

Abstract
Aims: Cardiovascular risk persists despite intensive lipid lowering therapy using statins. Serum levels of lipoprotein (a) [Lp(a)] can be a residual cardiovascular risk for adverse events. Aim of the present study was to evaluate the impact of Lp(a) on long-term clinical outcomes in patients treated with statin after percutaneous coronary intervention.
Methods: We prospectively enrolled 3507 consecutive CAD patients who underwent a first percutaneous coronary intervention (PCI) between 1997 and 2011 at our institution. We identified 1768 patients (50.4%) who had treated with statin during PCI. Eligible 1336 patients were stratified to two groups according to Lp(a) levels (median Lp (a) 21.5 mg/dL). The primary outcome was major adverse cardiac events (MACE) including cardiac death and non-fatal acute coronary syndrome.
Results: MACE occurred 144 (10.8%) including 34 (2.5%) cardiac death and 110 (8.7%) non-fatal ACS during median follow-up period of 1920 days. The cumulative rate of MACE was significantly higher in group with high Lp(a) group (log-rank p = 0.0460). Multivariate Cox regression analysis showed a significant correlation between Lp (a) levels treated as a natural logarithm-transformed continuous variable and increased MACE (adjusted HR for MACE 1.28, 95%CI 1.04–1.58, p = 0.0184)
Conclusion: Elevated levels of Lp(a) is significantly associated with long-term adverse clinical outcomes among CAD patients who received statin therapy after PCI.
Keywords: Lipoprotein (a), Coronary artery disease, Statin, Percutaneous coronary intervention
Introduction
Cardiovascular disease is the leading cause of mortality and morbidity worldwide. Clinical trials have shown that statins significantly reduce cardiovascular events in patients with coronary artery disease (CAD)1, 2) and statins are the most widely used for individuals with CAD. However, cardiovascular risk persists despite intensive lipid lowering therapy with statins and has been called residual risk3, 4). The mechanisms underlying this residual risk are uncertain and identification of these factors is important for more effective tailoring of risk reduction strategies.
A number of additional lipid risk factors have been proposed to be independently associated with cardiovascular disease (CVD). Lipoprotein (a) [Lp(a)] is a low-density lipoprotein like particle with apolipo-protein (apo) B-100, linked by a disulfide bond to apolipoprotein (a)5). Lp(a) has enhanced atherogenic and thrmbogenic properties and Lp(a) has been identified as an independent, casual risk factor for CVD6–9). Elevated serum Lp(a) are consequently associated with an increased adverse clinical events in patients with CAD after coronary intervention10, 11). However, its prognostic utility as a marker of risk in this setting of secondary prevention treated with statin after percutaneous coronary intervention (PCI) is not well established. The association between Lp(a) and cardiovascular events requires ongoing exploration in different population cohorts in order to elucidate its potential role as a marker of clinical risk and target for therapeutic manipulation, especially in such high risk patients. From this perspective, we evaluated the impact of Lp(a) on long-term clinical outcomes in patients with CAD treated with statin after coronary intervention.
Methods
Study Population and Data Collection
We analyzed data from a single-center, observational study of patients who underwent PCI at our institution between January 1997 and December 2011. We included only data from CAD patients with CAD who were treated with statin at the time of PCI. Missing Lp(a) data were excluded from the study. The patients were assigned to two groups based upon Lp(a) values. The cut-offs for Lp(a) were median Lp(a) of 21.5 mg/dL (Fig. 1).
Fig. 1.
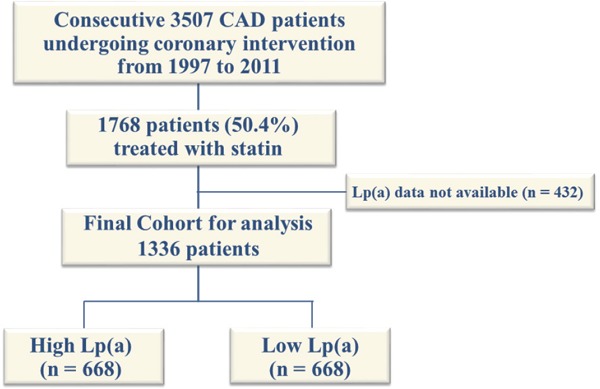
Study flow chart
Demographic data as well as information about coronary risk factors, medications, revascularization procedure-related factors and comorbidities were prospectively collected and analyzed. Blood samples were collected during the early morning after an overnight fast and blood pressure (BP) was measured at the time of admission. Patients with BP > 140/90 mmHg or under anti-hypertensive medication were considered hypertensive. Diabetes mellitus was defined as either hemoglobin A1c (HbA1c) ≥ 6.5% or under medication with insulin or oral hypoglycemic drugs. The estimated HbA1c (%) was calculated as the National Glycohemoglobin Standardization Program (NGSP) equivalent value (%) using the formula HbA1c (%) = 1.02 × HbA1c (JDS; %) + 0.25%12). We defined renal impairment as an estimated glomerular filtration rate (eGFR) of < 60 mL/min/1.73 m2, and calculated the eGFR based on the modification of diet in renal disease equation modified with a Japanese coefficient using baseline serum creatinine13). Levels of Lp(a) were measured using latex agglutination immunoassays (Special Reference Laboratories, Hachioji, Japan).
Written informed consent was obtained from all patients before undergoing coronary intervention. This study proceeded under the approval of our Institutional Review Board in accordance with the Declaration of Helsinki.
Primary Endpoint
The primary outcomes of this study were cardiac events defined as a composite of cardiac death and non-fatal acute coronary syndrome. Clinical follow-up comprised analyses of office visit charts and responses to questionnaires sent to patients or their families and telephone contact. Mortality data were collected from the medical records of patients who died or who were treated at our institution and details and causes of death were obtained from other hospitals where patients had been admitted. Cardiac death was defined as death from CAD, cardiogenic shock and sudden death. We defined acute coronary syndrome (ACS) as unstable angina pectoris (UAP), non-ST segment elevation myocardial infarction (NSTEMI) or STEMI requiring emergency hospital admission for either PCI or coronary artery bypass grafting. Unstable angina was diagnosed as angina at rest or in an accelerating pattern with negative cardiac biomarkers, with or without electrocardiographic (ECG) changes indicative of myocardial ischemia. Myocardial infarction was defined as troponin T positivity.
Statistical Analysis
Quantitative data are expressed as means ± SD and categorical variables are presented as frequencies. Continuous variables across groups were compared using unpaired t test or Mann-Whitney U test for continuous variables and the chi-square test or Fisher's exact probability test for categorical variables. Unadjusted cumulative event rates were estimated using Kaplan-Meier curves and compared between the groups using log-rank test. Hazard ratios and 95% confidence intervals for each variable were calculated using a Cox proportional hazards model. Predictors of cardiovascular events were determined by multivariate Cox regression analysis. Variables with P < 0.05 in the univariable model were included in the multivariable analysis as well as age and gender. We also performed multivariable Cox regression analysis that included Lp(a) levels treated as a natural logarithm-transformed continuous variable to determine whether findings differed from the cutoff points. Models were initially adjusted for age and gender (Model 1). Thereafter, Model 2 was adjusted for lipid parameters including low-density lipoprotein cholesterol, high-density lipoprotein cholesterol and triglyceride in addition to Model 1. Model 3, adjusted for variables in model 1 plus covariates with P < 0.05 in univariable analysis comprised multivessel disease, ACS and diabetes. Values with P < 0.05 were considered to indicate statistically significant difference. All data were analyzed using JMP version 10.0 for Windows (SAS Institute, Cary, NC, USA) and SPSS v.18.0 (Chicago, IL, USA).
Results
Among 3507 patients who underwent scheduled PCI during the study period, we analyzed data from 1768 eligible patients with CAD who were treated with statins. Fig. 1 shows the flow chart of study population. Fig. 2 shows the distribution of Lp(a) levels. Table 1 shows the baseline characteristics of patients. The median follow-up period was 1620 (interquartile range, 737 to 2840) days and prognostic data were fully documented during the entire follow-up period. During the follow-up, 144 (10.8 %) cardiac events occurred that included 41 (3.1 %) cardiac deaths, 103 (7.7 %) non-fatal ACS. Fig. 3 shows the cumulative event free survival curves for all-cause death among the groups. The incidence of cardiovascular events was significantly higher in the group with high Lp(a) than with low Lp(a) (log-rank test, p = 0.0460). Table 2 shows univariate and multivariate Cox hazard regression analyses. Variables with p < 0.05 in univariate analysis comprised diabetes, multivessel disease, ACS with Lp(a) values. Table 3 summarizes the findings of univariate and multivariate Cox hazard regression analyses in which Lp(a) values were treated as natural logarithm-transformed continuous variables. Logarithm-transformed Lp(a) was significantly associated with higher rate of cardiovascular events even after adjustment for other covariates.
Fig. 2.
The distribution of Lp(a) levels
Table 1. Baseline clinical characteristics of the study population.
| High Lp(a) Group | Low Lp(a) Group | P value | |
|---|---|---|---|
| (n = 668) | (n = 668) | ||
| Age, years | 64.3 ± 10.2 | 64.7 ± 10.2 | 0.57 |
| Men, n (%) | 523 (78.3) | 572 (85.6) | < 0.001 |
| Hypertension, n (%) | 474 (71.0) | 471 (70.5) | 0.86 |
| Diabetes mellitus, n (%) | 307 (46.0) | 290 (43.4) | 0.35 |
| Metabolic syndrome, n (%) | 265 (48.3) | 317 (51.8) | 0.23 |
| Current Smoking, n (%) | 172 (25.8) | 172 (25.8) | 0.98 |
| Family History, n (%) | 223 (33.5) | 197 (29.6) | 0.13 |
| Multivessel Disease, | 408 (61.8) | 382 (57.6) | 0.56 |
| ACS, n (%) | 149 (22.3) | 173 (25.9) | 0.13 |
| Renal impairment, n (%) | 180 (26.9) | 155 (23.2) | 0.11 |
| LVEF, % | 61.3 ± 12.9 | 62.2 ± 11.9 | 0.38 |
| BMI, kg/m2 | 24.4 ± 3.5 | 24.7 ± 3.3 | 0.14 |
| SBP, mmHg | 132.8 ± 21.9 | 133.2 ± 21.9 | 0.82 |
| DBP, mmHg | 72.2 ± 13.0 | 72.8 ± 12.4 | 0.34 |
| LDL-C, mg/dL | 116.6 ± 36.9 | 105.4 ± 32.9 | < 0.001 |
| HDL-C, mg/dL | 45.3 ± 14.9 | 44.7 ± 11.6 | 0.4 |
| TG, mg/dL | 132.4 ± 69.1 | 146.1 ± 80.0 | 0.002 |
| FBG, mg/dL | 115.8 ± 41.9 | 118.1 ± 45.1 | 0.54 |
| eGFR, ml/min/1.73 m2 | 73.1 ± 11.1 | 72.4 ± 17.1 | 0.43 |
| Medication | |||
| Aspirin, n (%) | 635 (95.6) | 636 (95.8) | 0.89 |
| Ca-channel blockers, n (%) | 239 (36.1) | 252 (38.0) | 0.46 |
| ACE-I/ARBs, n (%) | 353 (53.1) | 357 (53.2) | 0.83 |
| β-Blockers, n (%) | 333 (50.2) | 353 (53.1) | 0.27 |
| Culprit of vessels | |||
| LMT or LAD prox, n(%) | 145 (21.7) | 151 (22.6) | 0.89 |
ACE inhibitors, angiotensin converting enzyme inhibitors; ACS, acute coronary syndrome; ARBs, angiotensin receptor blockers; BMI, body mass index;; DBP, diastolic blood pressure; FBG, fasting blood glucose; eGFR, estimated glomelular filtration rate; high-density lipoprotein cholesterol; LAD, left anterior descending coronary artery; LDL-C, low-density lipoprotein cholesterol; LMT, left main trunk; LVEF, left ventricular ejection fraction; SBP, systolic blood pressure; TG, triglyceride
Fig. 3.
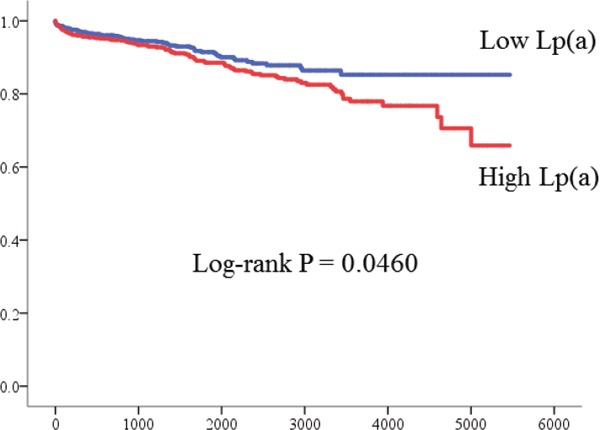
Kaplan-Meier curves for MACE
Table 2. Univariate and multivariate Cox hazard model predicting clinical events.
| Univariate |
Multivariate |
|||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| Age | 1.01 | 0.99–1.03 | 0.22 | 1.89 | 0.67–5.43 | 0.23 |
| Male Sex | 0.93 | 0.63–1.42 | 0.73 | 0.99 | 0.64–1.49 | 0.97 |
| Diabetes Mellitus | 1.45 | 1.05–2.02 | 0.026 | 1.38 | 0.99–1.92 | 0.054 |
| Hypertension | 1.08 | 0.76–1.55 | 0.67 | |||
| Current Smoking | 0.72 | 0.48–1.07 | 0.1 | |||
| Renal Impairment | 1.09 | 0.74–1.56 | 0.66 | |||
| BMI | 0.98 | 0.93–1.03 | 0.44 | |||
| SBP | 1.005 | 0.99–1.01 | 0.2 | |||
| DBP | 0.99 | 0.98–1.01 | 0.35 | |||
| Low density lipoprotein cholesterol | 1.001 | 0.99–1.01 | 0.81 | |||
| High density lipoprotein cholesterol | 0.99 | 0.98–1.01 | 0.31 | |||
| Triglycerides | 1.001 | 0.997–1.002 | 0.48 | |||
| Lipoprotein(a) high/low | 1.41 | 1.01–1.98 | 0.012 | 1.28 | 1.04–1.58 | 0.018 |
| ACE-I·ARB | 1.01 | 0.73–1.41 | 0.93 | |||
| βblocker | 0.83 | 0.60–1.15 | 0.26 | |||
| Ca-channel blocker | 1.09 | 0.77–1.53 | 0.62 | |||
| Multivessel disease | 1.74 | 1.23–2.50 | 0.002 | 1.75 | 1.23–2.53 | 0.002 |
| LAD proximal or LMT lesion | 1.12 | 0.74–1.64 | 0.58 | |||
| Acute coronary syndrome at presentation | 1.54 | 1.09–2.16 | 0.015 | 1.8 | 1.27–1.63 | 0.006 |
HR, hazard ratio; 95%CI, 95% confidential interval.
Table 3. Hazard ratios of Lp(a) levels as a natural logarithm-transformed continuous variable for cardiac events.
| HR | 95% CI | P | |
|---|---|---|---|
| Unadjusted | 3.5 | 1.31–9.62 | 0.012 |
| Model 1 | 1.29 | 1.06–1.59 | 0.011 |
| Model 2 | 1.31 | 1.07–1.61 | 0.009 |
| Model 3 | 1.32 | 1.08–1.63 | 0.006 |
Model 1, age and gender adjusted
Model 2, adjusted for variables in model 1 plus lipid parameters including LDL-C, HDL-C and TG.
Model 3, adjusted for variables in model 1 plus variables with p < 0.05 in univariable analysis comprised diabetes, multivessel disease and ACS.
ACS, acute coronary syndrome; CI, confidence interval; HDL-C, high-density lipoprotein cholesterol; HR, hazard ratio; LDL-C, low-density lipoprotein cholesterol; TG, triglyceride; MACE, major adverse cardiac events.
Discussion
This observational study demonstrated that elevated Lp(a) levels were associated with long-term adverse clinical outcomes in CAD patients treated with statin after PCI. A higher logarithm-transformed Lp(a) value were associated with a greater risk of cardiac events, and the association remained significant even after adjustment for other independent variables. To our knowledge, the present study showed this is the first study to show the clinical impact of Lp(a) levels on long-term clinical outcomes in patients with CAD who underwent PCI and statin therapy.
Lipoprotein (a) has been identified as an independent, casual risk factor for CVD which is continuous in shape without a threshold and does not depend of high levels of LDL-C14, 15). A previous meta-analysis also reported the association between elevated Lp(a) and increased CVD risk using 18 prospective studies of general population16). Recent mendelian randomization studies reported that genetic polymorphisms associated with elevated Lp(a) level and the risk of CVD suggesting a direct role for Lp(a) in atherothrombotic events17, 18).
Statins significantly reduce cardiovascular events in patients with coronary artery disease (CAD) and are the most widely used for individuals with CAD for secondary prevention19, 20), however residual risk still exist21). Previous studies have suggested statin may attenuate the cardiovascular risk associated with Lp(a)22). Although Lp (a) may prove to be a casual risk factor for the development of CVD, its clinical utility as a prognostic biomarker in secondary prevention remains uncertain. A recent study showed the association between Lp(a) concentration and further CVD events in patients with CAD on statin therapy from LIPID study (Long-Term Intervention with pravastatin in Ischaemic Disease)23). Another secondary prevention population study using 6708 subjects with CAD from 3 studies including two large statin trial, CARE (Cholesterol and Recurrent Event) and PROVE IT-TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction) trials, demonstrated Lp(a) is significantly associated with the risk of CV events in patients with established CAD24). In the present study using Japanese PCI cohort, we evaluated Lp(a) values as a natural logarithm-transformed continuous variable to determine whether findings differed from the cutoff points and we confirmed a higher logarithm-transformed lipoprotein(a) was associated with a greater risk of cardiac events in patients with CAD who underwent PCI and statin therapy which was consistent with the current secondary prevention trials. In a coronary angiographic study among 2769 patients being treated with statins, elevated Lp(a) defined as > 30 mg/dL was associated significantly with the severity of coronary atherosclerosis and a greater risk of CVD events, particularly revascularizations6). Another recent study evaluated the association between Lp(a) levels and vulnerable plaque phenotype using optical coherence tomography (OCT)25). Mitsuda T et al reported lipoprotein (a) levels predict adverse vascular events in 176 patients with ST-elevated myocardial infarction during 3-year follow-up after primary PCI26). In contrast to these previous studies which included coronary revascularization in the primary endpoint, we focused the primary end-point on cardiac events defined as cardiac death and non-fatal acute coronary syndrome reflecting more coronary atherogenic property of Lp(a) with longer follow-up period. Our study showed that patients with CAD who have a high level of Lp(a) may be at an increased risk of subsequent adverse events after PCI and may serve as a potential surrogate marker to predict the future clinical cardiac events after PCI.
Accumulated data suggest that new therapeutic lipid-modifying therapies tha reduce Lp(a)27), such as cholesterylester transfer protein (CETP) inhibitors28), proprotein covertase subtilisin/kexin type 9 (PCSK9) inhibitors29). In particular, evolocumab is a fully human monoclonal antibody to PCSK9, a protein synthesized and secreted by hepatocyte, which binds to LDL receptor, targeting it for lysosomal degradation. LAPLACE-TIMI 57 (the assessment with proprotein convertase subtilisin kexin type 9 monoclonal antibody inhibition combined with statin therapy-thrombolysis in myocardial infarction 57) trial demonstrated that evolocumab reduced Lp(a) up to 32% among subjects with hypercholesterolemia receiving statin therapy30). A pooled analysis of data from 4 clincal trials showed the inhibition of PCSK9 with evolocumab resulted in significant dose-related reductions in Lp(a)31). A further clinical trials would give us some clues for the Lp(a) targeted therapy in addition to statin in patients with CAD.
Several limitations of the present study require consideration. This single-center, observational study included a small sample cohort, and other unknown confounding factors might have affected outcomes regardless of adjustments in the statistical analyses. In addition, relatively few events occurred during the present study, which resulted in the absence of statistically significant differences in outcome measures. Thus, an observational study of a larger cohort is necessary. Furthermore, the choice or dose of statins depended on each physician and we had no information about the patient compliance with prescribed medical therapy during follow-up.
In conclusion, elevated Lp(a) values are significantly associated with long-term mortality in patients CAD treated with statin after PCI and might serve as residual risk.
Acknowledgements
The authors thank the staff of the Department of Cardiovascular Medicine at Juntendo University. We also thank Yumi Nozawa for secretarial assistance.
Disclosures
Dr Daida has received Speakers' Bureau Honoraria from MSD K.K., AstraZeneca K.K., Kowa Pharmaceutical Company LTD., Sanofi-Aventis K.K., GlaxoSmithKline K.K., Shionogi & Co., Ltd., Daiichi-Sankyo Company, Limited, Takeda Pharmaceutical Co., Ltd, Mitsubishi Tanabe Pharma Corp., Pfizer Co., Ltd., and Astellas Pharma Inc. and research funds from Kirin Co. Ltd., Kaken Pharmaceutical Co. Ltd., Abbott Japan Co., Ltd., Astellas Pharma Inc., Astrazeneca K.K., Bayer Yakuhin, Ltd., Boston Scientific Japan K.K., Bristol-Myers Squibb, Daiichi Sankyo Company, MSD K.K., Pfizer Inc., Philips Respironics, Sanofi K.K., and Takeda Pharmaceutical Co. Ltd.
Dr Miyauchi has received Speakers' Bureau Honoraria from MSD K.K., Sanofi-Aventis K.K., Daiichi-Sankyo Company, Limited, Takeda Pharmaceutical Co., Ltd, Amgen Astellas BioPharma K.K. and Bayer Yakuhin, LTd.
References
- 1).Scandinavian Simvastatin Survival Study Group: Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet, 1994; 344: 1383-1389 [PubMed] [Google Scholar]
- 2). Cannon CP, Braunwald E, McCabe CH, Rada DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM, for the Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarctioni 22 Investigators : Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Eng J Med, 2004; 350: 1495-1504 [DOI] [PubMed] [Google Scholar]
- 3). Mora S, Wenger NK, DeMicco DA, Breazna A, Boekholdt SM, Arsenault BJ, Deedwania P, Kastelein JJ, Waters DD: Determinants of residual risk in secondary prevention patients treated with high- versus low-dose statin therapy: the Treating to New Targets (TNT) study. Circulation, 2012; 125: 1979-1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Reith C, Armitage J: Management of residual risk after statin therapy. Atherosclerosis, 2016; 245: 161-170 [DOI] [PubMed] [Google Scholar]
- 5). Tsimikas S, Hall JL: Lipoprotein (a) as a potential casual genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol, 2012; 60: 716-721 [DOI] [PubMed] [Google Scholar]
- 6). Nicholls SJ, Tang WH, Scoffone H, Brennan DM, Hartiala J, Allayee H, Hazen SL: Lipoprotein(a) levels and long-term cardiovascular risk in the contemporary era of statin therapy. J Lipd Res, 2010; 51: 3055-3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7). Bennet A, Di Angelantonio E, Erqou S, Eiriksdottir G, Sigurdsson G, Woodward M, Rumley A, Lowe GD, Danesh J, Gudnason V: Lipoprotein(a) levels and risk of future coronary heart disease: large-scale prospective data. Arch Intern Med, 2008; 168: 598-608 [DOI] [PubMed] [Google Scholar]
- 8). Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG: Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA, 2009; 301: 2331-2339 [DOI] [PubMed] [Google Scholar]
- 9). Suk Danik J, Rafai N, Buring JE, Ridker PM: Lipoprotein(a), measured with an assay independent of apolipoprotein (a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA, 2006; 296: 1363-1370 [DOI] [PubMed] [Google Scholar]
- 10). Ikenaga H, Ishihara M, Ioue I, Kawagoe T, Shimatani Y, Miura F, Nakama Y, Dai K, Otani T, Ejiri K, Oda N, Nakamura M, Miki T: Usefulness of lipoprotein (a) for predicting progression of non-culprit coronary lesions after acute myocardial infarction. Circ J, 2011; 75: 2847-2852 [DOI] [PubMed] [Google Scholar]
- 11). Kardys I, Oemrawsingh RM, Kay IP, Hones GT, McCormick SP, Daemen J, Van Geuns RJ, Boersma E, Van Domburg RT, Serruys PW: Lipoprotein (a), interleukin-10, C-reactive protein, and 8-year outcome after percutaneous coronary intervention. Clin Cardiol, 2012; 35: 482-489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).The Committee of Japan Diabetes Society on the diagnostic criteria of diabetes mellitus: Report of the Committee on the classification and diagnostic criteria of diabetes mellitus. J Jpn Diabetes Soc, 2010; 53: 450-467 [Google Scholar]
- 13). Matsuo S, Imai E, Horio M, Yasuda Y, Tomita K, Nitta K, Yamagata K, Tomino Y, Yokoyama H, Hishida A, Collaborators developing the Japanese equation for estimated GFR : Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidey Dis, 2009; 53: 982-992 [DOI] [PubMed] [Google Scholar]
- 14). Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reniner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A, for the European Athersclerosis Society Consensus Panel : Lipoprotein(a) as a cardiovascular risk factor; current status. Eur Heart J, 2014; 35: 1683-1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Erqous S, Kaptoge S, Perry PL, Di AE, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J: Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA, 2009; 302: 412-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Bennet A, Di AE, Erqou S, Eiriksdottir G, Sigurdsson G, Woodward M, Rumlley A, Lowe GD, Danesh J, Gudnason V: Lipoprotein(a) levels and risk of future coronary heart disease: large-scaler prospective data. Arch Intern Med, 2008; 168: 598-608 [DOI] [PubMed] [Google Scholar]
- 17). Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Helth SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveria A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M, PROCARDIS Consortium : Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med, 2009; 361: 2518-2528 [DOI] [PubMed] [Google Scholar]
- 18). Kamstrup PR, Tybjaerg-Hansen A, Steffense R, Nordestgaard BG: Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA, 2009; 301: 2331-2339 [DOI] [PubMed] [Google Scholar]
- 19).Cholesterol Treatment Trialists' (CCT) Collaborators: Efficacy and safty of cholesterol-lowering treatment: prospective meta-analysis of data from 90056 participants in 14 randomised trials of statins. Lancet, 2005; 366: 1267-1278 [DOI] [PubMed] [Google Scholar]
- 20). Wakabayashi K, Nozue T, Yamamoto S, Tohyama S, Fukui K, Umezawa S, Onishi Y, Kunishima T, Sato A, Miyake S, Morino Y, Yamauchi T, Muramatsu T, Hibi K, Terashima M, Suzuki H, Michishita I, TRUTH investigators : Efficacy of Statin Therapy in Inducing Coronary Plaque Regression in Patients with Low Baseline Cholesterol Levels. J Atheroscler Thromb, 2016; 23: 1055-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Reith C, Armitage J: Management of residual risk after statin therapy. Atherosclerosis, 2016; 245: 161-170 [DOI] [PubMed] [Google Scholar]
- 22). Maher VM, Brown BG, Marcovina SM, Hillger LA, Zhao XQ, Albers JJ: Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein (a). JAMA, 1995; 274: 1771-1774 [PubMed] [Google Scholar]
- 23). Nestel PJ, Bares EH, Tonkin AM, Simes J, Fournier M, White HD, Colquhoun DM, Blankenberg S, Sullivan DR: Plasma Lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol, 2013; 33: 2902-2908 [DOI] [PubMed] [Google Scholar]
- 24). O'Donoghue ML, Morrow DA, Tsimikas S, Sloan S, Ren AF, Hoffman EB, Desai NR, Solomon SD, Domanski M, Arai K, Chiuve SE, Cannon CP, Sacks FM, Sabatine MS: Lipoprotein(a) for risk assessment in patients with established coronary artery disease. J Am Coll Cardiol, 2014; 63: 520-527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Niccoli G, Cin D, Scalone G, Panebianco M, Abbolito S, Cosentino N, Jacoangeli F, Refaat H, Gallo G, Volpe M, Crea F, De Biase L: Lipoprotein(a) is related to coronary atherosclerotic burden and a vulnerable plaque phenotype in angiographically obstructive coronary artery disease. Atherosclerosis, 2016; 246: 214-210 [DOI] [PubMed] [Google Scholar]
- 26). Mitsuda T, Uemura Y, Ishii H, Takemoto K, Uchikawa T, Koyasu M, Ishikawa S, Miura A, Imai R, Iwamiya S, Ozaki Y, Kato T, Shibata R, Watarai M, Murohara T: Lipoprotein(a) levels predict adverse vascular events aftter acute myocardial infarction. Heart Vessels, 2016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 27). Kolski B, Tsimikas S: Emerging therapeutic agents to lower lipoprotein(a) levels. Curr Opin Lipid, 2012; 23: 560-568 [DOI] [PubMed] [Google Scholar]
- 28). Cannnon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Asharf TB, Zafarino J, Mitchel Y, Barter P: Safty of anacetrapib in patients with or at high risk for coronary heart disease. N Eng J Med, 2010; 363: 2406-2415 [DOI] [PubMed] [Google Scholar]
- 29). McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA: Safty and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receving ongoing stable atorvastatin therapy. J Am Coll Cardiol, 2012; 59: 2344-2353 [DOI] [PubMed] [Google Scholar]
- 30). Desai NR, Kohli P, Giugliano RP, O'Donoghue ML, Somaratne R, Zhou J, Hoffman EB, Huang F, Rogers WJ, Wasserman SM, Scott R, Sabatine MS: AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduce lipoprotein(a) I hypercholesterolemic patiens receiving statin therapy: an analaysis from the LAPLACE-TIMI 57 trial. Circulation, 2013; 128: 962-969 [DOI] [PubMed] [Google Scholar]
- 31). Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Langslet G, Bays H, Blom D, Eriksson M, Dent R, Wasserman SM, Huang F, Xue A, Albizem M, Scott R, Stein EA: Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG145). J Am Coll Cardiol, 2014; 63: 1278-1288 [DOI] [PubMed] [Google Scholar]