

Abstract
Human WBSCR22 gene is involved in tumor metastasis, cell growth and invasion, however, its role in chemosensitivity to antitumor agents remains unknown. In this study, we analyzed the TCGA cohort and found the expression of WBSCR22 was significantly elevated in human colorectal cancer (CRC) tissue. WBSCR22 could be served as an independent risk predictor for overall survival (OS), and up-regulated WBSCR22 could predict unfavorable OS for CRC patients. Knockdown of WBSCR22 significantly sensitized CRC cells to oxaliplatin in vitro and in vivo, while overexpression of WBSCR22 led to cellular resistance to oxaliplatin treatment. Although WBSCR22 knockdown did not change cell cycle, it increased the oxaliplatin-induced cellular apoptosis. WBSCR22 knockdown augmented the oxaliplatin-induced intracellular reactive oxygen species (ROS) production and ROS-induced 8-oxoguanine (8-oxoG) oxidative lesion accumulation, likely sensitizing oxaliplatin treatment. These results demonstrate that WBSCR22 is involved in CRC resistance to oxaliplatin, suggesting WBSCR22 may represent a novel oxaliplatin resistance biomarker as well as a potentail target for CRC therapeutics.
Introduction
Human WBSCR22 gene was initially identified as one of 26 genes deleted in Williams-Beuren syndrome characterized by dysmorphic facial features, congenital heart and vascular disease, unique cognitive1–3. While the mRNA was detected ubiquitously in all tissues, and the protein was markedly expressed in heart, skeletal muscle and kidney4,5. WBSCR22 contains a nuclear localization signal and a common S-adenosyl-L-methionine binding motif that is evolutionarily conserved in methyltransferases6. However, the specific cellular function of WBSCR22 is still poorly understood.
WBSCR22 was over-expressed in invasive breast cancer, and its ectopic expression in non-metastatic cells significantly promoted the metastasis formation by suppressing Zac1/p53-dependent apoptosis, but no effects on cell growth and motility7. In multiple myeloma, WBSCR22 was required for the tumor cells to survive8, and its product was up-regulated in both primary plasma cells and primary multiple myeloma tumor cells, indicating its roles in plasma cell biology8. Knockdown of WBSCR22 attenuated cell growth and invasive abilities in multiple cells9,10. However, WBSCR22 was significantly down-regulated in lung inflammatory and neoplastic pathologies11, suggesting its diverse functions in the context of cellular environment. WBSCR22 was also identified as a novel glucocorticoid receptor (GR) co-modulator by regulating GR recruitment to the genome as well as mediating subsequent histone modification through binding to the histone-associated proteins and protein kinases11.
Human colorectal cancer (CRC) is continuously the third most common cancer and the third most common cause of cancer-related death worldwide12. Oxaliplatin, a third-generation platinum-based antitumor agent, is widely used as the standard first-line chemotherapy for CRC. However, the development of chemoresistance limits its effectiveness in clinical practice. While many mechanisms were identified for oxaliplatin resistance13–16, recent studies showed that DNA hypermethylation, histone post-translational modifications and microRNAs were also involved in chemoresistance15,17,18.
In the present study, we analyzed the TCGA cohort and found WBSCR22 was significantly expressed in human CRC tissue. We further investigated the effects of WBSCR22 on oxaliplatin sensitivity in CRC, showed that WBSCR22 knockdown significantly sensitized CRC cell to oxaliplatin in vitro and in vivo. WBSCR22 knockdown also induced the cell apoptosis and increased oxaliplatin-induced intracellular ROS production and ROS-induced 8-oxoG oxidative lesion accumulation. The results show that WBSCR22 induces the chemosensitivity to oxaliplatin in CRC, suggesting it may represent a novel resistance biomarker as well as a potential target for colorectal cancer therapy.
Results
WBSCR22 mRNA was elevated in human CRC and served as an independent prognostic factor for CRC patients
We analyzed the mRNA expression profile of WBSCR22 in an independent TCGA cohort, and found WBSCR22 had more than 2-fold higher expression in 13% (46/348) of CRC tissues than 32 cases of normal controls (Fig. 1A). It was significantly elevated in CRC tissues, however, there was no significant differences across the TNM stages (Fig. 1B). The association between CRC lymphatic invasion and high WBSCR22 expression was statistically significant (p = 0.011) (Table 1). Kaplan-Meier analyses showed that all CRC patients with high WBSCR22 expression had a significantly shorter overall survival(OS) than low expression, therefore, WBSCR22 could predict significantly unfavorable OS for patients with high WBSCR22 exporession level (Fig. 1C). Subsequent univariate and multivariate Cox regression analyses were performed to determine the independence of the prognostic value of WBSCR22. After correction for clinical characteristics, high WBSCR22 expression was found to be an independent risk predictor of OS (p = 0.005, HR = 2.391, 95% CI = 1.310–4.365) for CRC patients (Table 2).
Figure 1.

WBSCR22 gene was over-expressed and predicted a poor clinical outcome in human CRC of the TCGA cohort. (A) The expression of WBSCR22 in 348 clinical CRC specimens in the TCGA cohort. The red bars represented CRC samples having a more than 2-fold higher WBSCR22 expression than normal samples. (B) WBSCR22 levels were compared between normal samples and different NMT stages of CRC samples. A single spot represented the WBSCR22 expression value of an individual sample, and the results were expressed as the mean ± SE. ***p < 0.001. (C) Kaplan-Meier survival curves were plotted according to the different WBSCR22 mRNA level of all CRC patients. p values were obtained from log-rank test, while hazard ratio (HR) and 95% confidence interval (CI) were determined by univariate Cox regression model.
Table 1.
The clinicopathological characteristics of the CRC patients in the TCGA cohort used in this study.
| Characteristics | Cases (n = 348) | WBSCR22 mRNA expression | p | |
|---|---|---|---|---|
| No. (%) | Low (n = 302) | High (n = 46) | ||
| Gender | ||||
| Male | 191(54.9) | 164(54.3) | 27(58.7) | 0.577 |
| Female | 157(45.1) | 138(45.7) | 19(41.3) | |
| Age | ||||
| ≥60 | 227(65.2) | 196(64.9) | 31(67.4) | 0.741 |
| <60 | 121(34.8) | 106(35.1) | 15(32.6) | |
| Primary site | ||||
| Colon | 266(76.4) | 234(77.5) | 32(69.6) | 0.238 |
| Rectum | 82(23.6) | 68(22.5) | 14(30.4) | |
| Histological type | ||||
| Adenocarcinoma | 307(88.2) | 266(88.1) | 41(89.1) | 0.837 |
| Mucinous adenocarcinoma | 41(11.8) | 36(11.9) | 5(10.9) | |
| NMT stage | ||||
| Stage I + II | 187(53.7) | 164(54.3) | 23(50.0) | 0.585 |
| Stage III + IV | 161(46.3) | 138(45.7) | 23(50.0) | |
| Venous invasion | ||||
| Negative | 232(66.7) | 207(68.5) | 25(54.3) | 0.071 |
| Positive | 68(19.5) | 58(19.2) | 10(21.7) | |
| Unkonw | 48(13.8) | 37(12.3) | 11(24.0) | |
| Lymphatic invasion | ||||
| Negative | 213(61.2) | 192(63.6) | 21(45.7) | 0.011 |
| Positive | 94(27.0) | 80(26.5) | 14(30.4) | |
| Unkonw | 41(11.8) | 30(9.9) | 11(23.9) | |
Table 2.
Univariate and multivariate Cox proportional hazards analysis of OS for the CRC patients in the TCGA cohort (HR: hazard ratio, CI: confidence interval).
| Characteristics | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| p | HR (95% CI) | p | HR (95% CI) | |
| Gender | ||||
| Male | 0.197 | 1.000 | ||
| Female | 0.720(0.437–1.186) | |||
| Age | ||||
| ≥60 | 0.018 | 1.000 | 0.003 | 1.000 |
| <60 | 0.479(0.261–0.879) | 0.376(0.199–0.713) | ||
| Primary site | ||||
| Colon | 0.157 | 1.000 | ||
| Rectum | 0.599(0.295–1.217) | |||
| Histological type | ||||
| Adenocarcinoma | 0.001 | 1.000 | 0.036 | 1.000 |
| Mucinous adenocarcinoma | 0.405(0.234–0.699) | 2.075(1.050–4.101) | ||
| TNM stage | ||||
| Stage I + II | 0.00035 | 1.000 | 0.002 | 1.000 |
| Stage III + IV | 2.518(1.517–4.179) | 2.419(1.376–4.252) | ||
| Venous invasion | ||||
| Negative | 0.002 | 1.000 | 0.235 | 1.000 |
| Positive | 2.604(1.498–4.527) | 1.380(0.607–3.138) | ||
| Unkonw | 1.823(0.939–3.542) | 0.401(0.089–1.805) | ||
| Lymphatic invasion | ||||
| Negative | 0.006 | 1.000 | 0.471 | 1.000 |
| Positive | 2.314(1.350–3.965) | 1.373(0.594–3.173) | ||
| Unkonw | 1.970(0.965–4.020) | 2.679(0.516–13.895) | ||
| WBSCR22 | ||||
| Low | 0.001 | 1.000 | 0.005 | 1.000 |
| High | 2.471(1.432–4.265) | 2.391(1.310–4.365) | ||
The effects of WBSCR22 knockdown on cell proliferation of CRC cells
To evaluate the significance of WBSCR22 in CRC, we examined the endogenous WBSCR22 expression in human CRC cell lines by Western blot. The results showed that most CRC cells expressed high WBSCR22 protein, consistent with the TCGA data. We also measured the WBSCR22 expression in CRC cells with different oxaliplatin sensitivity. Interestingly, the WBSCR22 expression level was correlated with their oxaliplatin sensitivity (Supplementary Fig. S1).
We next knocked down WBSCR22 gene in HT-29, Caco-2 and RKO cell lines. Western blot and RT-qPCR showed shRNA1 significantly decreased the expression of WBSCR22 at both mRNA and protein levels (60–70% decrease) (Fig. 2A,B). We then assessed the effects of WBSCR22 knockdown on cell proliferation. For RKO cells, WBSCR22 knockdown significantly increased the cell proliferation (p < 0.01), and the result was further confirmed by colony formation assay (Fig. 2C,D). For HT-29 and Caco-2 cells, however, there were no significant difference in the cell prolifreation dected by either MTT assay (Fig. 2C) or colony formation (Fig. 2D). Cell flow cytometry, however, showed WBSCR22 knockdown did not change the cell cycle distributions in all three cell lines (Fig. 2E).
Figure 2.

The effects of WBSCR22 knockdown on cell proliferation and cell cycle of human CRC cells. (A) Western blot of WBSCR22 in wild-type (WT), control (shCon), and WBSCR22-knockdown (shRNA1) CRC cells. The β-actin was used as an internal control. (B) RT-qPCR analysis of WBSCR22 expression in WT, shCon, and shRNA1 treated CRC cells. **p < 0.01 (shRNA1 v.s. either WT or shCon). The results were from three independent experiments in triplicate and the data were expressed as the mean ± SD. (C) Cell proliferation analysis of the shCon and shRNA1 treated cells by MTT assays. **p < 0.01 (shRNA1 v.s. shCon). The results were representative of three independent experiments in quadruplicate and the results were expressed as the mean ± SD. (D) Colony formation assay of the shCon and shRNA1 treated cells. (E) Cell cycle distribution by flow cytometry analysis. Percentages of cell populations in each phase of the cell cycle were indicated.
The effects of WBSCR22 on oxaliplatin sensitivity in vitro
To investigate the effect of WBSCR22 on oxaliplatin sensitivity, MTT and colony formation assays were performed. The cells pretreated with shCon and shRNA1 were incubated with oxaliplatin at various concentrations for 72 hours, and IC50 values were determined. The IC50 values for oxaliplatin in the shCon and shRNA1 treated cells were 12.39 ± 0.66 and 3.78 ± 0.14 μM for HT-29 cells; 43.31 ± 2.67 and 14.05 ± 1.09 μM for Caco-2 cells; 5.76 ± 0.20 and 1.41 ± 0.21 μM for RKO cells, respectively. In all three lines, the IC50 for the shRNA1 treated cells were significantly reduced compared to the shCon cells (3.28-fold, 3.08-fold and 4.09-fold decrease for HT-29, Caco-2 and RKO cells, respectively) (Fig. 3A), indicating that WBSCR22 knockdown could sensitize CRC cells to oxaliplatin. Indeed, colony formation assays showed that the number of colonies upon oxaliplatin treatment for 48 hours was significantly decreased after WBSCR22 knockdown (Fig. 3B,C). We also performed the assays in Caco-2 cells using an additional shRNA2. The efficiency of knockdown and the effects on oxaliplatin sensitivity were comparable to shRNA1 (Supplementary Fig. S2).
Figure 3.

WBSCR22 knockdown sensitized CRC cells to oxaliplatin. (A) Cell proliferation assays of the shCon and shRNA1 treated CRC cells upon oxaliplatin treatment. The IC50 value of each cell line was indicated. The results were the representative of at least three independent experiments and the results were expressed as the mean ± SD. (B) Colony formation of the shCon and shRNA1 treated cells in the presence of oxaliplatin. (C) Quantification of the colony formation of the shCon and shRNA1 treated cells. The results were expressed as the mean ± SD.
Importantly, overexpression of WBSCR22 in Caco-2 cells (Caco-2-Wbscr22) led to a higher chemoresistance to oxaliplatin than the control (Caco-2-vetor). The IC50 values for oxaliplatin in Caco-2-Wbscr22 and Caco-2-vetor cells were 98.39 ± 8.79 and 42.41 ± 3.02 μM, respectively (Fig. 4A,B).
Figure 4.

The expression of WBSCR22 affected Caco-2 cell sensitivity to oxaliplatin. (A) Western blot analysis of WBSCR22 in Caco-2 cells transfected by Wbscr22 or vector constructs, respectively. (B) Proliferation study of the Caco-2-Wbscr22 and Caco-2-vector cells upon oxaliplatin treatment at the indicated concentration. The results were representative of three independent experiments and the results were expressed as the mean ± SD. (C) Western blot of WBSCR22 in the shCon or shRNA1 treated Caco-2 cells transfected by either Myc-DDK-WBSCR22 or vector constructs, respectively. (D) Cell proliferation of Caco-2 shRNA1-vector, shRNA1-Wbscr22, shCon-vector and shCon-Wbscr22 cells upon oxaliplatin treatment. The IC50 value of each cell line was indicated. The results were representative of three independent experiments and the results were expressed as the mean ± SD.
To further confirm that WBSCR22 affects oxaliplatin sensitivity, we rescued the WBSCR22 expression by transiently transfecting Myc-DDK-WBSCR22 construct or vector control into the Caco-2 shRNA1 cells (shRNA1-Wbscr22, shRNA1-vector) or shCon cells (shCon-Wbscr22, shCon-vector). Interestingly, the restoration of WBSCR22 protein decreased the sensitivity of shRNA1-Wbscr22 cell to oxaliplatin, indicating the elevated WBSCR22 conferred the resistance of CRC cell to oxaliplatin (Fig. 4C,D).
WBSCR22 konckdown promoted the oxaliplatin-induced apoptosis in CRC cells
We further investigated whether WBSCR22 knockdown affects the oxaliplatin-induced apoptosis. The shRNA1 treatd cells showed an increased rate of apoptosis upon oxaliplatin treatment at 48 hours (Fig. 5A,B). The apoptosis markers, both cleaved PARP and cleaved caspase-3 fragments induced by oxaliplatin, were detected in the shRNA1 treated cells but not in the shCon treated cells (Fig. 5C), indicating that the oxaliplatin-induced apoptosis was enhanced by knockdown of WBSCR22 in CRC cells.
Figure 5.

WBSCR22 knockdown increased the oxaliplatin-induced apoptosis in CRC cells. (A,B) Cellular apoptosis rate was detected by annexinV-PI assay after oxaliplatin treatment for 48 hours. The rate of apoptosis was indicated. *p < 0.05 (shRNA1 v.s. shCon). The results were from three independent experiments and the data were expressed as the mean ± SD. (C) Western blot of cleaved PARP and cleaved caspase-3 in CRC cells after oxaliplatin treatment for 48 hours.
WBSCR22 knockdown increased the CRC sensitivity to oxaliplatin in vivo
To assess whether WBSCR22 knockdown affects CRC sensitivity to oxaliplatin in vivo, we compared the tumor growth inhibitory activity of oxaliplatin in the Caco-2-shCon and -shRNA1 tumor xenograft models. All of the nude mice bearing the tumors survived during the treatment. As shown in Fig. 6A,B, the relative mean tumor volume (RMTV) was significantly smaller in the treatment group of Caco-2-shRNA1 model (481.50 ± 84.49 mm3) at day 25, as compared to the control group (772.70 ± 66.27 mm3; p < 0.05) with the inhibition rate (IR) of 45.08%. There were no differences in the Caco-2-shCon model between the treatment group (962.70 ± 73.28 mm3) and the control group (949.61 ± 56.50 mm3) (Fig. 6C). The results demonstrated WBSCR22 knockdown could sensitize the Caco-2-shRNA1 xenograft tumor to oxaliplatin. There were no significant differences for the Caco-2-shCon and -shRNA1 xenograft mice in the control groups (p > 0.05) (Fig. 6D).
Figure 6.

WBSCR22 knockdown increased oxaliplatin sensitivity in vivo. The mice in the treatment groups were treated with oxaliplatin by intraperitoneal injection, on a schedule of two injections every week at 7.5 mg/kg per injection. The mice in the control groups were injected with solvent (aqueous glucose 5% solution [5% GS]). (A) Macroscopic images of xenografted tumors excised at day 25. (B) The relative mean tumor volume (RMTV) curve of the Caco-2 shRNA1 xenograft models. The data were expressed as the mean ± SE. *p < 0.05, (Caco-2 shRNA1 + oxaliplatin [treatment group] vs. Caco-2 shRNA1 + 5% GS [control group]). (C) The relative mean tumor volume (RMTV) curve of the Caco-2 shCon xenograft models. The results were expressed as the mean ± SE. (D) The mean tumor volume (MTV) curve of the Caco-2 shCon and shRNA1 xenograft models in the control groups. The data were expressed as the mean ± SE. (E) Immunohistochemical analysis of WBSCR22 protein in the excised tumor tissues from the shCon and shRNA1 control groups (×40, ×200). (F) Immunohistochemical analysis of cleaved caspase-3 in the excised tumor tissues from the shCon and shRNA1 treatment groups (×200). The data were expressed as the mean ± SD. ***p < 0.001 (shRNA1 + oxaliplatin [treatment group] vs. shCon + oxaliplatin [treatment group]).
Immunohistochemical analysis of the xenografted tumor tissues at day 25 showed that the Caco-2-shRNA1 tumor had a lower level of WBSCR22 protein compared with the Caco-2-shCon tissues (Fig. 6E). The Caco-2-shRNA1 tissue in the treatment group had more cleaved caspase-3-positive cells than the control group (p < 0.001) (Fig. 6F), further confirming that WBSCR22 knockdown increased the CRC cell sensitivity to oxaliplatin in vivo.
WBSCR22 knockdown increased the oxaliplatin-induced intracellular ROS generation in CRC cells
ROS generation is known to be a critical event in the oxaliplatin-induced cell death. To investigate whether WBSCR22 knockdown regulates intracellular ROS generation, we measured the ROS levels. The results showed no significant changes for the ROS levels in the shRNA1 treated cells as compared to the shCon treated cells. Treatment of these cells with nonspecific free radical scavenger N-Acetyl-cysteine (NAC) for 30 minutes did not significantly reduce the ROS production (Fig. 7A), suggesting that WBSCR22 gene was not protective against the ROS generation in CRC cell lines.
Figure 7.

WBSCR22 knockdown increased oxaliplatin-induced ROS generation in CRC cells. (A) Comparison of ROS levels in the CRC cells treated by shRNA1 and shCon. Cells were pretreated with 20 mM NAC or serum–free DMEM for 30 minutes. (B) HT-29, Caco-2 and RKO cells were treated with oxaliplatin at the indicated concentrations for 1 hour, and DCFH fluorescence was quantified immediately by a fluorescence microplate reader. *p < 0.05, **p < 0.01, (shRNA1 v.s. shCon). (C) Comparison of the oxaliplatin-induced cell proliferation in the shCon and shRNA1 treated CRC cells. Cells were pretreated with 5 mM NAC or serum-free DMEM for 1 hour, followed by oxaliplatin treatment for 24 hours. All results were representative of three independent experiments and the data were expressed as the mean ± SD.
We also quantified the levels of the oxaliplatin-induced intracellular ROS in CRC cells. As shown in Fig. 7B, there was a marked increase in the ROS level in the shRNA1 treated cells after 1 hour treatment with oxaliplatin, as compared to the shCon treated cells (HT-29 and RKO cells at 10 μM or 20 μM concentrations of oxaliplatin, Caco-2 cells at 10 μM or 25 μM), consistent with the results from the fluorescence image (Supplementary Fig. S3). Pretreatment with 5 mM NAC for 1 hour significantly protected both shCon- and shRNA1-treated cells against the toxicity of oxaliplatin (Fig. 7C). Our results demonstrated that WBSCR22 knockdown increased the oxaliplatin-induced intracellular ROS generation, thus enhancing oxaliplatin sensitivity.
WBSCR22 knockdown increased the ROS-induced 8-oxoG accumulation in CRC cells
It was reported that oxaliplatin stimulates the intracellular ROS production, thus inducing the 8-oxoG oxidative lesion19. We therefore tested whether knockdown of WBSCR22 could increase the accumulation of the ROS-induced 8-oxoG lesion by treating the cells with oxaliplatin. The intracellular 8-oxoG oxidative lesion was observed at the indicated concentrations for 24 hours by immunofluorescence staining (Fig. 8). Upon the oxaliplatin treatment, the shRNA1 treatd cells showed a brighter green fluorescence density (8-oxoG) compared to the the shCon treated cells, indicating that WBSCR22 knockdown increased the oxaliplatin-induced 8-oxoG accumulation. The results were consistent with the elevated oxaliplatin-induced ROS level in the shRNA1 treated cells.
Figure 8.
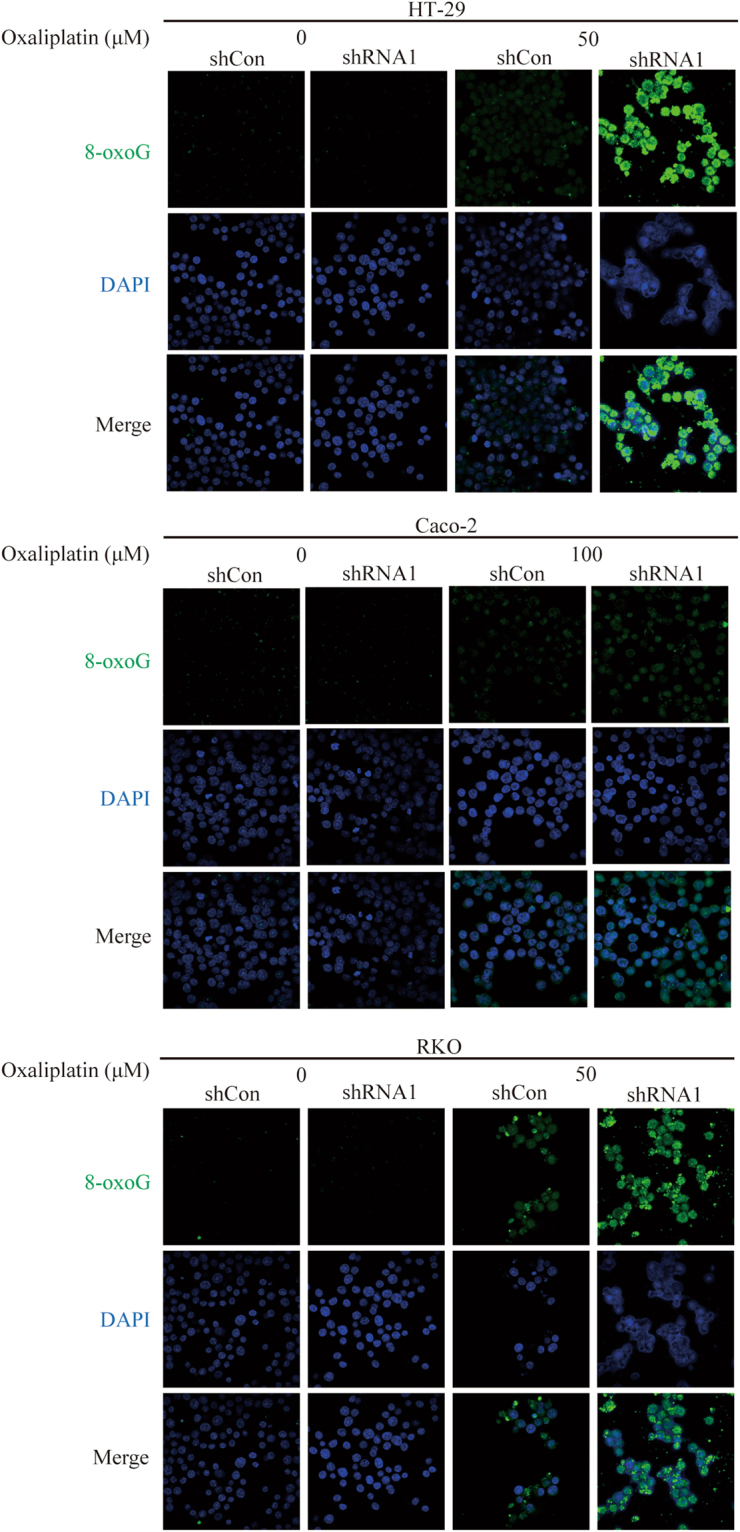
WBSCR22 knockdown increased ROS-induced 8-oxoG accumulation in CRC cells. HT-29, Caco-2 and RKO cells were treated with oxaliplatin at the indicated concentrations for 24 hours. The intracellular 8-oxoG immunofluorescence (green) and cell nuclear DNA staining (blue) images were captured by a confocal laser scanning microscope (×400).
Discussion
Oxaliplatin, a third-generation platinum antitumor drug, prevents DNA replication and transcription by forming both inter- and intra-strand cross-links in DNA, thus resulting in cell death. Oxaliplatin is non-cross-resistant with cisplatin and carboplatin20. The oxaliplatin chemoresistance is caused by multiple mechanisms, including overexpression and/or depletion of drug resistance associated proteins12,21–23, mutated genes24, altered platinum accumulation and DNA-Pt adduct formation25–27, microRNAs28, and defect in signal transduction pathway29. Therefore, how to overcome oxaliplatin resistance will be a key issue for more effective individualized therapeutic strategies.
Human WBSCR22 gene is initially identified as a deleted gene in Williams-Beuren syndrome. Little is known about its biological function, particularly the impact on antitumor drug resistance. We have initially reported that WBSCR22 knockdown attenuates the sensitivity of non-small cell lung cancer cell lines H460, harboring wild type p53, to 7-Ethyl-10-hydroxycamptothecin (SN-38; the active metabolite of camptothecin) and 5-fluorouracil (5-FU), but not in p53-null H1299 cells30.
In the present study, we found that CRC cells expressed high WBSCR22 protein, and the gene expression level of WBSCR22 was correlated with the oxaliplatin sensitivity. Konckdown of WBSCR22 significantly sensitized CRC cell to oxaliplatin in vitro and in vivo by increasing oxaliplatin-induced rate of apoptosis. Knockdown of WBSCR22 significantly increased the production of oxaliplatin-induced ROS, thus simultaneously enhancing ROS-induced 8-oxoG oxidative lesion production. Pretreatment with NAC reversed the cytotoxicity of oxaliplatin in CRC parent cells as well as shRNA1 treated cells, indicating the oxaliplatin-induced ROS was a cytotoxic mechanism for oxaliplatin.
ROS are highly reactive oxygen free radicals or non-radical molecules that are generated by multiple mechanisms. ROS induce the 8-oxoG oxidative lesion production, as a pathogenic macromolecular lesion in the mechanism of platinum compound-induced cytotoxicity31. Oxaliplatin treatment destroys tumor cells in part through the induction of acute oxidative stress32,33. The production of ROS induces apoptosis and is involved in the oxaliplatin-induced cell death pathway34,35. Therefore, in our study the increased ROS and 8-oxoG level might sensitize human CRC cells to oxaliplatin. To our best knowledge, this study is the first demonstrating that WBSCR22 is involved in oxaliplatin chemosensitivity.
In the future, the clinical samples should be collected to provide further evidence that WBSCR22 is a potential prognostic biomarker and a promising therapeutic target for treating human CRC. These studies should focus on WBSCR22 signal transduction pathways to further understand the molecular mechanism of oxaliplatain resistance, therefore designing therapeutic inhibitors to reverse its resistant effects.
In conclusion, our data showed that WBSCR22 is involved in the CRC cell chemosensitivity to oxaliplatin, and CRC patients with lower WBSCR22 expression might benefit more from receiving first-line oxaliplatin-based regimens than the patients with high WBSCR22 expression. Our findings also strongly suggest that inhibition WBSCR22 may be a potential therapeutic strategy for reducing colorectal tumor resistance to oxaliplatin treatment.
Materials and Methods
Analysis of TCGA data
Expression profile, clinical significance and prognostic value of WBSCR22 were analyzed in the Caner Genome Atlas (TCGA)36 cohort. The data of gene expression and clinical information were obtained from TCGA data portal (http://tcga-data.nci.nih.gov/tcga/) and cBioPortal (http://www.cbioportal.org/public-portal/).
Cell lines and reagents
The human colon cancer cell lines HT-29, Caco-2, RKO, HCT116 and Colo205 were purchased from cell bank (Shanghai Institute of Biological Sciences, Shanghai, China). Colo205, HT-29 and RKO grew as monolayers in Dulbecco’s modified Eagles medium (DMEM) containing 10% fetal calf serum (Gibco, Grand Island, NY, USA) in a 5% CO2, 95% at 37 °C. Caco-2 grew in DMEM containing 20% fetal calf serum (Gibco). HCT116 grew in McCoy’5 A (Sigma, Saint Louis, MO, USA) containing 10% fetal calf serum (Gibco). Oxaliplatin was purchased from Meilun Biological Co., Ltd. (Dalian, China). Oxaliplatin was dissolved at 2000 μM in 5% GS, as stocks and stored at −20 °C. Oxaliplatin stocks were diluted at a series of concentrations in serum-free DMEM immediately prior to use in the in vitro experiments. For the in vivo studies, oxaliplatin was dissolved at 0.625 g/l in 5% GS and 5% GS was used as the solvent control. N-Acetyl-cysteine (NAC) was purchased from Sigma. NAC was dissolved at 100 mM in serum-free DMEM, as stocks and stored at −20 °C.
Plasmid construction and stable transfections
Plasmid construction and stable transfections were performed as described previously30. The shRNA sequence targeting human WBSCR22 were 5′-GCCCTGTTACCTGCTGGAT-3′ (shRNA1) and 5′-GTCAGATGAAGGGCACTAT-3′ (shRNA2)7. HT-29, Caco-2 and RKO were stably transfected with vectors containing the shRNA sequence targeting human WBSCR22 (shRNA1, shRNA2) or the negative control shRNA (shCon).
Expression of WBSCR22
The cells were transiently transfected with WBSCR22 plasmid (C-terminal Myc-DDK-Tagged) (OriGene, Rockville, MD, USA). The vector pCMV6-Entry (C-terminal Myc-DDK-Tagged) (OriGene) was used as the negative control. Transfections were performed using Lipofectamin™ 2000 (Gibco) according to the manufacturer’s instructions.
RT-qPCR
Total RNA isolation, first-strand cDNA synthesis and qPCR were performed as described previously30. β-actin was used as a reference gene for qPCR analysis.
Measurement of cell proliferation inhibition
The cell proliferation inhibition assay was carried out using MTT (Sigma). Cells were plated in 96-well plates at 1–5×103 cells per well. At 24 hours later, oxaliplatin was added at the indicated concentrations for 24–72 hours. The absorbance was measured on a microplate reader at 570 nm. The IC50 values were calculated using GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). The IC50 values were the means of at least three independent experiments.
Colony formation assays
Cells were plated in 6-well plates at 5 × 102 cells per well. After 24 hours, cells were treated with the indicated concentrations of oxaliplatin for 48 hours. Cells were then maintained in fresh medium for another 10 (HT-29 and Caco-2) or 5 (RKO) days. Colonies were stained with 0.1% crystal violet for 30 minutes and then counted.
Western blotting
Cell lysis and western blot analysis were performed as described previously30. Antibodies used were as follows: anti-human WBSCR22 (GeneTex, Irvine, CA, USA), anti-actin, anti-PARP and anti-caspase-3 (Cell Signaling Technology, Inc., Danvers, MA, USA).
Flow cytometry
The cell cycle distribution and apoptosis rate were analyzed by flow cytometry. For cell cycle distribution, 3 × 104 cells were analyzed using a CytomicsTM FC 500 instrument (Beckman Coulter, Inc., Brea, CA, USA). ModiFit LT 3.1 trial cell cycle analysis software was used to determine the percentage of cells in each phase of the cell cycle. For apoptosis analysis, cells were harvested and washed with cold PBS after oxaliplatin treatment for 48 hours. The cells were then stained with annexinV-FITC and PI (Invitrogen Life Technologies, Grand Island, NY, USA), according to the manufacturer’s instructions.
In vivo tumor growth inhibition assay
The animal study was approved by the Zhejiang Experimental Animal Center (Hangzhou, Zhejiang, China) under the project number: SCXK2008-0016, and the mice were maintained in accordance to the Institute Animal Ethical Committee guidelines approved by Zhejiang Academy of Medical Sciences (Hangzhou, Zhejiang, China). BALB/c nu/nu mice (male, 4–5 weeks) were housed in the laminar air-flow cabinets under pathogen-free conditions with a 14-hour light/10-hour dark schedule, and fed the autoclaved standard chow and water ad libitum. Both shCon treated and shRNA1 treated Caco-2 cells (2 × 106 cells in 200 μl of serum-free DMEM) were subcutaneously injected into the right flank of mice. After the tumor volumes (TV) reached to 100 to 300 mm3 on day 8, Caco-2-shCon and -shRNA1 tumor xenograft mice were randomized into two groups (control group and treatment group) with five animals for each group. The mice in the treatment groups were treated with oxaliplatin by intraperitoneal (i.p) injection at 10 a.m. twice a week for 3 weeks, at a dose of 7.5 mg/kg per injection, and the mice in the control groups received solvent (5% GS). The TV was measured every 5 days during the treatment period (25 days). The TV was calculated using the formula: π/6 × (length × width2), where length = longest diameter and width = diameter perpendicular to length. The MTV, RMTV and IR were calculated. RMTV was calculated using the formula: MTV on day n (MTVn)/MTV on day 0 (MTV0). The IR was calculated using the formula: (1-RMTV in treatment group/RMTV in control group) ×100.
Immunohistochemistry
Formalin fixed, paraffin embedded sections were deparaffinized by xylene and rehydrated. For antigen retrieval, the sections were placed in citrate buffer (pH 6.0) and heated in a microwave oven for 10 minutes. For immune peroxidase labeling, endogenous peroxidase was blocked by 0.3% H2O2 in methanol for 15 minutes at room temperature. The sections were then incubated overnight at 4 °C with primary antibody and washed with PBS containing 0.05% TrionX-100. Incubation with corresponding secondary antibody and the peroxidase-antiperoxidase complex were carried out for 30 minutes at room temperature. Immunoreactive site were visualized by 3, 3′-DAB. The slices were counterstained by hematoxylin. Antibodies were as follows: anti-human WBSCR22 (GeneTex) and anti-cleaved caspase-3 (Cell Signaling Technology, Inc.). Immunostaining were reviewed under a microscope at ×40 and ×200 magnification, and the images were captured using Leica DM2500 camera. The expressions of the cleaved caspase-3 were quantified by counting the number of immunopositive cells in three different fields per slide. All slides were reviewed independently by two experts.
Measurement of the ROS production
Intracellular ROS levels were measured using a DCFH-DA assay. Briefly, 1 × 104 cells/well (96-well plate) were allowed to adhere and treated with both DCFH-DA and oxaliplatin for 1 hour at 37 °C in 5% CO2. After treatment, the media was removed. The cells were washed with PBS, then added 100 μl/well PBS. ROS-dependent conversion of DCFH-DA to fluorescent product was quantified immediately by a fluorescence microplate reader (excitation 485 nm, emission 530 nm) and fluorescence images were captured by a fluorescence microscope.
8-oxoG damage assay
The cells grown in the glass bottom dishes were exposed to oxaliplatin for 24 hours then washed with PBS and fixed for 5 minutes at 4 °C with an ethanol: methanol (1:1; v/v) solution. Following three washes with PBS, cells were exposed to RNase A (DNase and Protease-free) solution (200 μg/ml; Thermo Scientific, Waltham, MA, USA) at 37 °C for 1 hour to digest RNAs. After a brief wash with PBS, fixed cells were treated with 5% bovine serum albumin in PBS for 1 hour at 20 °C and subsequently incubated overnight at 4 °C in primary antibody solution containing mouse anti-8-oxoG monoclonal IgM (1:2000; Abcam, Cambridge, UK), followed by a brief PBS washing and incubation with secondary antibody solution containing Alexa Fluor488-conjugated goat anti-mouse IgM (1:250, Sungenebiotech, Tianjin, China) for 2 hours. Cells nuclear DNA was stained using DAPI for 3 minutes. Images were captured using a confocal laser scanning microscope (Zeiss Lsm710; Carl Zeiss AG, Oberkochen, Germany). The excitation/emission wavelengths for Alexa Fluor488 and DAPI were 495 nm/519 nm and 340 nm/488 nm respectively.
Statistical analysis
The associations between WBSCR22 expression and clinicopathological parameters were analyzed using Chi-square tests. For survival analysis, OS was defined as the elapsed time between diagnosis and death or the last follow-up. Survival curves were estimated by the Kaplan-Meier method and compared using the log-rank test. To build a model for the prediction of survival, univariate and multivariate Cox proportional-hazards regression analyses were performed, in which clinical variables with p < 0.05 in univariate analysis were pooled into multivariate analysis. The data of the in vitro experiments were analyzed using the unpaired t-test and two-tailed t-test. The data of MTV in the xenograft models were analyzed using the repeated-measures analysis of variance. The data of RMTV at the end of treatment in the animal models were analyzed using the Mann-Whitney test. SPSS 22.0 software (SPSS Inc., Armonk, NY, USA) was used for all statistical analysis and p < 0.05 was considered statistically significant for all tests. Figures were plotted in SPSS 22.0 software (SPSS Inc.) or GraphPad Prism 5 (GraphPad Software, Inc.).
Electronic supplementary material
Acknowledgements
This study was funded by the Natural Science Foundation in Zhejiang Province (LY12H31009 and LY13H310004) and the Medical and Health Technology Project in Zhejiang Province (2014KYA045).
Author Contributions
Dongmei Yan, Xiaoliang Zheng and Xiaoju Wang conceived and designed the experiments; Dongmei Yan, Xiaoliang Zheng, Linglan Tu and Liyan Cheng performed the experiments; Haining Yuan and Jianfei Fang analysed the data; Dongmei Yan and Xiaoju Wang wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-15749-z.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xiaoliang Zheng, Email: zhengxl@zjams.com.cn.
Xiaoju Wang, Email: wangxj@zjams.com.cn.
References
- 1.Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 2.Bellugi U, Lichtenberger L, Mills D, Galaburda A, Korenberg JR. Bridging cognition, the brain and molecular genetics: evidence from Williams syndrome. Trends Neurosci. 1999;22:197–207. doi: 10.1016/S0166-2236(99)01397-1. [DOI] [PubMed] [Google Scholar]
- 3.Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL. Natural history of Williams syndrome: physical characteristics. J Pediatr. 1988;113:318–326. doi: 10.1016/S0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- 4.Merla G, Ucla C, Guipponi M, Reymond A. Identification of additional transcripts in the Williams-Beuren syndrome critical region. Hum Genet. 2002;110:429–438. doi: 10.1007/s00439-002-0710-x. [DOI] [PubMed] [Google Scholar]
- 5.Doll, A. & Grzeschik, K. H. Characterization of two novel genes, WBSCR20 and WBSCR22, deleted in Williams-Beuren syndrome. Cytogenet Cell Genet95, 20–27, 57012 (2001). [DOI] [PubMed]
- 6.Petrossian TC, Clarke SG. Uncovering the human methyltransferasome. Mol Cell Proteomics. 2011;10(M110):000976. doi: 10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakazawa Y, Arai H, Fujita N. The novel metastasis promoter Merm1/Wbscr22 enhances tumor cell survival in the vasculature by suppressing Zac1/p53-dependent apoptosis. Cancer Res. 2011;71:1146–1155. doi: 10.1158/0008-5472.CAN-10-2695. [DOI] [PubMed] [Google Scholar]
- 8.Tiedemann RE, et al. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012;72:757–768. doi: 10.1158/0008-5472.CAN-11-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ounap K, Kasper L, Kurg A, Kurg R. The human WBSCR22 protein is involved in the biogenesis of the 40S ribosomal subunits in mammalian cells. PLoS One. 2013;8:e75686. doi: 10.1371/journal.pone.0075686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefanska B, et al. Genome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targets. Clin Cancer Res. 2014;20:3118–3132. doi: 10.1158/1078-0432.CCR-13-0283. [DOI] [PubMed] [Google Scholar]
- 11.Jangani M, et al. The methyltransferase WBSCR22/Merm1 enhances glucocorticoid receptor function and is regulated in lung inflammation and cancer. J Biol Chem. 2014;289:8931–8946. doi: 10.1074/jbc.M113.540906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng ZL, et al. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin Cancer Res. 2014;20:1042–1052. doi: 10.1158/1078-0432.CCR-13-0171. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues AS, et al. Genomics and cancer drug resistance. Curr Pharm Biotechnol. 2012;13:651–673. doi: 10.2174/138920112799857549. [DOI] [PubMed] [Google Scholar]
- 14.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Yang BB. Friend or foe: the role of microRNA in chemotherapy resistance. Acta Pharmacol Sin. 2013;34:870–879. doi: 10.1038/aps.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bunz F. Cell death and cancer therapy. Curr Opin Pharmacol. 2001;1:337–341. doi: 10.1016/S1471-4892(01)00059-5. [DOI] [PubMed] [Google Scholar]
- 17.Rolfo C, et al. Impact of microRNAs in resistance to chemotherapy and novel targeted agents in non-small cell lung cancer. Curr Pharm Biotechnol. 2014;15:475–485. doi: 10.2174/1389201015666140519123219. [DOI] [PubMed] [Google Scholar]
- 18.Fojo T. Multiple paths to a drug resistance phenotype: mutations, translocations, deletions and amplification of coding genes or promoter regions, epigenetic changes and microRNAs. Drug Resist Updat. 2007;10:59–67. doi: 10.1016/j.drup.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Laurent A, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65:948–956. [PubMed] [Google Scholar]
- 20.Graham J, Mushin M, Kirkpatrick P. Oxaliplatin. Nat Rev Drug Discov. 2004;3:11–12. doi: 10.1038/nrd1287. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki S, Tanigawara Y. Forced expression of S100A10 reduces sensitivity to oxaliplatin in colorectal cancer cells. Proteome Sci. 2014;12:26. doi: 10.1186/1477-5956-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatch SB, et al. XPF protein levels determine sensitivity of malignant melanoma cells to oxaliplatin chemotherapy: suitability as a biomarker for patient selection. Int J Cancer. 2014;134:1495–1503. doi: 10.1002/ijc.28454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnould S, Hennebelle I, Canal P, Bugat R, Guichard S. Cellular determinants of oxaliplatin sensitivity in colon cancer cell lines. Eur J Cancer. 2003;39:112–119. doi: 10.1016/S0959-8049(02)00411-2. [DOI] [PubMed] [Google Scholar]
- 24.Lin YL, et al. KRAS mutation is a predictor of oxaliplatin sensitivity in colon cancer cells. PLoS One. 2012;7:e50701. doi: 10.1371/journal.pone.0050701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samimi G, et al. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin Cancer Res. 2004;10:4661–4669. doi: 10.1158/1078-0432.CCR-04-0137. [DOI] [PubMed] [Google Scholar]
- 26.Hector S, Bolanowska-Higdon W, Zdanowicz J, Hitt S, Pendyala L. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer Chemother Pharmacol. 2001;48:398–406. doi: 10.1007/s002800100363. [DOI] [PubMed] [Google Scholar]
- 27.Vaisman A, et al. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998;58:3579–3585. [PubMed] [Google Scholar]
- 28.Zhou Y, et al. miR-203 induces oxaliplatin resistance in colorectal cancer cells by negatively regulating ATM kinase. Mol Oncol. 2014;8:83–92. doi: 10.1016/j.molonc.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toscano F, et al. p53 dependent and independent sensitivity to oxaliplatin of colon cancer cells. Biochem Pharmacol. 2007;74:392–406. doi: 10.1016/j.bcp.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Yan D, et al. Knockdown of Merm1/Wbscr22 attenuates sensitivity of H460 non-small cell lung cancer cells to SN-38 and 5-FU without alteration to p53 expression levels. Mol Med Rep. 2015;11:295–302. doi: 10.3892/mmr.2014.2764. [DOI] [PubMed] [Google Scholar]
- 31.Preston TJ, Henderson JT, McCallum GP, Wells PG. Base excision repair of reactive oxygen species-initiated 7,8-dihydro-8-oxo-2′-deoxyguanosine inhibits the cytotoxicity of platinum anticancer drugs. Mol Cancer Ther. 2009;8:2015–2026. doi: 10.1158/1535-7163.MCT-08-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 33.Bratasz A, et al. Reversal to cisplatin sensitivity in recurrent human ovarian cancer cells by NCX-4016, a nitro derivative of aspirin. Proc Natl Acad Sci USA. 2006;103:3914–3919. doi: 10.1073/pnas.0511250103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–790. doi: 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- 35.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.