

Abstract
During the development of single particle cryo-EM in past five decades, icosahedral viruses have led the resolution progress owing to their large mass and high symmetry. Many technical advances in cryo-EM were first established with viruses. Since reaching ~4 Å resolution in 2008, it has become a relatively routine task to solve the atomic structure of isolated viruses. The future of structural virology will be increasingly focused on remaining challenges including solving structures of jumbo viruses, intermediate functional states during assembly, maturation, and infection, and in situ structures. Recent demonstrations of near-atomic resolution structure with electron tomography and sub-tomogram averaging opens a new direction for high resolution studies of pleomorphic viruses and the pleomorphic states of icosahedral viruses that have defied past efforts using the single particle cryo-EM approach.
Introduction
In recent years, single particle cryo-electron microscopy (cryo-EM) has matured into a revolutionary structural biology method that can determine near-atomic resolution (2–4 Å) structures of a wide range of macromolecular assemblies from ~64 kDa hemoglobin [1] to ~150 MDa adenovirus (Figure 1E) [2]. In the five decades since late 1960s, viruses, with their advantageous large mass and high symmetry, have played critical roles as model systems for the development of new techniques essential for high resolution imaging and 3-D reconstruction. With a single image containing sufficient 3-D information due to the helical symmetry, the T4 phage tail was the first 3-D structure determined from 2-D images in 1968 [3]. By computationally determining the relative views of multiple particle images of tomato bushy stunt virus in 1970 [4], the concept of single particle TEM imaging and 3-D reconstruction was demonstrated. In 1984, the high contrast image of Adenovirus embedded in vitreous ice by plunge freezing [5] signaled the arrival of cryo-EM. In 1997, cryo-EM progressed to 7.4 Å [6] at which the helices of the capsid protein of Hepatitis B virus core particles could be resolved. With multiple viruses solved at ~4 Å resolution in 2008 [7,8,9], single particle cryo-EM finally broke the barrier for near-atomic resolution. The 3.3 Å aquareovirus structure solved in 2010 (Figure 1F) [10] represents a major advance that allowed de novo full-atomic modeling utilizing the well resolved sidechain densities. With the arrival of direct electron detectors, single particle cryo-EM could also obtain sufficient signal to solve the structure of smaller, lower symmetry macromolecular complexes to near-atomic resolution [11,12]. However, viruses continued to play important roles in further technical developments in cryo-EM. With movie images of viruses, beam induced sample motion [13] was observed and optimal weighting of movie frames [14*] was developed to compensate radiation damages to later frames. Elliptic distortion of the TEM imaging lenses was characterized and computationally corrected [15*,16*], and affinity grid method was shown capable of reaching 2.6 Å resolution (Figure 1B) [17**].
Figure 1.
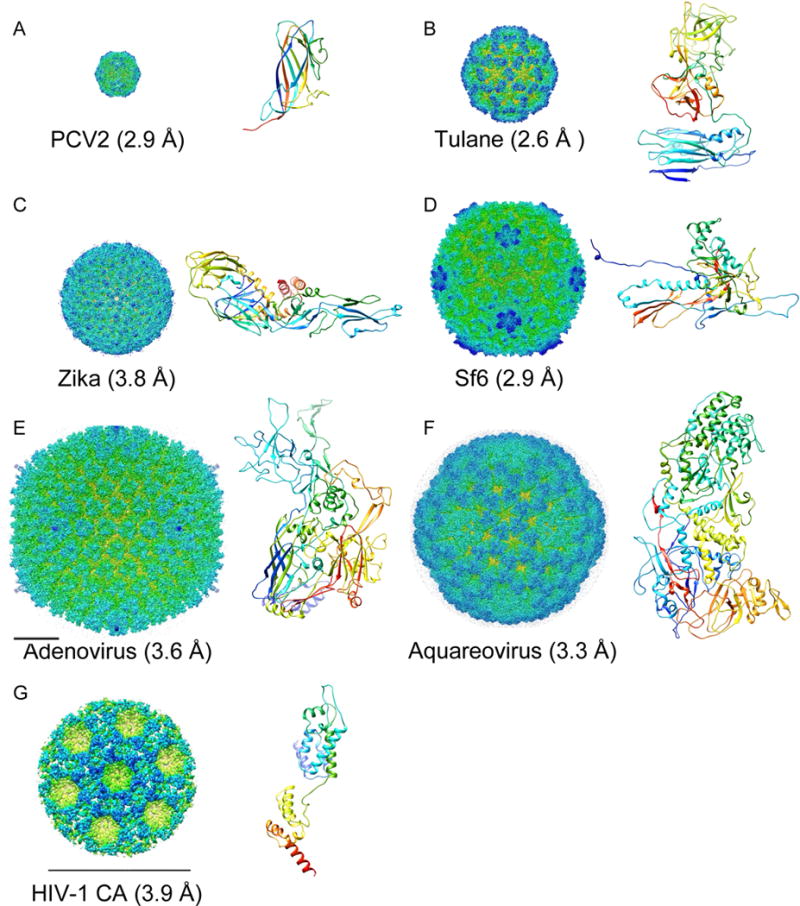
Cryo-EM maps of representative viruses (surface rainbow-colored radially) and atomic models of their major capsid proteins (N➔C rainbow-colored ribbon diagrams). Cryo-EM maps in (A-F) are in the same scale, with the bar in (E) corresponding to 200 Å. The bar in (G) is 200 Å. The capsid protein ribbon diagrams are in the same scale.
Cryo-EM is the method of choice for structural virology
In the 1970s and early 1980s, X-ray crystallography became the first method capable of determining 3-D structures of icosahedral viruses at atomic resolution [18,19]. Since then, X-ray crystallography has been used to solve atomic structures of over 300 icosahedral viruses (http://viperdb.scripps.edu). However, it remains difficult to achieve atomic resolution for large viruses. Virus-like particles (VLP) assembled from heterologously expressed capsid proteins or truncated/mutated versions were often used to facilitate assembly and crystallization. In contrast, cryo-EM can be utilized to image authentic virions and solve near atomic resolution structures within a considerably shorter time span, making cryo-EM the method of choice for structural studies of viruses. For example, an altered adenovirus with short fiber was necessary to obtain crystals diffracting to 3.5 Å resolution [20] while the cryo-EM structure of similar resolution was determined in much shorter time using a wild type virion (Figure 1E) [2]. A recent example is the structure of Zika virus solved by cryo-EM within months of outbreak (Figure 1C) [21**,22*]. Crystals have not yet been reported for any of the large number of tailed dsDNA phages. It took significant effort over a long period of time [23] to achieve 3.44 Å resolution crystal structure of the ~650 Å diameter bacteriophage HK97 VLP [24,25]. In contrast, a single cryo-EM session was used recently to reach a 2.9 Å resolution structure of the icosahedral capsid of bacteriophage Sf6 virion of similar size (Figure 1D) [26**]. This, together with other near atomic resolution cryo-EM structures of large bacterial viruses [27,28,29], has led to new and unbiased understanding enabled by more reliable atomic models.
It has essentially become a routine task to solve symmetric virus structures to near-atomic resolutions using cryo-EM. There has been an explosion of 4 Å and higher resolutions virus structures (Figure 2) across a wide range of sizes (Figure 3).
Figure 2.
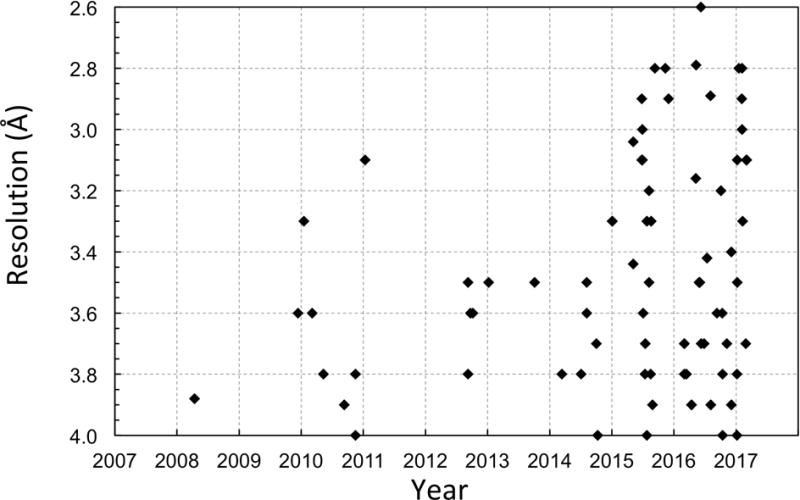
Cryo-EM structures of viruses at better than 4 Å resolution plotted against years.
Figure 3.
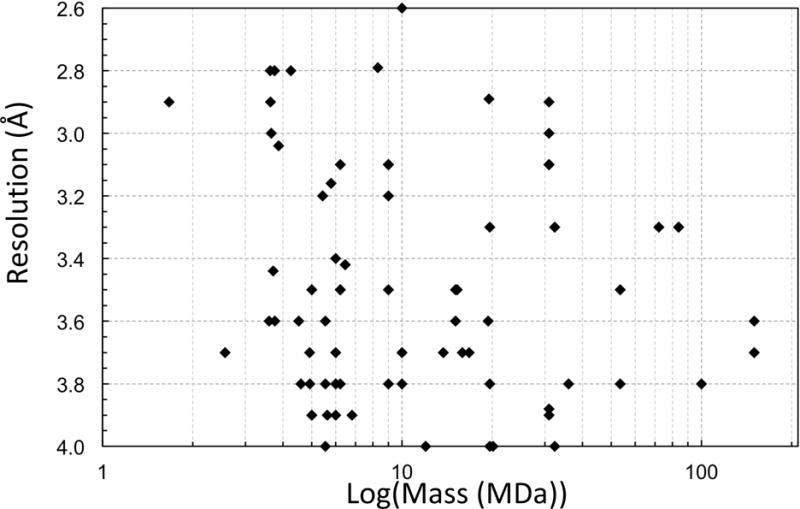
Cryo-EM structures of viruses at better than 4 Å resolution plotted against assembly mass.
Virus structures beyond icosahedral symmetry
The difficulty in crystalizing authentic phage virions is mainly due to tail structure that does not follow the symmetry of the icosahedral capsid [30,31]. Such asymmetric structural features have fallen beyond the reach of methods relying on icosahedral averaging. One ubiquitous asymmetric structural feature in viruses is the viral genome. X-ray crystallographic studies showed that fractions of viral genome were ordered as a result of interactions with capsid proteins, but most of the viral genome was not resolved due to non-crystallographic symmetry averaging and averaging during crystallization. In last two years, cryo-EM has dramatically contributed to our understanding of genome organization in dsRNA [32**,33**] and ssRNA viruses [34,35**]. In the recent 3.6 Å resolution cryo-EM asymmetric reconstruction of (+)RNA bacteriophage, MS2, ~80% of the 3,569 nucleotides in the viral genome could be traced [35**].
Moreover, cryo-EM has been successfully applied in high resolution structural studies of filamentous viruses [36] and HIV capsid structures [37**], which are refractory to crystallization. In the latter cases, the cone-shaped HIV capsid has variable sizes, preventing direct averaging of entire particles by single particle cryo-EM. Instead, cryo– electron tomography (cryo-ET) and subtomogram averaging were utilized to achieve 3.9 Å resolution (Figure 1G) [37**].
Future perspective of structural virology using cryo-EM
As summarized above, it has now essentially become routine to solve the 3-D structure of many small to intermediate sized (<100 nm diameter) icosahedral viruses to near-atomic resolutions using single particle cryo-EM. Nevertheless, many remaining challenges in structural virology remain. We will briefly discuss these challenges and comment on potential solutions.
Atomic structures of jumbo viruses (>100nm)
Currently, the largest virus solved to near-atomic resolution is adenovirus (~90 nm in diameter, 3.6 Å) [2]. Although there is no fundamental barrier to extending similar achievements to jumbo viruses, for example, Herpes (~125 nm) [38], PBCV (~190 nm) [39], and mimivirus (~500 nm) [40], large particle size does pose several practical challenges.
Geometric distortion of the TEM imaging lenses
Several Titan Krios microscopes were found to have 2–3% elliptic distortion which limits the structures of viruses of 70–80 nm in diameter to 7–8 Å resolutions [15*, 16*]. Computational estimation and correction of the distortion were essential to improve these virus structures to 2–4 Å resolutions. The jumbo viruses will be more sensitive to distortion and require more accurate correction. Small variations of magnification and distortion might require per-micrograph or per-particle level correction [16*,41 *]. Correction of additional distortions such as spiral, barrel, and pincushion [42] might become essential.
Focus gradient within a single virus particle
This is equivalent to the Ewald sphere problem. Current CTF correction methods, which assume a single constant defocus for the entire particle, might be insufficient for large, high resolution structures. Although several algorithms have been proposed [43,44,45,46], successful application to experimental data remains elusive. New algorithms will be needed to overcome this challenge. A practical, brutal force approach for addressing this issue is to use a larger number of particles and taking advantage of the icosahedral symmetry. Regardless of the orientation, the asymmetric units near the equator plane of a virus particle will provide accurate structural information consistent with the mean defocus. In contrast, the asymmetric units at the top or bottom of the particle will be out of focus. The effective symmetry of the 60-fold icosahedral symmetry is thus reduced to 30 or fewer, which can be compensated by a larger number of particles. While inelegant, this practical approach is expected to determine atomic structures of jumbo viruses of 100–200 nm in diameter in the near future.
Data collection
Collection of the same number of particles will take significantly more time for jumbo viruses than for smaller viruses. Larger area detectors, for example, the new 8kx8k DE-64 camera, which is about 4x of the size of other direct electron detectors, will help significantly increase data collection efficiency. As observed in the 2.9 Å resolution structure of PCV2 (Figure 1A) solved using a close-packed particle strategy [41*], grids with high particle densities can also be utilized to further improve data collection efficiency.
Non-crystallographic symmetry (NCS) averaging
Larger virus particles also have larger T-numbers, i.e. more subunits of the same protein localized at quasi-equivalent environments in an asymmetric unit [47]. Although the conformations of these subunits can vary significantly, for example, the subunits in the “T=2” inner shell of dsRNA viruses [10,48] and the T=7 dsDNA phages [9,25,26**,27,28], the conformations of these subunits in many viruses, for example, rotavirus VP6 trimers [7,14*], are indistinguishable even at near-atomic resolutions. NCS averaging of these subunits can effectively increase the total symmetry of the particles up to 60T and significantly reduce the number of particles required to reach high resolutions. The reduction of particle number requirement will be proportional to the T-number, potentially reducing the number of particles by several orders for viruses such as: T=16 for Herpes [38], T=169 for PBCV [39], and 972 ≤ T≤ 1200 for mimivirus [40]. By averaging all or a fraction of the subunits, it is possible to keep the data collection effort largely at similar levels for viruses of different sizes, which would allow the jumbo virus structures to be solved more easily.
Computational resources
Large image sizes needed for jumbo virus data analysis pose serious computational challenges. For example, the ~190 nm PBCV would require ~32GB memory for each 3D map of ~20003 voxels, which is beyond the memory of current GPUs used by Relion [49] and CryoSPARC [50] software and the per-core memory of almost all Linux cluster nodes used by jspr [51] and EMAN [52] software. Image processing programs will need to explicitly consider the symmetry and only store a fraction of the structure to reduce the memory requirement. Without algorithmic improvements, a period of 5+ years would be required for future computers to have sufficient resources for image processing of jumbo virus cryo-EM data assuming Moore’s Law.
Structural complexity and heterogeneity
It is a common misunderstanding that viruses are simple icosahedrons. Almost all viruses are more complex for which the complete structural solution is beyond an icosahedral reconstruction.
Symmetry mismatches
All viruses have structural components that have different symmetries than the icosahedral shell. Well known examples include the genome encapsulated inside the capsid (discussed above), the tail attached to the portal vertex of dsDNA phages [30,31], the fibers attached to Adenovirus vertices [53], the RNA polymerases attached to the inner surface of dsRNA viruses [32**,33**], and the nucleocapsid of Flaviviruses [21**,22*,54], etc. When there are three or more components with mismatched symmetries, such as the corestack in the T7 phage capsid I with 5, 12, 8, and 4-fold symmetries in tandem at one of the icosahedral vertices, the combinatory assembly of these components can create a large number, for example, 5*12*8*4=1920, of states that are imaged as a mixture [55]. A focused refinement strategy is needed to zoom in on individual components to resolve them separately [55,56]. The nucleocapsid of Flaviviruses, such as Zika and Dengue, remains elusive to structural studies [21**,22*,54,57]. It is unclear if the core has icosahedral symmetry but with a fixed or random orientation offset to the surrounding icosahedral glycoprotein layer, a different symmetry, or even without a defined structure. We hope the recently increased awareness of Zika virus will help bring sufficient attention to solve this puzzle.
Intermediate states
Many viruses undergo dramatic conformational changes in their assembly and maturation process. Most structural studies have focused on one or a small number of stable states that can be purified in large quantity [28,58,59]. These structures provide a few key frames for the dynamic process of virus assembly and maturation. It is time to extend structural studies to additional sub-states to address questions that cannot be answered by sparse key frames. For instance, how do the trimers in Flavivirus immature particles morph into a raft of dimers in mature particles before being released from host cells and then again into trimers triggered by low pH after infecting new host cells [58,60]? Structural studies of these intermediate states are challenged by the high level of structural heterogeneity of the particles [61,62]. It is conceivable that the paths between the major states represents one or even multiple trajectories and each of the particles can assume any points in the paths. Such a continuously varying conformational heterogeneity is unsolvable by current methods which are limited to a small number of discrete states. Development of new algorithms, such as manifold learning [63**], will be needed to solve this class of problems.
Pleomorphic state of icosahedral virus and inherently pleomorphic viruses
When an icosahedral virus is morphing between two major states, will the intermediate states maintain icosahedral symmetry? Current studies by cryo-EM [61,62] and molecular dynamics simulations typically assume preservation of icosahedral symmetry [64]. The symmetry was even explicitly embedded to accelerate computations [65]. However, more evidence has suggested that symmetry is not necessarily preserved through the maturation process as shown by the half immature and half mature Dengue virus particles [66]. We argue that deviations from symmetry are more likely as the triggers of conformational changes, such as receptor binding, are not likely to occur synchronously for all the 60T subunits and the propagation of the changes cannot be instantaneous. When the 60T subunits undergo independent conformational changes, the virus particles can assume an astronomic number of conformations, for example, 260 or 1018 states for the simplest system of 2 states per subunit for T=1 viruses. The symmetry has become a curse instead of benefit for structural studies and the overall structure is now better regarded as pleomorphic just as those inherently pleomorphic viruses such as influenza and HIV. New reconstruction methods have been proposed to solve this class of problem by only assuming symmetry for the ensemble average instead of individual particles [67]. It remains to be seen if such method can reach high resolution. The recently demonstrated HIV capsid protein at 3.9 Å resolution using sub-tomogram averaging [37**] suggests that tomography has now become a viable approach for high resolution studies of pleomorphic structures by local region classification and averaging.
Structural virology beyond purified virus particles
For viruses to propagate they need to successfully bind the receptor on the host cell surface, enter the cell, replicate the genome, assemble of progeny virions, and release the progeny virions for subsequent rounds of infection. High resolution cryo-EM studies will be needed to provide structural insights to these critical steps of viral infection.
Native state structures
Most structural studies have used highly purified virus particles. The short-lived states or low abundance states could easily be lost by the lengthy purification process. It is also a concern as to whether the virus particles, especially for those unstable enveloped viruses, such as Dengue virus, still maintain their native, functional states and if the cryo-EM structures do genuinely represent the functional states. This concern is corroborated by the discrepancy between the virus concentrations based on plaque assays (i.e. PFU) and particle counts. For example, the particle to PFU ratio for human herpes simplex virus 1 is ~10:1 [68] but ~40,000:1 for Varicella-zoster virus, another human herpes virus [69]. Affinity cryo-EM methods [70,71,72] capture target particles directly from cell lysate without pre-purification. The virus particles are more likely to resemble the native states. In one of the first applications of this approach, multiple capsid states of T7 phage captured from cell lysate were solved to 7–8 Å resolutions[73]. Using Tulane virus as a model system, the antibody affinity cryo-EM approach was shown capable of reaching 2.6 Å resolution [17**], the highest resolution published virus structure. An increase in use of the affinity cryo-EM strategy is anticipated to cross-validate the functional relevance of structures solved using purified samples, and more importantly, to discover novel functional states lost to purification. The affinity cryo-EM method will help reduce the health risk associated with structural studies of highly contagious or dangerous viruses by using smaller quantity samples. It will also help expedite structural solutions to new virus outbreaks by directly utilizing patient samples and forgoing the need of establishing a cell culture system which can take decades for some viruses, such as human noroviruses [74].
In situ structures
A truly native structure is ideally obtained from virus particles in the act of infecting a host cell or being assembled inside a host cell. Such structures are currently available at low resolutions by electron tomography of cell sections [75,76] or small bacterial cells [77,78]. As shown by the success with HIV [37**], sub-tomogram averaging of virus particles, either at the whole particle level or using local regions of the particles, is a promising method for extending tomographic studies to near-atomic resolutions. An alternative approach, which has not been reported yet, is to simply treat the virus particles in the cell sections as standard single particle cryo-EM projects. The crowded cytoplasmic background will be nosier than chemical buffers, however, the section can be set to a thickness just slightly larger than the particle diameter to reduce this effect. A partially cut virus particle should still have sufficient signals to allow reliable determination of its orientation owing to the high redundancy of the icosahedral symmetry. Once the technique barriers for high resolution in situ structures have been overcome, the entire dynamic process of receptor binding, cell entry, uncoating, assembly, and release processes can then be systematically investigated at atomic model resolutions.
Highlights.
Large, symmetric viruses played critical roles in developing cryo-EM techniques
Cryo-EM can routinely solve icosahedral virus structures to 2–4 Å resolution
Cryo-EM has become the method of choice for structural virology
Cryo-EM is poised to solve structures of jumbo viruses and pleomorphic viruses
Structural virology is moving towards solving atomic structures of intermediate functional states and viruses in situ
Acknowledgments
This work was supported by a sub-project of the Kansas COBRE grant P30 GM103326 (to L.T.), and grant R01AI111095 (to W.J.) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Khoshouei M, Radjainia M, Baumeister W, Danev R. Cryo-EM structure of haemoglobin at 3.2 A determined with the Volta phase plate. bioRxiv. 2016 doi: 10.1038/ncomms16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu HR, Jin L, Koh SBS, Atanasov I, Schein S, Wu L, Zhou ZH. Atomic Structure of Human Adenovirus by Cryo-EM Reveals Interactions Among Protein Networks. Science. 2010;329:1038–1043. doi: 10.1126/science.1187433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Rosier DJ, Klug A. Reconstruction of three dimensional structures from electron micrographs. Nature. 1968;217:130–134. doi: 10.1038/217130a0. [DOI] [PubMed] [Google Scholar]
- 4.Crowther RA, Amos LA, Finch JT, De Rosier DJ, Klug A. Three dimensional reconstructions of spherical viruses by fourier synthesis from electron micrographs. Nature. 1970;226:421–425. doi: 10.1038/226421a0. [DOI] [PubMed] [Google Scholar]
- 5.Adrian M, Dubochet J, Lepault J, Mcdowall AW. Cryo-Electron Microscopy of Viruses. Nature. 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- 6.Bottcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Settembre E, Xu C, Dormitzer PR, Bellamy R, Harrison SC, Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu X, Jin L, Zhou ZH. 3.88 A structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature. 2008;453:415–419. doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, Baker ML, Jakana J, Weigele PR, King J, Chiu W. Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature. 2008;451:1130–1134. doi: 10.1038/nature06665. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH. 3.3 angstrom Cryo-EM Structure of a Nonenveloped Virus Reveals a Priming Mechanism for Cell Entry. Cell. 2010;141:472–482. doi: 10.1016/j.cell.2010.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li XM, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng YF. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature Methods. 2013;10:584. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao MF, Cao EH, Julius D, Cheng YF. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brilot AF, Chen JZ, Cheng AC, Pan JH, Harrison SC, Potter CS, Carragher B, Henderson R, Grigorieff N. Beam-induced motion of vitrified specimen on holey carbon film. Journal of Structural Biology. 2012;177:630–637. doi: 10.1016/j.jsb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 angstrom reconstruction of rotavirus VP6. Elife. 2015:4. doi: 10.7554/eLife.06980. This work developed a weighting scheme to optimally compensate the radiation damages in different movie frames. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15*.Grant T, Grigorieff N. Automatic estimation and correction of anisotropic magnification distortion in electron microscopes. Journal of Structural Biology. 2015;192:204–208. doi: 10.1016/j.jsb.2015.08.006. This work dicovered significant elliptic distortion of a Titan Krios microscope and developed a pre-calibration method to computationally correct the distortion at micrograph level. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Yu GM, Li KP, Liu Y, Chen ZG, Wang ZQ, Yan R, Klose T, Tang L, Jiang W. An algorithm for estimation and correction of anisotropic magnification distortion of cryo-EM images without need of pre-calibration. Journal of Structural Biology. 2016;195:207–215. doi: 10.1016/j.jsb.2016.06.003. This work developed a method to estimate and correct elliptic distortions at particle level without the need of pre-calibration. With this method, several structures were improved to 2.4–2.6 Angstrom resolutions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17**.Yu G, Li K, Huang P, Jiang X, Jiang W. Antibody-Based Affinity Cryoelectron Microscopy at 2.6-Å Resolution. Structure. 2016;24:1984–1990. doi: 10.1016/j.str.2016.09.008. This work demonstrated the feasibility of reaching near-atomic resolution using antibody affinity grid method. The 2.6 Angstrom is currently the highest resolution for cryo-EM virus structures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abad-Zapatero C, Abdel-Meguid SS, Johnson JE, Leslie AG, Rayment I, Rossmann MG, Suck D, Tsukihara T. Structure of southern bean mosaic virus at 2.8 A resolution. Nature. 1980;286:33–39. doi: 10.1038/286033a0. [DOI] [PubMed] [Google Scholar]
- 19.Harrison SC, Olson AJ, Schutt CE, Winkler FK, Bricogne G. Tomato bushy stunt virus at 2.9 A resolution. Nature. 1978;276:368–373. doi: 10.1038/276368a0. [DOI] [PubMed] [Google Scholar]
- 20.Reddy VS, Natchiar SK, Stewart PL, Nemerow GR. Crystal Structure of Human Adenovirus at 3.5 angstrom Resolution. Science. 2010;329:1071–1075. doi: 10.1126/science.1187292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21**.Sirohi D, Chen ZG, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ. The 3.8 angstrom resolution cryo-EM structure of Zika virus. Science. 2016;352:467–470. doi: 10.1126/science.aaf5316. This work reported an atomic structure of Zika virus within a few months of outbreak. This is a good example showing that cryo-EM has become of the method of choice for structural virology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22*.Kostyuchenko VA, Lim EXY, Zhang SJ, Fibriansah G, Ng TS, Ooi JSG, Shi J, Lok SM. Structure of the thermally stable Zika virus. Nature. 2016;533:425. doi: 10.1038/nature17994. This work also reported an atomic structure of Zika virus using cryo-EM just a few months after the outbreak but a few weeks after the Science paper. [DOI] [PubMed] [Google Scholar]
- 23.Johnson JE. Confessions of an icosahedral virus crystallographer. Microscopy (Oxf) 2013;62:69–79. doi: 10.1093/jmicro/dfs097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gertsman I, Gan L, Guttman M, Lee K, Speir JA, Duda RL, Hendrix RW, Komives EA, Johnson JE. An unexpected twist in viral capsid maturation. Nature. 2009;458:646–650. doi: 10.1038/nature07686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. Topologically linked protein rings in the bacteriophage HK97 capsid. Science. 2000;289:2129–2133. doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- 26**.Zhao H, Li K, Lynn AY, Aron KE, Yu G, Jiang W, Tang L. Structure of a headful DNA-packaging bacterial virus at 2.9 A resolution by electron cryo-microscopy. Proc Natl Acad Sci U S A. 2017;114:3601–3606. doi: 10.1073/pnas.1615025114. The highest resolution and the first sub-3 Angstrom resolution structure of tailed dsDNA phages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hryc CF, Chen DH, Afonine PV, Jakana J, Wang Z, Haase-Pettingell C, Jiang W, Adams PD, King JA, Schmid MF, et al. Accurate model annotation of a near-atomic resolution cryo-EM map. Proc Natl Acad Sci U S A. 2017;114:3103–3108. doi: 10.1073/pnas.1621152114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo F, Liu Z, Fang PA, Zhang Q, Wright ET, Wu W, Zhang C, Vago F, Ren Y, Jakana J, et al. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc Natl Acad Sci U S A. 2014;111:E4606–4614. doi: 10.1073/pnas.1407020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai W, Hodes A, Hui WH, Gingery M, Miller JF, Zhou ZH. Three-dimensional structure of tropism-switching Bordetella bacteriophage. Proc Natl Acad Sci U S A. 2010;107:4347–4352. doi: 10.1073/pnas.0915008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science. 2006;312:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 31.Jiang W, Chang J, Jakana J, Weigele P, King J, Chiu W. Structure of epsilon15 bacteriophage reveals genome organization and DNA packaging/injection apparatus. Nature. 2006;439:612–616. doi: 10.1038/nature04487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Zhang X, Ding K, Yu X, Chang W, Sun J, Zhou ZH. In situ structures of the segmented genome and RNA polymerase complex inside a dsRNA virus. Nature. 2015;527:531–534. doi: 10.1038/nature15767. 3D organization of the 10 genomic dsRNA molecules and the viral RNA-dependent RNA polymerase molecules in a dsRNA virus in both quiescent and transcribing states were visualized at high resolution for the first time. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33**.Liu H, Cheng L. Cryo-EM shows the polymerase structures and a nonspooled genome within a dsRNA virus. Science. 2015;349:1347–1350. doi: 10.1126/science.aaa4938. 3D organization of the 10 genomic dsRNA molecules and the viral RNA-dependent RNA polymerase molecules in a dsRNA virus in both quiescent and transcribing states were visualized at high resolution for the first time. [DOI] [PubMed] [Google Scholar]
- 34.Koning RI, Gomez-Blanco J, Akopjana I, Vargas J, Kazaks A, Tars K, Carazo JM, Koster AJ. Asymmetric cryo-EM reconstruction of phage MS2 reveals genome structure in situ. Nat Commun. 2016;7:12524. doi: 10.1038/ncomms12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Dai X, Li Z, Lai M, Shu S, Du Y, Zhou ZH, Sun R. In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature. 2017;541:112–116. doi: 10.1038/nature20589. The majority of the ssRNA genome was traced in this 3.6 Å resolution cryoEM asymmetric reconstruction. Atomic models were built for sixteen RNA stem-loop elements. The structure revealed a stunning array of RNA:RNA, RNA:capsid and RNA:maturation protein interactions, providing rich insights into viral genome organization, genome packaging during virus assembly and genome delivery upon infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DiMaio F, Yu X, Rensen E, Krupovic M, Prangishvili D, Egelman EH. Virology. A virus that infects a hyperthermophile encapsidates A-form DNA Science. 2015;348:914–917. doi: 10.1126/science.aaa4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37**.Schur FK, Obr M, Hagen WJ, Wan W, Jakobi AJ, Kirkpatrick JM, Sachse C, Krausslich HG, Briggs JA. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science. 2016;353:506–508. doi: 10.1126/science.aaf9620. This work has demonstrated the feasibility of reaching 3.9 angstrom resolution using electron tomography and subtomogram averaging. It opens a new direction for high-resolution structural studies of pleomorphic viruses and for visualization of biological molecules in their native, cellular environments at atomic detail. [DOI] [PubMed] [Google Scholar]
- 38.Dai XH, Gong DY, Wu TT, Sun R, Zhou ZH. Organization of Capsid-Associated Tegument Components in Kaposi’s Sarcoma-Associated Herpesvirus. Journal of Virology. 2014;88:12694–12702. doi: 10.1128/JVI.01509-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang XZ, Xiang Y, Dunigan DD, Klose T, Chipman PR, Van Etten JL, Rossmann MG. Three-dimensional structure and function of the Paramecium bursaria chlorella virus capsid. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14837–14842. doi: 10.1073/pnas.1107847108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao C, Kuznetsov YG, Sun SY, Hafenstein SL, Kostyuchenko VA, Chipman PR, Suzan-Monti M, Raoult D, McPherson A, Rossmann MG. Structural Studies of the Giant Mimivirus. Plos Biology. 2009;7:958–966. doi: 10.1371/journal.pbio.1000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41*.Liu Z, Guo F, Wang F, Li TC, Jiang W. 2.9 A Resolution Cryo-EM 3D Reconstruction of Close-Packed Virus Particles. Structure. 2016;24:319–328. doi: 10.1016/j.str.2015.12.006. This works showed that close-packed particles can also lead to high resolution structures if a proper image processing method is used. It also showed the highest resolution and the only sub-3 Angstrom structure using images recorded on photographic film. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capitani GC, Oleynikov P, Hovmoller S, Mellini M. A practical method to detect and correct for lens distortion in the TEM. Ultramicroscopy. 2006;106:66–74. doi: 10.1016/j.ultramic.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 43.DeRosier DJ. Correction of high-resolution data for curvature of the Ewald sphere. Ultramicroscopy. 2000;81:83–98. doi: 10.1016/s0304-3991(99)00120-5. [DOI] [PubMed] [Google Scholar]
- 44.Wan Y, Chiu W, Zhou ZH. Communications, Circuits and Systems, 2004. IEEE; 2004. Full contrast transfer function correction in 3D cryo-EM reconstruction; pp. 960–964. ICCCAS 2004. 2004 International Conference on. [Google Scholar]
- 45.Wolf M, DeRosier DJ, Grigorieff N. Ewald sphere correction for single-particle electron microscopy. Ultramicroscopy. 2006;106:376–382. doi: 10.1016/j.ultramic.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 46.Leong PA, Yu X, Zhou ZH, Jensen GJ. Correcting for the Ewald Sphere in High-Resolution Single-Particle Reconstructions. Methods in Enzymology. 2010:369–380. doi: 10.1016/S0076-6879(10)82015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caspar DL, Klug A. Cold Spring Harbor symposia on quantitative biology. Cold Spring Harbor Laboratory Press; 1962. Physical principles in the construction of regular viruses; pp. 1–24. [DOI] [PubMed] [Google Scholar]
- 48.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. Atomic model of an infectious rotavirus particle. EMBO J. 2011;30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimanius D, Forsberg BO, Scheres SH, Lindahl E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife. 2016;5:e18722. doi: 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 51.Guo F, Jiang W. Single particle cryo-electron microscopy and 3-D reconstruction of viruses. Methods Mol Biol. 2014;1117:401–443. doi: 10.1007/978-1-62703-776-1_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 53.Yu X, Veesler D, Campbell MG, Barry ME, Asturias FJ, Barry MA, Reddy VS. Cryo-EM structure of human adenovirus D26 reveals the conservation of structural organization among human adenoviruses. Science Advances. 2017;3 doi: 10.1126/sciadv.1602670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang XK, Ge P, Yu XK, Brannan JM, Bi GQ, Zhang QF, Schein S, Zhou ZH. Cryo-EM structure of the mature dengue virus at 3.5-angstrom resolution. Nature Structural & Molecular Biology. 2013;20:105–U133. doi: 10.1038/nsmb.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo F, Liu Z, Vago F, Ren Y, Wu W, Wright ET, Serwer P, Jiang W. Visualization of uncorrelated, tandem symmetry mismatches in the internal genome packaging apparatus of bacteriophage T7. Proc Natl Acad Sci U S A. 2013;110:6811–6816. doi: 10.1073/pnas.1215563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ilca SL, Kotecha A, Sun XY, Poranen MM, Stuart DI, Huiskonen JT. Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nature Communications. 2015;6 doi: 10.1038/ncomms9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, Kostyuchenko VA, Rossmann MG. Structural analysis of viral nucleocapsids by subtraction of partial projections. J Struct Biol. 2007;157:356–364. doi: 10.1016/j.jsb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science. 2008;319:1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- 59.Prasad VM, Miller AS, Klose T, Sirohi D, Buda G, Jiang W, Kuhn RJ, Rossmann MG. Structure of the immature Zika virus at 9 angstrom resolution. Nature Structural & Molecular Biology. 2017;24:184. doi: 10.1038/nsmb.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kostyuchenko VA, Zhang Q, Tan JL, Ng TS, Lok SM. Immature and Mature Dengue Serotype 1 Virus Structures Provide Insight into the Maturation Process. Journal of Virology. 2013;87:7700–7707. doi: 10.1128/JVI.00197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lata R, Conway JF, Cheng NQ, Duda RL, Hendrix RW, Wikoff WR, Johnson JE, Tsuruta H, Steven AC. Maturation dynamics of a viral capsid: Visualization of transitional intermediate states. Cell. 2000;100:253–263. doi: 10.1016/s0092-8674(00)81563-9. [DOI] [PubMed] [Google Scholar]
- 62.Heymann JB, Cheng NQ, Newcomb WW, Trus BL, Brown JC, Steven AC. Dynamics of herpes simplex virus capsid maturation visualized by time-lapse cryo-electron microscopy. Nature Structural Biology. 2003;10:334–341. doi: 10.1038/nsb922. [DOI] [PubMed] [Google Scholar]
- 63**.Frank J, Ourmazd A. Continuous changes in structure mapped by manifold embedding of single-particle data in cryo-EM. Methods. 2016;100:61–67. doi: 10.1016/j.ymeth.2016.02.007. This paper proposed a manifold embedding method in machine learning to solve the structures of continously varying conformations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roy A, Post CB. Long-distance correlations of rhinovirus capsid dynamics contribute to uncoating and antiviral activity. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5271–5276. doi: 10.1073/pnas.1119174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cagin T, Holder M, Pettitt BM. A Method for Modeling Icosahedral Virions -Rotational Symmetry Boundary-Conditions. Journal of Computational Chemistry. 1991;12:627–634. [Google Scholar]
- 66.Plevka P, Battisti AJ, Sheng J, Rossmann MG. Mechanism for maturation-related reorganization of flavivirus glycoproteins. Journal of Structural Biology. 2014;185:27–31. doi: 10.1016/j.jsb.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu N, Doerschuk PC. Engineering in Medicine and Biology Society (EMBC), 2016 IEEE 38th Annual International Conference of the. IEEE; 2016. Statistical characterization of ensembles of symmetric virus particles: 3-D stochastic signal reconstruction from electron microscope images; pp. 3977–3980. [DOI] [PubMed] [Google Scholar]
- 68.Watson D, Russell W, Wildy P. Electron microscopic particle counts on herpes virus using the phosphotungstate negative staining technique. Virology. 1963;19:250–260. doi: 10.1016/0042-6822(63)90062-x. [DOI] [PubMed] [Google Scholar]
- 69.Carpenter JE, Henderson EP, Grose C. Enumeration of an extremely high particle-to-PFU ratio for Varicella-zoster virus. J Virol. 2009;83:6917–6921. doi: 10.1128/JVI.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kelly DF, Dukovski D, Walz T. Monolayer purification: a rapid method for isolating protein complexes for single-particle electron microscopy. Proc Natl Acad Sci U S A. 2008;105:4703–4708. doi: 10.1073/pnas.0800867105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu G, Li K, Jiang W. Antibody-based affinity cryo-EM grid. Methods. 2016;100:16–24. doi: 10.1016/j.ymeth.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benjamin CJ, Wright KJ, Hyun SH, Krynski K, Yu G, Bajaj R, Guo F, Stauffacher CV, Jiang W, Thompson DH. Nonfouling NTA-PEG-Based TEM Grid Coatings for Selective Capture of Histidine-Tagged Protein Targets from Cell Lysates. Langmuir. 2016;32:551–559. doi: 10.1021/acs.langmuir.5b03445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang C, Vago F, Guo F, Liu Z, Yu G, Serwer P, Jiang W. Affinity cryo-microscopy studies of viral particles captured directly from cell culture. Microscopy and Microanalysis. 2015;21:547–548. [Google Scholar]
- 74.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, et al. Replication of human noroviruses in stem cell-derived human enteroids. Science. 2016;353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang K, Strunk K, Zhao G, Gray JL, Zhang P. 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. Journal of structural biology. 2012;180:318–326. doi: 10.1016/j.jsb.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Romero-Brey I, Bartenschlager R. Viral infection at high magnification: 3D electron microscopy methods to analyze the architecture of infected cells. Viruses. 2015;7:6316–6345. doi: 10.3390/v7122940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dai W, Fu C, Raytcheva D, Flanagan J, Khant HA, Liu X, Rochat RH, Haase-Pettingell C, Piret J, Ludtke SJ, et al. Visualizing virus assembly intermediates inside marine cyanobacteria. Nature. 2013;502:707–710. doi: 10.1038/nature12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fu CY, Wang K, Gan L, Lanman J, Khayat R, Young MJ, Jensen GJ, Doerschuk PC, Johnson JE. In vivo assembly of an archaeal virus studied with whole-cell electron cryotomography. Structure. 2010;18:1579–1586. doi: 10.1016/j.str.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]