

Abstract
The pace at which cryo-EM is being adopted as a mainstream tool in structural biology has continued unabated over the past year. Initial successes in obtaining near-atomic resolution structures with cryo-EM were enabled to a large extent by advances in microscope and detector technology. Here, we review some of the complementary technical improvements that are helping sustain the cryo-EM revolution. We highlight advances in image processing that permit high resolution structure determination even in the presence of structural and conformational heterogeneity. We also review selected examples where biochemical strategies for membrane protein stabilization facilitate cryo-EM structure determination, and discuss emerging approaches for further improving the preparation of reliable plunge-frozen specimens.
Introduction
Whether the meteoric rise of cryo-EM is viewed as a revolution, or as an evolution that is a result of the steady growth of biological electron microscopy, there is little doubt that it is making a major impact in structural biology. The number of structures reported at near-atomic resolutions (resolutions better than 4 Å) in the EM Data Bank jumped from only 36 in 2014 to 114 and 240 in the years 2015 and 2016, respectively. The number of publications per year associated with EMDB entries almost tripled between 2011 and 2016 (123 to 325). An impressive spectrum of biological problems are beginning to be addressed, ranging from progress with more traditional cryo-EM favorites such as ribosomes [1,2] and icosahedral viruses [3,4], to previously uncharted territory in studies of membrane proteins [5,6] and a host of multi-protein complexes [7-11] of varying sizes. The advent of direct electron detectors is deservedly credited with being a key factor in driving recent advances in high-resolution structure determination by single particle cryo-EM [12-14]. However, it will be necessary for advances to also occur in other aspects of the cryo-EM workflow so that the hardware advances in microscope and detector architecture can be properly leveraged to ensure continued growth of the cryo-EM field.
Among the many challenges most frequently encountered, some can be readily identified as important current bottlenecks to progress. For example, during the process of specimen preparation, where a thin aqueous suspension is created and plunge-frozen, functionally relevant protein complexes that are stable in bulk solution may become dissociated [15-17]. In addition, proteins and protein complexes in aqueous solution are intrinsically flexible to varying extents [18], creating structural heterogeneity that can be even more pronounced in multi-domain proteins and multi-protein complexes [19,20]. These types of heterogeneity can confound the process of cryo-EM structure determination, since it relies on averaging projection images from tens of thousands of individual macromolecules [21,22]. Structurally homogeneous samples can sometimes be prepared by removal of flexible components or by introducing stabilizing mutations, but these may come at the price of steering the samples away from physiologically relevant conditions [23]. Finally, even when all of the biochemistry has been optimized, non-uniform distribution of proteins on substrate grids, including problems such as aggregation and preferred orientation, can still stymie progress [24]. While customized solutions are sometimes necessary to solve each of these problems, this review highlights advances on three specific fronts (3D classification of heterogeneous structures, protein stabilization, and specimen preparation) where a variety of novel strategies are being employed to tackle outstanding problems in the field.
Computational strategies to address conformational heterogeneity
The presence of multiple, coexisting conformations in a sample is a challenge for high resolution structure determination by both single particle cryo-EM and X-ray crystallography, as both depend on averaging information from multiple copies of the relevant biological entity. In the crystallographic context, conformational heterogeneity can impede crystal formation [25], and even when crystals are successfully obtained, this can lead to disorder in parts of the structure that are prone to greater mobiity. When functionally relevant conformational changes are introduced in pre-formed crystals, crystal disruption can occur [26]; historically, hemoglobin was the first instance where this phenomenon was reported [27]. In contrast, with cryo-EM structure determination, it is often possible to discern this heterogeneity computationally, either by focusing analysis only on structurally homogeneous regions of the complex [28], or by deriving an ensemble of structures when there are discrete subpopulations [29]. In most cases, conformational separation also provides invaluable clues to the functional mechanism, as illustrated in the examples discussed below.
Glutamate dehydrogenase (GDH), a highly conserved hexameric enzyme [30], plays a central role in glutamate metabolism by catalyzing the deamination of glutamate to generate α-ketoglutarate (AKG), coupled with the transfer of a pair of electrons to either NAD+ or NADP+. In its apo-form, GDH exists in an “open” conformation, with a stable core region and more mobile nucleotide binding domains. Whereas the highest resolution reported structure of this state by X-ray crystallography is at 2.7 Å resolution [31], single particle cryo-EM analysis that takes into account some of the heterogeneity of the periphery can achieve a resolution of ∼1.8 Å in the ordered core regions (Figure 1a) [11]. Crystallographic analysis of the closed state of GDH has always required the presence GTP and glutamate as well as NADH, leaving open the question of the individual effect of NADH binding to the protein conformation. Cryo-EM analyses show that binding of NADH alone results in an equilibrium between closed and open states, which differ with respect to the conformation of NADH in the regulatory site (Figure 1b) [32]. Cryo-EM approaches thus not only yielded higher resolution structural information for stable regions of the molecule, but also enabled the discovery of a potential role for NADH in regulation of GDH activity.
Figure 1.
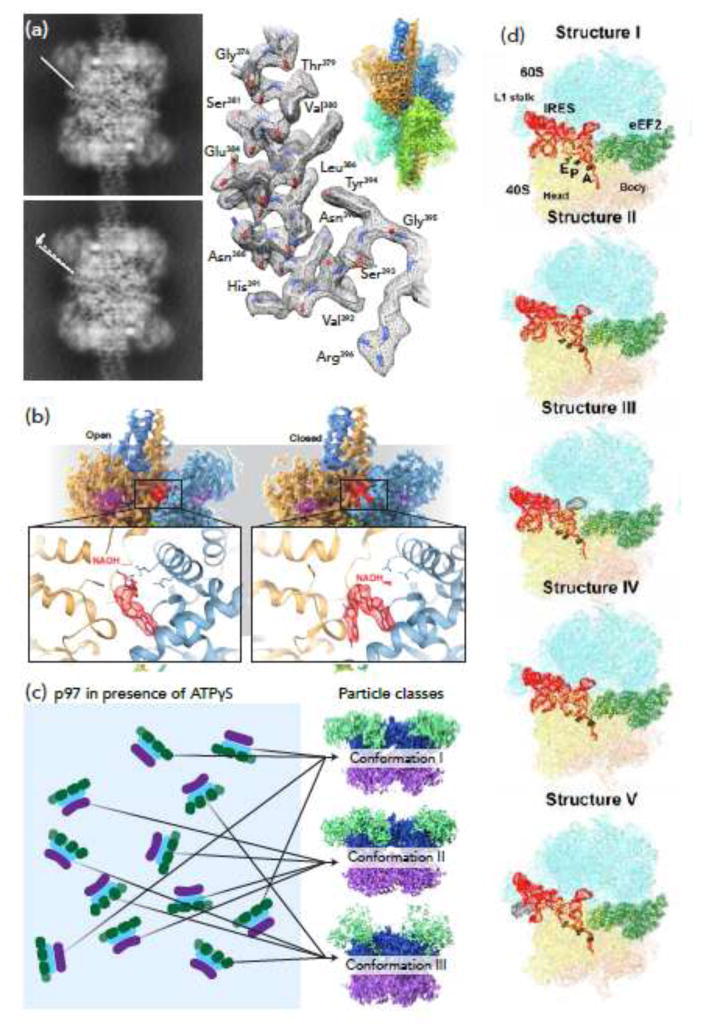
Using computational strategies to reveal conformational heterogeneity. (a) In its open conformation, the nucleotide binding domains (NBDs) of glutamate dehydrogenase (GDH) are significantly more mobile than the core region (sharpened map showing core overlaid with unsharpened map showing lower resolution NBDs, upper right). Left: projection views of 3D classes of apo GDH; center: region from GDH core that reaches 1.8 Å resolution. Adapted from [11]. (b) When bound by NADH alone, GDH displays two distinct conformations, open (left) and closed (right). The orientation of NADH in the regulatory site differs between these two states (insets). Adapted from [32]. (c) In the presence of ATPγS, the AAA+ ATPase p97 displays three distinct conformations (depending on ATPγS occupancy), which can be differentiated by distinct orientations of the D1 domain (purple) and the N domain (green) versus the D2 domain (blue). Adapted from [33]. (d) Computational methods have been used to separate 5 distinct conformations of the ribosome, tracking the translocation of viral IRES through the molecular machine. From [1].
A second example comes from structural studies of p97, a hexameric AAA+ ATPase and cancer target. p97 binds multiple factors, including ubiquitin-binding adaptors, ubiquitin ligases and deubiquitinating enzymes; as a multi-domain molecular machine, it is expected to adopt multiple conformational states. However, structural studies of full-length p97 by X-ray crystallography have been limited to low resolution (3.5 Å to 4.7 Å), precluding an understanding of structural changes under native conditions and in complex with enzymatic inhibitors including those of clinical interest. Recent single particle cryo-EM analyses have significantly advanced mechanistic understanding of p97 function by resolving three well-defined, co-existing conformational states that are simultaneously populated when ATPγS is added to p97 (Figure 1c) [33]. These three states reveal an unambiguous and step-wise set of conformational changes upon binding of ATPγS, providing both a “movie” of how p97 is activated and new mechanistic insights into how a conformation-selective inhibitor binds at the interface between the two nucleotide-binding domains to lock the enzyme in an inactive state.
A third example comes from structural studies of the yeast ribosome bound to a viral mRNA containing an internal ribosome entry site (IRES). To initiate translation, a structured IRES RNA moiety interacts with the 40S subunit or the 80S ribosome, without the need for additional host initiation factors. Translocation of the IRES mRNA, however, requires binding of the eukaryotic elongation factor 2 (eEF2). Using maximum-likelihood based classification approaches, five distinct IRES-eEF2- bound ribosome structures were identified within a single mixture (Figure 1d) [1], with differences in the conformations of ribosomal subunits, IRES RNA and the translocase eEF2. This collection of distinct structures suggests a plausible sequence of steps for the eF2-induced translocation of IRES and also revealed previously unresolved intermediate states, providing new insights into the structural basis for the mechanism of translocase action. Conformational sorting by cryo-EM of ribosomes engineered to stall the association of ribosomal components, has also yielded significant insights into the how these complex particles use multiple pathways to achieve assembly [29].
Obtaining reliable quantitation of the proportional occupancy of each of the conformational substates identified by computational separation can be difficult, but some interesting approaches using complementary structural methods are emerging. In a recent study of the archaeal AAA+ ATPase VAT, Huang et al. confirmed the presence of both a “stacked ring” (a planar, closed ring structure) and a “split ring” (a broken ring structure with a slight helical pitch) conformation of this hexameric ATPase using NMR, and further verified that the “stacked ring” represented the more common conformation [34].
Taming Membrane Proteins for Cryo-EM
Despite the abundance of membrane proteins in nature, relatively few of the crystal structures in the Protein Data Bank are of membrane proteins. Until the breakthrough demonstration that near-atomic resolution structures of membrane proteins could be obtained using single particle cryo-EM [5], the only membrane protein structures that were determined at high resolution using electron microscopy were obtained using electron crystallography of 2D crystals of the proteins embedded in a lipid bilayer [35].
Because removal of membrane proteins from their native lipid bilayer environment by detergent solubilization can destabilize them (reviewed in [24]), there is considerable interest in finding ways to generate stable, but essentially “aqueous” solutions of membrane proteins. Amphipols, whose application to stabilizing membrane proteins was recognized over two decades ago [36], are amphipathic polymers that can replace detergent molecules surrounding the transmembrane region and restrict overall conformational flexibility. Following the initial application of amphipols to obtain a high resolution structure for the TRPV1 channel [5], their use as a detergent alternative has grown significantly. Although there is concern that the use of amphipols may steer the conformation away from physiologically relevant conditions, many new structures of membrane proteins solubilized with amphipols have been reported, including structures of the ryanodine receptor [37], Trp channels [38-40], gamma secretase [41,42], as well as a V-type ATPase [43].
A growing number of membrane proteins are also being successfully studied by cryo-EM in detergent micelles. Recent examples of membrane proteins whose structures have been determined to high resolution in detergent include the ryanodine receptor [44,45], Trp channels [46,47], gamma secretase [48], glutamate receptor [49], the calcium channel Cav1.1 [50], and the mitochondrial complex [51-53]. While these successes demonstrate that it is possible to achieve high resolution in detergent with some classes of proteins, issues with both protein stability and the presence of excess detergent remain stumbling blocks to obtaining high resolution. A recently reported gradient-based method, GraDeR [54] (Figure 2a), removes excess boundary detergent, offering a novel approach to enhancing membrane protein stability in the context of the detergent-solubilized state. The GraDeR method achieves detergent reduction by slow removal of excess amphipathic detergents such as lauryl maltose-neopentyl glycol (LMNG) [54], and has been recently used to obtain cryo-EM structure for the innexin-6 channel [55].
Figure 2.
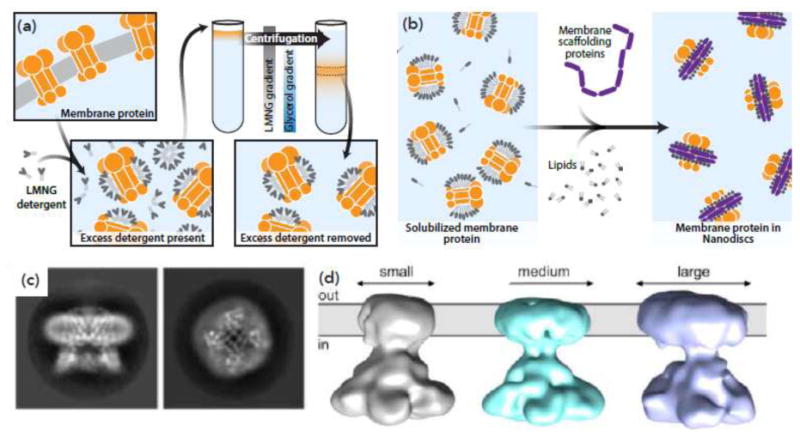
Novel techniques for visualizing integral membrane proteins by cryo-EM. (a) In the GraDeR technique [54], membrane proteins solubilized in an amphipathic detergent are run over a gradient, with increasing concentrations of glucose and decreasing concentrations of detergent. This removes excess detergent. (b) To assemble membrane proteins into nanodiscs, free phospholipids and membrane scaffold protein (MSP), which contains the lipids in a set size “disc”, are mixed with purified membrane protein. Detergent is subsequently removed. Based on [56]. (c) Three 3D classes of the membrane channel CorA in nanodiscs, showing that nanodisc size can vary significantly, precluding high resolution structure determination by 3D averaging. From [57]. (d) Side (left) and top (right) 2D classes of the membrane protein TRPV1 in nanodiscs. From [58].
Reconstitution of membrane proteins into nanodiscs has been proposed to offer the best compromise between maintaining a lipid-like environment at the boundary of the membrane protein, while also allowing the membrane proteins to be present as “single particles” that can be studied using cryo-EM [56]. Nanodiscs are essentially small discs of lipid bilayers bounded by a stable protein ring that is thought to form a belt around the periphery of the disc (Figure 2b). Variations in the size of the nanodisc can potentially preclude achieving high resolution structures [57] (Figure 2c), driving the need for improved methods to generate more homogeneously-sized nanodisc assemblies. Although there are only a few membrane proteins whose structures have been determined to high resolution in the nanodisc-embedded state [58,59] (Figure 2d), the fact that structures determined using this approach can also provide information on boundary lipids makes this an appealing alternative to be developed further.
Grid Modification Methods for Specimen Preparation
The principles underlying current methods for cryo-EM specimen preparation have remained largely unchanged from those developed in the early 80's. In the vast majority of cases, samples of purified protein are applied to an electron microscope grid, blotted with filter paper to leave behind a thin aqueous film, and then plunge-frozen in a cryogen such as liquid ethane cooled by liquid nitrogen. This process can result in significant interfacial forces on the suspended proteins, leading sometimes to dissociation of labile complexes, as well as other issues such as preferential orientation, aggregation, and non-homogeneous distribution over the grid, all of which can lower successful outcomes in cryo-EM imaging. A number of methods that attempt to improve the traditional grid preparation process are being reported [60], including the introduction of modifications to grid structure and the use of chemistry to improve protein distribution and cryo-EM image quality.
One interesting category of grid modification includes variations on the theme of “affinity grids”, where, for example, antibodies attached to a grid can capture untagged proteins or complexes out of lysate, without requiring prior purification [61,62]. This has successfully been used for the high resolution 3D reconstruction of an icosahedral virus [63]. A similar approach, using Ni-NTA modified grids to capture His-tagged proteins, has also been used to perform “on-grid” purification [64,65], including for smaller protein complexes (Figure 3a). However, both of these techniques can also have limitations. Not only does the presence of the antibody introduce an additional layer of complexity for 3D reconstruction, but can lead to preferential orientation of the bound macromolecule, which, in turn, leads to loss of structural information and lower resolution in the direction of the electron beam.
Figure 3.
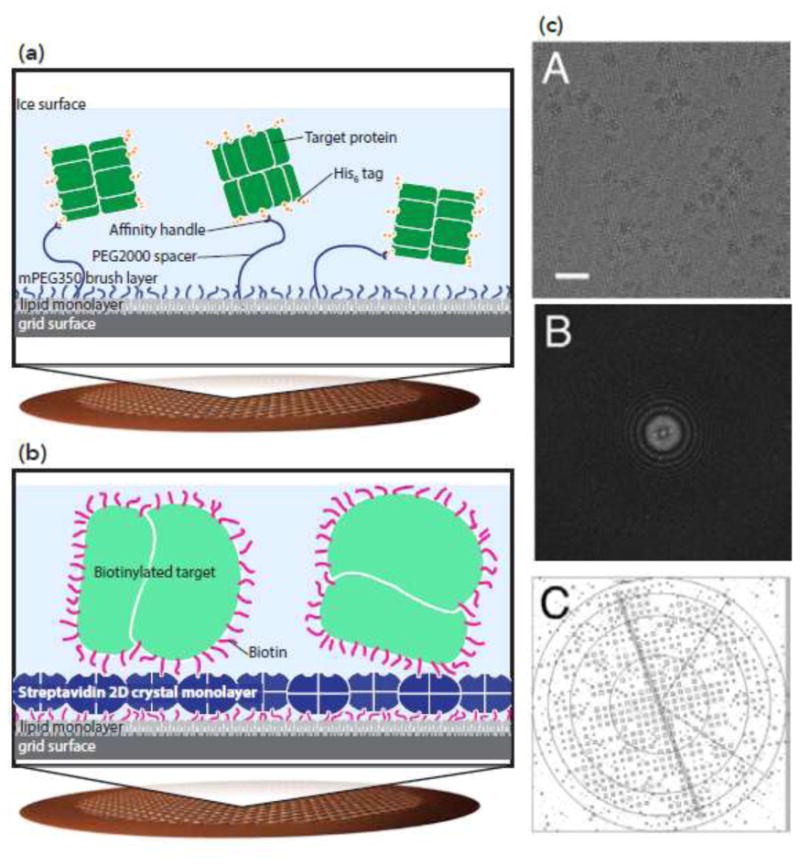
Affinity grids can control protein distribution and orientation. (a) Ni-NTA-based affinity grids. A copper mesh grid with holey carbon is functionalized with polyethylene glycol; long polymers with Ni-NTA are mixed with short polymers. Long polymers capture His-tagged proteins. Adapted from [65]. (b-c) Streptavidin-based affinity grids. (b) 2D crystals of streptavidin are layered on top of holey carbon grids. Biotinylated proteins stick to the streptavidin surface of the grid. (c) The streptavidin crystal can be seen on the surface of the grid, and density for the streptavidin crystal can be computationally removed. Top: cryo-EM micrograph of biotinylated ribosome on streptavidin affinity grid; center: Fourier transform of the image; bottom: IQ plot showing the diffraction pattern from the streptavidin crystal. From [67].
Another, similar method attempts to overcome some of the drawbacks of the antibody-based affinity grid approach. In this technique, a 2D crystal of streptavidin is deposited on the surface of a grid [66] (Figure 3b). Protein complexes of interest (purified in solution) are then biotinylated; because biotin binds non-specifically to the protein surface, this should in theory not result in preferential distribution of orientations. However, because the streptavidin coat is crystalline, the density for the streptavidin can be computationally removed from the micrograph (Figure 3c). Glaeser and colleagues have demonstrated that it is possible to determine high resolution structures of the ribosome using this method [67]. It remains to be seen if this technique can successfully yield high resolution structures also of smaller protein complexes.
An assortment of other methods are also emerging to tackle the general problem of improving specimen quality. The quality of crystallographic structures is generally higher under conditions where the protein is thermally stable [68], and extending this to cryo-EM is being explored in approaches such as the “Proteoplex” screen devised by Stark and colleagues [17]. The use of surface modification to enhance the proportion of protein complexes that distribute into the “holes” of holey carbon grids is another strategy that helps offset preferential binding of protein complexes to carbon film substrates [69]. A more radical approach (“Spotiton”) that potentially eliminates the need for the process of blotting prior to plunge-freezing has been proposed [24, 70]. In this procedure, picoliter sized amounts of protein solution are delivered to individual grid squares, and can be used in conjunction with grids specially modified with carbon nanowires to facilitate better spreading of the aqueous film by capillary action [71].
Conclusion
The application of electron microscopy for protein structure determination, as embodied in single particle cryo-EM, is now firmly established as a cutting-edge method for studying protein complexes spanning a wide range of sizes. The use of single particle cryo-EM to obtain structures of dynamic protein complexes with sizes smaller than 500 kDa has been especially transformative in structural biology. In 2012, prior to the recent advances in detector technology, there were only four single particle cryo-EM structures available in the EMDB of protein complexes with sizes smaller than 500 kDa [72]. None of these structures were at resolutions better than ∼ 8 Å, and few expected then that it would only be four years before numerous single particle cryo-EM structures at resolutions better then 3 Å would be reported for proteins in this size range. As discussed in this review, there is increasing recognition that in addition to the availability of better detectors and improved image processing tools, customized approaches for protein stabilization and specimen preparation will also be necessary to further advance the reach of cryo-EM in the coming years.
Highlights.
The number of structures determined by cryo-EM is continuing to grow rapidly
Computational methods are helping resolve conformational heterogeneity
Improved biochemical methods are driving advances in cryo-EM of membrane proteins
Novel approaches for cryo-EM specimen preparation are being explored
Acknowledgments
We wish to thank Drs. Alberto Bartesaghi and Erin Tran for helpful comments on the manuscript, and Dr. Soojay Banerjee for helpful discussions. This work was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (NIH).
Footnotes
Conflict of interest for Earl, Falconieri, Milne and Subramaniam (2017): Nothing declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1*.Abeyrathne PD, Koh CS, Grant T, Grigorieff N, Korostelev AA. Ensemble cryo-EM uncovers inchworm-like translocation of a viral IRES through the ribosome. Elife. 2016;5 doi: 10.7554/eLife.14874. Computational approaches were used to identify 5 distinct conformational states, from a single sample, for the ribosome during translocation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray J, Savva CG, Shin BS, Dever TE, Ramakrishnan V, Fernandez IS. Structural characterization of ribosome recruitment and translocation by type IV IRES. Elife. 2016;5 doi: 10.7554/eLife.13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Hill MG, Klose T, Chen Z, Watters K, Bochkov YA, Jiang W, Palmenberg AC, Rossmann MG. Atomic structure of a rhinovirus C, a virus species linked to severe childhood asthma. Proc Natl Acad Sci USA. 2016;113:8997–9002. doi: 10.1073/pnas.1606595113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou ZH. Structures of viral membrane proteins by high-resolution cryoEM. Curr Opin Virol. 2014;5:111–119. doi: 10.1016/j.coviro.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5*.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. In this study, the authors determined the structure of the TRPV1 receptor, solubilized in amphipols, to high resolution. This was the first high resolution structure of a membrane protein determined by single particle cryo-EM analysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong X, Qian H, Zhou X, Wu J, Wan T, Cao P, Huang W, Zhao X, Wang X, Wang P, et al. Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell. 2016;165:1467–1478. doi: 10.1016/j.cell.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen TH, Galej WP, Fica SM, Lin PC, Newman AJ, Nagai K. CryoEM structures of two spliceosomal complexes: starter and dessert at the spliceosome feast. Curr Opin Struct Biol. 2016;36:48–57. doi: 10.1016/j.sbi.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agafonov DE, Kastner B, Dybkov O, Hofele RV, Liu WT, Urlaub H, Luhrmann R, Stark H. Molecular architecture of the human U4/U6. U5 tri-snRNP Science. 2016;351:1416–1420. doi: 10.1126/science.aad2085. [DOI] [PubMed] [Google Scholar]
- 9.Yan C, Wan R, Bai R, Huang G, Shi Y. Structure of a yeast activated spliceosome at 3.5 A resolution. Science. 2016;353:904–911. doi: 10.1126/science.aag0291. [DOI] [PubMed] [Google Scholar]
- 10.Nogales E, Louder RK, He Y. Cryo-EM in the study of challenging systems: the human transcription pre-initiation complex. Curr Opin Struct Biol. 2016;40:120–127. doi: 10.1016/j.sbi.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JL, et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell. 2016;165:1698–1707. doi: 10.1016/j.cell.2016.05.040. This study reported the first single particle cryo-EM structure at resolutions better than 2 Å and the first near-atomic resolution structure of a protein smaller than 100 kDa in size. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grigorieff N. Direct detection pays off for electron cryo-microscopy. Elife. 2013;2:e00573. doi: 10.7554/eLife.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai XC, Fernandez IS, McMullan G, Scheres SH. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Elife. 2013;2:e00461. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takizawa Y, Binshtein E, Erwin AL, Pyburn TM, Mittendorf KF, Ohi MD. While the revolution will not be crystallized, biochemistry reigns supreme. Protein Sci. 2017;26:69–81. doi: 10.1002/pro.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stark H, Chari A. Sample preparation of biological macromolecular assemblies for the determination of high-resolution structures by cryo-electron microscopy. Microscopy (Oxf) 2016;65:23–34. doi: 10.1093/jmicro/dfv367. [DOI] [PubMed] [Google Scholar]
- 17.Chari A, Haselbach D, Kirves JM, Ohmer J, Paknia E, Fischer N, Ganichkin O, Moller V, Frye JJ, Petzold G, et al. ProteoPlex: stability optimization of macromolecular complexes by sparse-matrix screening of chemical space. Nat Methods. 2015;12:859–865. doi: 10.1038/nmeth.3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uversky VN. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J Biol Chem. 2016;291:6681–6688. doi: 10.1074/jbc.R115.685859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 20.Aylett CH, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, Maier T. Architecture of human mTOR complex 1. Science. 2016;351:48–52. doi: 10.1126/science.aaa3870. [DOI] [PubMed] [Google Scholar]
- 21.Meyerson JR, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A, Mayer ML, Subramaniam S. Structural mechanism of glutamate receptor activation and desensitization. Nature. 2014;514:328–334. doi: 10.1038/nature13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonic S. Computational methods for analyzing conformational variability of macromolecular complexes from cryo-electron microscopy images. Curr Opin Struct Biol. 2017;43:114–121. doi: 10.1016/j.sbi.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 23.Wlodawer A, Minor W, Dauter Z, Jaskolski M. Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 2013;280:5705–5736. doi: 10.1111/febs.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker MR, Fan G, Serysheva II. Single-Particle Cryo-EM of the Ryanodine Receptor Channel in an Aqueous Environment. Eur J Transl Myol. 2015;25:4803. doi: 10.4081/ejtm.2015.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith JL, Skiniotis G, Sherman DH. Architecture of the polyketide synthase module: surprises from electron cryo-microscopy. Curr Opin Struct Biol. 2015;31:9–19. doi: 10.1016/j.sbi.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shumilin IA, Cymborowski M, Chertihin O, Jha KN, Herr JC, Lesley SA, Joachimiak A, Minor W. Identification of unknown protein function using metabolite cocktail screening. Structure. 2012;20:1715–1725. doi: 10.1016/j.str.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haurowitz F. The balance between hemoglobin and oxygen. Hoppe-Seylers Zeitschrift Fur Physiologische Chemie. 1938;254:266–274. [Google Scholar]
- 28.Quade N, Boehringer D, Leibundgut M, van den Heuvel J, Ban N. Cryo-EM structure of Hepatitis C virus IRES bound to the human ribosome at 3.9-A resolution. Nat Commun. 2015;6:7646. doi: 10.1038/ncomms8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis JH, Tan YZ, Carragher B, Potter CS, Lyumkis D, Williamson JR. Modular Assembly of the Bacterial Large Ribosomal Subunit. Cell. 2016;167:1610–1622 e1615. doi: 10.1016/j.cell.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudson RC, Daniel RM. L-glutamate dehydrogenases: distribution, properties and mechanism. Comp Biochem Physiol B. 1993;106:767–792. doi: 10.1016/0305-0491(93)90031-y. [DOI] [PubMed] [Google Scholar]
- 31.Smith TJ, Schmidt T, Fang J, Wu J, Siuzdak G, Stanley CA. The structure of apo human glutamate dehydrogenase details subunit communication and allostery. J Mol Biol. 2002;318:765–777. doi: 10.1016/S0022-2836(02)00161-4. [DOI] [PubMed] [Google Scholar]
- 32.Borgnia MJ, Banerjee S, Merk A, Matthies D, Bartesaghi A, Rao P, Pierson J, Earl LA, Falconieri V, Subramaniam S, et al. Using Cryo-EM to Map Small Ligands on Dynamic Metabolic Enzymes: Studies with Glutamate Dehydrogenase. Mol Pharmacol. 2016;89:645–651. doi: 10.1124/mol.116.103382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 2016;351:871–875. doi: 10.1126/science.aad7974. With cryo-EM and computational approaches to separate multiple conformations from a single sample, this study showed that these methods can be used to identify novel mechanisms for small molecular inhibitor action at high resolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Huang R, Ripstein ZA, Augustyniak R, Lazniewski M, Ginalski K, Kay LE, Rubinstein JL. Unfolding the mechanism of the AAA+ unfoldase VAT by a combined cryo-EM, solution NMR study. Proc Natl Acad Sci USA. 2016;113:E4190–4199. doi: 10.1073/pnas.1603980113. In this study, the authors used both cryo-EM and complementary approaches including NMR to examine the presence of two distinct conformations of an archaeal ATPase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stahlberg H, Biyani N, Engel A. 3D reconstruction of two-dimensional crystals. Arch Biochem Biophys. 2015;581:68–77. doi: 10.1016/j.abb.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Tribet C, Audebert R, Popot JL. Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc Natl Acad Sci USA. 1996;93:15047–15050. doi: 10.1073/pnas.93.26.15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei R, Wang X, Zhang Y, Mukherjee S, Zhang L, Chen Q, Huang X, Jing S, Liu C, Li S, et al. Structural insights into Ca(2+)-activated long-range allosteric channel gating of RyR1. Cell Res. 2016;26:977–994. doi: 10.1038/cr.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkes M, Madej MG, Kreuter L, Rhinow D, Heinz V, De Sanctis S, Ruppel S, Richter RM, Joos F, Grieben M, et al. Molecular insights into lipid-assisted Ca2+ regulation of the TRP channel Polycystin-2. Nat Struct Mol Biol. 2017 doi: 10.1038/nsmb.3357. [DOI] [PubMed] [Google Scholar]
- 39.Paulsen CE, Armache JP, Gao Y, Cheng Y, Julius D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature. 2015;520:511–517. doi: 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zubcevic L, Herzik MA, Jr, Chung BC, Liu Z, Lander GC, Lee SY. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat Struct Mol Biol. 2016;23:180–186. doi: 10.1038/nsmb.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH. Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife. 2015;4 doi: 10.7554/eLife.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, et al. Three-dimensional structure of human gamma-secretase. Nature. 2014;512:166–170. doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazhab-Jafari MT, Rohou A, Schmidt C, Bueler SA, Benlekbir S, Robinson CV, Rubinstein JL. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature. 2016;539:118–122. doi: 10.1038/nature19828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.des Georges A, Clarke OB, Zalk R, Yuan Q, Condon KJ, Grassucci RA, Hendrickson WA, Marks AR, Frank J. Structural Basis for Gating and Activation of RyR1. Cell. 2016;167:145–157 e117. doi: 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SR, Yan N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science. 2016;354 doi: 10.1126/science.aah5324. [DOI] [PubMed] [Google Scholar]
- 46.Grieben M, Pike AC, Shintre CA, Venturi E, El-Ajouz S, Tessitore A, Shrestha L, Mukhopadhyay S, Mahajan P, Chalk R, et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2) Nat Struct Mol Biol. 2016 doi: 10.1038/nsmb.3343. [DOI] [PubMed] [Google Scholar]
- 47.Huynh KW, Cohen MR, Jiang J, Samanta A, Lodowski DT, Zhou ZH, Moiseenkova-Bell VY. Structure of the full-length TRPV2 channel by cryo-EM. Nat Commun. 2016;7:11130. doi: 10.1038/ncomms11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun L, Zhao L, Yang G, Yan C, Zhou R, Zhou X, Xie T, Zhao Y, Wu S, Li X, et al. Structural basis of human gamma-secretase assembly. Proc Natl Acad Sci USA. 2015;112:6003–6008. doi: 10.1073/pnas.1506242112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyerson JR, Chittori S, Merk A, Rao P, Han TH, Serpe M, Mayer ML, Subramaniam S. Structural basis of kainate subtype glutamate receptor desensitization. Nature. 2016;537:567–571. doi: 10.1038/nature19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, Yan N. Structure of the voltage-gated calcium channel Cav1.1 at 3.6 A resolution. Nature. 2016;537:191–196. doi: 10.1038/nature19321. [DOI] [PubMed] [Google Scholar]
- 51.Fiedorczuk K, Letts JA, Degliesposti G, Kaszuba K, Skehel M, Sazanov LA. Atomic structure of the entire mammalian mitochondrial complex I. Nature. 2016;538:406–410. doi: 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu J, Vinothkumar KR, Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536:354–358. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vinothkumar KR, Zhu J, Hirst J. Architecture of mammalian respiratory complex I. Nature. 2014;515:80–84. doi: 10.1038/nature13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54*.Hauer F, Gerle C, Fischer N, Oshima A, Shinzawa-Itoh K, Shimada S, Yokoyama K, Fujiyoshi Y, Stark H. GraDeR: Membrane Protein Complex Preparation for Single-Particle Cryo-EM. Structure. 2015;23:1769–1775. doi: 10.1016/j.str.2015.06.029. The GraDeR approach allows gentle lowering of the concentration of detergent in solubilized membrane proteins, potentially improving their suitability for high resolution structure determination with cryo-EM. [DOI] [PubMed] [Google Scholar]
- 55.Oshima A, Tani K, Fujiyoshi Y. Atomic structure of the innexin-6 gap junction channel determined by cryo-EM. Nat Commun. 2016;7:13681. doi: 10.1038/ncomms13681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56*.Denisov IG, Sligar SG. Nanodiscs for structural and functional studies of membrane proteins. Nat Struct Mol Biol. 2016;23:481–486. doi: 10.1038/nsmb.3195. This review discusses the formation and use of nanodiscs for a variety of structural and biochemical methods, highlighting how these nanodiscs have been used for cryo-EM structural analyses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matthies D, Dalmas O, Borgnia MJ, Dominik PK, Merk A, Rao P, Reddy BG, Islam S, Bartesaghi A, Perozo E, et al. Cryo-EM Structures of the Magnesium Channel CorA Reveal Symmetry Break upon Gating. Cell. 2016;164:747–756. doi: 10.1016/j.cell.2015.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58*.Gao Y, Cao E, Julius D, Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature. 2016;534:347–351. doi: 10.1038/nature17964. The near-atomic resolution structure of the TRPV1 channel in nanodiscs represents one of the few high resolution structures determined of a nanodisc-embedded membrane protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gatsogiannis C, Merino F, Prumbaum D, Roderer D, Leidreiter F, Meusch D, Raunser S. Membrane insertion of a Tc toxin in near-atomic detail. Nat Struct Mol Biol. 2016;23:884–890. doi: 10.1038/nsmb.3281. [DOI] [PubMed] [Google Scholar]
- 60.Russo CJ, Passmore LA. Progress towards an optimal specimen support for electron cryomicroscopy. Curr Opin Struct Biol. 2016;37:81–89. doi: 10.1016/j.sbi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. Ultrastructural analysis of hepatitis C virus particles. Proc Natl Acad Sci USA. 2013;110:9505–9510. doi: 10.1073/pnas.1307527110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu G, Li K, Jiang W. Antibody-based affinity cryo-EM grid. Methods. 2016;100:16–24. doi: 10.1016/j.ymeth.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu G, Li K, Huang P, Jiang X, Jiang W. Antibody-Based Affinity Cryoelectron Microscopy at 2.6-A Resolution. Structure. 2016;24:1984–1990. doi: 10.1016/j.str.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benjamin CJ, Wright KJ, Bolton SC, Hyun SH, Krynski K, Grover M, Yu G, Guo F, Kinzer-Ursem TL, Jiang W, et al. Selective Capture of Histidine-tagged Proteins from Cell Lysates Using TEM grids Modified with NTA-Graphene Oxide. Sci Rep. 2016;6:32500. doi: 10.1038/srep32500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benjamin CJ, Wright KJ, Hyun SH, Krynski K, Yu G, Bajaj R, Guo F, Stauffacher CV, Jiang W, Thompson DH. Nonfouling NTA-PEG-Based TEM Grid Coatings for Selective Capture of Histidine-Tagged Protein Targets from Cell Lysates. Langmuir. 2016;32:551–559. doi: 10.1021/acs.langmuir.5b03445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, Ounjai P, Sigworth FJ. Streptavidin crystals as nanostructured supports and image-calibration references for cryo-EM data collection. J Struct Biol. 2008;164:190–198. doi: 10.1016/j.jsb.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67*.Han BG, Watson Z, Kang H, Pulk A, Downing KH, Cate J, Glaeser RM. Long shelf-life streptavidin support-films suitable for electron microscopy of biological macromolecules. J Struct Biol. 2016;195:238–244. doi: 10.1016/j.jsb.2016.06.009. Streptavidin 2D crystals attached to cryo-EM grid are used to control the distribution and orientation of biotinylated ribosomes. The authors report a high resolution reconstruction of the ribosome using this method. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deller MC, Kong L, Rupp B. Protein stability: a crystallographer's perspective. Acta Crystallogr F Struct Biol Commun. 2016;72:72–95. doi: 10.1107/S2053230X15024619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meyerson JR, Rao P, Kumar J, Chittori S, Banerjee S, Pierson J, Mayer ML, Subramaniam S. Self-assembled monolayers improve protein distribution on holey carbon cryo-EM supports. Sci Rep. 2014;4:7084. doi: 10.1038/srep07084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jain T, Sheehan P, Crum J, Carragher B, Potter CS. Spotiton: a prototype for an integrated inkjet dispense and vitrification system for cryo-TEM. J Struct Biol. 2012;179:68–75. doi: 10.1016/j.jsb.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Razinkov I, Dandey VP, Wei H, Zhang Z, Melnekoff D, Rice WJ, Wigge C, Potter CS, Carragher B. A new method for vitrifying samples for cryoEM. J Struct Biol. 2016;195:190–198. doi: 10.1016/j.jsb.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Milne JL, Borgnia MJ, Bartesaghi A, Tran EE, Earl LA, Schauder DM, Lengyel J, Pierson J, Patwardhan A, Subramaniam S. Cryo-electron microscopy--a primer for the non-microscopist. FEBS J. 2013;280:28–45. doi: 10.1111/febs.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]