

Abstract
It is now possible to routinely determine atomic resolution structures by electron cryo-microscopy (cryoEM), facilitated in part by the method known as micro electron-diffraction (MicroED). Since its initial demonstration in 2013, MicroED has helped determine a variety of protein structures ranging in molecular weight from a few hundred Daltons to several hundred thousand Daltons. Some of these structures were novel while others were previously known. The resolutions of structures obtained thus far by MicroED range from 3.2 Å to 1.0 Å, with most better than 2.5 Å. Crystals of various sizes and shapes, with different space group symmetries, and with a range of solvent content have all been studied by MicroED. The wide range of crystals explored to date presents the community with a landscape of opportunity for structure determination from nano crystals. Here we summarize the lessons we have learned during the first few years of MicroED, and from our attempts at the first ab initio structure determined by the method. We re-evaluate theoretical considerations in choosing the appropriate crystals for MicroED and for extracting the most meaning out of measured data. With more laboratories worldwide adopting the technique, we speculate what the first decade might hold for MicroED.
MicroED a cryo-EM method
MicroED is a new method of electron cryo-microscopy (cryo-EM) for determining the atomic structures of proteins from sub-micron sized protein crystals using a conventional electron cryo microscope. The practical and theoretical aspects of MicroED have recently been reviewed elsewhere [1•,2•,3•]. Here we briefly summarize the emergence of MicroED and take measure of what this new technique has achieved since its first demonstration in 2013.
Electron diffraction of proteins has a storied past, being used throughout the last half a century to solve the structures of proteins from two-dimensional crystals [4,5•]. The introduction of cryogenic techniques in electron microscopy greatly benefitted the study of proteins by preserving their structure in the face of radiation damage [6]. While no physical limitation impeded the determination of protein structures from 3D crystals by electron diffraction, the approach was not demonstrated successfully until 2013 when the model protein lysozyme was determined anew using a transmission electron microscope operating at 200 keV [7••]. The crystals of lysozyme were kept at cryogenic temperatures and diffracted using an extremely low dose electron beam that imparted less than 0.01 electrons per Ångstrom squared per second on the crystal allowing 90° worth of rotation data to be collected per crystal (total accumulated dose was <9 electrons per Å2). Presently, data collection by MicroED is achieved by continuously rotating the sample unidirectionally as it is exposed to the electron beam and diffraction movies are acquired [8••,1•]. Diffraction movies are converted [2•,9•] to a format readable by modern crystallographic processing programs including XDS, DIALS, MOSFLM, and HKL [2•]. Structure determination and refinement proceeds as with X-ray structures using programs such as Phenix, Refmac, and SHELX, making explicit use of tabulated electron scattering factors [2•].
Properties of structures determined by MicroED
Structures of various macromolecules have now been determined by MicroED (Figure 1). While the array of structures determined is broad, most structures are of proteins with a monomeric mass of less than 50 kDa. The largest protein assemblies determined to date include catalase (a 240 kDa tetramer) and the calcium ATP-ase (~110 kDa) [10•,11•]. Some of the smallest structures determined include protein segments in the amyloid state, as short as six residues (~800 Da) [12••]. Structures as small as these remain a major challenge for other cryo-EM methods, which typically struggle to extract meaningful signal from tiny proteins. MicroED has also been used to determine the structure of a protein complex: TGF-βm bound to its receptor, TβR II [36•]. With improvements to processing and data collection, the range of sizes of molecules whose structures are determined by MicroED is expected to grow. However, because larger molecules are likely to form larger unit cells, fewer can fit into a sub-micron sized crystal. A small crystal with few unit cells may produce diffraction that is difficult to measure, is of poor quality, or is low in resolution and may necessitate better detectors than the ones presently used. Moreover, as the unit cell grows, so might its solvent content, creating difficulties for cryogenic preparations without cryo protectants. The structure with the greatest solvent content, determined by MicroED contained approximately 60% solvent.
Figure 1.
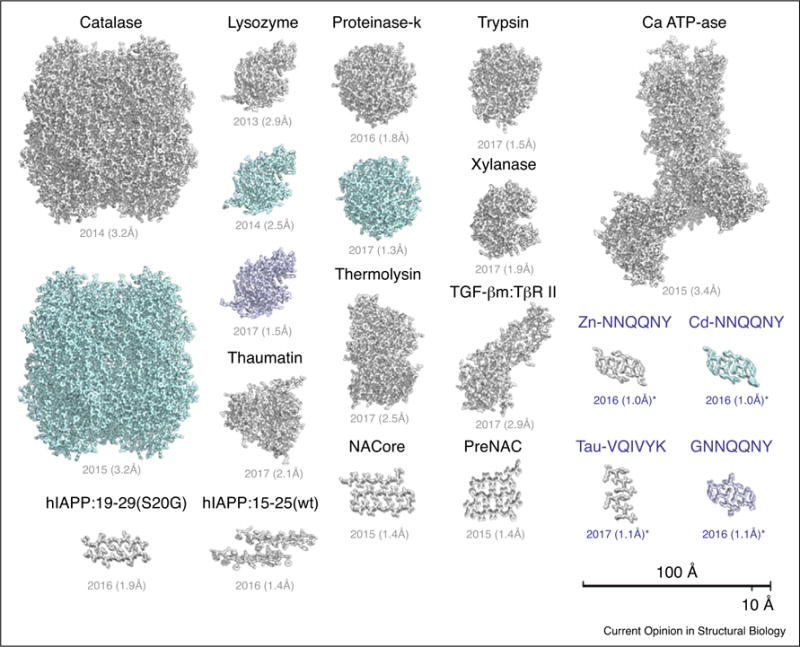
Atomic models are shown for each protein structure determined by MicroED. Also listed are the date the model was published and their reported resolution. Where a protein was determined multiple times, each copy is shown. Structures determined ab initio are shown in blue. Further details for each of the structures are listed in Table 1. Scale bars, 10 and 100 Å.
Screening of sub-micron crystals with conventional tools, including light microscopes, remains inefficient. Many conditions that contain what appears to be amorphous precipitate also contain nano-scale crystals [1•]. In addition, breaking of large, imperfect crystals can produce sub-micron crystallites (or crystal domains) suitable for structure determination by MicroED and free of the pathologies inherent to their larger parent crystal [36••]. This approach has now been demonstrated for various crystals formed by different proteins, specifically: lysozyme, proteinase K, xylanase, thaumatin, trypsin, thermolysin, the TGF-βm:TβRII complex, and a segment of the protein Tau (Table 1). Further development of approaches to crystal preparation and screening are important to help make MicroED more accessible.
Table 1.
A survey of protein structures determined by MicroED. All structures have been determined within the last four years. Listed for each are the protein name, PDB code, Rwork, and Rfree values, citation, method of phasing, resolution (in Ångstrom), space group, and unit cell dimensions.
| Sample | PDB code | Rwork | Rfree (%) | Ref | Phasing method | Res. (Å) | Space group | Cell dimensions a, b, c (Å) | α, β, γ (°) |
|---|---|---|---|---|---|---|---|
| Lysozyme | 3J4G | 25.5 | 27.8 | [7••] | MR | 2.9 | P 43 21 2 | 77, 77, 37 | 90, 90, 90 |
| Lysozyme | 3J6K | 22.0 | 25.5 | [8••] | MR | 2.5 | P 43 21 2 | 76, 76, 37 | 90, 90, 90 |
| Lysozyme | 5K70 | 23.9 | 28.4 | [36••] | MR | 1.5 | P 43 21 2 | 76, 76, 37 | 90, 90, 90 |
| Catalase | 3J7B | 26.2 | 30.8 | [10•] | MR | 3.2 | P 21 21 21 | 68, 172, 182 | 90, 90, 90 |
| Catalase | 3J7U | 27.2 | 31.7 | [11•] | MR | 3.2 | P 21 21 21 | 69, 174, 206 | 90, 90, 90 |
| Ca-ATPase | 3J7T | 27.7 | 31.5 | [11•] | MR | 3.4 | C 2 | 166, 64, 147 | 90, 98, 90 |
| Proteinase K | 519S | 19.7 | 25.6 | [9•] | MR | 1.8 | P 43 21 2 | 67, 67, 102 | 90, 90, 90 |
| Proteinase K | 5K7S | 22.4 | 25.5 | [36••] | MR | 1.3 | P 43 21 2 | 67.6, 67.6, 101.4 | 90, 90, 90 |
| Thermolysin | 5K7T | 28.7 | 30.6 | [36••] | MR | 1.6 | P 61 2 2 | 90.8, 90.8, 126 | 90, 90, 120 |
| Trypsin | 5K7R | 24.8 | 28.1 | [36••] | MR | 1.5 | P 21 21 21 | 53.1, 56.1, 64.4 | 90, 90, 90 |
| Thaumatin | 5K7Q | 24.5 | 29.2 | [36••] | MR | 2.1 | P 41 21 2 | 57.8, 57.8, 150 | 90, 90, 90 |
| Xylanase | 5K7P | 22.1 | 26.2 | [36••] | MR | 1.9 | P 21 21 21 | 49.1, 59, 70 | 90, 90, 90 |
| TGFβ:TβRII | 5TY4 | 29.2 | 32.8 | [36••] | MR | 2.9 | P 21 21 21 | 41.5, 71.3, 79.5 | 90, 90, 90 |
| NACore | 4RIL | 24.8 | 27.5 | [14••] | MR | 1.4 | C 2 | 70.8, 4.8, 16.8 | 90, 106, 90 |
| PreNAC | 4ZNN | 23.5 | 28.2 | [14••] | MR | 1.4 | P 21 | 17.9, 4.7, 33 | 90, 94, 90 |
| hIAPP 19-29 (S20G) | 5KNZ | 22.8 | 27.5 | [35•] | MR | 1.9 | P 21 21 21 | 4.8, 18.6, 70.8 | 90, 90, 90 |
| hIAPP 15-25 (WT) | 5KO0 | 22.4 | 25.9 | [35•] | MR | 1.4 | P1 | 11.7, 18.2, 19.9 | 63, 89, 88 |
| Tau (VQIVYK) | 5K7N | 21.0 | 22.4 | [36••]a | DM | 1.1 | C 2 | 29.3, 4.97, 37.6 | 90, 112, 90 |
| Sup35 (Zn-NNQQNY) | 5K2E | 15.6 | 19.4 | [12••]a | DM | 1.0 | P 21 | 21.5, 4.9, 23.9 | 90, 104, 90 |
| Sup35 (Cd-NNQQNY) | 5K2F | 22.0 | 24.2 | [12••]a | DM | 1.0 | P 21 | 22.1, 4.9, 23.5 | 90, 104, 90 |
| Sup35 (GNNQQNY-1) | 5K2G | 18.7 | 22.4 | [12••]a | DM | 1.1 | P 21 | 22.9, 4.9, 24.2 | 90, 108, 90 |
| Sup35 (GNNQQNY-2) | 5K2H | 17.7 | 18.6 | [12••]a | DM | 1.05 | P 21 21 21 | 23.2, 4.9, 40.5 | 90, 90, 90 |
Denotes protein structures determined ab initio by direct methods.
Improvements in MicroED have already been demonstrated using model proteins, including lysozyme and proteinase K [9•,13]. The resulting atomic models have gained in resolution because of advancements in data collection and processing. The most recent structure of lysozyme determined by MicroED (PDB ID 5K70) is in the same space group (P43212) as that determined in 2013 and is obtained from seven sub-micron crystals, but now features 1.5 Å resolution compared to the initial 2.9 Å model (Table 1). This was facilitated in part by the introduction of a scheme for continuous rotation of crystals within the electron beam. The method of continuous rotation minimizes artifacts that arise from processing of still diffraction images, particularly for electron diffraction where the short DeBroglie wavelength of the electron produces a large Ewald sphere. Rapid and sensitive CMOS detectors allow for continuous data collection, producing diffraction movies that facilitate fine sampling of reciprocal space. Pipelines for data processing have been streamlined [2•] to convert proprietary data formats produced by electron detectors into those amenable for processing by conventional X-ray crystallography software. To facilitate their dissemination, annual workshops take place at the Janelia Research Campus, where students from around the world gather to collect diffraction from tiny crystals of proteinase K and determine its structure [1•]. The workshops have been heavily oversubscribed and allow participants to experience all aspects of MicroED: crystal harvesting, grid preparation, cryo transfer into the microscope, setup of the microscope, data collection in continuous rotation data analysis and structure determination.
MicroED as a tool for studying amyloid structures
Many proteins enter the amyloid state, converting to elongated unbranched fibrils, usually between 5 and 20 nm in width, and up to several microns long. These include proteins found associated with Alzheimer’s, Parkinson’s, ALS and other diseases as well as many denatured proteins, and functional protein assemblies in both prokaryotic and eukaryotic cells. As far as is known, each amyloid fibril contains only one type of protein molecule. The molecules stack on one another to form β-sheets that run the entire length of the fibrils, with each molecule forming amide hydrogen bonds to the molecules immediately above and below it in the stack.
Where they have been crystallized, amyloid fibrils form crystals much smaller than conventional protein crystals. As has long been known, β-sheets generally show a gentle left-handed twist. The result is that amyloid fibrils are twisted. Hence, like DNA, they do not readily crystallize, where translational symmetry is required. However, it was learned in 2001 that short protein segments that form amyloid fibrils can be crystallized [37]. In these crystals the β-sheets are held untwisted by the lattice. Apparently as the crystals grow, the propensity of the β-sheets to twist builds up a strain, which eventually limits the crystal size. The first amyloid crystals were of amyloid-forming peptides 4–7 residues in length and were micron sized. They could be mounted on narrow glass pins, and the development of synchrotron microbeams permitted collection of X-ray diffraction data and determination of structures [38].
As longer amyloid-forming peptide segments of interest were crystallized, crystal sizes diminished. For example, the 11-residue core of the Parkinson’s disease-related protein, α-synucein formed crystals with dimensions of only a few hundred nanometers. These crystals of what was termed NACore could be visualized only by electron microscopy, and their miniscule size precluded mounting for X-ray data collection. Fortunately, MicroED revealed atomic structure of NACore [14••]. The crystal of NACore used to yield a structure by MicroED was ~10 000 000 000 times smaller in volume than the crystals of hemoglobin that yielded one of the first protein structures by X-ray diffraction.
Since then, informative structures of segments of other amyloid proteins have been determined by microED. These include segments from IAPP [35•] and Tau [36••], as well as FUS, TDP-43, and PrP [all in preparation for publication]. It seems unlikely that these structures could have been determined by technologies other than MicroED.
Lessons from ab initio structure determination in MicroED
While most proteins determined by MicroED have been phased by the method of molecular replacement, not all have been model proteins. The first unknown structure determined by MicroED was that of a segment of the amyloid forming protein alpha synuclein [14••]. The structures of two segments of amyloid forming segments from alpha synuclein were determined by MicroED and used to create a model of the toxic core of synuclein aggregates, the cause of Parkinson’s disease. These structures served as a first demonstration that entirely new protein structures could be accessible from sub-micron crystals by MicroED. They followed demonstrations of the accuracy of intensities measured by MicroED, based on refinement of starting models from standard proteins where ligands of the known model had been omitted [10•].
Historically, the potential to determine structures of complex molecules by electron diffraction had been tempered by concerns over dynamical scattering, which can reduce the accuracy of measured diffraction intensities from thick 3D crystals [15–17]. However, the accuracy of measured intensities in MicroED has been further demonstrated by the ab initio determination of four structures of prion forming segments of the yeast protein Sup35 from sub-micron sized crystals [12••]. In these four experimental examples, a complex structure was obtained in the absence of any preconceived notion of the atomic arrangement, relying only on the measured diffraction intensities. A recent study demonstrating the structure of 8 different proteins from crystals that vary in thickness [36••] indicate that if continuous rotation is employed for data collection and the crystals are a few hundred nanometers thick, accurate data can be faithfully collected. A key limitation to the achievement of high-resolution diffraction from small protein crystals is the dose required to produce sufficient signal. MicroED measurements must be performed at doses below those that may significantly damage a crystal lattice and reduce its diffraction quality [12••]. All MicroED structures published to date have been obtained from crystals that were dosed less than 10 e−/Å2. Global intensity decay has been observed in cases where a 5 e−/Å2 was dose imparted on a crystal [8••]. All structures determined ab initio were obtained from crystals that received a dose of less than 5 e−/Å2, suggesting that this dose may act as a threshold for loss of atomic resolution detail [12•]. We note that other cryo-EM techniques often deposit doses larger than 5 electrons per Ångstrom squared on their samples [18]. While dose fractionation is often implemented, the dose required to achieve atomic resolution detail for many cryo-EM methods remains under investigation. Here, MicroED measurements could serve as a guide, indicating with acute precision when molecular changes manifest in response to dose induced damage.
The achievement of ab initio structure determination necessitated accurate intensities to a resolution of at least 1.2 Å. This requirement is placed by the approach used for the ab initio search during phasing [19,20]. While the prion structures determined by MicroED dispel any concern over non-kinematical diffraction phenomena inhibiting phasing by direct methods, the extension of this approach to larger proteins has yet to be realized. There is no fundamental limitation to this extension, but experimental hurdles include the need for very well ordered crystals capable of producing near 1 Å diffraction. Recent efforts in ab initio structure determination have succeeded in phasing lower resolution structures, and may be applicable to MicroED [21,22].
Theoretical considerations
Some of the structures determined recently by MicroED are the highest resolution cryo-EM structures to date and can be used to inform or refine aspects of structure determination previously estimated from experiments with inorganic or non-protein materials [23–25]. Model peptides have been used to facilitate these efforts. Structures with resolutions near 1 Å are now publicly available to benchmark improvements in theory [12••]. The quality of these models is exceptional, revealing the arrangement of hydrogen atoms with unprecedented detail. Two theoretical assumptions already being reconsidered given recent experimental demonstrations by MicroED are the scattering factor tables presently used during refinement, and the guidelines for crystal thickness required achieving high quality kinematic scattering.
The scattering of electrons by charged atoms differs from that of uncharged atoms, particularly at low resolution [26]. Several efforts have attempted to account for these effects but without guidance from atomic resolution protein structures [11•,26]. An improvement in the present scattering tables for electrons could facilitate the identification and refinement of complex chemistry for atomic resolution models, perhaps revealing the locations of ions in maps produced by MicroED.
Deviations from kinematic scattering are a greater concern for electron diffraction than they are for experiments with X-rays with similarly sized crystals [16,27]. This is due to the shorter mean free path of electrons traveling through a protein crystal. At the electron energies typically used in MicroED (200–300 keV), the interactions between electrons and matter are strong enough to produce substantial inelastic scattering from samples over a micron thick [28]. The multiple scattering of electrons by crystals is also a concern [27]. However, the effect of these phenomena has not precluded the determination of protein structures from crystals almost half a micron thick, even for ab initio solutions. Nearly two dozen structures have been determined by MicroED in high and low symmetry space groups, and from crystals with varying thickness (Table 1). Most of the datasets collected from these crystals show minimal evidence of dynamical diffraction, particularly for the amyloid segments whose shortest unit cell (4.9 Å) is smaller than those of some small molecules [12••,29,30]. We attribute this incongruence to limitations in the models presently used to estimate the effects of dynamical diffraction. Future models could be refined to include deviations from perfect crystals such as crystal bending, nano-scale mosaicity, inelastic scattering, and the effects of bulk solvent.
Forecast: the first decade of MicroED
Much has been learned from the first handful of structures determined by MicroED (Figure 1; Table 1; Figure 2). Advances to the method during its first three years include the approach of data collection by continuous rotation, the use of molecular replacement and ab initio methods for structure determination, and the preparation of suitably small crystallites from large imperfect crystals (Figure 3). We speculate on how benefits brought by future advancements to MicroED could impact structural biology:
The field of intrinsically disordered proteins is rich with potential targets for MicroED due to their inability to form large, well-ordered crystals. Two amyloid forming proteins, alpha synuclein and amylin, have already benefitted from structure determination by MicroED [14••]. The first, alpha synuclein, remains a challenge to structurally characterize. Several new NMR models have suggested structures for synuclein fibrils, while MicroED has provided an atomic view of a potential structure for the toxic core of synuclein aggregates with structures for two segments from its NAC region, preNAC and NACore (Figure 1). The second, amylin or human islet amyloid polypeptide (hIAPP), is associated with type 2 diabetes and pancreatic beta cell toxicity, and remains out of reach of conventional crystallographic methods. Two structures of amylin segments have now been determined by MicroED (Figure 1) from overlapping regions of the protein [35•]. One of the segments contains a familial mutation (S20G) that forms rapid aggregates and leads to early onset type 2 diabetes. These and other structures can now be used to inform the rational design of therapeutic agents against presently incurable diseases.
MicroED could facilitate the direct localization of charged ions within protein structures in a way that broadly impacts membrane protein biophysics. However, the only membrane protein structure determined by MicroED to date is that of calcium ATPase. Obtaining structures of membrane proteins is in part limited by challenges in extracting crystallites from the sometimes thick or extremely viscous environments in which they grow, such as lipidic cubic phase (LCP), and the fact that membrane protein crystals in detergents can be more fragile than crystals of soluble proteins. Screening for sub-micron crystals within such heterogeneous environments is also difficult.
The use of extremely low electron doses by MicroED opens the possibility for determination of nearly damage-free protein structures. Intact disulfide bonds have been observed in structures determined by MicroED [36••]. Future efforts could produce intact structures of more radiation sensitive features, possibly including the acutely radiation-sensitive active sites in metalloenzymes [31]. If crystals of these were to diffract at high enough resolution, the assignment of hydrogens within their active site environment could also be possible, as is now with amyloid segments. This may necessitate the development of fully automated pipelines for efficient ultra-low dose data collection and the adaptation of better hardware. More sensitive detectors, perhaps single electron counting, and energy filters could introduce greater clarity and sensitivity to patterns, allowing new algorithms to extract even the weakest of reflections at finer resolutions. Structural dynamics has been developed and demonstrated in electron crystallography of 2D crystals [32–34]; similar time resolution can be applied to MicroED samples.
Figure 2.
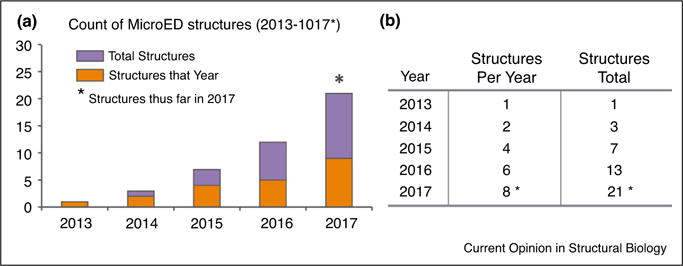
MicroED structures determined by year from 2013 to 2017. (a) Bar graph indicating the number of protein structures determined by MicroED on an annual basis. (b) A table summarizes the data presented (a), counting both the number of structures per year and the total structures to date per year. An asterisk marks 2017 indicating the count for the year is incomplete and includes only structures published at the start of the year.
Figure 3.
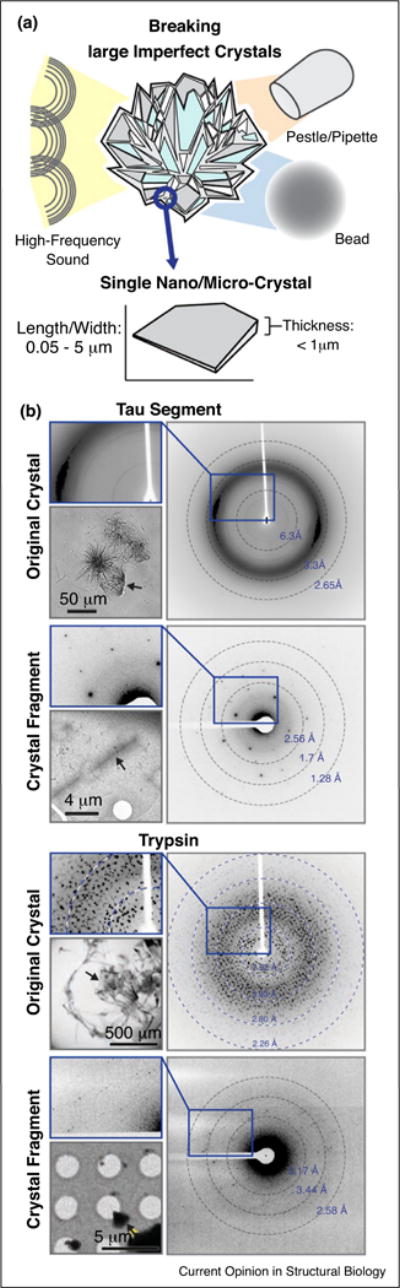
Breaking down large imperfect crystals for structural analysis by MicroED. (a) Diagram illustrating disruption of large imperfect crystals by three approaches: high-frequency sound, or crushing by use of a pestle, pipette tip, or a small rigid bead. A second diagram illustrates a small crystalline fragment, typical of those obtained when breaking large crystals. The dimensions of the fragment are listed as ranges. (b) Examples of the results obtained from the procedures outlined in (a). Two examples are shown, one of the protein Tau (top), and at right are crystals of Trypsin (bottom). For each, light micrographs of a large polycrystalline clusters are shown along with their X-ray diffraction patterns, below these are examples where single crystallites were obtained from each cluster by fragmentation, their corresponding MicroED pattern, and a magnified view of that pattern.
Acknowledgments
We thank Drs. Johan Hattne, Duilio Cascio, Michael Sawaya, and Michael Martynowycz for their input. Jose Rodriguez is supported by the Searle Scholars Program and Arnold and Mabel Beckman Foundation. This work has been supported by the Howard Hughes Medical Institute and the Janelia Research Campus Visiting Fellows program.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
•of special interest
••of outstanding interest
- 1•.Shi D, Nannenga BL, de la Cruz MJ, Liu J, Sawtelle S, Calero G, Reyes FE, Hattne J, Gonen T. The collection of MicroED data for macromolecular crystallography. Nat Protoc. 2016;11:895–904. doi: 10.1038/nprot.2016.046. http://dx.doi.org/10.1038/nprot.2016.046. This paper outlines a detailed protocol for setting up the cryo TEM for MicroED including data collection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2•.Hattne J, Reyes FE, Nannenga BL, Shi D, de la Cruz MJ, Leslie AGW, Gonen T. MicroED data collection and processing. Acta Crystallogr Sect Found Adv. 2015;71:353–360. doi: 10.1107/S2053273315010669. http://dx.doi.org/10.1107/S2053273315010669. This paper describes the processing of MicroED data. In combination with the paper above the two outline the complete protocol for MicroED data collection and analysis from start to end. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3•.Rodriguez JA, Gonen T. Chapter fourteen—high-resolution macromolecular structure determination by MicroED, a cryo-EM method. In: Crowther RA, editor. Methods Enzymol. Academic Press; 2016. pp. 369–392. This is an extensive review that discusses the theory behind electron diffraction, set up of the microscope for MicroED data collection and compares and contrasts MicroED with other cryo EM methods such as electron crystallography of 2D crystals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walz T, Grigorieff N. Electron crystallography of two-dimensional crystals of membrane proteins. J Struct Biol. 1998;121:142–161. doi: 10.1006/jsbi.1998.3945. http://dx.doi.org/10.1006/jsbi.1998.3945. [DOI] [PubMed] [Google Scholar]
- 5•.Gonen T. The collection of high-resolution electron diffraction data. In: Schmidt-Krey I, Cheng Y, editors. Electron Crystallogr Soluble Membr Proteins. Humana Press; 2013. pp. 153–169. http://dx.doi.org/10.1007/978-1-62703-176-9_9. (Accessed 30 November 2015). This review paper discusses detailed setup of the cryo TEM for electron diffraction and provides examples of good and bad diffraction patterns. While the paper is focused on 2D crystals many of the principles discussed apply to MicroED where 3D crystals are used. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor KA, Glaeser RM. Electron microscopy of frozen hydrated biological specimens. J Ultrastruct Res. 1976;55:448–456. doi: 10.1016/s0022-5320(76)80099-8. http://dx.doi.org/10.1016/S0022-5320(76)80099-8. [DOI] [PubMed] [Google Scholar]
- 7••.Shi D, Nannenga BL, Iadanza MG, Gonen T. Three-dimensional electron crystallography of protein microcrystals. eLife. 2013:2. doi: 10.7554/eLife.01345. http://dx.doi.org/10.7554/eLife.01345. This is the first paper describing the method of MicroED. It is the proof of principle work which outlines for the first time that an electron microscope can be used in diffraction mode for solving protein structure from vanishingly small crystals of dose sensitive samples. Here the term “MicroED” is first coined. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8••.Nannenga BL, Shi D, Leslie AGW, Gonen T. High-resolution structure determination by continuous-rotation data collection in MicroED. Nat Methods. 2014;11:927–930. doi: 10.1038/nmeth.3043. http://dx.doi.org/10.1038/nmeth.3043. In this paper the MicroED method is improved from “still diffraction” of the original paper to the new “continuous rotation” method that is now standard in MicroED. This method is analogous to the rotation method in x-ray crystallography. In this paper the authors outline the setup of the microscope and illustrate that excellent data can be collected by continuous rotation and for the first time x-ray data reduction software is used for structure solution. This was the first successful implementation of such an approach in cryo EM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9•.Hattne J, Shi D, de la Cruz MJ, Reyes FE, Gonen T. Modeling truncated pixel values of faint reflections in MicroED images. J Appl Crystallogr. 2016;49:1029–1034. doi: 10.1107/S1600576716007196. http://dx.doi.org/10.1107/S1600576716007196. In this paper Hattne and co-workers developed a simple algorithm to help extract more accurate data our of noisy images. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Nannenga BL, Shi D, Hattne J, Reyes FE, Gonen T. Structure of catalase determined by MicroED. eLife. 2014;3:e03600. doi: 10.7554/eLife.03600. http://dx.doi.org/10.7554/eLife.03600. Catalase was the second protein structure determined by MicroED. It was completed by continuous rotation from a single nano crystal only 180 nm thick—roughly 8 molecules of catalase thick. The resolution obtained was similar to what was obtained in the 1980 by xray crystallography from crystals billions of times larger. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11•.Yonekura K, Kato K, Ogasawara M, Tomita M, Toyoshima C. Electron crystallography of ultrathin 3D protein crystals: atomic model with charges. Proc Natl Acad Sci. 2015;112:3368–3373. doi: 10.1073/pnas.1500724112. http://dx.doi.org/10.1073/pnas.1500724112. In this work the Yonekura laboratory in Japan employs MicroED methods successfully for solving structures of lysozyme and catalase. Effects of charge are discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12••.Sawaya MR, Rodriguez J, Cascio D, Collazo MJ, Shi D, Reyes FE, Hattne J, Gonen T, Eisenberg DS. Ab initio structure determination from prion nanocrystals at atomic resolution by MicroED. Proc Natl Acad Sci. 2016:201606287. doi: 10.1073/pnas.1606287113. http://dx.doi.org/10.1073/pnas.1606287113. This is the first time that ab initio methods are used for solving the phase problem in MicroED. Moreover, currently this work represents the highest resolution ever achieved by any cryo EM method at ~1 Å. This work indicated that dynamical effects in MicroED are minimal and kinematic scattering can be assumes as long as crystals are a few hundred nanometers thick and continuous rotation is used. This work lays the foundation to future studies where larger proteins are studied. [DOI] [PMC free article] [PubMed]
- 13.Nannenga BL, Gonen T. Protein structure determination by MicroED. Curr Opin Struct Biol. 2014;27:24–31. doi: 10.1016/j.sbi.2014.03.004. http://dx.doi.org/10.1016/j.sbi.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, Jiang L, Arbing MA, Nannenga BL, Hattne J, Whitelegge J, Brewster AS, Messerschmidt M, Boutet S, Sauter NK, Gonen T, Eisenberg DS. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 2015;525:486–490. doi: 10.1038/nature15368. http://dx.doi.org/10.1038/nature15368. In this landmark study, Rodrigues and co-workers determine the first two novel structures by MicroED. The fragments of alpha synuclein protein formed crystals smaller than the wavelength of light. A decade of effort was put into trying to grow the crystals large enough for X-ray crystallography without success. However, using MicroED the crystals which were smaller than the wavelength of visible light diffracted to 1.4 Å resolution allowing the team to solve the structure and shed light on disease progression in Parkinsons patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glaeser RM, Ceska TA. High-voltage electron diffraction from bacteriorhodopsin (purple membrane) is measurably dynamical. Acta Crystallogr A. 1989;45:620–628. doi: 10.1107/s0108767389004599. http://dx.doi.org/10.1107/S0108767389004599. [DOI] [PubMed] [Google Scholar]
- 16.Grigorieff N, Henderson R. Comparison of calculated and observed dynamical diffraction from purple membrane: implications. Ultramicroscopy. 1996;65:101–107. http://dx.doi.org/10.1016/S0304-3991(96)00061-7. [Google Scholar]
- 17.Diaz-Avalos R, Long C, Fontano E, Balbirnie M, Grothe R, Eisenberg D, Caspar DLD. Cross-beta order and diversity in nanocrystals of an amyloid-forming peptide. J Mol Biol. 2003;330:1165–1175. doi: 10.1016/s0022-2836(03)00659-4. http://dx.doi.org/10.1016/S0022-2836(03)00659-4. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Grigorieff N, Penczek PA, Walz T. A primer to single-particle cryo-electron microscopy. Cell. 2015;161:438–449. doi: 10.1016/j.cell.2015.03.050. http://dx.doi.org/10.1016/j.cell.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauptman H. The direct methods of X-ray crystallography. Science. 1986;233:178–183. doi: 10.1126/science.233.4760.178. http://dx.doi.org/10.1126/science.233.4760.178. [DOI] [PubMed] [Google Scholar]
- 20.Usón I, Sheldrick GM. Advances in direct methods for protein crystallography. Curr Opin Struct Biol. 1999;9:643–648. doi: 10.1016/s0959-440x(99)00020-2. http://dx.doi.org/10.1016/S0959-440X(99)00020-2. [DOI] [PubMed] [Google Scholar]
- 21.Rodríguez DD, Grosse C, Himmel S, González C, de Ilarduya IM, Becker S, Sheldrick GM, Usón I. Crystallographic ab initio protein structure solution below atomic resolution. Nat Methods. 2009;6:651–653. doi: 10.1038/nmeth.1365. http://dx.doi.org/10.1038/nmeth.1365. [DOI] [PubMed] [Google Scholar]
- 22.Jorda J, Sawaya MR, Yeates TO. Progress in low-resolution ab initio phasing with CrowdPhase. Acta Crystallogr Sect Struct Biol. 2016;72:446–453. doi: 10.1107/S2059798316003405. http://dx.doi.org/10.1107/S2059798316003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorset DL. Electron diffraction intensities from bent molecular organic crystals. Acta Crystallogr A. 1980;36:592–600. http://dx.doi.org/10.1107/S0567739480001271. [Google Scholar]
- 24.Bethe HA, Rose ME, Smith LP. The multiple scattering of electrons. Proc Am Philos Soc. 1938;78:573–585. [Google Scholar]
- 25.Doyle PA, Turner PS. Relativistic Hartree–Fock X-ray and electron scattering factors. Acta Crystallogr Sect A. 1968;24:390–397. http://dx.doi.org/10.1107/S0567739468000756. [Google Scholar]
- 26.Zhong S, Dadarlat VM, Glaeser RM, Head-Gordon T, Downing KH. Modeling chemical bonding effects for protein electron crystallography: the transferable fragmental electrostatic potential (TFESP) method. Acta Crystallogr A. 2002;58:162–170. doi: 10.1107/s0108767301020256. http://dx.doi.org/10.1107/S0108767301020256. [DOI] [PubMed] [Google Scholar]
- 27.Subramanian G, Basu S, Liu H, Zuo J-M, Spence JCH. Solving protein nanocrystals by cryo-EM diffraction: multiple scattering artifacts. Ultramicroscopy. 2015;148:87–93. doi: 10.1016/j.ultramic.2014.08.013. http://dx.doi.org/10.1016/j.ultramic.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Leis A, Rockel B, Andrees L, Baumeister W. Visualizing cells at the nanoscale. Trends Biochem Sci. 2009;34:60–70. doi: 10.1016/j.tibs.2008.10.011. http://dx.doi.org/10.1016/j.tibs.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 29.Dorset DL, Hauptman HA. Direct phase determination for quasi-kinematical electron diffraction intensity data from organic microcrystals. Ultramicroscopy. 1976;1:195–201. doi: 10.1016/0304-3991(76)90034-6. [DOI] [PubMed] [Google Scholar]
- 30.van Genderen E, Clabbers MTB, Das PP, Stewart A, Nederlof I, Barentsen KC, Portillo Q, Pannu NS, Nicolopoulos S, Gruene T, Abrahams JP. Ab initio structure determination of nanocrystals of organic pharmaceutical compounds by electron diffraction at room temperature using a Timepix quantum area direct electron detector. Acta Crystallogr Sect Found Adv. 2016;72:236–242. doi: 10.1107/S2053273315022500. http://dx.doi.org/10.1107/S2053273315022500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen AE, Soltis SM, González A, Aguila L, Alonso-Mori R, Barnes CO, Baxter EL, Brehmer W, Brewster AS, Brunger AT, Calero G, Chang JF, Chollet M, Ehrensberger P, Eriksson TL, Feng Y, Hattne J, Hedman B, Hollenbeck M, Holton JM, Keable S, Kobilka BK, Kovaleva EG, Kruse AC, Lemke HT, Lin G, Lyubimov AY, Manglik A, Mathews II, McPhillips SE, Nelson S, Peters JW, Sauter NK, Smith CA, Song J, Stevenson HP, Tsai Y, Uervirojnangkoorn M, Vinetsky V, Wakatsuki S, Weis WI, Zadvornyy OA, Zeldin OB, Zhu D, Hodgson KO. Goniometer-based femtosecond crystallography with X-ray free electron lasers. Proc Natl Acad Sci. 2014;111:17122–17127. doi: 10.1073/pnas.1418733111. http://dx.doi.org/10.1073/pnas.1418733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowal J, Chami M, Baumgartner P, Arheit M, Chiu P-L, Rangl M, Scheuring S, Schröder GF, Nimigean CM, Stahlberg H. Ligand-induced structural changes in the cyclic nucleotide-modulated potassium channel MloK1. Nat Commun. 2014;5:3106. doi: 10.1038/ncomms4106. http://dx.doi.org/10.1038/ncomms4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramaniam S, Henderson R. Electron crystallography of bacteriorhodopsin with millisecond time resolution. J Struct Biol. 1999;128:19–25. doi: 10.1006/jsbi.1999.4178. http://dx.doi.org/10.1006/jsbi.1999.4178. [DOI] [PubMed] [Google Scholar]
- 34.Unwin N, Miyazawa A, Li J, Fujiyoshi Y. Activation of the nicotinic acetylcholine receptor involves a switch in conformation of the α subunits. J Mol Biol. 2002;319:1165–1176. doi: 10.1016/S0022-2836(02)00381-9. http://dx.doi.org/10.1016/S0022-2836(02)00381-9. [DOI] [PubMed] [Google Scholar]
- 35••.Krotee P, Rodriguez JA, Sawaya MR, Cascio D, Reyes FE, Shi D, Hattne J, Nannenga B, Oskarrson M, Griner S, Jiang L, Westermark G, Gonen T, Eisenberg DS. Atomic structures of fibrillar segments of hIAPP suggest tightly mated β-sheets are important for cytotoxicity. eLife. 2017;6:e19273. doi: 10.7554/eLife.19273. http://dx.doi.org/10.7554/eLife.19273. In this work Krotee and co-workers use MicroED to solve two novel structures further illustrating how the method can be used successfully. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36••.de la Cruz JM, Hattne J, Shi D, Seidler P, Rodriguez J, Reyes FE, Sawaya M, Cascio D, Eisenberg DS, Gonen T. Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED. Nat Methods. 2017;14:399–402. doi: 10.1038/nmeth.4178. Here the authors present 8 structures determined by MicroED between 2.9–1 Å resolution. Large crystals with various pathologies that render them useless for X-ray diffraction were fractionated and the resulting crystal fragments used for MicroED experiments. In all cases the crystal fragments diffracted as well as or better than the parent crystals. This work illustrates that even without crystal growth optimization, MicroED can be used to solve atomic structures. Moreover, this paper also illustrates the first structure of a protein complex determined by MicroED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balbirnie M, Grothe R, Eisenberg DS. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated b-sheet structure for amyloid. Proc Natl Acad Sci. 2001;5:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson R, Sawaya MR, Balbirnie M, Madsen A, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]