

Summary
Cancer cells consume glucose and secrete lactate in culture. It is unknown whether lactate contributes to energy metabolism in living tumors. We previously reported that human non-small cell lung cancers (NSCLC) oxidize glucose in the tricarboxylic acid (TCA) cycle. Here we show that lactate is also a TCA cycle carbon source for NSCLC. In human NSCLC, evidence of lactate utilization was most apparent in tumors with high 18fluorodeoxyglucose uptake and aggressive oncological behavior. Infusing human NSCLC patients with 13C-lactate revealed extensive labeling of TCA cycle metabolites. In mice, deleting monocarboxylate transporter-1 (MCT1) from tumor cells eliminated lactate-dependent metabolite labeling, confirming tumor-cell autonomous lactate uptake. Strikingly, directly comparing lactate and glucose metabolism in vivo indicated that lactate's contribution to the TCA cycle predominates. The data indicate that tumors, including bona fide human NSCLC, can use lactate as a fuel in vivo.
eTOC
Human non-small cell lung cancer preferentially utilizes lactate over glucose to fuel TCA cycle and sustain tumor metabolism in vivo.
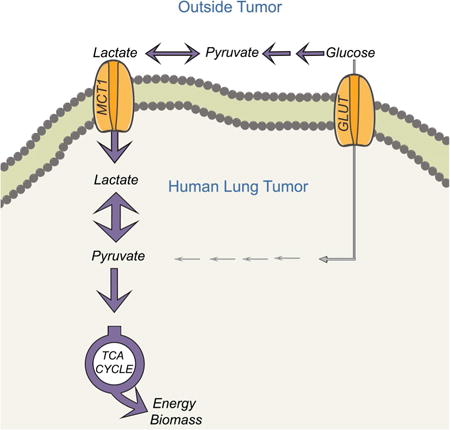
Introduction
Constitutive glucose uptake is a hallmark of cancer cells and provides energy and biosynthetic material for cancer cell proliferation (Hanahan and Weinberg, 2011). Accordingly, enhanced expression of glucose transporters and glycolytic enzymes is a common consequence of oncogenic signaling and transcriptional networks (Vander Heiden and DeBerardinis, 2017). Glucose uptake is also important for metabolic imaging and staging in cancer. 18fluoro-2-deoxyglucose positron emission tomography (FDG-PET) utilizes the propensity of many solid tumors to take up and retain glucose, providing imaging contrast with adjacent tissue (Fletcher et al., 2008). The metabolic fates of glucose in FDG-PET-positive tumors and the extent to which other fuels complement glucose utilization in these tumors are areas of active study (Davidson et al., 2016; Fan et al., 2009; Hensley et al., 2016; Mashimo et al., 2014).
In cultured cancer cells, most glucose carbon is converted to pyruvate via glycolysis, reduced to lactate via lactate dehydrogenase (LDH) and secreted (DeBerardinis et al., 2008). Disposal of this large carbon pool as lactate serves at least two functions. The NADH-dependent reduction of pyruvate to lactate by LDH recycles the NAD+ reduced to NADH during glycolysis, allowing glycolysis to persist. Excreting lactate through monocarboxylate transporters (e.g. MCT1, 4) eliminates protons arising from the glyceraldehyde 3-phosphate dehydrogenase reaction in glycolysis, thereby maintaining pH homeostasis inside the cell and acidifying the extracellular space. Evidence indicates that lactate and/or protons in the microenvironment modulate immune cell function and promote invasion and metastasis, implicating tumor glycolysis and lactate secretion in processes that impact cancer-related mortality (Brand et al., 2016; Rizwan et al., 2013; Xie et al., 2014).
However, other studies have proposed that tumors use lactate as a fuel, expanding its metabolic functions in cancer. Lactate circulates at levels of 1-2 mM and acts as an inter-organ carbon shuttle in mammals (Cori and Cori, 1929). Some cancer cells use lactate as a respiratory substrate and lipogenic precursor in culture (Chen et al., 2016). Blocking lactate uptake with an MCT1 inhibitor reduces respiration and promotes glycolysis in some cancer cell lines, and suppresses xenograft growth in mice (Pavlides et al., 2009; Sonveaux et al., 2008). In mouse models of breast cancer, the growth-promoting effect of stromal cells is impaired by glycolytic inhibition, suggesting that the stroma provides nutritional support to malignant cells by secreting lactate (Bonuccelli et al., 2010; Pavlides et al., 2009). Although pyruvate is less abundant than lactate, its circulating levels of 100-200 μM are comparable to other fuels like leucine. Pyruvate is rapidly taken up by malignant tissue, as indicated by the utility of hyperpolarized [1-13C]pyruvate as an imaging probe in human prostate cancer (Nelson et al., 2013). Alanine, at 200-400 μM in the plasma, is the most abundant amino acid other than glutamine and exchanges with pyruvate via alanine aminotransferase. Some cancer cells require alanine to supply central carbon metabolism (Sousa et al., 2016). These observations raise the possibility that lactate and/or the related 3-carbon intermediates pyruvate and alanine serve as fuels to complement glucose metabolism in tumors.
Recent work has begun to interrogate metabolic pathway flow in human tumors in vivo by providing intravenous 13C-glucose before or during surgical resection (Faubert and DeBerardinis, 2016). This approach reveals concomitant glycolysis and glucose oxidation in human NSCLC (Fan et al., 2009; Hensley et al., 2016). Although these tumors are metabolically heterogeneous, a consistent feature is that pyruvate from glycolysis enters the tricarboxylic acid (TCA) cycle through pyruvate dehydrogenase and pyruvate carboxylase (PDH and PC), with both enzymes demonstrating enhanced flow in tumors compared to adjacent benign lung (Hensley et al., 2016; Sellers et al., 2015). Furthermore, dilution of 13C labeling between glucose and TCA cycle intermediates extracted from the tumors implies oxidation of alternative fuels in addition to glucose, with lactate as one candidate (Hensley et al., 2016).
Here we provide the first direct evidence that circulating lactate contributes to central metabolism in human NSCLC in vivo. We also find that lactate's contribution as a respiratory fuel exceeds that of glucose, particularly in tumors growing in the lung.
Results
[U-13C]glucose infusion reveals two major patterns of 13C enrichment in human lung tumors
We used a previously-described protocol to characterize human NSCLC through pre-surgical imaging, intra-operative infusion of 13C-labeled nutrients, and post-surgical analysis of metabolic, molecular and histological features of tumor and adjacent lung (Hensley et al., 2016). Table S1 contains clinical information from the 35 patients in this cohort, including 9 from our previous study (Hensley et al., 2016). The tumors included adenocarcinoma (n=28), squamous cell carcinoma (n=3), large cell neuroendocrine carcinoma (n=1) and carcinoid tumors (n=3). A sequencing panel to assess oncogenes and tumor suppressor genes commonly mutated in NSCLC, including KRAS, EGFR, PIK3CA, BRAF, STK11 and TP53 confirmed molecular heterogeneity in the cohort (Table S1).
Thirty of these patients were infused with [U-13C]glucose. Metabolites extracted from benign lung revealed a progressive reduction in 13C enrichment downstream of glucose (Figure 1A). This pattern suggests dilution of 13C enrichment by the utilization of unlabeled nutrients. A similar pattern was observed in a subset of tumors, which as a group were practically indistinguishable from metabolite labeling in adjacent lung (Figure 1A). However, many tumors displayed a different pattern of 13C enrichment in which lactate had excess labeling relative to upstream glycolytic intermediates like 3-phosphoglycerate and phosphoenolpyruvate (3PG and PEP) (Figure 1A). Hereafter, we refer to these two phenotypes as Low and High Lactate/3PG labeling ratios. Labeling patterns in adjacent lungs did not differ between tumors with High and Low Lactate/3PG labeling ratios (Figure S1A). Complete 13C isotopologue distributions of all metabolites analyzed in each fragment are in Table S2.
Figure 1. Human tumors display heterogeneous patterns of 13C labeling in vivo.
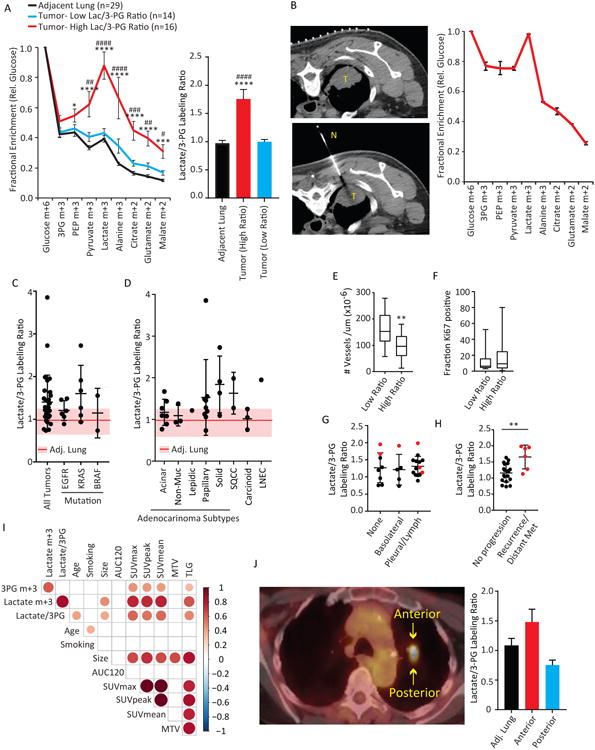
(A) 13C enrichments in adjacent lung (black, n=29), Low ratio tumors (blue, n=14) and High ratiotumors (red, n=16). Fractional enrichments of glycolytic and TCA cycle metabolites arenormalized to enrichment of glucose in the tissue. Lactate/3PG labeling ratios are summarized inthe graph to the right. Average values and S.E.M. are displayed. For High ratio vs. Adjacent Lung:**** p< 0.0001; *** p<0.001; *p<0.05. For High Ratio vs. Low Ratio: #### p< 0.0001; ###p<0.001; ## p<0.01; #p<0.05 (Left, Two-way ANOVA, Tukey post-hoc; Right, one-way ANOVA,Tukey post-hoc).
(B) Left: Computed tomography-guided biopsy of a human NSCLC before (top) and during (bottom)the biopsy. Right: Mass isotopologues from the tumor. Error bars represent S.D. of threetechnical replicates.
(C) Lactate/3PG labeling ratio in tumors of various oncogenotypes (n=30). The shaded regionindicates the mean (line) and S.E.M. of benign lung samples (n=29).
(D) Lactate/3PG labeling ratios in tumors of various histological types (n=30). The shaded regionindicates the mean (line) and S.E.M. of benign lung samples (n=29).
(E) Microvessel density (MVD) (n=12 per group). ** p<0.01 (Students t-test).
(F) Ki67 content by MIB1 staining (n=12 low; n=14 high).
(G) Lactate/3PG labeling ratios in Stage 1 or 2 tumors of various levels of invasiveness at time of infusion (n=25). Pleural/lymph indicates tumors with spread to the pleural cavity or regional lymph nodes. Tumors that recurred or developed distant metastases in the years after the infusions are in red.
(H) Lactate/3PG labeling ratios of Stage 1 or 2 tumors that progressed (distant metastasis or recurrence in the lung) or did not progress during the study. ** p<0.01 (Student's t-test).
(I) Correlation analysis of clinical and metabolic features. Each circle denotes a statistically significant (p<0.05) correlation, with size and color scaling with level of significance. Smoking was measured in pack-years (i.e. average number of packs per day x number of years smoking).
(J) Left: Axial view of patient K1051's FDG-PET scan. Regions of differing FDG-PET signal were sampled. Right: Labeling ratios from three regions of K1051's resection. Data are average and S.D. of three small fragments obtained from each site.
Abbreviations: 3PG, 3-phosphoglycerate; PEP, phosphoenolpyruvate; AUC120, area under the relative contrast enhancement curve after 120 seconds during dynamic contrast-enhanced MRI; SUV, standardized uptake value; MTV, metabolic tumor volume; TLG, total lesion glycolysis. T, tumor; N, needle.
Our protocol includes ligation of the pulmonary artery prior to surgical resection. Although tumor and adjacent lung tissue continue to receive blood flow from the bronchial artery and collateral vessels, occlusion of the pulmonary artery could in principle temporarily impact metabolism and 13C labeling. One of the tumors in our cohort could not be resected because it was affixed to the chest wall. In this patient, [U-13C]glucose was infused intravenously during a needle biopsy of the mass without first occluding blood flow. Metabolites extracted from the resulting 3 mg biopsy had a High Lactate/3PG labeling ratio similar to the ratio in tumors resected surgically (Figure 1B). These findings demonstrate that a) as previously suggested (Hensley et al., 2016), the labeling observed in these tumors is unrelated to perturbations of blood flow accompanying pulmonary artery clamping; and b) needle biopsies contain sufficient tissue for labeling analysis, thereby expanding the clinical contexts in which this type of data may be obtained.
Because the Lactate/3PG labeling ratio was heterogeneous, we sought to correlate this feature with biological factors that might influence tumor metabolism (Figure 1C-I). EGFR-mutant tumors had homogeneous Lactate/3PG labeling ratios similar to adjacent lung, whereas the KRAS-mutant group was more heterogeneous and included tumors with ratios far in excess of lung (Figure 1C). Similarly, adenocarcinomas with predominantly acinar and non-mucinous histology tended to be homogeneous with low ratios, whereas some tumors with papillary, solid and large cell neuroendocrine features had much higher ratios (Figure 1D). Histopathology revealed that high-ratio tumors had lower microvessel densities (Figure 1E), but there were no differences in MIB1/Ki67 staining or stromal content (Figure 1F, S1B).
At the time of infusion, the tumors differed in their degree of local invasiveness, with some tumors displaying no invasion and others having already spread to regional lymph nodes or the pleural cavity. None of the patients had evidence of distant metastasis, which would have contraindicated resection. The Lactate/3PG labeling ratio did not correlate with invasion or spread at the time of infusion (Figure 1G). Remarkably, however, this ratio did correlate with tumor progression over several years of follow up (Figure 1H). Stage I and stage II tumors that recurred in the lung or gave rise to distant metastases had, on average, higher Lactate/3PG labeling ratios at the time of the original [U-13C]infusion and surgery.
FDG-PET signal (SUVmax, SUVpeak, SUVmean) correlated positively with 13C enrichment in 3PG, lactate and the Lactate/3PG labeling ratio, whereas clinical factors like age and smoking were not correlated (Figure 1I). Many tumors displayed regional heterogeneity in FDG-PET signal (Figure 1J), and in some but not all cases, areas of higher signal correlated with higher Lactate/3PG labeling ratios (Figure 1J). Altogether, these data indicate heterogeneity of the Lactate/3PG labeling ratio among human NSCLC tumors and within individual tumors.
Labeled glycolytic products in the blood contribute to tumor metabolism
A high Lactate/3PG labeling ratio is consistent with uptake from the circulation of labeled 3-carbon intermediates distal to 3PG in glycolysis. As previously reported (Hensley et al., 2016), our infusions produce measurable levels of plasma [U-13C]lactate (Figure 2A). We also detected plasma [U-13C]pyruvate and [U-13C]alanine (Figure 2A). Because labeling of plasma lactate and pyruvate exceeded labeling of tumor 3PG, uptake of one or both could contribute to a high Lactate/3PG labeling ratio.
Figure 2. Evidence for lactate import in human NSCLC.
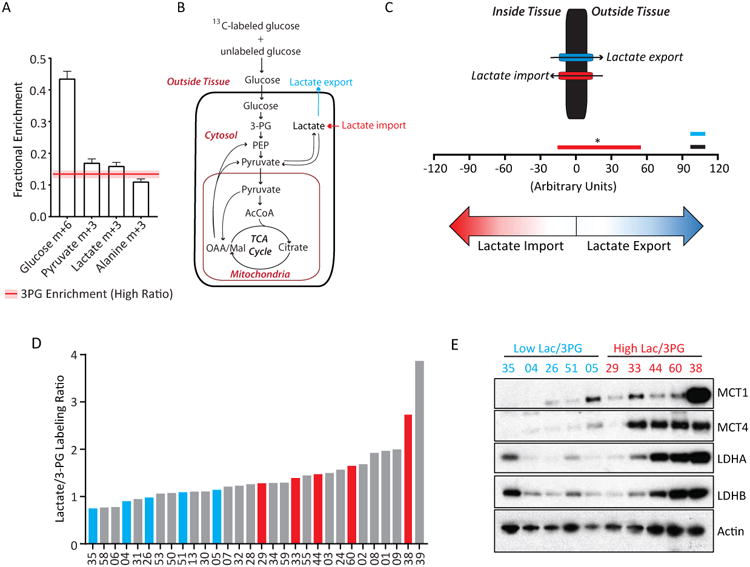
(A) Fractional enrichment in human plasma metabolites. 3PG enrichment in tumors with High Lactate/3PG labeling ratios is indicated by the red line (average) and shaded bar (S.E.M.). Data are average and S.E.M of all patients (n=30).
(B) Schematic of selected reactions from the MFA model.
(C) Net flux ranges for lactate transport in adjacent lung (black bar) and tumors with Low (blue bar) and High (red bar) Lactate/3PG labeling ratios. Bars are the lower and upper bounds of 95% confidence intervals. Data are considered significant (p<0.05) if the bounds do not overlap.
(D) All tumors infused with [U-13C]glucose ranked from lowest to highest Lactate/3PG labeling ratio.
(E) Immunoblot analysis of tumor MCT1, MCT4, LDHA and LDHB. Actin is used as a loading control.
We next used metabolic flux analysis (MFA) to explore the contribution of circulating lactate and related metabolites to the tumors. Quantitative MFA in cultured cells usually relies on definitive import/export rates (e.g. glucose import/lactate export) to constrain intracellular fluxes (Buescher et al., 2015). In vivo, estimating relative fluxes can provide an overview of metabolic activity even if definitive rates cannot be quantified (Hensley et al., 2016). We constructed a metabolic network to fit 13C mass isotopologues from plasma glucose and lactate, and tissue glucose, lactate, 3PG, PEP and TCA cycle intermediates (Figure 2B, Table S3). The model incorporates enzymes that convert 4-carbon TCA cycle intermediates to PEP and pyruvate to explain labeling patterns in those 3-carbon intermediates. Such enzymes could include malic enzyme (malate→pyruvate) and phosphoenolpyruvate carboxykinase (Oxaloacetate→PEP). The model does not exclude contributions from the pentose phosphate pathway, which could also produce complex labeling in 3-carbon intermediates. We also examined lactate exchanges between the plasma and tissue. In lungs and tumors with Low Lactate/3PG labeling ratios, these exchanges fit within a narrow range consistent with net lactate export (Figure 2C). However, tumors with high Lactate/3PG labeling ratios were more difficult to fit, with a much wider range of acceptable fluxes, including values consistent with net lactate import (Figure 2C). Although this model cannot define quantitative lactate exchange rates or the net direction of flow in vivo, it indicates that tumors with Low and High Lactate/3PG labeling ratios differ with respect to lactate transport, with 13C features of the latter group consistent with lactate import from plasma.
To assess expression of transporters and enzymes involved in taking up and metabolizing lactate, we ranked all tumors from the lowest to highest Lactate/3PG labeling ratio (Figure 2D). Several tumors below and above the median were chosen for western blot analysis. MCT1, MCT4, and lactate dehydrogenase isoforms A and B (LDHA and LDHB) were broadly expressed in these fragments, but the levels of all four proteins tended to be elevated in fragments with a high ratio (Figure 2E).
Metabolism of circulating lactate/pyruvate by human NSCLC
We next infused five NSCLC patients with 13C-lactate (one with [2-13C]lactate and four with [U-13C]lactate). The sterile, pyrogen-free [13C]lactate used in some of the infusions contained a small amount of [13C]pyruvate, which we estimated at 0.003% of the [13C]lactate concentration (Figure S2A). The material was introduced using a bolus followed by a continuous infusion during anesthesia, similar to the [U-13C]glucose infusions. Although the amount of lactate introduced was small relative to the total circulating pool, we monitored for signs of metabolic acidosis. Three patients were modestly acidotic, but in all cases the acidosis was respiratory in origin, as indicated by elevated partial pressure of CO2 in arterial blood (Figure 3A). The acidosis rapidly resolved with increased mechanical ventilation. Plasma lactate and pyruvate concentrations were not significantly perturbed (Figure S2B).
Figure 3. Lactate is a carbon source in human lung tumors.

(A) Arterial blood pH, partial pressure of CO2 and bicarbonate concentrations from patients infusedwith 13C-lactate. Reference ranges are displayed by the shaded bar.
(B) Plasma metabolite enrichment in five patients infused with 13C lactate. Data are average andS.E.M from all 5 patients. M+1 isotopologues were evaluated for the patient infused with [2-13C]lactate. M+3 isotopologues were evaluated for patients infused with [U-13C]lactate.
(C) Schematic illustrating routes of 13C entry from infused 13C-lactate.
(D) Metabolite enrichment in tumor samples from patients infused with 13C lactate. (11 fragments from n=5 patients). **** p< 0.0001, **p<0.001, **p<0.01. (One-way ANOVA, Dunnett post-hoc vs 3PG)
These infusions resulted in essentially steady state labeling of circulating lactate in the 20-25% range (Figure 3B). Pyruvate and alanine were also labeled to appreciable levels, as expected because lactate exchanges with pyruvate and pyruvate exchanges with alanine. Labeling in pyruvate modestly exceeded labeling in lactate. This likely arose from a combination of the infused 13C-pyruvate, which had a proportionally large effect on the small circulating pyruvate pool, and excess dilution of lactate by unlabeled lactate secreted from the muscle and other tissues. Labeled glucose was also detected, indicating that infused 13C-lactate was used for gluconeogenesis (Figure 3B, S2C).
In the tumors, comparing labeling in 3PG to labeling in lactate, pyruvate, alanine and TCA cycle intermediates allowed us to infer whether 13C entry occurred predominantly through uptake of glucose or of three-carbon intermediates (Figure 3C). Inspection of tumor metabolites revealed barely detectable labeling in 3PG and PEP, consistent with the scarce labeling in circulating glucose (Figure 3D). However, the tumors contained measurable label in lactate, pyruvate, alanine and TCA cycle intermediates, most of which were more enriched than 3PG (Figure 3D). These findings indicate the propensity of these tumors to take up one or more 3-carbon glycolytic products from the circulation and use them to supply the TCA cycle.
Subcutaneous and orthotopic NSCLC xenografts use plasma lactate as a carbon source
We next turned to mouse models to more directly assess the role of lactate and related metabolites in live tumors. Mice bearing orthotopic lung or subcutaneous xenografts from H460 NSCLC cells were infused with [U-13C]glucose using a protocol analogous to the human studies (Marin-Valencia et al., 2012). These infusions resulted in labeling of 40% of circulating glucose as [U-13C]glucose (Figure 4A; isotopologues from mouse experiments are in Table S4). Plasma glucose contained m+1, m+2, m+3 and m+5 labeling in addition to the expected m+6, indicating that gluconeogenesis from labeled 3-carbon intermediates occurred during the experiment (Figure S3A). Plasma lactate, pyruvate and alanine were also labeled (Figure 4A). Healthy lung tissue from these mice displayed a low Lactate/3-PG labeling ratio, similar to human lung (Figure 4B,C). In contrast, the tumors displayed enhanced labeling in lactate, pyruvate and alanine relative to 3PG, regardless of the site of the tumor (Figure 4B,C). Subcutaneous xenografts of HCC827 and HCC15 NSCLC cells also displayed high Lactate/3PG labeling ratios after infusion with [U-13C]glucose (Figure 4C).
Figure 4. Lactate is a fuel in mouse NSCLC xenografts.

(A) Plasma metabolite enrichment in mice infused with [U-13C]glucose. Data are average and S.E.M.(n=6 mice).
(B) Tissue metabolite enrichment in H460 tumor-bearing mice infused with [U-13C]glucose.Enrichments in normal lung, flank tumor and lung tumor are normalized to glucose enrichmentin each tissue. Data are average and S.E.M. **** p< 0.0001, *** p< 0.001, ** p< 0.01, (Two-wayANOVA, Tukey post-hoc) (n=3-4 mice per group).
(C) Left: Lactate/3PG labeling ratios of tissues from (B). Right: Mice bearing HCC827 and HCC15flank xenografts were infused as in (B). Lactate/3PG labeling ratios are displayed as average andS.E.M. The Lactate/3PG labeling ratio of HCC15 tumors are derived from Vector control tumorsin Fig. 5F. **** p< 0.0001, *** p< 0.001, ** p< 0.01 (One-way ANOVA, Dunnett post-hoc vs.Adjacent lung; n=5-6 mice per group).
(D) Mice bearing H460 tumors in the flank or lung were infused with [3-13C]lactate. Enrichmentvalues are relative to 3PG. Data are average and S.E.M. ****p< 0.0001, ***p<0.001, *p<0.05 vs.adjacent lung. (Two-way ANOVA, Tukey post-hoc; n=4-5 mice per group).
(E) Left: Lactate/3PG labeling ratios of tissues in (D). Right: Mice bearing HCC827 and HCC15 flank xenografts were infused as in (D). Data are average and S.E.M. ** p<0.01, * p< 0.05 (One-way ANOVA, Dunnett post-hoc vs. 3PG; n=4-5 mice per group). The Lac/3PG ratio of the HCC15 mice is derived from the vector control tumors in panel 5E.
(F) Schematic of [2-2H]lactate metabolism.
(G) Plasma and tumor metabolite enrichments in mice infused with [2-2H]lactate. Data are average and S.D. (n=3 mice).
We next infused mice with [13C]lactate, [13C]pyruvate or [13C]alanine to assess the contribution of each nutrient. In mice bearing H460 tumors, infusion with [3-13C]lactate resulted in high levels of 13C labeling in tumor lactate and TCA cycle intermediates relative to 3PG, particularly in tumors grown in the lung (Figure 4D,E). These infusions did not significantly increase plasma lactate concentrations (Figure S3B) and resulted in steady-state plasma lactate enrichments of 23.5 ± 0.01%. HCC827 and HCC15 subcutaneous xenografts also displayed high Lactate/3PG labeling ratios under this protocol (Figure 4E). Infusion of either [U-13C]pyruvate or [U-13C]alanine also labeled tumor metabolites, but in neither case did labeling differ as dramatically from labeling in the lung as for infusions with [13C]lactate (Figure S3C,D).
Lactate is much more abundant in plasma than either pyruvate or alanine, so we focused further effort on documenting its utilization by these tumors. When lactate exchanges with pyruvate via LDH, the proton attached to lactate carbon 2 is transferred to NAD+, producing NADH (Figure 4F). We obtained [2-2H]lactate as a tracer for infusion studies, because LDH converts this tracer to unlabeled pyruvate. If the tumor takes up [2-2H]lactate, lactate m+1 appears in the tumor. On the other hand, no label is transferred to the tumor if the route of entry is via pyruvate or alanine derived elsewhere from [2-2H]lactate. Infusion of [2-2H]lactate resulted in 8-10% enrichment in the plasma lactate pool (Figure 4G). This low enrichment compared to labeling by [13C]lactate likely reflects loss of 2H by peripheral LDH-catalyzed lactate/pyruvate exchanges; during [13C]lactate infusions, 13C is retained during these exchanges. We detected enrichment in tumor lactate, but as expected, no labeling in 3PG, pyruvate or alanine (Figure 4G). The TCA cycle intermediate malate was also labeled, presumably through transfer of 2H from labeled NADH arising from LDH activity (Figure 4G). Although this experiment does not rule out uptake of pyruvate and alanine, it confirms that the tumors take up lactate from the circulation.
MCT1-dependent lactate uptake by HCC15 tumors in vivo
We explored the mechanism of lactate transport by NSCLC xenografts. Both MCT1 and MCT4 are expressed in human NSCLC (Figure 2E, S4A). For functional analysis of MCT1 and MCT4, we used HCC15 cells, an NRAS and STK11-mutant NSCLC cell line expressing both transporters. We used CRISPR/Cas9 editing to obtain clones of HCC15 lacking either MCT1 or MCT4 (Figure 5A). In culture, proliferation persisted after loss of either transporter, with no major effect on the doubling time (Figure S4B). MCT1 loss resulted in increased oxygen consumption and decreased extracellular acidification, whereas loss of MCT4 had no significant effect on either (Figure 5B,C). Direct measurements of glucose utilization and lactate secretion revealed a large suppression of glycolysis after MCT1 loss and a modest effect after MCT4 loss (Figure 5D), indicating that MCT1 is required for the glycolytic phenotype of HCC15 cells in culture.
Figure 5. MCT1 regulates lactate uptake in HCC15 xenografts.
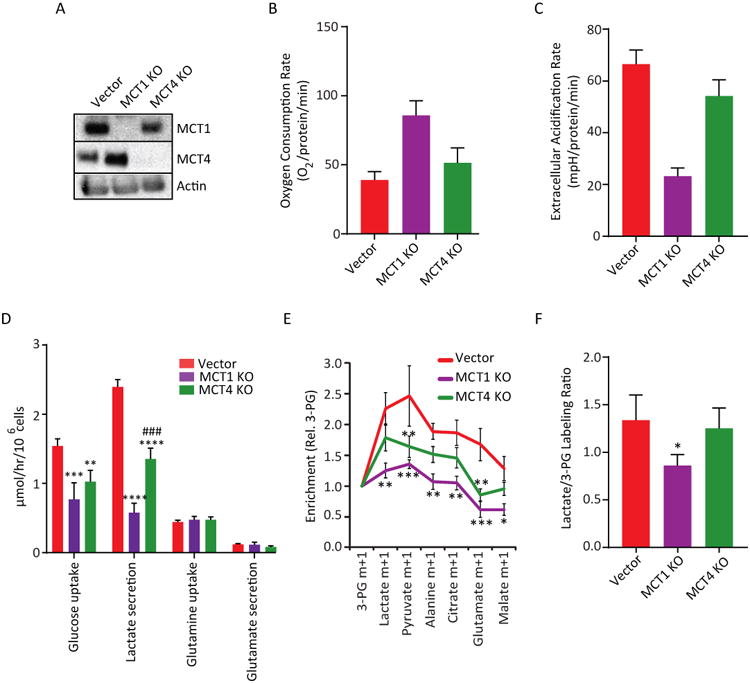
(A) Immunoblot of HCC15 cells expressing a control vector or CRISPR-mediated knockoutof MCT1 or MCT4. Actin is used as a loading control.
(B,C) Oxygen consumption and extracellular acidification rates in Control, MCT1 KO and MCT4 KOcells. Data are average ± SD from a representative experiment (n= 6).
(D) Metabolic rates from cultures of Vector control HCC15 cells and sub-lines with knockout of MCT1 or MCT4. **** p< 0.0001; *** p<0.001, **p<0.01 vs. Vector. ### p<0.001 vs MCT1 KO(Two-way ANOVA, Tukey post-hoc, n=3 separate experiments).
(E) Mice bearing HCC15 Vector control, MCT1 KO, or MCT4 KO tumors were infused with [2-13C]lactate. Enrichment values are normalized to 3PG. *** p< 0.001, ** p< 0.01, *p<0.05. (Two-way ANOVA, Tukey post-hoc; n=3-4 mice per group).
(F) Mice bearing HCC15 Vector control, MCT1 KO, or MCT4 KO tumors were infused with [U-13C]glucose. The Lactate/3PG labeling ratio is plotted for each tumor type. Data are average andS.EM. * p< 0.05 (One-way ANOVA, Tukey post-hoc; n=3-5 mice per group).
To examine the role of MCT1 and MCT4 in vivo, subcutaneous xenografts were established from vector control, MCT1-deficient and MCT4-deficient HCC15 lines, then tumor-bearing mice were infused with [2-13C]lactate. MCT1 deficiency reduced labeling in tumor metabolites, nearly eliminating the elevated Lactate/3PG labeling ratio observed in control tumors (Figure 5E). Reduced labeling was not an artifact of a large accumulation of unlabeled lactate, because the total lactate pool was not significantly altered by MCT1 deficiency (Figure S4C). Because the MCT1-deficient tumors had nearly equivalent 13C enrichment in lactate and 3PG, it is conceivable that labeling in both metabolites arose largely from import of 13C-glucose labeled via gluconeogenesis. MCT4 deletion only modestly reduced labeling of tumor metabolites (Figure 5E). Infusing these tumors with [U-13C]glucose also revealed a prominent effect of MCT1 loss, which suppressed the Lactate/3PG labeling ratio and resulted in a pattern similar to normal lung, whereas MCT4 loss had no effect on labeling from [U-13C]glucose (Figure 5F and S4D). Thus, in HCC15 cells and tumors, MCT1 is the predominant isoform involved in lactate transport. In culture under high-glucose conditions, it promotes a glycolytic phenotype and in vivo, it permits entry of lactate carbon from the circulation. These data also demonstrate cancer cell-autonomous lactate uptake in HCC15 xenografts and indicate that MCT1 is required for the high Lactate/3PG labeling ratio observed during infusion of either 13C-lactate or 13C-glucose.
Lactate is preferred to glucose as a fuel for the TCA cycle in vivo
We next sought to compare the contributions of lactate and glucose to tumor TCA cycle metabolism. To do this, we established subcutaneous HCC15 tumors and infused mice concurrently with [U-13C]glucose and [3-13C]lactate. In this scheme, pyruvate and lactate derived from glucose are initially uniformly labeled (m+3) and TCA cycle intermediates arising downstream of PDH are labeled as m+2 (Figure 6A). Metabolites arising from lactate are labeled as m+1. Short (30 min) infusions were used to limit labeling complexity. Glucose and lactate concentrations in the plasma were not altered (Figure 6B). At the end of these infusions, the plasma lactate m+1 and m+3 fractions were equivalent (Figure 6C), indicating that some of the glucose-derived labeling in the tumors likely arises from peripheral conversion of glucose to lactate followed by entry of labeled lactate. Thus, the method underestimates lactate's contribution and may somewhat overestimate the contribution of glycolysis within the tumor. 13C-Lactate was also converted to 13C-glucose during the infusion, but the glucose m+6 far exceeded glucose m+1 in the circulation (Figure 6C). Tumor metabolite labeling was normalized to labeling of the precursor (glucose or lactate) in the plasma so that relative contributions of each nutrient could be estimated. Although glucose and lactate made similar contributions to tumor pyruvate, lactate made a much larger relative contribution to the TCA cycle (Figure 6D). MCT1-deficient tumors displayed reduced lactate-derived labeling in pyruvate and TCA cycle intermediates, consistent with MCT1's role in lactate import in HCC15-derived tumors (Figure 6E). Interestingly, MCT1 deficiency blunted glucose's relative contributions and reduced the absolute enrichment in tumor lactate from either precursor (Figure 6E,F). This suggests that MCT1 enables lactate uptake from the circulation and also supports glycolytic metabolism in vivo as it does in culture.
Figure 6. Lactate is preferred to glucose as a fuel for the TCA cycle.

(A) Schematic for co-infusions with [U-13C]glucose and [3-13C]lactate. In short infusions, glucose-derived pyruvate and lactate are primarily m+3 and TCA cycle metabolites are primarily m+2,whereas lactate-derived metabolites are primarily m+1.
(B) Abundance of glucose and lactate pre- and post-infusion. N.S., not significant. (Student's t-test;n=3-5 samples per group)
(C) Plasma fractional enrichment of glucose and lactate during co-infusion of [U-13C]glucose and [3-13C]lactate. Data are average and S.E.M (n=8 co-infused mice).
(D) Mice with flank xenografts of HCC15 control cells were co-infused with [U13C]glucose and [3-13C]lactate. Enrichments are normalized to the enrichment of each precursor in plasma. Data areaverage and S.E.M. (n=4 mice).
(E) Mice bearing flank xenografts of HCC15 MCT1 KO cells were co-infused with [U13C]glucose and[3-13C]lactate as in (C). Enrichments are normalized to the enrichment of each precursor inplasma. Data are average and S.D. (n=2 mice).
(F) Effect of MCT1 knockout on enrichments of tumor lactate derived from circulating glucose orcirculating lactate. Data are from the infusions shown in (D) and (E) and are expressed asaverage and S.E.M.
(G) Mice bearing flank (left) and lung (right) H460 tumors were co-infused with [U-13C]glucose and[3-13C]lactate. Enrichments are normalized to enrichment of each precursor in plasma. Data areaverage and S.E.M. (n=3 mice).
Finally, we compared glucose and lactate metabolism within tumors at two different sites in the same mice, where both tumors were exposed to the same supply of labeled glucose and lactate. H460-derived tumors were grown in the subcutaneous space and orthotopically in the lung, then the mice were co-infused with [U-13C]glucose and [3-13C]lactate. As with the HCC15 tumors, glucose and lactate made comparable contributions to pyruvate, but lactate made a more prominent contribution to the TCA cycle (Figure 6G). This difference was much more dramatic in lung tumors, where there was little dilution of label between circulating lactate and TCA cycle intermediates (Figure 6G). Thus, NSCLC-derived xenografts consume glucose and lactate concomitantly, with lactate predominating as a carbon source for the TCA cycle. However, the extent to which lactate is used as a respiratory fuel is at least partially related to the anatomic site of the tumor.
Discussion
One of the most fundamental challenges in cancer metabolism is to identify nutrients that fuel production of energy and macromolecular precursors in aggressive tumors. These nutrients and the pathways they supply are candidate targets for cancer imaging and therapy. Several lines of evidence indicate that solid tumors, including NSCLC, display higher levels of glucose metabolism than adjacent benign tissue. The broad utility of FDG-PET indicates that many tumors have an enhanced ability to take up glucose in vivo. Recurrent genomic alterations, including mutations in KRAS, PIK3CA, STK11, TP53 and others enhance glycolytic flux, indicating that reprogrammed glucose metabolism is a shared, cell-autonomous consequence of these mutations (Faubert et al., 2014; Hu et al., 2016; Matoba et al., 2006; Whang et al., 2016; Ying et al., 2012). Examination of carbon fates by infusing NSCLC patients with 13C-glucose confirmed both glycolysis and glucose oxidation within the tumors (Fan et al., 2009; Hensley et al., 2016). However, careful inspection of 13C labeling strongly suggested that the majority of carbon flow through the TCA cycle in tumors does not arise directly from glucose. The current work presents evidence from both 13C-glucose and 13C-lactate infusions that 3-carbon products of glycolysis, including lactate, are consumed by human NSCLC in vivo.
Lactate is as an inter-organ carbon shuttle supplying aerobic metabolism and gluconeogenesis (Brooks, 2002). It is used as a fuel in the heart, brain and skeletal muscle (Gladden, 2004). Although most cancer metabolism studies have considered lactate primarily as a waste product secreted as the terminal component of the Warburg effect, recent work has implicated lactate as a potential fuel. In breast cancer cells, estrogen-related receptor alpha (ERRα) induces expression of genes involved in oxidative metabolism, promotes lactate oxidation and allows lactate to support cell survival during glucose depletion (Park et al., 2016). Xenografts from breast cancer cell lines take up lactate from the circulation and convert some of it to glutamate, indicating its use as a fuel in these models (Kennedy et al., 2013).
Human NSCLC exhibits heterogeneity in the extent to which glucose carbon supplies the TCA cycle (Hensley et al., 2016). We find here that lactate metabolism is also heterogeneous between and within tumors. The Lactate/3PG labeling ratio varied across our cohort but tracked with several molecular and clinical features. While no histological subtypes displayed exclusively high ratios, we note that tumors with predominantly acinar, non-mucinous and carcinoid features tended to have low ratios similar to the lung, whereas higher ratios were observed in tumors with papillary, solid and squamous cell histology. This may indicate that tissue architecture is related to the relative preference or propensity to use lactate as a fuel. A direct assessment of lactate metabolism in mice indicated that the tumor's anatomic location also influences lactate handling. Tumors localized in the lung display a particular predilection to oxidize lactate in the TCA cycle, perhaps because of the high level of perfusion and oxygenation in the lung.
In humans, tumors with high Lactate/3PG labeling ratios also tended to have high FDG uptake, implying that glucose and lactate are consumed concurrently. High FDG uptake is associated with poor overall survival in NSCLC (Berghmans et al., 2008). In our cohort, tumors with high Lactate/3PG labeling ratios were more likely to progress during the study, even though the Lactate/3PG labeling ratio was measured at the time of resection of the primary tumor, in some cases years before recurrence or metastases were observed. Expanding our cohort and extending longitudinal follow-up will allow us to further explore the relationship between metabolic activities and survival. We note that 11C-lactate has been developed as a PET probe to image cardiac metabolism, and perhaps could be used to assess rates of lactate uptake in human NSCLC (Herrero et al., 2007).
It should be emphasized that the Lactate/3PG labeling ratio in tumors infused with 13C-glucose is an imperfect surrogate for lactate utilization. Low Lactate/3PG labeling ratios do not rule out lactate utilization, because normal lung displayed ratios close to 1.0 yet still demonstrated metabolism of lactate carbon during 13C-lactate infusions (data not shown). It is unlikely that tumors with Low Lactate/3PG labeling ratios have a particular preference to oxidize glucose because they tend to have low FDG-PET signal and low 13C enrichment in TCA cycle intermediates after infusion with [U-13C]glucose. Additional markers, such as quantitation of informative metabolites, may help complement 13C-labeling features to better predict fuel preferences. A limitation of the study is that because of the high degree of reversibility of both lactate transporters and lactate dehydrogenases, it is difficult to say whether the transfer of lactate carbon from the plasma into the tumor pyruvate pool represents a net flux. It was not possible in these human lung tumors to cannulate afferent and efferent blood vessels specifically supplying the tumor to calculate rates of glucose extraction/lactate secretion, as Warburg attempted to do in rats (Warburg et al., 1927). Furthermore, it is likely that some label entering the tumor is carried as pyruvate rather than lactate, as MCT1 transports both molecules (Garcia et al., 1994; Hong et al., 2016). It is also clear that the relative importance of MCT1 and MCT4 in lactate transport, the net direction of transport, and the metabolic impact of inhibiting each isoform varies among models and contexts (Doherty et al., 2014; Hong et al., 2016; Marchiq et al., 2015). However, even considering these caveats, the 2H labeling experiment indicates that lactate enters NSCLC xenografts without first being converted to pyruvate, and 13C labeling of human tumor TCA cycle metabolites after 13C-lactate infusions demonstrate that a substantial fraction of this carbon arises from circulating lactate. Altogether, the data provide an explanation for the surprisingly low contribution of glucose-derived 13C to the TCA cycle in human NSCLC (Hensley et al., 2016).
It is interesting that glucose and lactate are consumed concomitantly. It was previously suggested in experimental tumor models that lactate oxidation occurs in well-oxygenated regions as part of symbiotic metabolite exchanges in which hypoxic cancer cells produce lactate and better oxygenated cells take up lactate to fuel respiration (Sonveaux et al., 2008). Although we cannot resolve whether such exchanges occur in human NSCLC, our data indicate that lactate from the circulation is also used as a respiratory substrate. We do not know whether glycolysis and lactate oxidation occur within the same cell, but it is interesting to consider the implications of such a phenotype. Converting pyruvate from glycolysis into lactate resolves the cytosolic redox costs of glycolysis and allows the pathway to persist. Importing lactate from outside the cell and using it as a respiratory fuel could presumably operate as a parallel pathway to support the energetic and synthetic functions of the TCA cycle. In tumors with high FDG-PET signal, the availability of extracellular lactate as a respiratory fuel might allow glucose to supply other growth-supporting pathways like the pentose phosphate pathway and synthesis of ribose and hexosamines. In culture, cancer cells can use lactate as a carbon source for the TCA cycle and citrate-dependent fatty acid synthesis even when glucose is available (Chen et al., 2016). In our experiments, co-infusing mice with 13C-glucose and 13C-lactate demonstrated equivalent labeling of tumor pyruvate, but a striking preference for lactate-dependent rather than glucose-dependent labeling of TCA cycle intermediates. This implies incomplete mixing of the pyruvate pools generated from 13C-glucose and lactate. Although several mechanisms could explain this phenomenon, a straightforward one is that mitochondria within these tumor cells contain LDH, thereby channeling lactate carbons into the mitochondrial pyruvate pool destined for the TCA cycle. Such an arrangement could also facilitate NADH generated by LDH to be passed to Complex I of the electron transport chain, maximizing the energy yield. Although cancer cell LDH has generally been viewed as a cytosolic activity, there is evidence for mitochondrial LDH in mammals, including some cancer cells (Chen et al., 2016; Passarella et al., 2008).
Analysis of metabolic flux in living human tumors should stimulate a reanalysis of traditional concepts in cancer metabolism, particularly the role of aerobic glycolysis or the Warburg effect. The major components of this pathway, as rendered by Warburg, are the preferential conversion of glucose to lactate, the suppression of glucose oxidation and the secretion of lactate into the extracellular space. We cannot in human experiments make a full accounting of the quantitative fates of glucose carbon, because we do not know how much lactate and other by-products of glucose metabolism are secreted from the tumor into the bloodstream. Nevertheless, in human NSCLC, there is no evidence for suppressed glucose oxidation, and the reported data rather suggest activated glucose oxidation relative to adjacent lung (Hensley et al., 2016; Sellers et al., 2015); lactate is taken up and seems to be a prominent fuel for the TCA cycle; and TCA cycle activity occurs in regions with high Ki67 content, suggesting that fuel oxidation does not counteract tumor cell proliferation (Hensley et al., 2016). The availability of multiple fuels for central carbon metabolism, including glucose, lactate and others, and the marked heterogeneity of fuel choice between and within tumors, will make it challenging to develop universal therapies based on targeting single fuels feeding core pathways like the TCA cycle.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ralph J. DeBerardinis (Ralph.DeBerardinis@UTSouthwestern.edu).
Experimental Model and Subject Details
Human Subjects
Thirty-five patients with NSCLC were enrolled in an IRB-approved protocol after obtaining informed consent. Patients were considered eligible for the study if they had solitary pulmonary masses measuring 1 cm or more in diameter. Uncontrolled diabetes was a contraindication to participation. Most patients consented for infusions with [U-13C]glucose. When we developed the hypothesis about lactate utilization, the IRB protocol was amended to allow us to infuse five patients with 13C-lactate. No specific clinical criteria were applied to choose the labeled substrate; 13C-lactate infusions were performed more or less in the next 5 patients recruited to the study, as long as 13C-lactate was available in the laboratory. Clinical details about patients participating in the study and their tumors are summarized in Table S1.
Animal Studies
All procedures were approved by UT Southwestern's Animal Care and Use Committee in accordance with the Guide for the Care and Use of Laboratory Animals. Healthy male and female Ncr nude mice were xenografted at approximately 8-10 weeks of age. All mice were housed in pathogen free conditions prior to use. Prior to the described studies, mice were monitored regularly and determined to be healthy by the veterinary staff.
Cell lines
Cell lines were identified using DNA fingerprinting and confirmed to be mycoplasma free using the e-Myco kit (Boca Scientific). H460, HCC15 and HCC827 cells were provided by J. D. Minna, UT Southwestern. Notable information about these cell lines included the following: H460: Large cell lung cancer cell line from male donor with mutations in KRAS (Q61H), LKB1 (Q37*) and PIK3CA (E545K).
HCC827
Adenocarcinoma cell line from female donor with mutation in EGFR (E746-A750 deletion).
HCC15
Squamous cell carcinoma cell line from male donor with mutations in NRAS (C186F), LKB1 (homozygous deletion spanning nucleotides 1 to 1,252 of cDNA sequence) and p53 (D259V). All cells were maintained in RPMI-1640 supplemented with penicillin/streptomycin, L-glutamine (4 mM) and 5% fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2 and 95% air.
Method Details
Patient Infusions
On the day of surgery, sterile, pyrogen-free [U-13C]glucose (Cambridge Isotope, Isotec) was administered as a bolus of 8g over 10 minutes followed by 4 or 8g/hr infusion through a peripheral intravenous line (PIV). Sterile, pyrogen-free [U-13C]lactate (Cambridge Isotope Laboratories or Isotec) or [2-13C]lactate (Isotec) was administered as a bolus of 150μmol/kg over 5 minutes followed by a 10μmol/kg continuous infusion through a PIV. Infusions generally proceeded for at least two hours prior to tumor resection. A second PIV in the contralateral arm was used to obtain blood to analyze 13C enrichment. Standard surgical procedures were followed, with the majority of cases being robotic lobectomies. Based on pre-operative imaging and gross inspection at resection, viable fragments of tumor and lung were sampled. In cases where metabolic heterogeneity was assessed, the resected lobe was oriented anatomically to identify regions of the tumor pre-selected by imaging. Tissue fragments were washed in ice-cold saline and immediately frozen in liquid nitrogen. Tumors were stratified to metabolic groups based on the Lactate/3PG labeling ratio. Ratios of 1.25 and above were considered ‘high’, and ratios of 1.24 and below were considered ‘low’. The value of 1.25 is one standard deviation above the average Lactate/3PG labeling ratio in all benign lung samples.
Multi-parametric Magnetic Resonance Imaging (mpMRI)
MRI scans occurred within 5 days before surgery on a 3T dual-transmit Achieva MR scanner (Philips Healthcare, Best, The Netherlands) with a 16-channel SENSE-XL Torso Coil or on a 3T Philips Ingenia scanner with a 28-channel dStream Anterior-Posterior coil (Philips Healthcare, Best, The Netherlands). Coronal and axial T2-weighted half-Fourier single-shot turbo spin-echo images were acquired for anatomic reference to localize the lesion. Dynamic contrast enhanced (DCE) MRI was performed using a 2D or 3D T1-weighted spoiled gradient-echo sequence before, during, and after administration of a bolus of 0.01 mmol/kg gadobutrol (Gadavist; Bayer Healthcare Pharmaceuticals) using a power injector at a rate of 2 cc/sec followed by a 20 cc saline flush at the same rate. DCE images were acquired with free-breathing for at least 4 minutes using the following parameters: TE/TR = 1.14-1.65/2.5-4.5 ms, flip angle = 10o, field of view = 200-300 × 200-300 mm2, in-plane resolution = 0.8-1.7 × 0.8-1.7 mm2, and temporal resolution = 0.5-5.2 sec. One or more imaging slices were acquired from the lesion with careful planning to avoid artifacts from the heart and major blood vessels. Tumor volume measurements were based on T2-weighted images by ROI segmentation in an Open-Source DICOM Viewer, Osirix. DCE MRI was analyzed by manually selecting regions of interest (ROIs) in Osirix. Average ROI values were plotted at every available time point through the whole series, and subtracted by the baseline signal. Contrast enhancement curves were generated using Excel Solver to calculate constants of a gamma variate function that satisfy a minimum among all data points of a DCE scan and the points of a gamma variate function. Initial area under the curve measurements were approximated with the trapezoidal rule over the first 120 seconds post-contrast injection.
FDG-PET Scans
PET-CT examinations performed at UT Southwestern used a Siemens Biograph 64 (2007) PET/CT Scanner (Siemens Healthcare). Patients were injected intravenously with a weight-based dose of 18F-FDG of 5.7 MBq/kg, with a minimum dose of 370 MBq and a maximum dose of 740 MBq. Images were acquired approximately 60 minutes later. For the noncontrast CT images that were concurrently acquired for attenuation correction and anatomic correlation with PET images, the parameters were 120 kVp, fixed 200 mAS, 3 mm slice thickness in the head and neck and 5 mm slice thickness in the body, pitch 0.8, rotation time 0.5 sec. Standardized uptake values (SUV) were calculated with the formula SUV = maximum tissue activity of FDG / (injected dose of FDG x patient body weight). The patients' FDG-PET/CT data was analyzed using a Syngo.Via station using automated gradient-based segmentation method, and the SUV max, mean, peak, metabolic tumor volume (40% threshold), and total lesion glycolysis (40% threshold) were obtained.
Metabolic Flux Analysis (MFA)
Steady state metabolic fluxes were calculated by 13C mass isotopologue distributions (MIDs) for glucose, 3-phosphoglycerate, phosphoenolpyruvate, pyruvate, lactate, alanine, citrate, glutamate, malate, aspartate and glutamine using the INCA software package (http://mfa.vueinnovations.com/), which applies an elementary metabolite unit framework to simulate MIDs. We developed reaction networks describing the stoichiometry and carbon transitions of central carbon metabolism with assumptions that are summarized below. To ensure that a global minimum of fluxes was identified, flux estimations were initiated from random values and repeated a minimum of 50 times. A chi-square test was applied to test goodness-of-fit, and accurate 95% confidence intervals were calculated by assessing the sensitivity of the sum of squared residuals to flux parameter variations. Data Table S3 contains the degrees of freedom and sum-of-squared residuals (SSR) for the best fit model and the lower and upper bounds of 95% confidence intervals for all fluxes.
MFA procedures and assumptions:
13CO2 produced during oxidation reactions is not reincorporated via carboxylation reactions.
The metabolites succinate and fumarate are symmetrical and their metabolism through the TCA cycle does not produce a particular orientation.
The metabolites pyruvate, acetyl-CoA, citrate, α-ketoglutarate, malate, fumarate and oxaloacetate are metabolically active in both the cytosol and mitochondria. Malate, citrate and α-ketoglutarate are allowed to freely mix between the compartments.
The extraction process homogenizes distinct pools of metabolites existing within distinct compartments. Therefore GC-MS analysis of the isotopic enrichment of these metabolites reflects the mixture of distinct metabolic pools. By employing the INCA platform to perform metabolic flux analysis, it is possible to extract meaningful information from these mixed pools. To do this, the model employs parameters to account for the mixing of mitochondrial and cytosolic metabolites and the existence of intracellular and extracellular metabolite pools.
All values are made relative to citrate synthase (CS), which was set at an arbitrary value of 100.
In order to ensure that the model did not become under-constrained, we only included a small number of carbon sources (plasma glucose, plasma glutamine, and tumor glycogen) and carbon sinks (plasma lactate, CO2, and lipogenic AcCoA) in the reaction network. These were hypothesized to be the major routes of carbon input and output based on our prior studies and others reported in the literature. We could not obtain direct measurements of these source/sink fluxes in vivo but were able to use the model to estimate their best-fit values through regression of the isotope labeling measurements. While not all fluxes could be precisely quantified using this approach, we were able to determine relative flux ranges (normalized to CS) that were consistent with the isotope labeling measurements (indicated by the 95% confidence bounds in Table S3). This allowed for rigorous statistical comparisons to be made between experimental groups, even when some fluxes in the model were not uniquely determined.
Immunohistochemistry
H&E slides were scanned in an Aperio scanoscope and analyzed by Aperio Imagescope software. Slides were stained with MIB-1 or CD31 and quantified. Tumor and stromal content was determined using pancytokeratin AE1/AE3 stain. Positive stain corresponds to tumor and negative stain corresponds to stroma. All histological analyses were conducted by surgical pathologists blinded to the results of the metabolic study.
Mutation Analysis
Patient DNA was isolated from blood or FFPE specimens after microdissection. This clinical next-generation sequencing assay employs the Ampliseq Cancer Hotspot Panel v2 run on the Ion Torrent Person Genome Instrument. Software includes Torrent Variant Caller, Intergrated Genomic Viewer, and Geneticist Assistant software.
Mouse Xenografts and Infusions
Cells (HCC827, H460 or HCC15) were suspended in RPMI, mixed 1:1 with Matrigel (Becton Dickinson), then 1×106 cells were subcutaneously injected into the flanks of Ncr nude mice. For lung xenografts, 1×106 H460 cells were suspended in saline solution, and injected into the tail veins of Ncr nude mice. Infusions occurred when tumors were 0.5-1.0cm diameter in the flank. Lung tumors were confirmed by MRI or by the onset of weight loss. Mice were fasted for 16 hours, then at close to 9 AM, 25 gauge catheters were placed in the lateral tail vein under anesthesia. Isotope infusions started immediately after implantation of the catheter and continued for approximately 3 hours, also under anesthesia. In the glucose infusions, the total dose of glucose was 2.16g/kg dissolved in 750μl saline. The glucose solution was administered as a bolus 125μl/min (1min) followed by a continuous rate of 2.5μl/min for 3 hours. The total dose of lactate was 1.44g/kg dissolved in 750μl saline. This was administered as a bolus of 15μl/min (10 mins) followed by a continuous rate of 2μl/min for 3 hours. Pyruvate infusions also used a dose of 1.44g/kg in 750μl saline, with a bolus of 15μl/min (10 mins) followed by a continuous rate of 2.5μl/min for 3 hours. The dose of alanine was 0.72g/kg dissolved in 750μl saline. This solution was administered as a bolus of 125μl/min (1min) followed by a continuous rate of 2.5μl/min for 3 hours. Blood samples of ∼20μl were obtained every 30 minutes via retro-orbital bleed. Animals were euthanized at the end of the infusion, then tumors were harvested, rinsed briefly in cold saline and frozen in liquid nitrogen.
Mass Spectrometry
Blood was obtained prior to and approximately every 30 minutes during infusion until tissue was removed from the patient or mouse. Whole blood was chilled on ice and centrifuged to separate and freeze the plasma. Aliquots of 25-50μl of plasma were added to 80:20 methanol:water for extraction. Frozen tissue fragments weighing 5-15mg were added to 80:20 methanol:water and extracted to analyze 13C enrichment. Samples were subjected to three freeze-thaw cycles, then centrifuged at 16,000xg for 15 minutes to precipitate macromolecules. The supernatants, with 1μl of d27-myristic acid added as an internal control, were evaporated, then re-suspended in 40μL anhydrous pyridine and added to a pre-prepared GC/MS autoinjector vials containing 80μl N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide (MTBSTFA) derivatization reagent. The samples were incubated at 70oC for 1 hour following which aliquots of 1μl were injected for analysis. Each sample was injected in triplicate to establish technical reproducibility. Samples were analyzed using either an Agilent 6890 or 7890 gas chromatograph coupled to an Agilent 5973N or 5975C Mass Selective Detector, respectively. The observed distributions of mass isotopologues were corrected for natural abundance.
CRISPR-mediated deletion of MCT1 or MCT4
CRISPR guides were created using CRISPR Design (http://crispr.mit.edu) from sequences available at the UCSC Genome Browser (http://genome.ucsc.edu/). gRNAs were cloned into the lentiCRISPR V2 system (Shalem et al., 2014). These viruses were then infected into HCC15 cells, and puromycin selection was used to obtain multiple control (Vector) clones, and multiple clones deficient for SLC16A1 (MCT1) or SLC16A3 (MCT4)-deficient HCC15. Three independent clones of each type were combined to create small pools used in the experiments.
Immunobloting
Cells and tissue samples were lysed in modified CHAPS buffer (10 mM Tris-HCl, 1 mM MgCl2, 1 mM EGTA, 0.5mM CHAPS, 10% glycerol, 5mM NaF) supplemented with the following additives: protease and phosphatase tablets (Roche), DTT (1 μg/ml), and benzamidine (1 μg/ml). Cleared lysates were resolved by SDS-PAGE, transferred to nitrocellulose, and incubated with primary antibodies against MCT1, MCT4, LDHA or LDHB. Actin was used as a loading control.
MCT isoform expression in human NSCLC
Differences in MCT1 or MCT4 mRNA expression were determined in lung adenocarcinoma tumors from The Cancer Genome Atlas (TCGA) (https://cancergenome.nih.gov/). Methods for data generation, normalization, and bioinformatic analyses were previously described in the TCGA LUAD publication (The Cancer Genome Atlas Research (Network, 2014). The present analysis downloaded and analyzed data from this cohort using cBioPortal (www.cbioportal.org).
Seahorse XF96 Respirometry
Cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience). In brief, cells were plated at 1×104/well in 100ml non-buffered DMEM containing 12.5mM glucose and 4mM glutamine. Cells were incubated in a CO2-free incubator at 37°C for 1 hr to allow for temperature and pH equilibration prior to loading into the XF96 apparatus. XF assays consisted of 7 pause (1 min), and measurement (3 min) cycles, allowing for determination of OCR/ECAR every 4 minutes.
Metabolite measurements
Extracellular glucose, lactate, glutamine and glutamate in culture medium were measured using a Flex Bioanalyzer (NOVA Bio-medical). Absolute glucose concentrations were measured using a colorimetric assay (Microdialysis, 000023). Lactate and pyruvate were measured using an NADH-linked enzymatic assay. Tissue samples were homogenized in 80% methanol:water, then dried down via O2 stream. Samples were re-suspended at 300mg/ml in water. Serum samples were added directly to the assay plate. The lactate assay solution (0.08M glutamate buffer, carbonate buffer, 10mg NAD, glutamate pyruvate transaminase and lactate dehydrogenase, 195μL) was mixed with 5μl of sample and absorbance was read at 340nm. The pyruvate assay solution (1.5M Tris-HCL with NADH, 100μl) was mixed with 100μl of serum sample. Absorbance measurements at 340nm occurred pre- and post- addiction of lactate dehydrogenase.
Cell Growth
Growth curves for all cell lines were determined by cell counting using trypan blue exclusion and a TC10 Automated Cell Counter (Bio-Rad).
Quantification and statistical analysis
Patient-matched tumor and non-cancerous lung samples, as well as intra-tumoral samples processed on the same day, were analyzed as described in the figure legends. All data were considered significant if p < 0.05. Statistics were calculated using PRISM software. The correlation analysis in Figure 1I was performed using the corrplot package in R Studio (Figure1I). Statistical details can be found in the figure legends of each experiment.
Supplementary Material
Supplementary Figure 1 (related to Figure 1). Isotope enrichment and stromal content from human samples.
(A) 13C enrichment in metabolites from benign lung tissue adjacent to tumors from the Low and High Lactate/3PG labeling groups. The fractional enrichment of glycolytic and TCA cycle metabolites is made relative to the enrichment of glucose in the tissue. Average values and S.E.M. are displayed.
(B) Estimated stromal content from each tumor fragment with Low and High Lactate/3PG labeling ratios. Data are average and S.E.M (n=12 per group).
Supplementary Figure 2 (related to Figure 3). Infusions of 13C-lactate in human subjects with NSCLC.
(A) The abundance of 13C-lactate and 13C-pyruvate in a commercial preparation of [2-13C]lactate was calculated using an enzymatic assay. Data are average and S.D. of triplicate measurements.
(B) Lactate (left) and pyruvate (right) concentrations before and after [13C]lactate infusions were calculated using an enzymatic assay. Each line represents the values from a single patient. In one subject, the post-infusion sample was hemolyzed, preventing the use of this colorimetric assay.
(C) Fractional enrichment of plasma glucose and lactate during infusion of [U-13C]glucose or [U-13C]lactate in patients. Data are average and S.E.M. (n=30 patients infused with [U-13C]glucose; n=4 patients infused with [U-13C]lactate).
Supplementary Figure 3 (related to Figure 4). Glucose, lactate, pyruvate and alanine metabolism in NSCLC xenografts.
(A) Mass isotopologues of glucose in mouse plasma after infusion with [U13C]glucose. Data are average and S.E.M (n=6).
(B) Plasma lactate concentrations were calculated using an enzymatic assay. Data are average and S.E.M. of samples from 3 infused mice.
(C) Mice bearing HCC15 flank xenografts were infused with [U-13C]pyruvate. Enrichment values were made relative to 3PG. Data are average and S.E.M of 3 mice.
(D) Mice bearing HCC827 flank xenografts were infused with [U-13C]alanine. Enrichment values were made relative to 3PG. Data are average and S.E.M. of 4 mice.
Supplementary Figure 4 (related to Figure 5). MCT1 and MCT4 expression and function.
(A) Expression levels of SLC16A1 (MCT1) and SLC16A3 (MCT4) from The Cancer Genome Atlas (TCGA) lung adenocarcinoma dataset. **** p< 0.0001 (Two-way ANOVA, Tukey post-hoc).
(B) Population doubling rate of HCC15 vector control cells and sub-lines with knockout of MCT1 or MCT4. Data are expressed as average and S.E.M. of three biological replicates in one experiment. The entire experiment was performed twice.
(C) Tissue lactate concentrations of Vector control, MCT1 KO and MCT4 KO xenografts infused with either [U-13C]glucose or [2-13C]lactate. Data are expressed as average and S.D. (n=2-3 tumors per condition).
(D) Mice bearing flank xenografts of Vector control, MCT1 KO, or MCT4 KO HCC15 cells were infused with [U-13C]glucose. Enrichment values are made relative to the tissue glucose enrichment. Data are average and S.E.M. Data are from the same mice shown in Figure 5F.
Supplementary Table 1 (Related to Figures 1-3). Clinical and pathological data from 35 NSCLC patients. Tumor diameter is reported in cm measured on T2-weighted MR imaging or pathology. Key for TNM staging: T1a, greatest dimension ≤2cm; T1b, greatest dimension >2cm but ≤3cm; T2a, greatest dimension >3cm but ≤5cm; N0, regional node metastases absent; N1, ipsilateral peribronchial and/or ipsilateral hilar lymph nodes metastases present; MX, distant metastasis unknown; M0, distant metastases absent. Key for tumor grade: G1, well differentiated; G2, moderately differentiated; G3, poorly differentiated; G4, undifferentiated. FDG-PET parameters: standardized uptake value-maximum, -peak and -mean (SUVmax, SUVpeak, and SUVmean); MTV, metabolic tumor volume; TLG, total lesion glycolysis. Histological subtypes include adenocarcinoma (ADC) with subtypes of acinar, papillary, non-mucinous, lepidic and solid; adenosquamous carcinoma; large cell neuroendocrine carcinoma (LCNEC); squamous cell carcinoma (SQCC); and carcinoid. Smoking history is reported in pack-years (i.e. average packs/day x years smoking). MIB-1 is the fraction of cells with nuclear Ki67 staining by IHC. %Stroma was determined using histopathological stains and examination by a pathologist. Microvessel density (MVD) is reported as number of vessels*10-6/μm2 after detecting vessels by staining for CD31. ND, no mutations detected. NA, tissue not available. NT, not tested. Patients coded K1 were infused with 13C-glucose; patients coded K2 were infused with 13C-lactate.
Supplementary Table 2 (Related to Figure 1-3). Human Isotopologue Data. Data include mass isotopologue distributions for metabolites detected during human infusions with 13C-glucose or 13C-lactate. Mass isotopologues from all infused patients are reported. Isotopologues reported previously (K1001-K1009) are shaded in grey; these metabolites used a different derivatization method, as described (Hensley et al., 2016). In some cases there was insufficient tissue to re-analyze using the updated method. Samples coded K1 were infused with 13C-glucose; patients coded K2 were infused with 13C-lactate.
Supplementary Table 3 (Related to Figure 2). INCA equations and calculations. Data include net and exchange flux estimates for all human normal lungs and tumors. Abbreviations: SSR, sum of squared ratios. DOF, degrees of freedom. NaN, Not a Number (i.e. the confidence interval algorithm could not be completed). LB, lower bound. UB, upper bound.
Supplementary Table 4 (Related to Figures 4-6). Mouse Isotopologue Data. Data include mass isotopologue distributions for metabolites analyzed during mouse infusions with 13C-labeled nutrients. Distributions for all metabolites reported in the Main and Supplemental figures are included in the Table.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MCT1 | Thermo Fisher | MA5-18288AB_2539662 |
| MCT4 | Santa Cruz | 50329 AB_2189333 |
| LDHA | Cell Signaling | C4B5AB_2066887 |
| LDHB | Novus | 3EP1565Y,AB_1109158 |
| B-Actin | Sigma-Aldrich | A3854AB_262011 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Blood samples | This study | N/A |
| Adjacent lung tissue | This study | N/A |
| Tumor tissue | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| [U-13C]glucose | Cambridge Isotopes | CLM-1396 |
| [U-13C]lactate | Cambridge Isotopes | CLM-1579 |
| [3-13C]lactate | Cambridge Isotopes | CLM-1578 |
| [U-13C]lactate | Sigma- Isotech | 660817 |
| [2-13C]lactate | Sigma- Isotech | 589209 |
| [2-2H]lactate | Sigma- Isotech | 693987 |
| [U-13C]pyruvate | Sigma- Aldritch | 490717 |
| [U-13C]alanine | Cambridge Isotopes | CLM-2184 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: H460 | Laboratory of Dr. J.D. Minna | N/A |
| Human: HCC827 | Laboratory of Dr. J.D. Minna | N/A |
| Human: HCC15 | Laboratory of Dr. J.D. Minna | N/A |
| Experimental Models: Organisms/Strains | ||
| Ncr nude mice | Taconic | NCRNU |
| Oligonucleotides | ||
| MCT1 CRISPRFWD: CACCGCGTATAGTCATGATTGTTGG | This paper | N/A |
| MCT1 CRISPR REV: AAACCCAACAATCATGACTATACGC | This paper | N/A |
| MCT4 CRISPRFWD: CACCGAAGAAGACACTGACGGCCTT | This paper | N/A |
| MCT4 CRISPR REV: AAACAAGGCCGTCAGTGTCTTCTTC | This paper | N/A |
| Recombinant DNA | ||
| LentiCRISPRv2 | Addgene | 49535 |
| Software and Algorithms | ||
| INCA | Vanderbilt | https://mfa.vueinnovations.com/ |
| The Cancer Genome Atlas | NIH | https://cancergenome.nih.gov/ |
| Other | ||
Highlights.
Lactate is metabolized by human lung tumors in vivo
Lactate use correlates with high FDG-PET signal and occurs in diverse oncogenotypes
MCT1 enables lactate consumption by some lung cancer xenografts.
Lactate's contribution to the TCA cycle exceeds that of glucose in vivo
Acknowledgments
We thank the DeBerardinis lab and Aron Jaffe for helpful discussions. BF is supported by the Canadian Institutes of Health Research (MFE 140911). JK is supported by an American Lung Association Fellowship (RT-306212). JDY is supported by NIH grant R01 DK106348. RJD is supported by grants from the NIH (R35CA220449), Welch Foundation (I-1733) and V Foundation (Translational Award). We acknowledge support from NIH grants P50 CA70907, P41 EB015908 and UL1 TR001105 and from the UT Southwestern Small Animal Imaging Resource, which is supported in part through an NCI Cancer Center Support Grant (1P30 CA142543-01). RJD is an advisor for Agios Pharmaceuticals.
Footnotes
Author Contributions. RJD, BF and KK designed the research. LC and MN performed bioinformatics. REL, QY and TW designed and implemented MRI acquisition techniques. QD and BF performed mouse MRI. DO analyzed tumors for mutations. GH managed the clinical protocol and consented patients. HL provided nursing support. JWW analyzed FDG-PET scans. KK performed the surgeries. DB, YB and JT analyzed histopathological specimens. BF and SD created MC1/MCT4-deficient cell lines. CY and BF performed enzymatic assays. JK and LZ provided metabolic expertise. BF, KL, CTH and RJD processed, analyzed and interpreted the tracer data. JDY designed the 13C modelling approach. BF and RJD wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berghmans T, Dusart M, Paesmans M, Hossein-Foucher C, Buvat I, Castaigne C, Scherpereel A, Mascaux C, Moreau M, Roelandts M, et al. Primary tumor standardized uptake value (SUVmax) measured on fluorodeoxyglucose positron emission tomography (FDG-PET) is of prognostic value for survival in non-small cell lung cancer (NSCLC): a systematic review and meta-analysis (MA) by the European Lung Cancer Working Party for the IASLC Lung Cancer Staging Project. J Thorac Oncol. 2008;3:6–12. doi: 10.1097/JTO.0b013e31815e6d6b. [DOI] [PubMed] [Google Scholar]
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell cycle (Georgetown, Tex) 2010;9:3506–3514. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24:657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Brooks GA. Lactate shuttles in nature. Biochem Soc Trans. 2002;30:258–264. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, DeBerardinis RJ, Feron O, Frezza C, Ghesquiere B, et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Jr, Mahieu NG, Huang X, Singh M, Crawford PA, Johnson SL, Gross RW, Schaefer J, Patti GJ. Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol. 2016;12:937–943. doi: 10.1038/nchembio.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cori CF, Cori GT. Glycogen formation in the liver from d- and l-lactic acid. J Biol Chem. 1929;81:389–403. [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O'Brien JP, Pierce KA, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23:517–528. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metabolism. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014;74:908–920. doi: 10.1158/0008-5472.CAN-13-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM. Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM) Mol Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, DeBerardinis RJ. Analyzing Tumor Metabolism In Vivo. Annual Review of Cancer Biology. 2016;1 null. [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JW, Djulbegovic B, Soares HP, Siegel BA, Lowe VJ, Lyman GH, Coleman RE, Wahl R, Paschold JC, Avril N, et al. Recommendations on the Use of 18F-FDG PET in Oncology. Journal of Nuclear Medicine. 2008;49:480–508. doi: 10.2967/jnumed.107.047787. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Goldstein JL, Pathak RK, Anderson RG, Brown MS. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: implications for the Cori cycle. Cell. 1994;76:865–873. doi: 10.1016/0092-8674(94)90361-1. [DOI] [PubMed] [Google Scholar]
- Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. 2004;558:5–30. doi: 10.1113/jphysiol.2003.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164:681–694. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero P, Dence CS, Coggan AR, Kisrieva-Ware Z, Eisenbeis P, Gropler RJ. L-3-11C-lactate as a PET tracer of myocardial lactate metabolism: a feasibility study. J Nucl Med. 2007;48:2046–2055. doi: 10.2967/jnumed.107.044503. [DOI] [PubMed] [Google Scholar]
- Hong CS, Graham NA, Gu W, Espindola Camacho C, Mah V, Maresh EL, Alavi M, Bagryanova L, Krotee PAL, Gardner BK, et al. MCT1 Modulates Cancer Cell Pyruvate Export and Growth of Tumors that Co-express MCT1 and MCT4. Cell reports. 2016;14:1590–1601. doi: 10.1016/j.celrep.2016.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, Varma G, Hung YP, Ullas S, Lauring J, et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell. 2016;164:433–446. doi: 10.1016/j.cell.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, Landon CD, Chi JT, Pizzo S, Schroeder T, et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One. 2013;8:e75154. doi: 10.1371/journal.pone.0075154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchiq I, Le Floch R, Roux D, Simon MP, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer research. 2015;75:171–180. doi: 10.1158/0008-5472.CAN-14-2260. [DOI] [PubMed] [Google Scholar]
- Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell metabolism. 2012;15:827–837. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science (New York, N Y) 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I, et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-(1)(3)C]pyruvate. Sci Transl Med. 2013;5:198ra108. doi: 10.1126/scitranslmed.3006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network T.C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Chang CY, Safi R, Liu X, Baldi R, Jasper JS, Anderson GR, Liu T, Rathmell JC, Dewhirst MW, et al. ERRalpha-Regulated Lactate Metabolism Contributes to Resistance to Targeted Therapies in Breast Cancer. Cell reports. 2016;15:323–335. doi: 10.1016/j.celrep.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarella S, de Bari L, Valenti D, Pizzuto R, Paventi G, Atlante A. Mitochondria and L-lactate metabolism. FEBS Lett. 2008;582:3569–3576. doi: 10.1016/j.febslet.2008.09.042. [DOI] [PubMed] [Google Scholar]
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell cycle (Georgetown, Tex) 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- Rizwan A, Serganova I, Khanin R, Karabeber H, Ni X, Thakur S, Zakian KL, Blasberg R, Koutcher JA. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin Cancer Res. 2013;19:5158–5169. doi: 10.1158/1078-0432.CCR-12-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M, 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 2015;125:687–698. doi: 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science (New York, NY) 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–483. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang YM, Park SI, Trenary IA, Egnatchik RA, Fessel JP, Kaufman JM, Carbone DP, Young JD. LKB1 deficiency enhances sensitivity to energetic stress induced by erlotinib treatment in non-small-cell lung cancer (NSCLC) cells. Oncogene. 2016;35:856–866. doi: 10.1038/onc.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014;19:795–809. doi: 10.1016/j.cmet.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 (related to Figure 1). Isotope enrichment and stromal content from human samples.
(A) 13C enrichment in metabolites from benign lung tissue adjacent to tumors from the Low and High Lactate/3PG labeling groups. The fractional enrichment of glycolytic and TCA cycle metabolites is made relative to the enrichment of glucose in the tissue. Average values and S.E.M. are displayed.
(B) Estimated stromal content from each tumor fragment with Low and High Lactate/3PG labeling ratios. Data are average and S.E.M (n=12 per group).
Supplementary Figure 2 (related to Figure 3). Infusions of 13C-lactate in human subjects with NSCLC.
(A) The abundance of 13C-lactate and 13C-pyruvate in a commercial preparation of [2-13C]lactate was calculated using an enzymatic assay. Data are average and S.D. of triplicate measurements.
(B) Lactate (left) and pyruvate (right) concentrations before and after [13C]lactate infusions were calculated using an enzymatic assay. Each line represents the values from a single patient. In one subject, the post-infusion sample was hemolyzed, preventing the use of this colorimetric assay.
(C) Fractional enrichment of plasma glucose and lactate during infusion of [U-13C]glucose or [U-13C]lactate in patients. Data are average and S.E.M. (n=30 patients infused with [U-13C]glucose; n=4 patients infused with [U-13C]lactate).
Supplementary Figure 3 (related to Figure 4). Glucose, lactate, pyruvate and alanine metabolism in NSCLC xenografts.
(A) Mass isotopologues of glucose in mouse plasma after infusion with [U13C]glucose. Data are average and S.E.M (n=6).
(B) Plasma lactate concentrations were calculated using an enzymatic assay. Data are average and S.E.M. of samples from 3 infused mice.
(C) Mice bearing HCC15 flank xenografts were infused with [U-13C]pyruvate. Enrichment values were made relative to 3PG. Data are average and S.E.M of 3 mice.
(D) Mice bearing HCC827 flank xenografts were infused with [U-13C]alanine. Enrichment values were made relative to 3PG. Data are average and S.E.M. of 4 mice.
Supplementary Figure 4 (related to Figure 5). MCT1 and MCT4 expression and function.
(A) Expression levels of SLC16A1 (MCT1) and SLC16A3 (MCT4) from The Cancer Genome Atlas (TCGA) lung adenocarcinoma dataset. **** p< 0.0001 (Two-way ANOVA, Tukey post-hoc).
(B) Population doubling rate of HCC15 vector control cells and sub-lines with knockout of MCT1 or MCT4. Data are expressed as average and S.E.M. of three biological replicates in one experiment. The entire experiment was performed twice.
(C) Tissue lactate concentrations of Vector control, MCT1 KO and MCT4 KO xenografts infused with either [U-13C]glucose or [2-13C]lactate. Data are expressed as average and S.D. (n=2-3 tumors per condition).
(D) Mice bearing flank xenografts of Vector control, MCT1 KO, or MCT4 KO HCC15 cells were infused with [U-13C]glucose. Enrichment values are made relative to the tissue glucose enrichment. Data are average and S.E.M. Data are from the same mice shown in Figure 5F.
Supplementary Table 1 (Related to Figures 1-3). Clinical and pathological data from 35 NSCLC patients. Tumor diameter is reported in cm measured on T2-weighted MR imaging or pathology. Key for TNM staging: T1a, greatest dimension ≤2cm; T1b, greatest dimension >2cm but ≤3cm; T2a, greatest dimension >3cm but ≤5cm; N0, regional node metastases absent; N1, ipsilateral peribronchial and/or ipsilateral hilar lymph nodes metastases present; MX, distant metastasis unknown; M0, distant metastases absent. Key for tumor grade: G1, well differentiated; G2, moderately differentiated; G3, poorly differentiated; G4, undifferentiated. FDG-PET parameters: standardized uptake value-maximum, -peak and -mean (SUVmax, SUVpeak, and SUVmean); MTV, metabolic tumor volume; TLG, total lesion glycolysis. Histological subtypes include adenocarcinoma (ADC) with subtypes of acinar, papillary, non-mucinous, lepidic and solid; adenosquamous carcinoma; large cell neuroendocrine carcinoma (LCNEC); squamous cell carcinoma (SQCC); and carcinoid. Smoking history is reported in pack-years (i.e. average packs/day x years smoking). MIB-1 is the fraction of cells with nuclear Ki67 staining by IHC. %Stroma was determined using histopathological stains and examination by a pathologist. Microvessel density (MVD) is reported as number of vessels*10-6/μm2 after detecting vessels by staining for CD31. ND, no mutations detected. NA, tissue not available. NT, not tested. Patients coded K1 were infused with 13C-glucose; patients coded K2 were infused with 13C-lactate.
Supplementary Table 2 (Related to Figure 1-3). Human Isotopologue Data. Data include mass isotopologue distributions for metabolites detected during human infusions with 13C-glucose or 13C-lactate. Mass isotopologues from all infused patients are reported. Isotopologues reported previously (K1001-K1009) are shaded in grey; these metabolites used a different derivatization method, as described (Hensley et al., 2016). In some cases there was insufficient tissue to re-analyze using the updated method. Samples coded K1 were infused with 13C-glucose; patients coded K2 were infused with 13C-lactate.
Supplementary Table 3 (Related to Figure 2). INCA equations and calculations. Data include net and exchange flux estimates for all human normal lungs and tumors. Abbreviations: SSR, sum of squared ratios. DOF, degrees of freedom. NaN, Not a Number (i.e. the confidence interval algorithm could not be completed). LB, lower bound. UB, upper bound.
Supplementary Table 4 (Related to Figures 4-6). Mouse Isotopologue Data. Data include mass isotopologue distributions for metabolites analyzed during mouse infusions with 13C-labeled nutrients. Distributions for all metabolites reported in the Main and Supplemental figures are included in the Table.