

Abstract
Backgrounds & Aims
Bacterially-derived factors from the gut play a major role in the activation of inflammatory pathways in the liver and in the pathogenesis of alcoholic liver disease. The intestinal brush-border enzyme intestinal alkaline phosphatase (IAP) detoxifies a variety of bacterial pro-inflammatory factors and also functions to preserve gut barrier function. The aim of this study was to investigate whether oral IAP supplementation could protect against alcohol-induced liver disease.
Methods
Mice underwent acute binge or chronic ethanol exposure to induce alcoholic liver injury and steatosis +/− IAP supplementation. Liver tissue was assessed for biochemical, inflammatory and histopathological changes. An ex vivo co-culture system was used to examine the effects of alcohol and IAP treatment in regard to the activation of hepatic stellate cells and their role in the development of alcoholic liver disease.
Results
Pretreatment with IAP resulted in significantly lower serum alanine aminotransferase (ALT) compared to the ethanol alone group in the acute binge model. IAP treatment attenuated the development of alcohol-induced fatty liver, lowered hepatic pro-inflammatory cytokine and serum LPS levels, and prevented alcohol-induced gut barrier dysfunction. Finally, IAP ameliorated the activation of hepatic stellate cells and prevented their lipogenic effect on hepatocytes.
Conclusions
IAP treatment protected mice from alcohol-induced hepatotoxicity and steatosis. Oral IAP supplementation could represent a novel therapy to prevent alcoholic-related liver disease in humans.
Keywords: Intestinal Alkaline Phosphatase, Alcoholic Liver Disease, Fatty Liver, Stellate cells
INTRODUCTION
Alcohol consumption is one of the leading causes of end-stage liver disease and the second most common cause of liver transplantation in the United States and Europe [1, 2]. Excessive alcohol drinking results in liver disorders that range from fatty liver (steatosis) to alcoholic hepatitis, cirrhosis, and in some, hepatocellular carcinoma, with a mortality rate of 30–50% in some cases of acute alcoholic hepatitis [3]. Steatosis, characterized by excessive fat deposition in hepatocytes, develops in the majority of heavy drinkers and is the first alteration associated with acute and chronic alcohol liver disease (ALD) [4].
Activation of inflammatory pathways in the liver cells plays a major role in the pathogenesis of ALD. Chronic ethanol consumption increases (the) production of inflammatory mediators from (in) liver cells, in particular TNF-α, Interleukin-1 β and reactive oxygen species (ROS), in some cases progressing to fatty liver, inflammation and fibrosis [5,6,7]. TNF-α levels are increased in patients with alcoholic steatohepatitis [8], its levels correlating with disease severity [9]. Activation of an immune response in liver cells occurs through the pattern recognition receptor toll like receptor (TLR4), upregulating NFκB signaling and the subsequent production of cytokines, playing a crucial role in the development of ALD [7,10, 11]. Indeed, alcohol-related liver damage is diminished in animals lacking TLR4 or its coreceptors [11].
Bacterial endotoxin (lipopolysaccharide, LPS), a Gram-negative bacterial cell wall component, is a potent activator of innate immune responses through its binding to the TLR4 complex. LPS plays a major role in the alcohol-induced liver inflammation and the pathogenesis of ALD [12, 13]. Furthermore, LPS enhances ethanol-induced hepatic microvascular dysfunction, liver injury and apoptosis [14, 15, 16]. Acute and chronic alcohol intake increases portal and systemic LPS in patients and animal models of ALD and has a positive correlation with the severity of the disease [13,17]. Studies comparing the effects of portal and systemic LPS in ethanol-exposed animals strongly suggest a gut source of endotoxins in the pathogenesis of ALD [18].
The small intestinal brush border enzyme intestinal alkaline phosphatase (IAP) is known to play a major role in the complex interactions between the host and the gut microbiota [19,20]. We and others have shown that IAP detoxifies and prevents the inflammation related to many bacterial pro-inflammatory factors, including LPS, flagellin, and CpG DNA [21,22]. In addition, IAP appears to directly promote the intestinal barrier, enhancing the tight junctions and preventing bacterial translocation [23]. Nakano et al has shown that the disruption of the murine IAP gene (Akp3) resulted in hepatic steatosis due to non-alcoholic liver disease [24]. Furthermore, we have found that oral IAP supplementation prevents the development of the metabolic syndrome and fatty liver due to high fat diet in mice [25]. Interestingly, ethanol has been associated with decreased IAP activity and excretion during and for few days after ethanol exposure in acute and chronic alcohol animal models [26,27,28]. Based upon the above data, we hypothesized that oral supplementation with IAP will prevent alcoholic liver disease by detoxifying bacterially derived inflammatory factors in the gut and preventing their translocation into the portal system that can result in hepatic inflammation. Indeed, in this study we show that oral supplementation of IAP prevents alcoholic fatty liver in mice. Furthermore, we demonstrate the role of hepatic stellate cells in alcohol-induced fat accumulation in hepatocytes. Accordingly, we believe that oral IAP supplementation could represent a novel therapy to prevent the development of alcoholic liver disease in humans.
Materials and Methods
Animals
10–12 week old C57BL/6 female mice were purchased from Charles River Laboratories, Wilmington, MA. The animals were maintained under a 12-hour light-dark cycle with free access to standard laboratory chow diet and water in accordance with the guidelines of the Committee on Animals of Harvard Medical School (Boston, MA) and those prepared by the IACUC at MGH based on the Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council (Department of Health, Education and Human Services, publication no. 85e23 (National Institute of Health), revised 1985). All animal work protocols (Protocol: 2008N000023) were reviewed and approved by the IACUC at MGH.
Alcohol Supplementation
After acclimation for 1 week, acute liver injury was induced with a model of binge drinking described by Carson and Pruett [29] and Zhou et al. [30]. Mice were divided into 3 groups (n = 5) and treated with a binge ethanol dose 5 g/kg by gastric gavage +/− pre-, simultaneous, or post-treatment with varying doses of IAP (100 U or 200 U). In a second set of experiments, C57BL/6 mice were gavaged with a binge ethanol dose 5 g/kg every 12 hours for a total of 3 doses +/− pre-treatment with IAP 200 U by gastric gavage. In the chronic alcohol experiment, mice were divided into 3 groups (n=6) and fed a liquid diet containing EtOH +/− IAP at a dose of 200 U/ml for 10 days. In all experiments, control mice were fed a liquid diet with an equal volume of an isocaloric solution of dextran maltose.
Serum ALT measurement
After euthanizing the mice, blood was obtained by cardiac puncture, placed on ice, and then centrifuged at 2,500 g for 10 minutes. The serum was collected and assayed for ALT using a Colorimetric Activity Assay Kit from Cayman (Ann Arbor, MI).
Liver histopathology and hepatic steatosis
Frozen liver tissues were submitted to the Histology Laboratory at MGH (Boston, MA) to prepare Oil Red O stained slides, which were then examined by an independent investigator (pathologist) in a blinded fashion. Hepatic steatosis was graded on a scale of 0 to 4+ depending on the size and distribution of fat droplets: 0 (none/minimal); 1+ (mild); 2+ (moderate); 3+ (moderate to marked); and 4+ (marked).
Hepatic triglycerides measurement
100 mg of frozen liver tissue was homogenized in 2 ml chloroform: methanol (2:1) as described by Folch et al [31]. The homogenate was filtered and 0.4 ml of 50 mM NaCl was added to each sample. The mixture was centrifuged for 20 min at room temperature, the lower chloroform phase collected and dried, and the pellets dissolved in PBS containing 1% Triton X-100. The triglyceride contents were determined by using an enzymatic reagent (Sigma-Aldrich, St. Louis, MO) and expressed as mg/gm tissue.
Limulus amebocyte lysate (LAL) assay
Serum LPS concentrations were determined using a kit (GenScript, Piscataway, NJ) based on the Limulus amebocyte lysate following the manufacturer’s instructions. Briefly, samples were diluted 1/10 and 1/100 with endotoxin-free water, adjusted to recommended pH, and heated for 10 min at 70°C to minimize interferences in the reaction. LAL reagents were added and incubated at 37°C for 45 min, and the absorbance was read at 545 nm. LPS was measured as endotoxin units/ml.
Liver Tissue cytokine levels
Frozen liver tissue were homogenized with 5 volumes of ice-cold RIPA buffer containing 150 mM NaCl, 10 mM Tris-HCl, pH 7.5, 1% sodium deoxycholate, 1% NP-40, 10 mM EDTA, 0.1% SDS, including both protease and phosphatase inhibitors (Thermo Scientific, IL), followed by incubation on ice for 30 minutes. Thereafter, the homogenates were centrifuged twice at 4°C, 15000 g for 15 min, and the supernatants were collected to determine protein concentration using the Coomassie (Bradford) Protein Assay Kit (Thermo Scientific, IL). TNF-α and IL-1β levels were measured using a commercially available sandwich enzyme immunoassay technique (enzyme-linked immunosorbent assay [ELISA]; eBioscience, San Diego, CA). TNF-α and IL-1β were measured in pg/mg protein.
In vivo intestinal permeability assay
Six hours after the last dose of the 3 binge alcohol experiment, mice were anesthetized using Florane and ketamine (50mg/kg). A midline incision was made in the abdominal wall and a 5 cm loop was constructed in the terminal ileum. A 100 μL of phosphate buffer saline (PBS, pH 7.2) containing 1 mg FITC-dextran (4 kDa) (Sigma-Aldrich, St. Louis, MO) was instilled into the loop using 25G needle and the abdominal wall incision closed in two layers using 5-0 silk suture. One and a half hours later, while still under anesthesia, blood was collected from the right ventricle and serum assayed for FITC-dextran concentration as described [25].
Quantitative Real-Time PCR
The liver and small intestine tissues were isolated, flushed with ice cold PBS and snap frozen in liquid nitrogen. The tissue was then processed with TrIzol to isolate total RNA. High Capacity cDNA Reverse Transcription kit (BIO-RAD, Hercules, CA) was used for the generation of cDNA for all samples. Quantitative real-time PCR (qRT-PCR) was performed with a Mastercyclerrealplex instrument (Eppendorf) using iQ SYBR Green Supermix kit (BIO-RAD, Hercules, CA). See supplementary data for primer list. For each sample, real-time PCR reactions were performed in triplicate, and the average threshold cycle (Ct) was calculated. Expression of target gene mRNA was normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression. Expression levels were calculated using the ΔΔCt method after correcting for differences in PCR efficiencies and expressed relative to control levels. The average copy number of mRNA expression in control samples was set to 1.0.
Hepatic stellate cells and hepatocyte isolation
Hepatocytes were isolated from control C57BL/6 mice and hepatic stellate cells isolated from control and ethanol +/− IAP-treated mice by enzymatic digestion and Percoll density gradient centrifugation with modifications [32]. The portal vein was perfused in situ with 30 mL of HBSS (without Ca2+ and Mg2+) and 30 mL of 0.05% collagenase B (Roche Diagnostics, Indianapolis, IN), respectively, at 37 °C with a flow rate of 6 ml/min. After perfusion, the partially digested liver was excised and incubated with 20 mL of 0.05% collagenase and DNase I 10 ug/mL, (Roche Diagnostics, Indianapolis, IN), at 37 °C for 30 minutes. The tissue was passed through a 70 μm nylon mesh to remove undigested materials and suspended in washing buffer (PBS containing DNase I). Hepatocytes were separated from the non-parenchymal cells and debris by centrifugation at 4°C in the following sequence: twice for 5 minutes at 50 g, and twice again for 5 minutes at 20 g. The supernatant was collected for stellate cell isolation and the hepatocytes present in the pellet were re-suspended in DMEM. Primary mouse HSCs were purified from the remainder of non-parenchymal cells. Cells were centrifuged at 635 g for 10 minutes and resuspended in washing buffer followed by pass through a 70 μm nylon mesh. The pellets were resuspended in 10 ml of 35 % Percoll (GE Healthcare, Pittsburgh, PA) with an overlay of 1 ml PBS. After centrifugation at 1130 g for 30 minutes, HSCs are the in the layer located between the PBS and 35% Percoll. Cells were counted using a hemocytometer (Neubauer chamber) and 0.4% trypan blue (Sigma-Aldrich, St. Louis, MO). The purity was determined by a UV sorting with BD FACS Aria II SORP Cell Sorter (BD Biosciences, San Jose, CA).
Co-culture of primary mouse hepatocytes and hepatic stellate cells
Isolated hepatocytes were re-suspended in DMEM medium containing 10% FBS, plated onto collagen-coated six-well plates at a density of 5×105 cells/well in 1.5 ml culture medium, and cultured for 4 hours. The medium was then changed into serum-free medium and the cells were co-cultured with hepatic stellate cells and loaded onto cell-culture inserts of 3 μm pore size (Corning, Corning, NY).
BODIPY staining of lipid droplets
Hepatocytes with the co-cultured stellate cells were washed with PBS, fixed with 4% formaldehyde for 15 minutes, and stained with BODIPY (1 μg/ml, Invitrogen, Carlsbad, CA) for 15 min at room temperature. Cells were then washed 3 times with PBS and stained with DAPI.
Statistics
All data were expressed as mean ± SEM. Statistical analysis was performed using Student’s t test and ANOVA where appropriate. Differences between groups were considered to be statistically significant at p<0.05.
Results
Oral supplementation with IAP prevents alcohol induced liver damage
Liver damage is associated with elevated serum ALT levels. To study the effect of IAP treatment on alcohol induced liver toxicity, we assayed serum ALT activity in mice after acute and chronic alcohol intake +/− different modalities of oral IAP supplementation. In the alcohol single dose experiment, acute EtOH administration caused an elevation in serum ALT levels (Figure 1A). However, pretreatment with IAP attenuated the EtOH-induced ALT increases in a dose dependent fashion; there was a 38% inhibition with the high dose of IAP, 200 U (Figure 1A). Simultaneous treatment with IAP (100 U/gavage) also lowered serum ALT (EtOH vs. EtOH + IAP = 110 ± 6.6 vs. 89.8 ± 5.7 U/L, p =0.04). In contrast, when IAP was gavaged (100 U) 60 min after EtOH gavage, there was no beneficial impact in regard to ALT levels. In the 3-dose EtOH binge model, pretreatment with IAP (200 U) significantly lowered serum ALT (Figure 1B) compared to ethanol alone. Daily oral IAP supplementation at a dose of 200 U/ml in the chronic alcohol mouse model where EtOH was fed for 10 days resulted in minimal changes in serum ALT levels (EtOH vs. EtOH + IAP, 237.7 ± 0.42 vs. 182.9 ± 0.34, p=0.10). These results suggest that the oral IAP supplementation inhibits alcohol binge-induced liver damage, as long as the IAP is administered before or during the alcohol.
Figure 1.
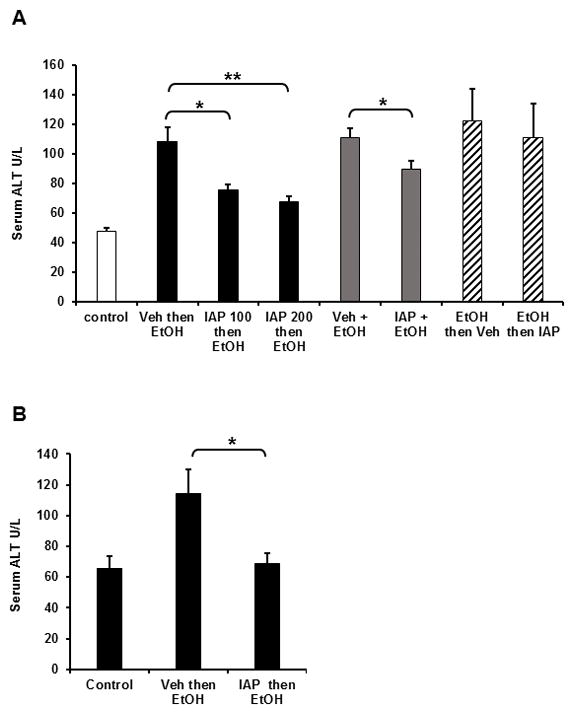
Oral IAP supplementation inhibits alcohol induced liver damage. Serum ALT Levels: (A) single alcohol dose, (B) 3-dose alcohol binge model. Values are expressed as mean ± SEM. Statistical significance between the groups was tested using one-way analysis of variance with Tukey’s multiple comparison posttests. *p < 0.05, **p < 0.01.
Oral IAP Supplementation ameliorates alcohol-induced steatosis and reduces fat accumulation in the liver
Fatty infiltration is the first alcohol-induced change in the liver and its development is required for the progression to more advanced stages including steatohepatitis, fibrosis and carcinoma [4]. To assess the effect of IAP supplementation on alcohol induced fatty liver, we used the 3-binge alcohol gavage model with pretreatment with 200 U of IAP before every EtOH dose. Figure 2A shows prominent steatosis in liver sections from mice treated with 3 doses of EtOH binge gavages. Pretreatment with IAP 200 U before each EtOH gavage resulted in significantly lower steatosis levels (Figure 2A). Hepatic steatosis scores were calculated for the liver sections as mentioned in the Materials and Methods (Figure 2B) and showed that the steatosis was less extensive in the IAP treated group compared to the livers from mice receiving EtOH alone (EtOH vs. EtOH + IAP = 4 vs. 2, Hepatic Steatosis Score) (Figure 2B).
Figure 2.
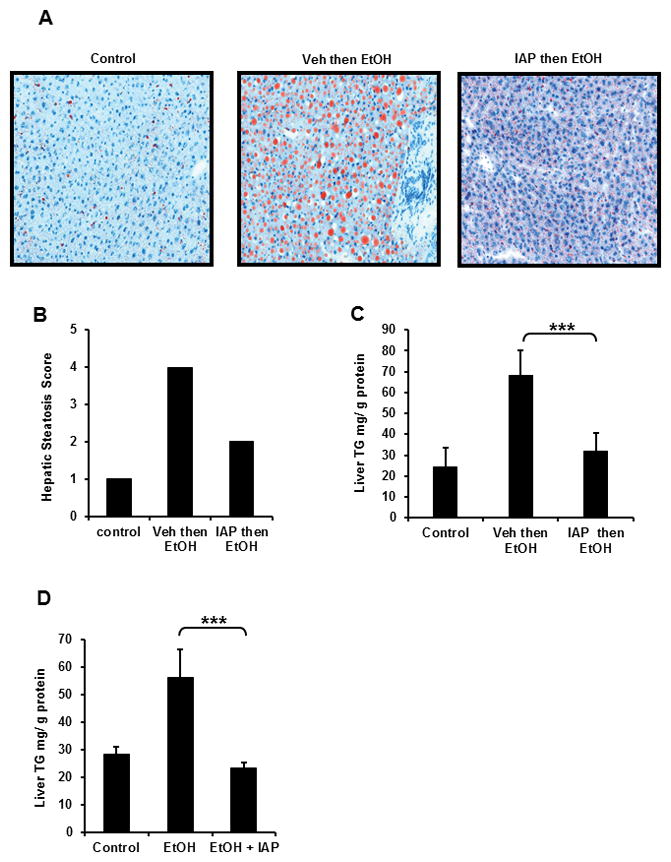
Oral IAP supplementation ameliorates alcohol-induced liver steatosis. In the 3-dose alcohol binge model, frozen liver tissues were obtained to determine (A) Oil Red O stain, (B) Hepatic steatosis score (0–4+). Liver triglyceride content was measured in (C) 3-dose alcohol binge model, (D) chronic alcohol model. Results were compared to mice untreated with alcohol and alcohol-alone treated mice. Values are expressed as mean ± SEM. Statistical significance between the groups was tested using one-way analysis of variance with Tukey’s multiple comparison posttests. ***p < 0.001.
Based on the result above, we sought to study the effect of IAP supplementation on the triglyceride (TG) content in livers from alcohol +/− IAP treated mice. Figure 2C shows that EtOH intake in the 3-dose model resulted in significantly higher TG liver contents, whereas pretreatment (Figure 2C) or simultaneous treatment (Figure 2D) with IAP significantly ameliorated the alcohol-induced increase in liver TG levels. These results demonstrate that oral IAP supplementation protects the mice from the development of fatty liver due to alcohol consumption.
IAP administration prevents liver inflammation due to alcohol intake
Activation of inflammatory pathways in liver cells plays a major role in the pathogenesis of ALD. Ethanol consumption increases production of inflammatory mediators from liver cells, in particular TNF-α and interlukin-1 β, leading to the fatty liver, inflammation and fibrosis. Furthermore, given that IAP functions to detoxify bacterially-derived factors and prevent inflammation in many disease models, we sought to determine the effect of IAP supplementation on alcohol induced liver inflammation in mice. Figure 3A & B show that EtOH supplementation in the 3-binge alcohol model resulted in elevated TNF-α and Il-1β cytokine levels in liver lysates. Furthermore, IAP pre-treatment ameliorated the EtOH effects, significantly reducing the levels of these cytokines. Additionally, EtOH supplementation resulted in elevated TNF-α and Il-1β cytokine mRNA levels in livers of mice in both the 3-binge and chronic alcohol mouse model. Oral IAP supplementation resulted in lower cytokines mRNA levels in the livers when compared to mice that received alcohol alone (Figure 3C, D).
Figure 3.
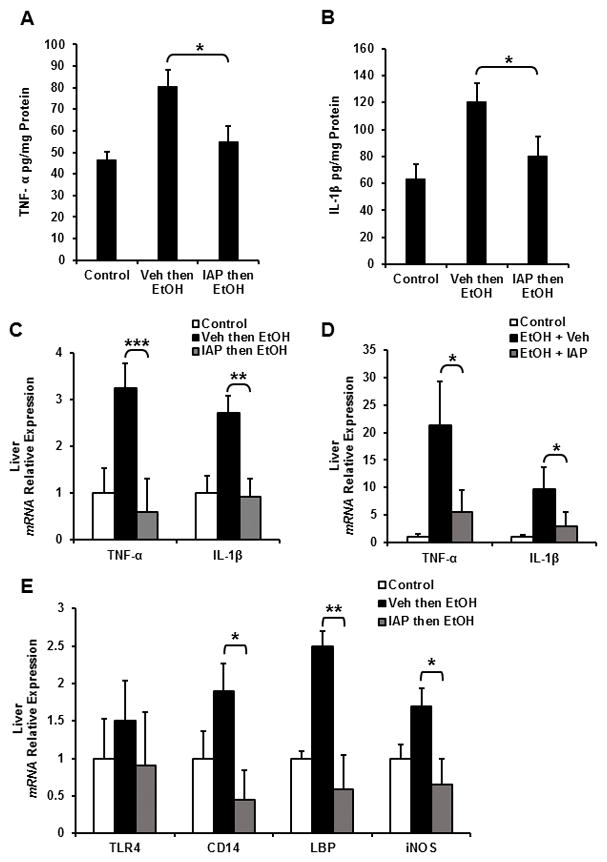
IAP treatment prevents alcohol-induced liver inflammation and the subsequent sensitivity to pro-inflammatory stimuli. In the 3-dose alcohol binge model, liver inflammatory cytokine levels: (A) TNF-α and (B) IL-1β were measured using ELISA. Liver inflammatory cytokine mRNA levels were determined by qPCR: (C) 3-dose alcohol binge model and (D) chronic alcohol model. (E) Liver Tlr4 and co-receptor mRNA expression levels from mice using the 3-dose alcohol binge model. Values are expressed as mean ± SEM. Statistical significance between the groups was tested using one-way analysis of variance with Tukey’s multiple comparison posttests; *p < 0.01, **p < 0.01, ***p < 0.001.
Alcohol intake increases the sensitivity of liver cells to inflammatory mediators by upregulating the expression of TLR4 and its co-receptors CD14, MD-2 and LPS-binding protein (LBP) mRNA and the absence of TLR4 pathway activation diminishes liver injury and inflammation due to alcohol intake [11]. Therefore, we sought to elucidate the effect of IAP treatment on TLR4, CD14, LBP and iNOS gene expression in alcohol-fed mice. Figure 3E shows that alcohol intake indeed upregulated the mRNA levels of TLR4, CD14, LBP, and iNOS in mice liver, whereas IAP supplementation largely prevented these increases. These results suggest that IAP treatment inhibits alcohol-induced liver inflammation and the subsequent sensitivity to pro-inflammatory stimuli.
Treatment with oral IAP prevents alcohol-induced endotoxemia
We next wanted to explore the mechanism by which IAP protects the liver from alcohol. Given the fact that IAP detoxifies LPS and other bacterial toxins, as well as preventing the translocation of bacterial endotoxins into the blood stream, we investigated the effect of IAP treatment on the levels of serum LPS in mice exposed to either 3 doses or 10 days of EtOH. Both binge doses of EtOH and chronic EtOH intake for 10 days resulted in a substantial increase in serum LPS levels (Figures 4A and 4B). We found that IAP supplementation by gastric gavage 1.5 h before the EtOH administration resulted in significantly lower serum LPS levels in both the binge and chronic alcohol models (Figures 4A and 4B). Given the gut source of endotoxins in the pathogenesis of ALD [27], we sought to assess the effect of IAP supplementation on endotoxin levels in portal vein blood. Figure 4C shows that EtOH gavage resulted in higher LPS levels in the portal system and that pretreatment with IAP significantly lowered these levels. These results suggest that IAP prevents alcohol-induced liver inflammation, damage, and steatosis by blocking LPS absorption across the intestinal barrier into the portal system.
Figure 4.
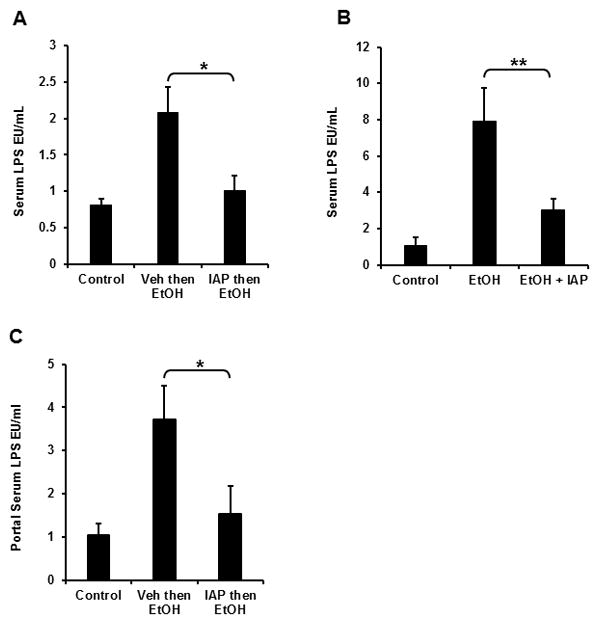
Treatment with oral IAP prevents alcohol-induced endotoxemia. Serum LPS in (A) 3-dose alcohol binge model and (B) chronic alcohol model. (C) Portal serum LPS in 3-dose alcohol binge model. Values are expressed as mean ± SEM. Statistical significance between the groups was tested using one-way analysis of variance with Tukey’s multiple comparison posttests; *p < 0.05, **p < 0.01.
Oral supplementation with IAP prevents alcohol-induced gut barrier dysfunction and junctional protein losses in mice
Alcohol intake is known to induce gut barrier dysfunction in mice and humans. Furthermore, we have shown the beneficial effects of exogenous IAP in the context of a variety of gut barrier dysfunction conditions [25,33]. We therefore sought to determine the effect of exogenous IAP supplementation on gut barrier function in the context of alcohol exposure. For these experiments we employed an isolated intestinal loop model in which the mice received 3 doses of EtOH gavages +/− IAP pretreatment and then the ileal segments were injected with FITC-Dextran. Blood was collected after 1.5 h to measure and assess for alterations in gut permeability. Figure 5A shows that EtOH gavages disrupted the gut barrier and resulted in higher serum FITC levels, whereas pretreatment with IAP resulted in significantly lower serum FITC levels (Figure 5A). We next quantified the mRNA levels of junctional proteins in the ileal tissue and found that alcohol intake significantly decreased the expression of ZO-1, ZO-2 and Occludin. Furthermore, supplementation with IAP prevented the downregulation of tight junction proteins ZO-1 and Occludin (Figure 5B). These changes in the junctional proteins were specific in that there were no changes seen in the cases of Claudin 1 and Claudin 3.
Figure 5.
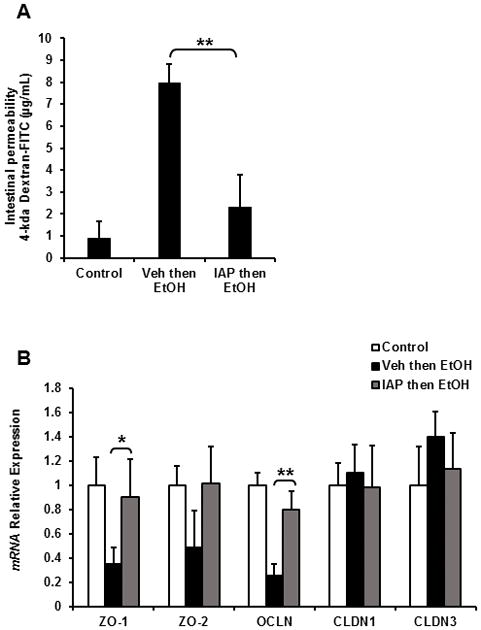
Oral supplementation with IAP prevents alcohol-induced gut barrier dysfunction and junctional protein losses. In the 3-dose alcohol binge model (A) 4-kda Dextran-FITC permeation in in vivo intestinal loop model and (B) Intestinal mRNA levels of junctional proteins measured by qPCR. Values are expressed as mean ± SEM. Statistical significance between the groups was tested using one-way analysis of variance with Tukey’s multiple comparison posttests; *p < 0.05, **p < 0.01.
Oral supplementation with IAP prevented alcohol induced intestinal inflammation
Based on our results above and the fact that inflammatory pathways are known to exert a major impact on gut barrier dysfunction, we sought to determine the effects of IAP treatment on intestinal inflammation due to alcohol intake. TNF-α and IL-1β mRNA levels were measured in ileal tissue from mice receiving either 3 doses of EtOH binge doses or 10 days of alcohol intake. Figures 6A & 6B show that alcohol intake significantly upregulated the expression levels of both TNF-α and IL-1β in both the 3-dose binge and chronic alcohol models. Furthermore, IAP supplementation significantly reduced the levels of these inflammatory cytokines in both the binge and chronic alcohol models.
Figure 6.
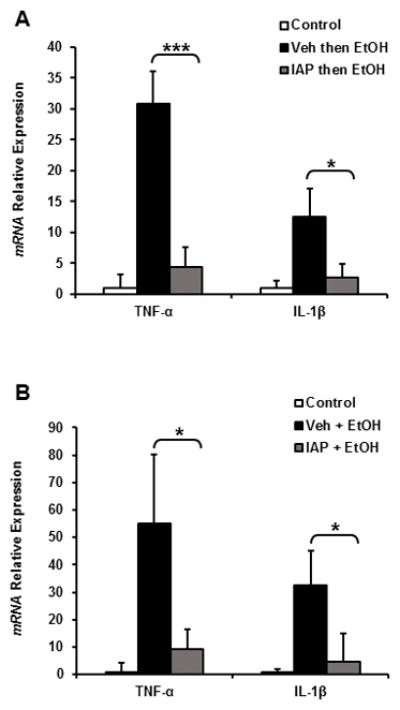
Oral supplementation with IAP prevents alcohol-induced intestinal inflammation. Inflammatory cytokine mRNA levels: TNF-α and IL-1β in both the (A) 3-dose binge and (B) chronic alcohol models. Values are expressed as mean ± SEM. Statistical significance between the groups was tested using one-way analysis of variance with Tukey’s multiple comparison posttests; *p < 0.05, ***p < 0.001.
IAP treatment prevents alcohol induced stellate cell activation and the lipogenic effect ex vivo
Activation of inflammatory pathways in liver Kupffer cells, hepatic stellate cells (HSCs), and hepatocytes play a major role in the pathogenesis of ALD. More specifically, it has recently been shown that alcohol activation of stellate cells plays an important role in the development of steatosis [34]. Therefore we sought to study the effect of IAP treatment on stellate cell activation and their lipogenic effect on hepatocytes. In this study, we isolated primary hepatocytes and hepatic stellate cells from mouse livers (Figures 7A and 7B). Primary HSCs were isolated from mice after 3 doses of binge EtOH gavages +/− pretreatment with IAP and co-cultured with primary hepatocytes from control mice. Fat accumulation in co-cultured hepatocytes was assessed using BODIPY staining. Figures 7C and 7D show that healthy hepatocytes co-cultured with HSCs from mice unexposed to alcohol show very little lipid droplets whereas healthy hepatocytes co-cultured with stellate cells from mice challenged with alcohol show significant amounts of intracellular lipid (Figures 7C and 7D). Remarkably, the stellate cells from alcohol-fed mice that were pretreated with IAP induced much less fat accumulation in the co-cultured hepatocytes (Figures 7C and 7D). Finally, we sought to test whether stellate cells from alcohol exposed mice induce hepatocytes to initiate inflammatory pathways that can lead to hepatitis. Figure 7E shows that healthy hepatocytes co-cultured with hepatic stellate cells from mice challenged with alcohol express higher levels of several inflammatory cytokines and chemokines, suggesting a key role for hepatic stellate cells in mediating the development of steatosis and hepatitis seen in ALD, expanding their known function as a main driver of hepatic fibrogenesis.
Figure 7.
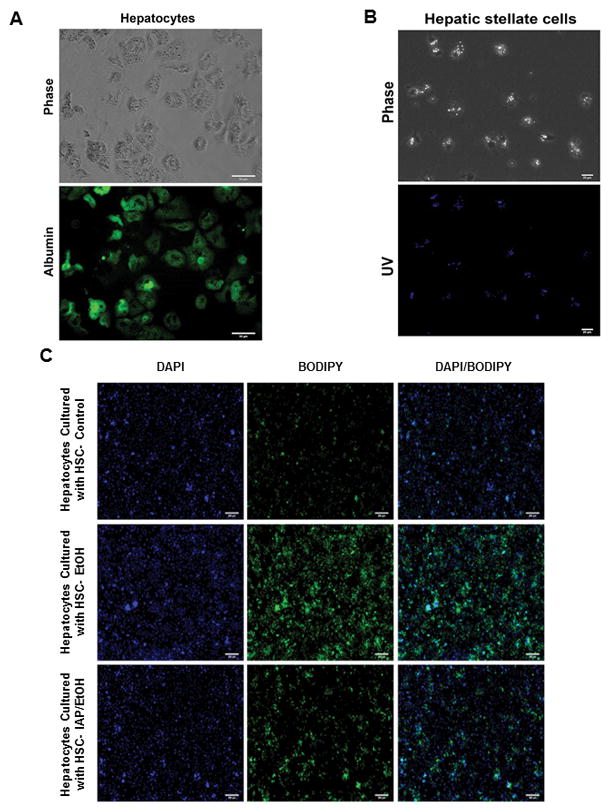
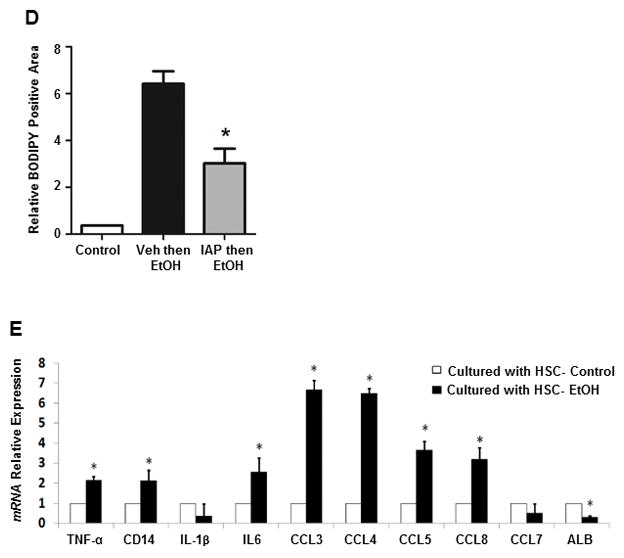
IAP treatment prevents alcohol-induced stellate cell activation and the lipogenic effect on hepatocytes ex vivo. Primary hepatocytes and stellate cells were isolated from mouse liver (A and B). Fat accumulation in hepatocytes were assessed with BODIPY staining (C and D) after being co-cultered with Hepatic stellate cells from mice exposed to 3 doses of binge EtOH gavages +/− pretreatment with IAP. qRT-PCR was performed to determine inflammatory cytokine and chemokine mRNA levels in co-cultured hepatocytes (E). Values are expressed as mean ± SEM. Statistical significance between two groups was tested using the two-tailed Student’s t test.; *p < 0.05.
Discussion
Alcohol consumption is one of the leading causes of end-stage liver disease and the second most common cause of liver transplantation in the United States and Europe [1, 2]. Acute alcoholic hepatitis has been associated with a mortality rate of 30–50% [3]. Steatosis, characterized by excessive fat deposition in hepatocytes and developing in the majority of heavy drinkers, is the first liver change evident in acute and chronic ALD [4].
Ethanol consumption increases production of inflammatory mediators from liver cells, in particular TNF-α, Interleukin-1 β and ROS, leading to the progression of fatty liver, inflammation and fibrosis [5,6,7]. TNF-α levels are increased in patients with alcoholic steatohepatitis [8], and its levels are correlated with the severity of the hepatitis [9]. Alcohol causes an activation of the immune response in liver cells, largely through toll like receptors that activate NFκB inflammatory signaling pathways and the subsequent production of cytokines [7,10]. Alcohol intake upregulates the expression of TLR4 and its coreceptors CD14, MD-2 and LPS-binding protein (LBP) in liver cells [11]. It is clear that bacterial endotoxin (LPS) plays a major role in alcohol-related liver damage. Acute heavy drinking caused endotoxemia in normal subjects and also in patients with mild forms of alcoholic hepatitis [35,36]. Ethanol also causes delayed endotoxin clearance from the circulation by Kupffer cells, intestinal bacteria overgrowth and increased gut permeability to macromolecules leading to elevated LPS levels [37,38,39]. Studies comparing the effect of portal and systemic LPS in ethanol animal models strongly suggest a gut source of endotoxins in the pathogenesis of ALD [18]. Interestingly, antibiotic treatment or altering the gut microflora with lactobacillus reduce LPS levels in the portal circulation and ameliorate the hepatotoxicity caused by ethanol [40,41].
Intestinal alkaline phosphates (IAP) is expressed in the enterocytes of the proximal small intestine and bidirectionally secreted into the intestinal lumen and the systemic circulation [42]. IAP detoxifies many bacterial pro-inflammatory factors, such as LPS, flagellin, and CpG DNA [22]. While others have shown the susceptibility of IAP knockout mice to high fat diet induced hepatic steatosis [24], we have recently shown the beneficial effect of oral IAP supplementation in preventing metabolic syndrome and fatty liver due to a high fat diet [25]. The fact that alcohol has been shown to decrease the secretion of IAP in rats and mice exposed to alcohol and its implications for thiamin absorption [43] may further represent part of the pathophysiology of alcohol-induced liver diseases.
Based on the known functions of IAP and the fact that its expression is lost with alcohol intake in rodents [26, 43], we sought to explore a potential role for this enzyme in the prevention of alcohol induced liver diseases. Our findings suggest that oral supplementation of IAP can prevent liver injury due to alcohol intake in mice. Alcohol induced liver cell damage results in elevated levels of serum ALT levels in human and rodents [32] and in clinical settings ALT levels are proportional to the degree of liver injury [33]. Both pre- and simultaneous IAP treatment with alcohol significantly lowered the serum ALT levels, while giving the IAP after the alcohol failed to block the elevations in ALT. These results indicate that higher levels of IAP in the gut at the time of alcohol intake are crucial for preventing alcohol induced liver injury. IAP supplementation during the 10 day alcohol exposure resulted in only a borderline decrease in ALT levels, suggesting that IAP supplementation may be most effective when taken freshly before alcohol intake.
In the present study, we show that IAP supplementation prevented the development of fatty liver due to alcohol intake. Fatty liver, or hepatic steatosis, is the first liver change of acute and chronic ALD [4]. Although the exact molecular mechanism of alcohol induced fatty liver is still not well known, recent advances in the understanding of the pathogenesis of ALD include direct toxicity on various liver cells, activation of innate immunity by ethanol-induced-endotoxemia, and subsequent production of pro-inflammatory cytokines [44]. Inflammatory cytokine levels including TNF-α and IL-1B are increased in patients with alcoholic steatohepatitis [8], the levels correlating with the severity of alcoholic hepatitis [9]. Preventing liver inflammation and production of cytokines block the development of fatty liver in mice on an alcohol diet [11]. Indeed, IAP treatment prevented liver inflammation and lowered cytokine levels in the liver tissue. It is likely that IAP supplementation prevented alcohol induced sensitivity to endotoxins by reducing the expression levels of components of the TLR4 pathway (TLR4 and its coreceptors, CD14 and LBP) in liver tissue.
IAP detoxifies LPS and prevents its translocation to the circulation from the gut in the context of a high fat diet. In this study we have demonstrated the ability of IAP to prevent LPS from entering the portal and systemic circulations in the context of alcohol consumption. Ethanol causes delayed endotoxin clearance from the circulation by Kupffer cells, intestinal bacteria overgrowth, and increased gut permeability to macromolecules leading to elevated LPS levels [37,38,39]. IAP is known to play a critical role in preserving the gut barrier function in many experimental settings [22,45]. In this study, we show that IAP treatment prevented the alcohol induced increase in intestinal permeability in the ileum of mice receiving the 3-dose alcohol binge. It appears that IAP preserves gut barrier function in the alcohol intake model by maintaining expression of tight junction proteins which are important components of the paracellular intestinal permeability. Additionally, IAP treatment prevented alcohol induced intestinal inflammation and lowered intestinal cytokine levels. It is interesting that even a single alcohol exposure disrupted the gut barrier in humans [35,36]. Clearly, a very strong link exists between alcohol intake, mucosal inflammation, and intestinal barrier dysfunction. Inflammatory cytokines such as TNF-α and IL-1β are associated with down-regulation of the junctional proteins, occludin and ZO-1, and deletion of toll like receptors (TLRs) or the inhibition of their downstream signaling in the intestinal epithelium has been shown to prevent inflammation and improve intestinal barrier function in mouse alcohol models [11,46].
It is likely that IAP ameliorates the effects of alcohol intake on liver injury and inflammation by detoxifying LPS and preventing endotoxemia although other mechanisms may also be at play, i.e., detoxification of other pro-inflammatory mediators in the intestinal lumen and prevention of their access to the liver tissue. In ALD, LPS has been the most intensively studied gut-derived inflammatory mediator. Since alcohol increases permeability of the gut to all macromolecules, many other bacterial components can gain access to the portal circulation and induce liver inflammation. Gustot et al. [47] has shown that mice on an EtOH diet upregulate multiple TLRs in the liver and sensitivity to their ligands like lipoteichoic acid (LTA), peptidoglycan (PGN), lipopolysaccharide (LPS), loxoribine, flagellin and CpG DNA. More recently, studies have shown the role of CpG DNA in the development of acute alcohol liver injury [11, 48]. Furthermore, others have shown the effect of augmenting the inflammatory response to alcohol when combining different TLRs ligands together [49]. Indeed, IAP is known to detoxify many bacterial pro-inflammatory factors, such as, flagellin, CpG DNA and ATP [22, 50].
Although the focus of alcohol research in regard to steatohepatitis is mainly on hepatic macrophages and hepatocytes, little is known about the role of hepatic stellate cells in the development of fatty liver and inflammation due to alcohol intake. In this study, we demonstrate the potential importance of stellate cells on the development of steatosis and inflammation in hepatocytes. We have shown that alcohol exposure activates stellate cells that are, in turn, capable of inducing fat accumulation in normal hepatocytes. Our results provide further evidence supporting a key role of stellate cells in the development of ALD [34]. Importantly, we have demonstrated in the study that IAP treatment can prevent the activation of hepatic stellate cells by alcohol intake and ameliorates its lipogenic effect on hepatocytes.
In this study, IAP function in the gut attenuated ALD in mice mainly due to its anti-inflammatory effect. Further studies will be needed to determine the precise role that the gut and IAP play in the development of ALD. IAP is a naturally occurring gut enzyme and has been safely administered to some patients without any adverse effects. We therefore suggest that IAP may represent a novel supplementation to prevent the development of ALD in at-risk humans. Furthermore, understanding the mechanism that IAP functions in alcohol intake models could identify new therapeutic targets for the treatment and prevention of alcoholic liver disease, in addition to fundamentally advancing our knowledge of the underlying molecular mechanisms that modulate alcoholic liver disease development.
Acknowledgments
Grant Support: National Institute of Health grant NIH/NIDDK T32 DK007754, The Ellison foundation grant and Nutritional Obesity Research Center at Harvard (NORCH) NIH P30-DK040561 (RAH).
Footnotes
Author Contribution: Study concept and theory (RAH,MYC,MSM,SRH,BK)); research design (RAH,MYC,SRH,BK); data acquisition (SRH, BK, KK, SAM, TJT, QT, MMRM, JMR, AK, WL, DH, AT, SSG, KPE, AKB, MSM, MYC, RAH); data analyses and interpretation (SRH, BK, KK, SAM, TJT, QT, MMRM, JMR, AK, WL, DH, AT, SSG, KPE, AKB, MSM, MYC, RAH); statistical analyses (SRH, KB, SAM, KPE, MYC, RAH); drafting of the manuscript (SRH, KB, CYM, RAH); critical review of the manuscript for important intellectual content (all authors); obtained funding (RAH); approval of the manuscript (all authors); study supervision (RAH,MYC).
Conflict of interests: There are none to declare.
References
- 1.Miniño AM, Murphy SL, Xu J, et al. Deaths: final data for 2008. Natl Vital Stat Rep. 2011 Dec 7;59:1–126. [PubMed] [Google Scholar]
- 2.Varma V, Webb K, Mirza DF. Liver transplantation for alcoholic liver disease. World J Gastroenterol. 2010 Sep 21;16:4377–93. doi: 10.3748/wjg.v16.i35.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. HEPATOLOGY. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 4.Celli R, Zhang X. Pathology of Alcoholic Liver Disease. J Clin Transl Hepatol. 2014 Jun 15;2:103–109. doi: 10.14218/JCTH.2014.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurman RG. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol Gastrointest Liver Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 6.Lin HZ, Yang SQ, Zeldin G, et al. Chronic ethanol consumption induces the production of tumor necrosis factor-alpha and related cytokines in liver and adipose tissue. Alcohol Clin Exp Res. 1998;22:231S–237S. doi: 10.1097/00000374-199805001-00004. [DOI] [PubMed] [Google Scholar]
- 7.Inokuchi S, Tsukamoto H, Park E, et al. Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcohol Clin Exp Res. 2011;35:1509–1518. doi: 10.1111/j.1530-0277.2011.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bird GL, Sheron N, Goka AK, et al. Increased plasma tumor necrosis factor in sever alcoholic hepatitis. Ann Intern Med. 1990;112:917–920. doi: 10.7326/0003-4819-112-12-917. [DOI] [PubMed] [Google Scholar]
- 9.Felver ME, Mezey E, McGuire M, et al. Plasma tumor necrosis factor alpha predicts decreased long term survival in severe alcoholic hepatitis. Alcohol Clin Exp Res. 1990;14:255–259. doi: 10.1111/j.1530-0277.1990.tb00482.x. [DOI] [PubMed] [Google Scholar]
- 10.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 11.Petrasek J, Mandrekar P, Szabo G. Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract. 2010 doi: 10.1155/2010/710381. pii:710381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nanji AA, Khettry U, Sadrzadeh SM, et al. Severity of liver injury in experimental alcoholic liver disease. Correlation with plasma endotoxin, prostaglandin E2, leukotriene B4, and thromboxane B2. Am J Pathol. 1993;142:367–73. [PMC free article] [PubMed] [Google Scholar]
- 13.Fukui H. Relation of endotoxin, endotoxin binding proteins and macrophages to severe alcoholic liver injury and multiple organ failure. Alcohol Clin Exp Res. 2005;29:172S–179S. doi: 10.1097/01.alc.0000189278.30237.e9. [DOI] [PubMed] [Google Scholar]
- 14.Horie Y, Kato S, Ohki E, et al. Role of endothelin in endotoxin-induced hepatic microvascular dysfunction in rats fed chronically with ethanol. Journal of Gastroenterology and Hepatology. 2001;16:916–922. doi: 10.1046/j.1440-1746.2001.02544.x. [DOI] [PubMed] [Google Scholar]
- 15.Horie Y, Kato S, Ohki E, et al. Hepatic microvascular dysfunction in endotoxemic rats after acute ethanol administration. Alcohol Clin Exp Res. 2000;24:691–698. [PubMed] [Google Scholar]
- 16.Deaciuc IV, Nikolova-Karakashian M, Fortunato F, et al. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol enhanced lipopolysaccharide hepatotoxicity. Alcohol Clin Exp Res. 2000;24:1557–1565. [PubMed] [Google Scholar]
- 17.Sandahl TD, Grønbaek H, Møller HJ, et al. Hepatic macrophage activation and the LPS pathway in patients with alcoholic hepatitis: a prospective cohort study. Am J Gastroenterol. 2014 Nov;109:1749–56. doi: 10.1038/ajg.2014.262. [DOI] [PubMed] [Google Scholar]
- 18.Mathurin P, Deng QG, Keshavarzian A, et al. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 19.Malo MS, Alam SN, Mostafa G, et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut. 2010;59:1476–1484. doi: 10.1136/gut.2010.211706. [DOI] [PubMed] [Google Scholar]
- 20.Chen KT, Malo MS, Beasley-Topliffe LK, et al. A role for intestinal alkaline phosphatase in the maintenance of local gut immunity. Dig Dis Sci. 2011;56:1020–1027. doi: 10.1007/s10620-010-1396-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bates JM, Akerlund J, Mittge E, et al. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. 2007;2:371–382. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen KT, Malo MS, Moss AK, et al. Identification of specific targets for the gut mucosal defense factor intestinal alkaline phosphatase. Am J Physiol Gastrointest Liver Physiol. 2010;299:G467–475. doi: 10.1152/ajpgi.00364.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rentea RM, Liedel JL, Welak SR, et al. Intestinal alkaline phosphatase administration in newborns is protective of gut barrier function in a neonatal necrotizing enterocolitis rat model. J Pediatr Surg. 2012;47:1135–1142. doi: 10.1016/j.jpedsurg.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Nakano T, Inoue I, Koyama I, et al. Disruption of the murine intestinal alkaline phosphatase gene Akp3 impairs lipid transcytosis and induces visceral fat accumulation and hepatic steatosis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1439–1449. doi: 10.1152/ajpgi.00331.2006. [DOI] [PubMed] [Google Scholar]
- 25.Kaliannan K, Hamarneh SR, Economopoulos KP, et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc Natl Acad Sci USA. 2013 Apr 23;110(17):7003–8. doi: 10.1073/pnas.1220180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hufnagel H, Bode C, Bode JC, et al. Damage of rat small intestine induced by ethanol. Effect of ethanol on fecal excretion of intestinal alkaline phosphatase. Res Exp Med. 1980;178:65–70. doi: 10.1007/BF01856759. [DOI] [PubMed] [Google Scholar]
- 27.Eloy R, Battinger F, Bignon JY, et al. Intestinal brush border enzymes and chronic alcohol ingestion. Res Exp Med. 1979;175:257–269. doi: 10.1007/BF01851282. [DOI] [PubMed] [Google Scholar]
- 28.Iakovleva LM. Structural and functional characteristic of rat jejunum after long-term exposure to alcohol. Morfologiia. 2012;141:45–48. [PubMed] [Google Scholar]
- 29.Carson EJ, Pruett SB. Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcohol Clin Exp Res. 1996;20:132–8. doi: 10.1111/j.1530-0277.1996.tb01055.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Z, Sun X, Lambert JC, et al. Metallothionein-independent zinc protection from alcoholic liver injury. Am J Pathol. 2002;160:2267–2274. doi: 10.1016/S0002-9440(10)61174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.FOLCH J, LEES M, SLOANE STANLEY GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 32.Mohar I, Brempelis KJ, Murray SA, et al. Isolation of non-parenchymal cells from the mouse liver. Methods Mol Biol. 2015;1325:3–17. doi: 10.1007/978-1-4939-2815-6_1. [DOI] [PubMed] [Google Scholar]
- 33.Hamarneh S, Morrison SA, Tantillo TJ, et al. A novel approach to maintaining gut mucosal integrity using an oral enzyme supplement. Annals of Surgery. 2014 Oct;260(4):706–15. doi: 10.1097/SLA.0000000000000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reeves HL, Burt AD, Wood S, et al. Hepatic stellate cell activation occurs in the absence of hepatitis in alcoholic liver disease and correlates with the severity of steatosis. Journal of Hepatology. 1996;25:677–683. doi: 10.1016/s0168-8278(96)80238-8. [DOI] [PubMed] [Google Scholar]
- 35.Fukui H, Brauner B, Bode JC, et al. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. 1991;12:162–169. doi: 10.1016/0168-8278(91)90933-3. [DOI] [PubMed] [Google Scholar]
- 36.Bode C, Fukui H, Bode JC. “Hidden” endotoxin in plasma of patients with alcoholic liver disease. Eur J Gastroenterol Hepatol. 1993;5:257–262. [Google Scholar]
- 37.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamai H, Kato S, Horie Y, et al. Effect of acute ethanol administration on the intestinal absorption of endotoxin in rats. Alcohol Clin Exp Res. 2000;24:390–4. [PubMed] [Google Scholar]
- 39.Yan AW, Fouts DE, Brandl J, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adachi Y, Moore LE, Bradford BU, et al. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108:218–224. doi: 10.1016/0016-5085(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 41.Forsyth CB, Farhadi A, Jakate SM, et al. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol. 2009;43:163–72. doi: 10.1016/j.alcohol.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eliakim R, Mahmood A, Alpers DH. Rat intestinal alkaline phosphatase secretion into lumen and serum is coordinately regulated. Biochim Biophys Acta. 1991;1091:1–8. doi: 10.1016/0167-4889(91)90213-h. [DOI] [PubMed] [Google Scholar]
- 43.Schaller K, Höller H. Thiamine absorption in the rat. ii. intestinal alkaline phosphatase activity and thiamine absorption from rat small intestine in-vitro and in-vivo. Int J Vitam Nutr Res. 1975;45:30–38. [PubMed] [Google Scholar]
- 44.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001 Jun;15:1335–49. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 45.Goldberg RF, Austen WG, Jr, Zhang X, et al. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci U S A. 2008 Mar 4;105:3551–6. doi: 10.1073/pnas.0712140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson AJ, Hughes KR. TNF-α-induced intestinal epithelial cell shedding: implications for intestinal barrier function. Ann N Y Acad Sci. 2012 Jul;1258:1–8. doi: 10.1111/j.1749-6632.2012.06523.x. [DOI] [PubMed] [Google Scholar]
- 47.Gustot T, Lemmers A, Moreno C, et al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43:989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Wu X, Zhang Y, et al. Discrepant roles of CpG ODN on acute alcohol-induced liver injury in mice. Int Immunopharmacol. 2012;12:526–533. doi: 10.1016/j.intimp.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 49.Oak S, Mandrekar P, Catalano D, et al. TLR2- and TLR4-mediated signals determine attenuation or augmentation of inflammation by acute alcohol in monocytes. J Immunol. 2006;176:7628–7635. doi: 10.4049/jimmunol.176.12.7628. [DOI] [PubMed] [Google Scholar]
- 50.Lallès JP. Intestinal alkaline phosphatase: novel functions and protective effects. Nutrition Reviews. 2013;72:82–94. doi: 10.1111/nure.12082. [DOI] [PubMed] [Google Scholar]