

Abstract
Spinocerebellar ataxia type 6 (SCA6) is an autosomal-dominant neurodegenerative disorder that is caused by a CAG trinucleotide repeat expansion in the CACNA1A gene. As one of the few bicistronic genes discovered in the human genome, CACNA1A encodes not only the α1A subunit of the P/Q type voltage-gated Ca2+ channel CaV2.1 but also the α1ACT protein, a 75 kDa transcription factor sharing the sequence of the cytoplasmic C-terminal tail of the α1A subunit. Isoforms of both proteins contain the polyglutamine (polyQ) domain that is expanded in SCA6 patients. Although certain SCA6 phenotypes appear to be specific for Purkinje neurons, other pathogenic effects of the SCA6 polyQ mutation can affect a broad spectrum of central nervous system (CNS) neuronal subtypes. We investigated the expression and function of CACNA1A gene products in human neurons derived from induced pluripotent stem cells from two SCA6 patients. Expression levels of CACNA1A encoding α1A subunit were similar between SCA6 and control neurons, and no differences were found in the subcellular distribution of CaV2.1 channel protein. The α1ACT immunoreactivity was detected in the majority of cell nuclei of SCA6 and control neurons. Although no SCA6 genotype-dependent differences in CaV2.1 channel function were observed, they were found in the expression levels of the α1ACT target gene Granulin (GRN) and in glutamate-induced cell vulnerability.
Keywords: : iPSC-derived neurons, SCA6, polyQ, CaV2.1, α1ACT, glutamate-induced stress
Introduction
Spinocerebellar ataxia type 6 (SCA6) is a rare autosomal dominant neurodegenerative disorder with a prevalence of 50/100,000, and it is characterized by late-onset, progressive ataxia, dysarthria, nystagmus, and double vision [1,2]. Neuropathological analyses showed a predominant loss of Purkinje neurons in the cerebellum, although neurodegeneration was also observed in other brain regions, including frontal cortex, basal ganglia, thalamus, and inferior olive [1,3].
SCA6 is caused by an abnormal CAG repeat expansion (19–33 CAG repeats in SCA6 patients vs. 4–18 repeats in normal individuals) located within exon 47 of the bicistronic gene CACNA1A, which encodes the transmembrane α1A subunit of the P/Q type voltage-gated Ca2+ channel (VGCC) CaV2.1 (α1A) [4], and α1ACT, a soluble 75 kDa protein that shares the primary sequence with the cytoplasmic C-terminal domain of the α1A protein (Fig. 1A) [5]. α1ACT protein is translated via an internal ribosome entry site (IRES) located within the coding region of the CACNA1A mRNA [5]. As a result, the polyglutamine (polyQ) tract, encoded by the CAG repeats, is present in two independently translated proteins: within the cytoplasmic C-terminal domain of the long splice variants of the α1A channel subunit, or within the α1ACT protein (Fig. 1A).
FIG. 1.

CACANA1A bicistronic gene expression in SCA6 iPSC-derived neurons. (A) Schematic representation of the human CACNA1A gene encoding the α1A subunit of the CaV2.1 channel (α1A) and the α1ACT transcription factor. Translation of α1ACT depends on an IRES. The inclusion of the polyQ-encoding CAG repeat in the α1A protein is determined by the alternative splicing of exon 46 encoding a stop codon. 1*-3* mark the binding sites for three sets of primers designed to amplify: all α1A mRNA isoforms (primer pair 1*), the long splice variants (including the CAG repeats) of the α1A mRNAs, and the α1ACT (primer pairs 2* and 3*). 1#-3# mark the epitopes for the antibodies designed to detect: the α1A protein (1#), the polyQ-containing α1ACT and long α1A protein isoforms (2#), and the all-known CACNA1A-encoded proteins (3#) (see Material and Methods section). (B) Phase-contrast microscopy images of SCA6 patient-derived cells used in this study: iPSCs, NPCs, immature neurons after 2 weeks of neuronal differentiation (2w neurons), and mature neurons after 5 weeks of neuronal differentiation (5w neurons). Scale bars 75 μm. (C) PCR of genomic DNA from control iPSCs yields a single amplicon containing the normal CAG repeat whereas from heterozygous SCA6-1 and SCA6-2 iPSCs two bands are amplified, with the higher band containing the expanded CAG repeats. (D) CACNA1A gene transcripts are amplified with primer set 1*, which detects all mRNA isoforms of CACNA1A encoding the α1A protein. CACNA1A is already detected in NPCs, and expression levels are highly upregulated during neuronal differentiation. No differences were detected between the expression levels of α1A encoding CACNA1A in SCA6 and control cells at any stage of neuronal differentiation. PolyQ-encoding splice variants for α1A and for α1ACT are detected in SCA6 and control neuronal cultures (primer set 2*). No difference was observed between the expression level of polyQ-encoding CACNA1A in SCA6 and control 2w neurons (immature neurons); however, a significant difference was detected between SCA6-2 and control 5w neurons. One-way ANOVA test followed by Tukey's multiple-comparisons test were applied, *P < 0.05. (E) Transcripts of the CACNA1A polyQ-encoding splice variants determined by RT-PCR in mature (5w) neurons (using primer set 3*). A single band is detected in control cells, whereas two amplicons are obtained from SCA6 neurons. The higher bands contain the expanded CAG repeat. ANOVA, analysis of variance; iPSC, induced pluripotent stem cell; IRES, internal ribosome entry site; NPCs, neural progenitor cells; NTC, no template control; RT-PCR, real-time-polymerase chain reaction; Rt-, reverse transcription negative control; SCA6, spinocerebellar ataxia type 6.
Apart from SCA6, two other diseases are caused by mutations in the CACNA1A gene: familial hemiplagic migraine type 1 (FHM1) and episodic ataxia type 2 (EA2) [4,6,7]. For FHM1 and EA2, the molecular mechanisms are well defined as loss or gain of CaV2.1 channel function, whereas SCA6 seems to have a more complex pathogenesis that is not fully elucidated and no treatment is available [6,8,9].
Studies hypothesizing an altered CaV2.1 channel function as the main component for SCA6 pathogenesis yielded conflicting findings. SCA6 transgenic mouse models of CaV2.1 channels with hyper-expanded polyQ repeats (84 or 118Q) were developed [10–12]. Although no deficits were reported in the gating properties of the P/Q type VGCC CaV2.1 channel with pathological polyQ expansion [11,13–16], recent findings suggest a link between the SCA6 mutation and abnormal electrophysiological properties of Purkinje neurons [10,17].
Previous studies in both cellular and animal models show that the α1ACT protein acts as a transcription factor that can translocate into the nucleus and activates the expression of several genes that are proposed to play a role in neuronal survival, including Granulin (GRN), TATA-box binding protein associated factor 1 (TAF1), and B cell translocation protein 1 (BTG1) [5]. Interestingly, an α1ACT mutant carrying the SCA6 polyQ expansion was unable to activate the expression of these genes [5].
Although the length of the polyQ repeat expansion in SCA6 is shorter than in other polyQ diseases [18], the observation that insoluble protein aggregates containing α1ACT are present in Purkinje neurons and in SCA6 patients indicates that mutated α1ACT might act as a toxic polyQ protein [19].
Although the available animal models recapitulate several features of SCA6 and have set the basis for remarkable progress in the understanding of SCA6 pathology, they require either polyQ hyper-expansion or α1ACT overexpression to reveal phenotypic abnormalities.
To overcome these limitations, neurons derived from patient induced pluripotent stem cells (iPSCs) can be employed to explore the pathophysiology of neurological diseases in human neurons. For SCA6, Ishida et al. published the first study describing the generation of SCA6 iPSC-derived Purkinje neurons in a self-organizing 3D culture system that revealed cell type-specific SCA6 phenotypes, including α1ACT-related neurotoxicity [20]. We have now generated iPSCs and neurons in a 2D culture system from two heterozygous SCA6 patients, and we analyzed the expression of the α1A subunit of the P/Q type VGCC CaV2.1 channel protein as well as the expression the α1ACT protein. We then tested calcium channel function, expression of α1ACT target genes, as well as cell vulnerability to induced neurotoxic stress in culture.
Materials and Methods
Patient consent
This study (No. UN50501) was approved by the Ethics Committee of the Medical University of Innsbruck (Approval No. 324/4.11). All methods were performed in accordance with the relevant guidelines and regulations of the Medical University of Innsbruck. The isolation and use of human somatic cells was performed after the patients and controls had provided written informed consent.
Cell culture
The main steps of the cell culture work-flow are depicted in Fig. 1B. All cell culture reagents were purchased from Thermo Fischer Scientific, if not stated otherwise. Cells were incubated at 37°C, 5% CO2, except iPSCs that were cultured at 37°C, 5% CO2, and 5% O2 (Biospherix Incubator).
iPSC generation and culture
iPSCs were generated as previously described, with minor modifications [21]. Skin biopsies were obtained from two SCA6 patients (Table 1), and primary fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM)+Glutamax supplemented with 10% fetal calf serum. Fibroblast cultures were transduced by using a polycistronic lentiviral vector containing the cDNAs coding for OCT4, SOX2, c-MYC, and KLF4 (SCR510; Millipore), following the manufacturer's protocol. iPSCs were cultured on vitronectin-coated tissue culture dishes in Essential 8 Flex medium, with the medium changed every second day. Cultures were passaged manually at a 1:2–1:4 split ratio every 4–7 days. For spontaneous differentiation assays, iPSCs were cultured for 15 days in DMEM-F12+Glutamax, 1% nonessential amino acids (NEAA), 0.1 mM 2-mercaptoethanol, 20% knockout serum replacement (KSR), 25 U/mL penicillin, and 25 U/mL streptomycin (P/S).
Table 1.
Demographic, Clinical, and Genetic Information of Two SCA6 Patients and the Six iPSC Clonal Lines Generated from Their Fibroblasts
| Code | SCA6-1 | SCA6-2 | ||||
|---|---|---|---|---|---|---|
| Sex | F | M | ||||
| Age | 45 | 49 | ||||
| Age at onset | 41 | 44 | ||||
| Mutation (CAG repeats) | 13/23 | 14/23 | ||||
| Symptoms at onset | Blurred vision, gait difficulty, downbeat nystagmus | Vertigo, gait difficulty, downbeat nystagmus, horizontal nystagmus | ||||
| Clones selected | SCA6-1.2 | SCA6-1.B1 | SCA6-1.B2 | SCA6-2.4 | SCA6-2.B1 | SCA6-2.B2 |
| Pluripotency markers | Validated | Validated | Validated | Validated | Validated | Validated |
| Karyotype | 46, XX | 46, XX | 46, XX | 46, XY | 46, XY | 46, XY |
| In vitro differentiation | Validated | Validated | Validated | Validated | Validated | Validated |
| Mutation in iPSCs | Kept | Kept | Kept | Kept | Kept | Kept |
| Transgene silencing | Verified | Verified | Verified | Verified | Verified | Verified |
| NPCs | Validated | Validated | Validated | Validated | Validated | Validated |
All iPSC lines were tested for uniform expression of pluripotency markers, normal karyotype, capacity of spontaneous in vitro differentiation in derivatives of the three germ layers, capacity of differentiation in NPCs, silencing of the transgenic viral construct, and presence of the SCA6 mutation. NPCs differentiated from each line were analyzed for the expression of neural markers after induction of neuronal differentiation.
iPSCs, induced pluripotent stem cells; NPCs, neural progenitor cells; SCA6, spinocerebellar ataxia type 6.
Neural induction and neural progenitor cell culture
Neural progenitor cells (NPCs) were generated from control and SCA6 iPSCs via two different neural induction approaches. The first approach is based on the protocol published by Falk et al. [22], with minor modifications. iPSC colonies were manually reduced to small cell aggregates and plated on nonadhesive plastic in DMEM-F12+Glutamax, 1% NEAA, 0.1 mM 2-mercaptoethanol, 20% KSR, and 25 U/mL P/S. After 5 days, floating cell aggregates were plated on Geltrex-coated tissue culture plates in the same medium. Neural rosette-like structures containing neuroepithelial stem (NES) cells appeared 1 week after plating. These rosettes were manually picked under the microscope and cultured in a nonadhesive plastic culture plate containing DMEM-F12+Glutamax, 1% NEAA, 25 U/mL P/S, and 1% N2 supplement (N2 medium) for 3–5 days.
In the second approach, NES cells were obtained by dual inhibition of SMAD signaling [23]. iPSCs were cultured in DMEM-F12+Glutamax, 1% NEAA, 0.1 mM 2-mercaptoethanol, 20% KSR, and 25 U/mL P/S with the addition of 100 nM LDN-193189 and 10 μM SB431542 (both from Sigma-Aldrich) on days 0–5. Cells were fed daily, and N2 medium was added in 25% increases every second day starting on day 4. After days 10–11, rosette-like clusters were manually picked and cultured in a nonadhesive plastic culture plate in N2 medium for 3–5 days.
For the generation of NPCs, floating NES cell aggregates were dissociated with 0.05% trypsin/EDTA for 5–10 min at 37°C to obtain a single-cell suspension, followed by trypsin inhibitor addition. The cell suspension was plated on Geltrex-coated tissue culture plates containing N2 medium, with the addition of 10 ng/mL fibroblast growth factor 2 (FGF2), 10 ng/mL epithelial growth factor (EGF), and 10 μg/mL heparin. Complete medium was changed daily, and NPCs were passaged at a 1:2–1:3 split ratio every 3–4 days with accutase. Validated NPC samples (uniformly expressing the neural markers SOX2, Nestin, and N-cadherin, see Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/scd) were frozen in liquid nitrogen in complete N2 medium with 10% DMSO. NPCs at passages 10–12 were used for all reported differentiation experiments.
Neuronal differentiation and maturation
Neuronal differentiation was started from NPCs by removing FGF2 and EGF from the medium (considered neuronal differentiation day 0) and continued for 5 weeks. For the first 7 days, cells were cultured in a 1:1 mixture of DMEM-F12-glutamax, 1% NEAA, 1% N2 supplement and Neurobasal, 1% Glutamax, and 1:50 B27 supplement (neuronal differentiation medium), with medium change every second day. On day 7, cells were dissociated with 0.05% trypsin/EDTA for 5 min at 37°C and plated on polyornithine-laminin-coated culture dishes at a density of 50,000 cells/cm2. Culture medium was changed to Neurobasal, 1% Glutamax, 1:50 B27 supplement, and 20 ng/mL brain-derived neurotrophic factor (neuronal maturation medium). Cytosine arabinoside (Ara-C) 2.5 μM was added once at day 15, with medium replaced after 3 days. From day 18 until the end of the experiment (week 5), only half of the medium volume was replaced weekly.
Genotyping
Genomic DNA was extracted from iPSCs by using the DNeasy Blood&Tissue kit (Qiagen). The presence of the CAG expansion in the SCA6-mutated allele was determined by polymerase chain reaction (PCR), using 100 ng of genomic DNA, forward primer 5′-CAC GTG TCC TAT TCC CCT GTG ATC C-3′, and reverse primer 5′-TGG GTA CCT CCG AGG GCC GCT GGT GG-3′, as reported [3], in 40 μL of reaction volume.
Quantitative real-time PCR
mRNA was isolated by using Dynabeads Oligo (dT)25 (Invitrogen) by following the manufacturer's protocol. For the cDNA synthesis, we used the High-Capacity cDNA Reverse Transcription Kit and proceeded as recommended by the manufacturer (Applied Biosystems). For the PCR, 200 ng of cDNA was used as a template in 20 μL reaction volume. Gene expression was analyzed by quantitative real-time PCR (qRT-PCR) by using the TaqMan Master Mix and Gene Expression Assays (see Supplementary Table S1), following the manufacturer's protocol [20].
Silencing of the viral transgenes used for reprogramming was determined by qRT-PCR by using the Ampliflou Viral OCT-4 Primer Set kit (Millipore), according to the manufacturer's instructions.
For the quantification of the CACNA1A gene encoding the α1A subunit, including all known splice variants (NM_000068.3, NM_023035.2, NM_001127221.1, NM_001127222.1, NM_001174080.1), we used the Taqman gene expression assay Hs01579431_m1 CACNA1A (set of primers 1* in Fig. 1A). For the quantification of the polyQ-encoding CACNA1A mRNA, we used a customized Taqman gene expression assay spanning the exon 46-intron 46-exon 47 border region: Taqman probe (TAMRA) 5′-CAC CGG CAG GGC AGT AGT TCC G-3′, forward primer: 5′-GAG GGC CGA GAG CAC ATG-3′, and reverse primer 5′-GGA GTG CTG GTA CCA GAT GTT G-3′ (set of primers 2* in Fig. 1A), leading to the selective amplification of α1A long splice variants 2 and 4 (mRNAs NM_023035.2 and NM_001127222.1) and of α1ACT mRNAs.
Ct values from three to six experiments for every condition, each experiment performed with three technical replicates, were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-Actin gene expression, and they were expressed as ΔCt values. Relative expression ratios were calculated by using the ΔΔCt method.
RT-PCR for polyQ-encoding CAG expansion detection
A set of primers spanning the exon 46-intron 46-exon 47 region and including the polyQ-encoding CAG repeat of the CACNA1A gene was designed to selectively amplify the expanded CAG repeat: forward primer 5′-CAC CGG CAG GGC AGT AGT T-3′, reverse primer 5′-TCC GAG GGC CGC TGG TG-3′ (set of primers 3* in Fig. 1A).
Antibodies for the detection of CaV2.1 channel α1A subunit and α1ACT protein
For the detection of the α1A subunit of the CaV2.1 channel protein, we used the antibody CaV2.1 from Synaptic Systems (Cat. No. 152 103), which binds to an epitope within the intracellular loop between the second and third transmembrane domain of the α1A subunit (Fig. 1A, 1#). Hence, this antibody recognizes all isoforms of the α1A subunit of the CaV2.1 channel.
For detection of the α1ACT protein, we generated a polyclonal antibody as described in Ishiguro et al. [19] The antibody, referred to as α1ACT#2 (Fig. 1A, 2#), was produced by Biogenes (Berlin, Germany), using the MERRVPGPARSESPRAC peptide for rabbit immunization and for subsequent affinity purification of the IgG fraction. The validation of the detection of α1ACT by the antibody 2# is shown in the Supplementary Fig. S2. An additional antibody, referred to here as 3#, was purchased from Synaptic Systems (Cat. No. 152 203), recognizing an epitope located within the C-terminal intracellular domain of the α1A channel subunit encoded by exon 47 (information by manufacturer, the exact epitope sequence is undisclosed). The same epitope is contained as well in α1ACT (Fig. 1A, 3#).
Immunocytochemistry
Cells cultured on glass coverslips were fixed with ice-cold 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature (RT) and washed twice with PBS. After 30 min incubation with blocking solution (2% BSA, 0.1% Triton X-100 in PBS), cells were incubated overnight at 4°C in blocking solution containing the appropriate primary antibodies (see Supplementary Table S2). Cells were then washed three times for 5 min with PBS before secondary antibody incubation (anti-mouse-488 or ant-rabbit-555 AlexaFluor conjugated) for 1 h at RT. After three washes with PBS, cells were counter-stained with either 4′,6-diamidino-2-phenylindole (DAPI) or RedDot2 (Biotium) and mounted on microscopy slides by using Roti Mount Fluo Care (Roth) medium. Microscopy images were taken with a 510 Meta Axiovert 200M confocal microscope (Zeiss). To quantify fluorescence intensities, confocal Z-stacks were acquired and images were analyzed by using ImageJ software. Z-stacks were processed to generate average intensities and converted into 8-bit images before background subtraction and fluorescence intensity measurement. MAP2 and RedDot2 fluorescence was used to create a mask to define extranuclear and nuclear regions of interest for analyses.
Electrophysiology
Action potentials were recorded in current clamp configuration of whole-cell mode. Patch pipettes were filled with the following (in mM): 130 K-Gluconate, 1 MgCl2, 10 HEPES, 5 EGTA, 4 Mg-ATP, and 0.3 a-GTP (pH 7.2 with KOH). The bath solution contained the following (in mM): 137 NaCl, 3 KCl, 10 HEPES, 2 MgCl2, 1.8 CaCl2, and 10 Glucose (pH 7.4 with NaOH).
Calcium channel currents were recorded by using the whole-cell patch-clamp technique with 10 mM Ba2+ as a charge carrier as previously described [24]. Patch pipettes were pulled from borosilicate glass (Harvard Apparatus), fire-polished (Microforge MF-830; Narishige), and had resistances of 2.5–4 MΩ when filled with the following (in mM): 120 cesium methanesulfonate, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 5 EGTA, 4 Mg-ATP, and 0.3 a-GTP (pH 7.2 with CsOH). The bath solution contained the following (in mM): 10 BaCl2, 110 NaCl, 20 TEA-Cl, 5 4-AP, 10 HEPES, 2 MgCl2, 3 KCl, 10 Glucose, and 0.001 TTX (pH 7.4 with NaOH). Currents were recorded with an EPC 10 amplifier controlled by PatchMaster software (HEKA Elektronik Dr. Schulze GmbH, Germany). Linear leak and capacitive currents were digitally subtracted with a P/4 prepulse protocol. The current-voltage dependence was fitted according to Equation (1), where Gmax is the maximum conductance of calcium channels, Vrev is the extrapolated reversal potential of the calcium current, V1/2 is the potential for half-maximal conductance, and k is the slope:
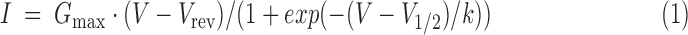 |
Neurons were held at −70 mV and were depolarized to 60 mV with a 10 mV step. CaV2.1 channels were blocked with 400 nM ω-Agatoxin IVA (Alomone Labs, Israel).
WST-1 cell viability assay under basal conditions and glutamate treatment
SCA6 and control neurons were plated after week 4 of differentiation on polyornithine-laminin-coated 96-well plates, at the density of 40,000 cells/well in 100 μL of neuronal maturation medium. The next day, half of the culture medium was changed. One week after plating, WST-1 reagent (Roche) was added to each well at 1:10 dilution. The colorimetric measurement was performed after incubation for 4 h at 37°C by using a Beckman Coulter DTX 880 plate reader at RT at 450 nm wavelength with a reference at 620 nm. For glutamate treatment, l-glutamate (Sigma) at a final concentration of 100 μM was added to each well 1 h before the WST-1 reaction. Glutamate treatment data are presented as relative absorbance intensities of the WST-1 reaction in the presence of glutamate normalized to the WST-1 reaction in the absence of glutamate.
Statistical analysis
Each differentiation experiment was repeated with three validated iPSC clonal lines for each individual (patients or control). qRT-PCR and immunostaining experiments were performed with three technical replicates; WST-1 assay was performed with four technical replicates. GraphPad software was used for statistical analysis. One-way analysis of variance was used to determine significant differences. Where significant differences were found, Tukey's multiple-comparisons test was applied, and P value <0.05 was considered significant.
Results
Generation and characterization of iPSCs from SCA6 patients
Dermal fibroblasts derived from two SCA6 patients (SCA6-1 and SCA6-2, clinical information in Table 1) and one unrelated healthy donor [21] were reprogrammed by using a polycistronic lentiviral vector to obtain iPSCs. Three clonal iPSC lines for each SCA6 patient and control were validated for the expression of pluripotency marker genes, lack of early differentiation markers, viral transgene silencing, potential to differentiate into derivatives of all three germ layers, and normal karyotypes (Table 1, Supplementary Fig. S3, and Supplementary Table S3). The presence of the mutated allele carrying the expanded CAG repeat was confirmed by PCR in all SCA6 iPSC lines (Table 1 and Fig. 1C).
Generation and characterization of SCA6 neurons
From each validated iPSC line, we generated NPCs and neurons (see Materials and Methods section and Fig. 1B). NPCs generated from SCA6 and control iPSC lines showed no significant differences in the expression of neural markers PAX6 at passage 1–2 and SOX2, Nestin, and NCAD at passage 10–12 (Supplementary Fig. S2). On initiation of neuronal differentiation of NPCs, mature neuronal markers TAU, SYNAPSIN, and SYNAPTOHPYSIN were upregulated with similar dynamics and at comparable levels in all lines (Fig. 2A). More than 75% MAP2-positive cells were detected at 5 weeks after growth factor removal. SCA6 and control neurons expressed glutamatergic or GABA-ergic markers (vGLUT1, vGLUT2, and GAD67) with no significant difference between genotypes (Fig. 2B, C), indicating that the SCA6 and control neuronal populations were composed of similar neuronal subtypes.
FIG. 2.

Neuronal phenotypes in SCA6 neuronal cultures. (A) mRNA expression levels of TAU, Synaptophysin (SYP), and Synapsin (SYN) in SCA6 and control neuronal cultures at 2 weeks (2w) and 5 weeks (2w) after starting NPC differentiation. Values are normalized to control NPCs. Each bar represents the mean ± s.e.m. from three independent experiments. One-way ANOVA test did not reveal any differences depending on genotype. (B) Immunostainings of SCA6 and control 5w neurons demonstrate expression of the pan-neuronal marker MAP2. The cultures contain a mixture of glutamatergic and GABAergic neurons expressing the neurotransmitter phenotype markers vesicular glutamate transporter 1 or 2 (vGLUT1/2) or glutamate decarboxylase 67 (GAD67). Scale bar 50 μm. (C) Quantification of neuronal marker expression in SCA6 and control 5w neuronal cultures. Bars represent the percentages of DAPI-positive cells expressing MAP2 in the upper panel, and the percentages of MAP2 neurons co-expressing GAD67, vGLUT1, and vGLUT2 in the lower panels. Each bar represents mean ± s.e.m. from three independent experiments. One-way ANOVA test was performed and failed to reveal genotype-dependent differences. (D) Typical action potentials recorded from 2w to 5w neuronal cultures. DAPI, 4′,6-diamidino-2-phenylindole; s.e.m, standard error of the mean.
Neuronal activity was recorded in human SCA6 and control neurons by whole-cell patch-clamping. Two weeks after growth factor withdrawal, only a minority of cells (18% ± 10%) were able to fire short trains of action potentials with quickly decreasing amplitudes on current injection, in both SCA6 and control neuronal cultures, suggesting a still immature neuronal phenotype. However, after 5 weeks of neuronal differentiation, the majority of neurons (65% ± 2%) exhibited sustained spontaneous action potential firing, in both SCA6 and control cultures, with increased amplitude and frequency (Fig. 2D).
Bicistronic CACNA1A gene expression in SCA6 neurons
We next analyzed the expression and distribution of CACNA1A gene products, the transmembrane α1A subunit of the CaV2.1 VGCC channel and the soluble α1ACT transcription factor (Fig. 1A), in SCA6 and control iPSC-differentiated cultures.
CACNA1A gene expression levels were determined by using two different sets of primers: the first designed to detect all five CACNA1A splice variants (primer set 1* in Fig. 1A), and the second designed to selectively detect transcripts containing the polyQ-encoding CAG repeats (primer set 2* in Fig. 1A). As shown in Fig. 1D, expression levels of the α1A-encoding CACNA1A mRNAs increased during neural differentiation and neuronal differentiation, with no significant difference between genotypes. Expression levels of the polyQ-encoding CACNA1A mRNAs were not different between SCA6 and control neurons after 2 weeks of neuronal differentiation, whereas the neurons from SCA6-2 showed a small but significant reduction in the amount of polyQ-encoding transcripts after 5 weeks compared with the control. We confirmed by RT-PCR the expression of the expanded CAG repeat in SCA6 neurons (Fig. 1E, primer set 3* in Fig. 1A). Control neurons expressed exclusively CACNA1A mRNA with the normal CAG repeat length whereas SCA6 neurons, consistent with their heterozygous genotype, expressed in addition the expanded allele.
Next, we analyzed the expression of the CaV2.1 channel α1A subunit protein. After 5 weeks of neuronal differentiation, CaV2.1 α1A immunoreactivity was detected in >75% of the neurons, in both SCA6 and control cultures (Fig. 3A, B, antibody epitope marked with 1# in Fig. 1A). Glutamatergic as well as GABAergic neurons were CaV2.1 positive, as shown in Fig. 3C. No α1A protein immunoreactivity was detected in cell nuclei (Fig. 3D).
FIG. 3.

α1A protein expression in SCA6 neurons. (A) α1A protein is detected in SCA6 as well as in control 5w neurons by immunostaining with a CaV2.1 antibody that binds to all splice variants of the α1A channel subunit (the epitope recognized by this antibody is marked with 1# in Fig. 1A). The same antibody was used in (B–F). (B) Percentages of CaV2.1-expressing cells in SCA6 and control cultures within the neuronal population. Each bar represents mean ± s.e.m. from three independent experiments. One-way ANOVA test was performed and did not reveal genotype-dependent differences. (C) α1A subunit of CaV2.1 protein is expressed in GABA-ergic GAD67-positive neurons as well as in glutamatergic vGLUT1-positive neurons. Representative confocal Z-stack projection images of neurons co-expressing CaV2.1 and MAP2 demonstrate that CaV2.1 staining is predominantly localized in dendrites and cell soma. Quantification of CaV2.1 immunofluorescence intensity within the MAP2-positive compartment in soma and dendrites. Each bar represents the mean ± s.e.m. from three independent experiments. One-way ANOVA test was performed and did not reveal differences in CaV2.1 subcellular localization depending on genotype. (D) CaV2.1 staining does not overlap with the DAPI-positive nuclear compartment. Scale bars (A, C, D, F) 50 μm.
The subcellular distribution of CaV2.1 α1A protein was determined by confocal microscopy analysis. Immunoreactivity was detected in both soma and neurites, and the distribution of CaV2.1 α1A protein between both cellular compartments was not different between SCA6 and control neurons (Fig. 3E, F).
Taken together, these results indicate that the α1A subunit of the CaV2.1 channel is expressed in neuronal cultures in GABA-ergic as well as glutamatergic neuronal subtypes and its expression levels and subcellular distribution are not affected by the SCA6 mutation.
Subsequently, we analyzed the expression of the secondary gene product of the CACNA1A gene, the α1ACT protein, by using the antibody 2# (Fig. 1A). This antibody is predicted to also recognize the long isoforms of α1A. Immunoreactivity was detected in the majority of the differentiated neurons (80%), in both patients and control (Fig. 4A, B). Immunostaining experiments revealed a subcellular distribution pattern consistent with staining of the α1ACT transcription factor rather than the α1A subunit of the CaV2.1 channel protein. The channel subunit was detected by antibody 1# in the soma and the dendrites outside of the cell nucleus, whereas staining with antibody 2# showed a predominant nuclear staining pattern (Fig. 4C). However, almost all control neurons also presented limited extra-nuclear punctate immunoreactivity (#2), which was absent in SCA6 neurons (Fig. 4C). Quantification by confocal microscopy analysis showed no difference between genotypes in nuclear immunofluorescence intensities after α1ACT transcription factor staining (Fig. 4D). The presence of nuclear α1ACT in neurons was confirmed by results obtained with a second independent antibody directed at the C-terminal epitope that was predicted to be present in all proteins encoded in CACNA1A (3# in Fig. 1A), which also revealed a prominent nuclear staining pattern in differentiated neurons, in combination with staining outside of the nucleus. From the combined results of our immunocytochemical analyses with antibodies 1#-3#, we conclude that SCA6 and control neurons express nuclear α1ACT along with extranuclear α1A, which appears to be mostly the short isoform.
FIG. 4.

α1ACT transcription factor expression in SCA6 neurons. (A) Nuclear α1ACT is detected in the nuclei of SCA6 and control 5w neurons by immunostaining with antibody 2#. The same antibody was also used in (B–D). Scale bar 50 μm. (B) Quantification of the MAP2-positive neurons expressing α1ACT detected with antibody #2. Each bar represents the mean ± s.e.m. from three independent experiments. One-way ANOVA test performed; no differences were found between genotypes. (C) Representative confocal Z-stack projection images of neurons immunostained with antibody 2#. α1ACT transcription factor immunoreactivity is detected in nuclei. RedDot2 was used for nuclear staining. Scale bar 20 μm. (D) Quantification of nuclear α1ACT immunofluorescence. Graph bar of relative α1ACT immunofluorescence intensity in nuclei. Each bar represents mean ± s.e.m. from three independent experiments. One-way ANOVA test was performed and did not indicate differences between genotypes. (E) The nuclear localization of α1ACT in neurons is confirmed by immunostaining with antibody 3#, designed to detect all known proteins encoded by the CACNA1A gene (Fig. 1A, see Materials and Methods section). Scale bar 20 μm.
Functional assessment of the bicistronic CACNA1A gene products in SCA6 neurons
CaV2.1 channel electrophysiology
The function of the CaV2.1 channels in SCA6 neurons was analyzed in patch-clamp recording. First, total calcium channel-current amplitude in SCA6 and control neuronal cultures was determined after 5 weeks of neuronal differentiation. Neurons from all genotypes displayed voltage-dependent Ca2+ currents with a maximum inward current at around 0 mV and a maximum normalized current density amplitude of around −50 pA/pF, with no significant differences between SCA6 and control neurons (Fig. 5A).
FIG. 5.

Cav2.1 and α1ACT functions in SCA6 neurons. (A) Cultured human neurons of all genotypes express functional VGCCs. Ba2+ I/V curves in SCA6 and control 5w neurons. Currents are normalized to cell capacitance (pA/pF). (B) Cultured human neurons of all genotypes express functional P/Q type channels. Representative traces (300 ms depolarization from −70 to 0 mV) of Ba2+ currents before and after application of 400 nM ω-Agatoxin IVA, specific blocker of P/Q type currents in SCA6 and control 5w neurons. Traces represent normalized to Imax without ω-Agatoxin IVA currents (I/Imax), showing current amplitude reduction on toxin perfusion, demonstrating the expression of ω-Agatoxin IVA-sensitive Cav2.1 channels. (C) mRNA expression levels of the α1ACT target genes GRN, BTG1, and TAF1 in SCA6 and control 5w neuronal cultures. Expression levels are normalized to control. GRN gene transcripts are significantly reduced in cultures of SCA6 neurons compared with the control. Each bar represents mean ± s.e.m. from three independent experiments. One-way ANOVA test, Tukey's multiple-comparisons test: *P < 0.05. (D) SCA6 neurons show increased vulnerability to glutamate compared with the control. Graph bar indicating WST-1 colorimetric absorbance intensity as a readout for cell viability in SCA6 and control 5w neuronal cultures treated for 1 h with 100 μM glutamate (relative to nontreated cells, indicated by the dashed line). Each bar represents the mean ± s.e.m. from three independent experiments. One-way ANOVA test, Tukey's multiple-comparisons test: *P < 0.05. NS, not significant.
We then confirmed that SCA6 neurons express functional CaV2.1 channels by applying the spider toxin ω-Agatoxin IVA to selectively block currents mediated by CaV2.1 channels. The addition of 400 nM ω-Agatoxin IVA reduced the calcium current amplitude, in both SCA6 and control neurons, indicating the presence of a P/Q type current component dependent on CaV2.1 channels (Fig. 5B).
Transcriptional regulation of α1ACT target genes in SCA6 neurons
The polyQ-encoding α1ACT protein functions as a transcription factor activating the expression of a set of genes that are relevant for neuronal survival (BTG1, GRN, and TAF1) [5]. We tested for differential mRNA expression of α1ACT target genes in neurons cultured for 5 weeks and found that GRN mRNA levels were significantly decreased in neuronal cultures derived from SCA6 patients, whereas no significant differences were observed for TAF1 and BTG1 mRNA expression levels in SCA6 neurons compared with controls (Fig. 5C).
SCA6 neurons show increased vulnerability to glutamate
No differences in cell viability between SCA6 and control neurons were detected under standard culture conditions with the WST-1 colorimetric assay (Fig. 5D). When neuronal cultures were exposed to cellular stress by treatment with 100 μM glutamate for 1 h, a reduction of cell viability/metabolic activity was observed in both SCA6 and control neurons compared with untreated cultures. This reduction was significantly enhanced in SCA6 neurons compared with the control (Fig. 5D), suggesting that SCA6 mutations increase neuronal vulnerability to glutamate-induced cellular stress.
Discussion
Since the first description of the CAG repeat expansion within the CACNA1A gene causing SCA6 [4], several research groups developed cellular and animal models for the study of the complex pathophysiological polyQ-dependent mechanisms of the disease [2,8,7].
Remarkable progress was achieved with the discovery of the bicistronic nature of the human CACNA1A gene (Fig. 1A) and of the transcription factor function of α1ACT protein [5].
Investigations of the Cav2.1 channel in vitro and in vivo yielded contradictory results [13,14,16], whereas other studies suggested an independent role for the α1ACT transcription factor in the pathogenesis of SCA6 [5,25].
The potential pathogenic mechanisms proposed for α1ACT addressed dysregulated gene expression [5], cytotoxicity of the protein containing expanded polyQ stretches [25], and increased cellular vulnerability to harmful stimuli [26].
The hypothesis of the mutant α1ACT as a cytotoxic polyQ protein is supported by pathological analyses of SCA6 patient brains, where nuclear inclusions were detected in Purkinje neurons, as well as in other brain areas, resembling the intracellular aggregates that characterize other polyQ disease [3].
In a recent study, Ishida et al. studied cellular pathomechanisms of SCA6 in iPSCs-derived SCA6 Purkinje neurons [20]. In this remarkable work, the authors described SCA6-related phenotypes, including dysregulation of α1ACT target genes expression and increased cell vulnerability to oxidative stress. However, in contrast to our study, this previous study did not address the function of the mutated CaV2.1 channel.
Another difference between our study and the one by Ishida et al. [20] is that instead of focusing on in vitro differentiated Purkinje neurons, we employed a more diverse population of mainly glutamatergic and GABA-ergic neurons with non-Purkinje phenotype (Fig. 2 and Supplementary Table S4). Purkinje neurons are the main cell type being affected early by SCA6 disease progression, whereas brain areas outside the cerebellum are also affected by the disease and CACNA1A gene expression is not limited to cerebellar Purkinje neurons. On the contrary, CACNA1A gene products (mRNA and protein) are abundant in several other brain regions [3]. Therefore, although certain SCA6 phenotypes might be specific for or more pronounced in Purkinje neurons, other pathogenic effects of SCA6 polyQ mutation are predicted to affect a broad spectrum of CNS neuronal subtypes of which our cultures are a useful model.
Our cellular models allowed us the parallel investigation of the three main hypotheses for SCA6 pathology: channelopathy, dysregulation of gene expression, and polyQ toxicity. In our experiments, iPSCs from all genotypes generated similar percentages of neurons during 5 weeks of neuronal differentiation and maturation and no differences were observed between SCA6 and control neuronal cultures with respect to the expression of neuronal markers (Fig. 2 and Supplementary Table S4).
To test the hypothesis of alterations in CaV2.1 channel function, we generated electrophysiologically active human neurons of both genotypes that exhibited increasing capability over time to fire sustained action potentials, reflecting a dynamic cellular maturation process. The observed time course of maturation was comparable with previous studies [27], where passive and active electrophysiological properties of iPSC-derived human neurons were analyzed in detail. The measurement of the total voltage-dependent calcium currents in both control and SCA6 neurons did not reveal significant differences between genotypes. The amplitude and I-V relationship of iPSC-derived neurons was comparable with published studies from adult mouse brain neuronal cultures [24,28], suggesting that advanced neuronal maturation was achieved. In addition, by adding a specific CaV2.1 channel blocker, ω-Agatoxin IVA, we were able to show the presence of functional CaV2.1 channels in both control and SCA6 neurons. However, our findings do not provide evidence for alterations of the CaV2.1 channel in human SCA6 iPSC-derived neurons. Taking into account the late onset of SCA6, the neurons in our cultures were potentially insufficiently matured to reveal late cellular phenotypes. In future studies, paradigms of artificially increased in vitro stress will have to be applied before testing channel pathophysiology, as was shown to be necessary for revealing other late-onset disease phenotypes in iPSC-derived human neurons [29,30].
The polyQ-encoding CACNA1A mRNAs were expressed at similar levels in SCA6 and control cultures, and the α1A channel subunit was expressed by the majority of differentiated neurons. A staining pattern typical for transmembrane proteins was observed outside of the cell nucleus, in the soma and neurites with an antibody directed against an epitope specific for the α1A channel subunit. With two antibodies directed against C-terminal epitopes that can detect the α1ACT transcription factor, we observed a clear and predominant nuclear staining pattern, although both antibodies bind to epitopes that are also present in long isoforms of the transmembrane α1A subunit of the CaV2.1 channel. This nuclear localization the α1ACT transcription factor detected with such antibodies is in accordance with previous studies [19,20]. Our immunostaining results could be explained by a model where most of the α1A channel subunit proteins in our neuronal cultures are short isoforms and, hence, in contrast to α1ACT, are not detected by the antibody 2#.
The wild-type α1ACT transcription factor activates the expression of BTG1, GRN, and TAF1, whereas the SCA6-mutated α1ACT was unable to activate these genes [5]. Differential expression of α1ACT target genes in our heterozygous model was observed for GRN, which showed a significant decrease in SCA6 neuronal cultures. Hence, our study does provide further evidence for altered gene expression in neurons carrying SCA6 mutations, although no significant differences were observed for BTG1 and TAF1.
Previous research suggests that polyQ-expanded α1ACT may act as a toxic polyglutamine protein [5,25]. SCA6 neurons in our experiments did not show differences in cell viability under baseline culture conditions compared with controls. Hence, we tested whether a cytotoxic effect of polyQ-expanded α1ACT could be observed in the presence of cellular stressors. Glutamate-induced stress has been hypothesized to play a role in numerous neurodegenerative diseases, including amyotrophic lateral sclerosis, Alzheimer's disease, and Huntington's disease [31]. Using glutamate to produce an acute insult, we found an increased vulnerability to glutamate treatment in SCA6 neurons as compared with the control. However, further experiments have to reveal whether this vulnerability is related to the polyQ expansion in α1ACT or in the long splice variants of the α1A channel subunit.
In conclusion, we have generated a new cellular model based on patient-derived SCA6 neurons where the disease-relevant proteins are expressed in a functional form in a human neuronal environment. Although the SCA6 neurons did not show differences in the expression levels or subcellular distribution of CACNA1A gene products, they differentially expressed an α1ACT target gene and presented increased vulnerability to cytotoxic stress induced by acute glutamate treatment.
Taking advantage of efficient and reproducible neuronal differentiation protocols to generate SCA6 patient-derived neurons, we demonstrated that this cellular disease model is useful for addressing the complex phenotypes associated with the SCA6 pathophysiology by electrophysiological, molecular, and cellular studies.
Supplementary Material
Acknowledgments
The authors thank the SCA6 patients for their willingness to participate in this study. They also thank the Department of Human Genetics of the Medical University of Innsbruck for the karyotyping analysis of iPSCs and Veronika Schuchter for linguistic revision. This study was supported by the Medical University of Innsbruck (project numbers SFB-F44 ST712600S442 and MUI-START ST201506015) and the Austrian Science Fund (FWF, SFB-F44150, “Cell signalling in chronic CNS disorders”).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Rub U, Schols L, Paulson H, Auburger G, Kermer P, Jen JC, Seidel K, Korf HW. and Deller T. (2013). Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol 104:38–66 [DOI] [PubMed] [Google Scholar]
- 2.Solodkin A. and Gomez CM. (2012). Spinocerebellar ataxia type 6. Handb Clin Neurol 103:461–473 [DOI] [PubMed] [Google Scholar]
- 3.Soga K, Ishikawa K, Furuya T, Iida T, Yamada T, Ando N, Ota K, Kanno-Okada H, Tanaka S, et al. (2017). Gene dosage effect in spinocerebellar ataxia type 6 homozygotes: a clinical and neuropathological study. J Neurol Sci 373:321–328 [DOI] [PubMed] [Google Scholar]
- 4.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY. and Lee CC. (1997). Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 15:62–69 [DOI] [PubMed] [Google Scholar]
- 5.Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC, Hansel C. and Gomez CM. (2013). Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 154:118–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guida S, Trettel F, Pagnutti S, Mantuano E, Tottene A, Veneziano L, Fellin T, Spadaro M, Stauderman K, et al. (2001). Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am J Hum Genet 68:759–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pietrobon D. (2010). CaV2.1 channelopathies. Pflugers Arch 460:375–393 [DOI] [PubMed] [Google Scholar]
- 8.Giunti P, Mantuano E, Frontali M. and Veneziano L. (2015). Molecular mechanism of Spinocerebellar Ataxia type 6: glutamine repeat disorder, channelopathy and transcriptional dysregulation. The multifaceted aspects of a single mutation. Front Cell Neurosci 9:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C. and Pietrobon D. (2002). Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci U S A 99:13284–13289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayabal S, Chang HH, Cullen KE. and Watt AJ. (2016). 4-aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci Rep 6:29489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watase K, Barrett CF, Miyazaki T, Ishiguro T, Ishikawa K, Hu Y, Unno T, Sun Y, Kasai S, et al. (2008). Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc Natl Acad Sci U S A 105:11987–11992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Unno T, Wakamori M, Koike M, Uchiyama Y, Ishikawa K, Kubota H, Yoshida T, Sasakawa H, Peters C, Mizusawa H. and Watase K. (2012). Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc Natl Acad Sci U S A 109:17693–17698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuyama Z, Wakamori M, Mori Y, Kawakami H, Nakamura S. and Imoto K. (1999). Direct alteration of the P/Q-type Ca2+ channel property by polyglutamine expansion in spinocerebellar ataxia 6. J Neurosci 19:RC14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Restituito S, Thompson RM, Eliet J, Raike RS, Riedl M, Charnet P. and Gomez CM. (2000). The polyglutamine expansion in spinocerebellar ataxia type 6 causes a beta subunit-specific enhanced activation of P/Q-type calcium channels in Xenopus oocytes. J Neurosci 20:6394–6403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saegusa H, Wakamori M, Matsuda Y, Wang J, Mori Y, Zong S. and Tanabe T. (2007). Properties of human Cav2.1 channel with a spinocerebellar ataxia type 6 mutation expressed in Purkinje cells. Mol Cell Neurosci 34:261–270 [DOI] [PubMed] [Google Scholar]
- 16.Toru S, Murakoshi T, Ishikawa K, Saegusa H, Fujigasaki H, Uchihara T, Nagayama S, Osanai M, Mizusawa H. and Tanabe T. (2000). Spinocerebellar ataxia type 6 mutation alters P-type calcium channel function. J Biol Chem 275:10893–10898 [DOI] [PubMed] [Google Scholar]
- 17.Mark MD, Krause M, Boele HJ, Kruse W, Pollok S, Kuner T, Dalkara D, Koekkoek S, De Zeeuw CI. and Herlitze S. (2015). Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J Neurosci 35:8882–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frontali M, Novelletto A, Annesi G. and Jodice C. (1999). CAG repeat instability, cryptic sequence variation and pathogeneticity: evidence from different loci. Philos Trans R Soc Lond B Biol Sci 354:1089–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishiguro T, Ishikawa K, Takahashi M, Obayashi M, Amino T, Sato N, Sakamoto M, Fujigasaki H, Tsuruta F, et al. (2010). The carboxy-terminal fragment of alpha(1A) calcium channel preferentially aggregates in the cytoplasm of human spinocerebellar ataxia type 6 Purkinje cells. Acta Neuropathol 119:447–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishida Y, Kawakami H, Kitajima H, Nishiyama A, Sasai Y, Inoue H. and Muguruma K. (2016). Vulnerability of Purkinje cells generated from spinocerebellar ataxia type 6 patient-derived iPSCs. Cell Rep 17:1482–1490 [DOI] [PubMed] [Google Scholar]
- 21.Eigentler A, Boesch S, Schneider R, Dechant G. and Nat R. (2013). Induced pluripotent stem cells from friedreich ataxia patients fail to upregulate frataxin during in vitro differentiation to peripheral sensory neurons. Stem Cells Dev 22:3271–3282 [DOI] [PubMed] [Google Scholar]
- 22.Falk A, Koch P, Kesavan J, Takashima Y, Ladewig J, Alexander M, Wiskow O, Tailor J, Trotter M, et al. (2012). Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS One 7:e29597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M. and Studer L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Etemad S, Obermair GJ, Bindreither D, Benedetti A, Stanika R, Di Biase V, Burtscher V, Koschak A, Kofler R, et al. (2014). Differential neuronal targeting of a new and two known calcium channel 4 subunit splice variants correlates with their regulation of gene expression. J Neurosci 34:1446–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kordasiewicz HB, Thompson RM, Clark HB. and Gomez CM. (2006). C-termini of P/Q-type Ca2+ channel alpha1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum Mol Genet 15:1587–1599 [DOI] [PubMed] [Google Scholar]
- 26.Li L, Saegusa H. and Tanabe T. (2009). Deficit of heat shock transcription factor 1-heat shock 70 kDa protein 1A axis determines the cell death vulnerability in a model of spinocerebellar ataxia type 6. Genes Cells 14:1253–1269 [DOI] [PubMed] [Google Scholar]
- 27.Prè D, Nestor MW, Sproul AA, Jacob S, Koppensteiner P, Chinchalongporn V, Zimmer M, Yamamoto A, Noggle SA. and Arancio O. (2014). A time course analysis of the electrophysiological properties of neurons differentiated from human induced pluripotent stem cells (iPSCs). PLoS One 9:e103418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanika R, Campiglio M, Pinggera A, Lee A, Striessnig J, Flucher BE. and Obermair GJ. (2016). Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Sci Rep 6:34528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim J, et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13:691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen J-Y, Wei C-Y, Shah K, Wong J, Wang C. and Chen H-SV. (2015). Maturation-based model of arrhythmogenic right ventricular dysplasia using patient-specific induced pluripotent stem cells. Circ J 79:1402–1408 [DOI] [PubMed] [Google Scholar]
- 31.Lewerenz J. and Maher P. (2015). Chronic glutamate toxicity in neurodegenerative diseases-What is the evidence? Front Neurosci 9:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.