Ndc80 kinetochore complex binds on the crest of the PF [20,21], while the CH domain of EB proteins binds in the crevasse between two PFs [8,17]. On the other hand, the MT binding region of doublecortin, which is not a CH domain, binds almost the same site on MTs as EB proteins [22]. Another example just described, of two totally different domains that interact with a common pocket on the MT surface using the same key amino acid, is that of the motor domain of kinesin, an ATPase related to G proteins, and the spectrin domain of PRC1 [18]. These examples demonstrate that the presence of a protein domain in a certain MAP does not dictate how it binds the MT based on what is known for other MAPs, invalidating what would have seemed reasonable predictions. This fact only adds to the fascinating complexity of MT binding motifs and the versatility of the MT surface as a landing pad for different cellular factors.
Structure of MTs from alternative sources
Most structural studies of tubulin and MTs to date have been carried out using tubulin purified from mammalian brain, a very rich source that, nevertheless, contains a complex mixture of tubulin isoforms and post-translational modifications [23]. With the development of new purification strategies [24] and expression systems [25,26], it has recently become possible to study more pure tubulin samples from alternative sources. A number of recent structural studies have now used recombinant human tubulin [25,27] and purified yeast tubulin [28] (which is significantly simpler in terms of tubulin isoforms). The latter opens the possibility to carry out parallel in vitro and in vivo studies in the characterization of tubulin mutants, as well as to explore, given the evolutionary distance between yeast and mammalian tubulin, the conservation (or not) of different structural properties between these two MT systems. Indeed, we have identified a number of surprising differences between yeast and mammalian MT concerning structure and interaction with binding partners. In our in vitro assembled MT studies, yeast MTs systematically have fewer PFs than their mammalian counterparts. Sequence differences around the lateral contacts may explain this observation. Both the number and nature of these sequence differences (a significant number of mammalian residues at the lateral contact correspond to serines in yeast tubulin) have the potential to alter slightly the geometry of the contacts, as well as their strength [28].
More intriguingly, the conformational change in α-tubulin that gives rise to a lattice compaction upon GTP hydrolysis [12,17] (as observed by comparing mammalian MTs bound to the slowly hydrolysable GTP-analog GMPCPP versus those bound to GDP), is not seen in yeast MTs [28]. For yeast, both GMPCPP stabilized MTs and MT grown in the presence of GTP appear extended. However, the binding of Bim1, a homolog of the human EB3, causes compaction, as does binding of GTPγS. Our previous mammalian studies showed that the GTPγS MT lattice is indeed compacted, leading us to propose that it may correspond to a GDP-Pi state [17]. We have also observed that EB3 speeds up the hydrolysis of GMPCPP, leading to a model in which EB proteins promote GTP hydrolysis [17]. In the context of yeast MTs, our observations suggest that GTP hydrolysis is delayed or partial in yeast MTs, but significantly promoted by Bim1. Interestingly, binding of Bim1 to yeast MTs was also distinct from the binding of EB3 to mammalian MTs. While the latter binds MTs with a tubulin dimer repeat, as most MAPs do, Bim1 binds yeast MTs with a tubulin monomer repeat, that is, double the stoichiometry [28]. A similar difference in binding pattern has also been previously observed for the Ndc80 kinetochore complex, which binds with the classical tubulin dimer repeat in C elegans, but with a tubulin monomer repeat in humans [7,20,21,29]. These observations indicate that the propensity of a MAP to distinguish or not α and β tubulin monomers can be exquisitely tuned by just a few amino acid changes. The biological significance for these binding differences across species is a fascinating subject in its own right.
Cryo-EM in the study of antimitotic agents
Tubulin is the target of a number of antimitotic, anticancer drugs that bind to tubulin and affect MT dynamics. While some of these agents have been shown to destabilize MT in vitro, others act as MT stabilizers. In the latter group is Taxol, a broadly used drug in the treatment of solid tumors. The binding site of this drug was described in the electron crystallographic structure of zinc-induced tubulin sheets [30,31]. This site is utilized by other agents, including zampanolide, which binds to it covalently. The binding of zampanolide and other taxane-site binders (e.g. epothilones) has been described in X-ray crystal structures of assembly-inhibited tubulin [32]. A novel class of MT stabilizers include peloruside, which binds to an alternative site on tubulin as described in another crystallographic study [33]. The increase in resolution afforded by direct detectors and the improvement in image analysis now allows the visualization of small molecules bound to the MT as well as any changes that they cause to the MT structure. Our lab has recently reported the structures for Taxol-, zampanolide- and peloruside-bound MTs at resolutions of 4.2 to 3.9 Å [34] (Fig 1D–F). These structures confirm that the site and mode of binding described in the previous electron and X-ray crystallographic studies are the same in the context of the assembled MT. Surprisingly, the effect of taxane and non-taxane site binder appears to be opposed. We have now described how the binding of Taxol and zampanolide, at a pocket on tubulin that faces the MT lumen, makes the MT lattice more irregular through deformations at lateral contacts, such that the MT cross section deviates from a perfect circle (Fig. 2). These irregularities explain why it has been more challenging to obtain high resolution cryo-EM reconstructions in the presence of this agent. Thus, the intrinsic disorder of the Taxol-bound MT lattice is a factor that should be taken into account when designing and interpreting structural and biophysical studies of MTs.
Fig. 2. – Flexible deformation of the microtubule lattice in the presence of drugs that bind the Taxol site.
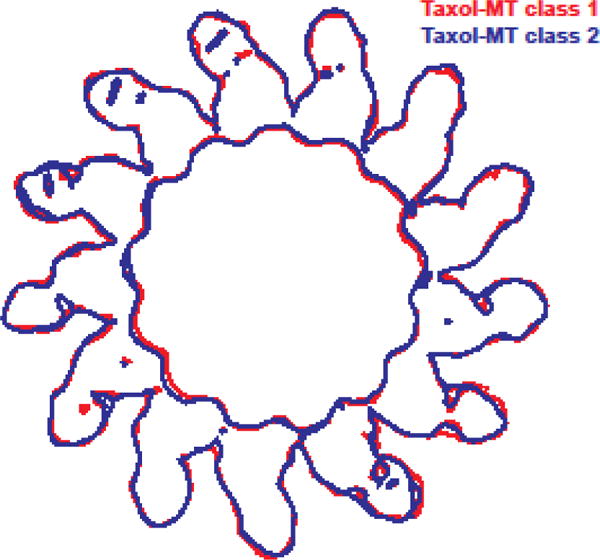
End-on outlines of two representative cryo-EM maps (each color represents a different “class” out of the five classes in which the full data set was separated during 3D classification (see [34]): red for class 1, blue for class 2) illustrating the variability observed among Taxol-bound microtubules.
In contrast to the disorder introduced by drugs that bind at the Taxol site, peloruside, which binds on the outside surface of the MT, right at the point of contact between PFs, regularizes the MT lattice. We have previously shown that most MTs, including dynamic MTs not bound by drugs, deviate from a cylindrical shape at the seam, where a small rotation (2–3°) of the PFs at the seam make the interface separate slightly. This slight deformation at the seam is best visualized by comparing the cryo-EM density map obtained without any symmetrization (i.e. C1 reconstruction), to one in which the PFs have been averaged by imposing pseudo-helical symmetry [17]. Generally we found that the deformation is larger for less stable MTs (e.g. GDP versus GMPCCP). We also observed previously that EB3, which binds across PFs, except at the seam, regularizes the lattice and closes up the seam, likely by serving as a wedge that fixes the angle across the non-seam contacts to make the seam interface come into perfect register [17]. Our recent drug study shows that this is the case also for peloruside (Fig 3) [34]. In the case of this stabilizing drug, which binds by the lateral interface between PFs, our C1 reconstruction shows very clearly that peloruside binding is maintained at the seam. Thus, peloruside appears to regularize the MT lattice both by promoting the appropriate angle between PFs, but likely also by “stapling” the seam. When MTs are bound to both Taxol and peloruside, the effect of peloruside on lateral interfaces is imposed and the MT lattice is regularized.
Fig. 3. – Microtubule lattice regularization by peloruside and EB3.
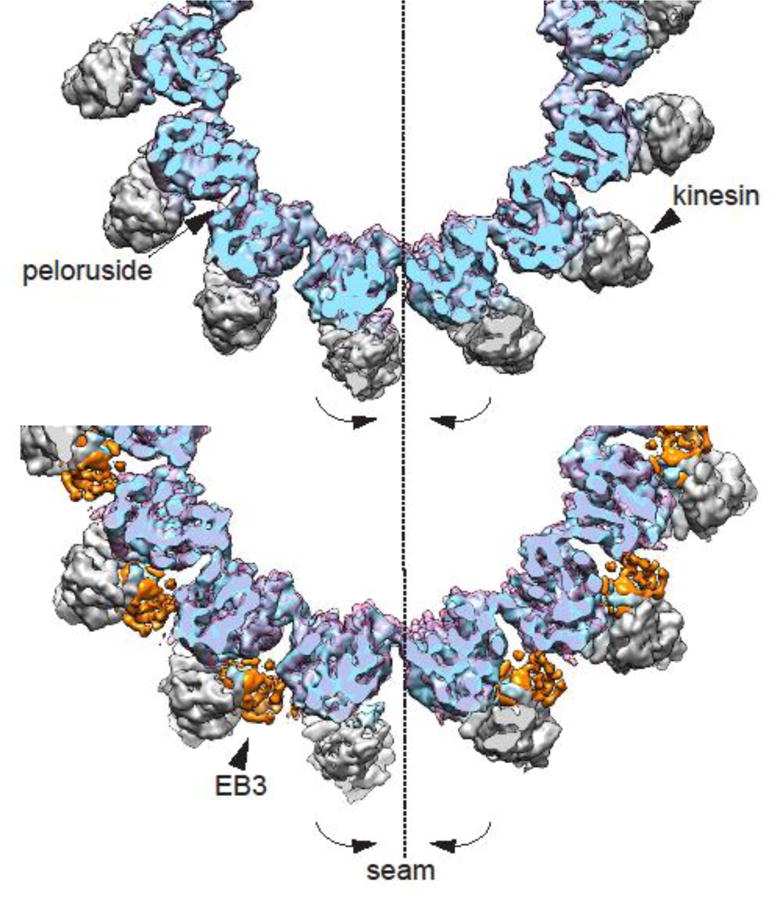
End-on view of the un-symmetrized (C1) 3D reconstruction of the peloruside-bound microtubule (top panel, in transparent pink) showing the closing and regularization of the seam with respect to C1 3D reconstruction of the drug-free microtubule (tubulin in blue and kinesin is gray). The same closure at the seam is observed when the later is compared with the EB3-bound microtubule C1 reconstruction (bottom panel, transparent pink, with EB3 density highlighted in orange).
These cryo-EM studies are an example of how the resolution obtained by this methodology now allows the visualization of drug binding and opens the door to the use of cryo-EM in drug development and optimization. By carrying out the studies on the full assembly that the drug naturally binds to, the studies go beyond identifying the binding site and provide information of its global effect on the tubulin assembly, just as it applies to the binding of a drug to a protein within a large macromolecular complex.
Conclusions and perspectives
Cryo-EM studies of MTs and their associated cellular factors or binding drugs are now producing high resolution structures, often allowing the generation of atomic models, and leading to a wealth of new biological information for a cytoskeletal system critical for the life of the cell and of significant medical value. Given the large number of interacting partners, many levels of regulation, and the myriad of cellular functions MT are involved with, the number of important biological questions that remain unanswered will keep structural biologists busy for a while. Those questions include not only how does the MT interact specifically with such a diverse array of known MAPs and small molecules, but how or whether these binding events are integrated on the MT surface to affect each other and the dynamic fate of the MT. The cryo-EM studies of MTs in the years to come will undoubtedly contribute to building a mechanistic understanding of this complex and fascinating biological system.
Highlights.
Technical progress leads to high-resolution cryo-EM structures of microtubules (MTs)
Atomic details of MT-binding proteins and antimitotic drugs bound to MTs
Characterization of the MT seam: a possible target of MT stability regulation
Plasticity of structural modules in the binding of MTs with their cellular partners
Cryo-EM of MTs shows high potential for drug development and improvement
Acknowledgments
This work was funded by NIGMS (GM63072 to EN). EN is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Nogales E. Structural insights into microtubule function. Ann Rev Biochem. 2000;69:277–302. doi: 10.1146/annurev.biochem.69.1.277. [DOI] [PubMed] [Google Scholar]
- 2.Arnal I, Metoz F, DeBonis S, Wade RH. Three-dimensional structure of functional motor proteins on microtubules. Current Biology. 1996;6:1265–1270. doi: 10.1016/s0960-9822(02)70712-4. [DOI] [PubMed] [Google Scholar]
- 3.Sosa H, Dias DP, Hoenger A, Whittaker M, Wilson-Kubalek E, Sablin E, Fletterick RJ, Vale RD, Milligan RA. A model for the microtubule-Ncd motor protein complex obtained by cryo-electron microscopy and image analysis. Cell. 1997;90:217–224. doi: 10.1016/s0092-8674(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 4.Hirose K, Amos WB, Lockhart A, Cross RA, Amos LA. Three-dimentional cryoelectron microscopy of 16-protofilament microtubules: structure, polarity and interaction with motor proteins. J Struc Biol. 1997;118:140–148. doi: 10.1006/jsbi.1997.3840. [DOI] [PubMed] [Google Scholar]
- 5.Li H, DeRosier DJ, Nicholson WV, Nogales E, Downing KH. Microtubule structure at 8 A resolution. Structure. 2002;10:1317–1328. doi: 10.1016/s0969-2126(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 6.Sindelar CV, Downing KH. An atomic-level mechanism for activation of the kinesin molecular motors. Proc Natl Acad Sci U S A. 2010;107:4111–4116. doi: 10.1073/pnas.0911208107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alushin GM, Ramey VH, Pasqualato S, Ball DA, Grigorieff N, Musacchio A, Nogales E. The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature. 2010;467:805–810. doi: 10.1038/nature09423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurer SP, Fourniol FJ, Bohner G, Moores CA, Surrey T. EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell. 2012;149:371–382. doi: 10.1016/j.cell.2012.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 10.Egelman EH. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J Struct Biol. 2007;157:83–94. doi: 10.1016/j.jsb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Sindelar CV, Downing KH. The beginning of kinesin’s force-generating cycle visualized at 9-A resolution. J Cell Biol. 2007;177:377–385. doi: 10.1083/jcb.200612090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in alphabeta-tubulin upon GTP hydrolysis. Cell. 2014;157:1117–1129. doi: 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiMaio F, Tyka MD, Baker ML, Chiu W, Baker D. Refinement of protein structures into low-resolution density maps using rosetta. J Mol Biol. 2009;392:181–190. doi: 10.1016/j.jmb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang R, Nogales E. A new protocol to accurately determine microtubule lattice seam location. J Struct Biol. 2015;192:245–254. doi: 10.1016/j.jsb.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nogales E, Zhang R. Visualizing microtubule structural transitions and interactions with associated proteins. Curr Opin Struct Biol. 2016;37:90–96. doi: 10.1016/j.sbi.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17**.Zhang R, Alushin GM, Brown A, Nogales E. Mechanistic Origin of Microtubule Dynamic Instability and Its Modulation by EB Proteins. Cell. 2015 doi: 10.1016/j.cell.2015.07.012. First microtubule structures obtained at better than 4 Å resolution, it allowed building and refinement of the structure of tubulin and EB3 into the cryo-EM map. This study revealed the details of the conformational change in α-tubulin upon GTP hydrolysis. It also identified the distortion of the circular cross section of the microtubule at the seam, where the two protofilaments involved in the heterotypic contacts are further apart from each other than those at the homotypic contacts. EB3, which does not bind at the seam, regularizes the lattice by closing up the seam. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Kellogg EH, Howes S, Ti SC, Ramirez-Aportela E, Kapoor TM, Chacon P, Nogales E. Near-atomic cryo-EM structure of PRC1 bound to the microtubule. Proc Natl Acad Sci U S A. 2016;113:9430–9439. doi: 10.1073/pnas.1609903113. High-resolution cryo-EM structure of the microtubule bound to a very small structural motif, a spectrin domain, which was sufficient to align the seam. The study visualized the loop in PRC1 that interacts with α-tubulin site shared with kinesin and dynein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian R, Wilson-Kubalek EM, Arthur CP, Bick MJ, Campbell EA, Darst SA, Milligan RA, Kapoor TM. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 2010;142:433–443. doi: 10.1016/j.cell.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alushin GM, Musinipally V, Matson D, Tooley J, Stukenberg PT, Nogales E. Multimodal microtubule binding by the Ndc80 kinetochore complex. Nat Struct Mol Biol. 2012;19:1161–1167. doi: 10.1038/nsmb.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Wilson-Kubalek EM, Cheeseman IM, Milligan RA. Structural comparison of the Caenorhabditis elegans and human Ndc80 complexes bound to microtubules reveals distinct binding behavior. Mol Biol Cell. 2016;27:1197–1203. doi: 10.1091/mbc.E15-12-0858. High-resolution structure of both human and C. elegans Ndc80 kinetochore complexes bound to the microtubule. This study conclusively established the different binding stoichimetry for the two species on the microtubule surface that have been proposed in two previous and separate studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fourniol FJ, Sindelar CV, Amigues B, Clare DK, Thomas G, Perderiset M, Francis F, Houdusse A, Moores CA. Template-free 13-protofilament microtubule-MAP assembly visualized at 8 A resolution. J Cell Biol. 2010;191:463–470. doi: 10.1083/jcb.201007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magiera MM, Janke C. Post-translational modifications of tubulin. Curr Biol. 2014;24:R351–354. doi: 10.1016/j.cub.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 24.Widlund PO, Podolski M, Reber S, Alper J, Storch M, Hyman AA, Howard J, Drechsel DN. One-step purification of assembly-competent tubulin from diverse eukaryotic sources. Mol Biol Cell. 2012;23:4393–4401. doi: 10.1091/mbc.E12-06-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25*.Ti SC, Pamula MC, Howes SC, Duellberg C, Cade NI, Kleiner RE, Forth S, Surrey T, Nogales E, Kapoor TM. Mutations in Human Tubulin Proximal to the Kinesin-Binding Site Alter Dynamic Instability at Microtubule Plus- and Minus-Ends. Dev Cell. 2016;37:72–84. doi: 10.1016/j.devcel.2016.03.003. First structure of microtubules assembled from recombinant human tubulin, both wild type and a mutant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Garnham CP, Vemu A, Wilson-Kubalek EM, Yu I, Szyk A, Lander GC, Milligan RA, Roll-Mecak A. Multivalent Microtubule Recognition by Tubulin Tyrosine Ligase-like Family Glutamylases. Cell. 2015;161:1112–1123. doi: 10.1016/j.cell.2015.04.003. The use of post-translationally unmodified tubulin resulted in better binding of the tubulin glutamylase and allowed cryo-EM studies of its interaction with the microtubule. The structure allowed the visualization of the C-terminal tails of tubulin as they are engaged by the enzyme. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Vemu A, Atherton J, Spector JO, Szyk A, Moores CA, Roll-Mecak A. Structure and Dynamics of Single-isoform Recombinant Neuronal Human Tubulin. J Biol Chem. 2016;291:12907–12915. doi: 10.1074/jbc.C116.731133. High-resolution structure of microtubules assembled from the purest tubulin source to date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Howes SC, LaFrance B, Zhang R, Kellogg EH, Geyer EA, Westermann S, Rice LM, Nogales E. Structural and functional differences between yeast and mammalian microtubules revealed by cryo-EM. under submission. First structural characterization at high resolution of yeast microtubules. A number of structural differences with mammalian microtubules are described, including the different stoichiometry of binding of Bim1, an EB3 homolog, on yeast microtubules, from that of Bim1 or EB3 on mammalian microtubules. [Google Scholar]
- 29.Wilson-Kubalek EM, Cheeseman IM, Yoshioka C, Desai A, Milligan RA. Orientation and structure of the Ndc80 complex on the microtubule lattice. J Cell Biol. 2008;182:1055–1061. doi: 10.1083/jcb.200804170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nogales E, Wolf SG, Downing KH. Structure of the ab tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 31.Lowe J, Li H, Downing KH, Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. Journal of Molecular Biology. 2001;V313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 32.Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 33.Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann KH, Miller JH, Diaz JF, Steinmetz MO. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew Chem Int Ed Engl. 2014;53:1621–1625. doi: 10.1002/anie.201307749. [DOI] [PubMed] [Google Scholar]
- 34**.Kellogg EH, Hejab NM, Howes S, Northcote P, Miller JH, Diaz JF, Downing KH, Nogales E. Insights into the Distinct Mechanisms of Action of Taxane and Non-Taxane Microtubule Stabilizers from Cryo-EM Structures. J Mol Biol. 2017 doi: 10.1016/j.jmb.2017.01.001. High-resolution cryo-EM structures showing the binding of antimitotic drugs and their effect on microtubule structure. The findings indicate a distinct mode of microtubule stabilization for drugs that bind at the Taxol site and for peloruside. Whereas taxane-binders result in microtubule lattice deformations, peloruside structurally regularizes the seam in a way reminiscient of EB3. [DOI] [PMC free article] [PubMed] [Google Scholar]