

Abstract
Activation of G protein‐coupled receptor 40/Free fatty acid receptor 1 (GPR40/FFAR1), which is highly expressed in pancreatic β cells, is considered an important pharmacologic target for the treatment of type 2 diabetes mellitus. The aim of this study was to determine the effect of MR1704, a novel GPR40/FFAR1 agonist, on glucose homeostasis in rats. MR1704 is a highly potent and selective, orally bioavailable agonist with similar in vitro potencies among humans, mice, and rats. Treatment of rat islets with MR1704 increased glucose‐dependent insulin secretion. Augmentation of glucose‐dependent insulin secretion was abolished by adding a GPR40/FFAR1 antagonist. In mouse, insulinoma MIN6 cells, palmitic acid induced the activity of caspase 3/7 after a 72‐h exposure, while pharmacologically active concentrations of MR1704 did not. In an oral glucose tolerance test in normal Sprague‐Dawley rats, orally administered MR1704 (1–10 mg·kg−1) reduced plasma glucose excursion and enhanced insulin secretion, but MR1704 did not induce hypoglycemia, even at 300 mg·kg−1, in fasted Sprague‐Dawley rats. In addition, orally administered MR1704 reduced plasma glucose excursion and enhanced insulin secretion in diabetic Goto‐Kakizaki rats. Oral administration of MR1704 once daily to Goto‐Kakizaki rats reduced their blood glucose levels during a 5‐week treatment period without reducing pancreatic insulin content; as a result, hemoglobin A1C levels significantly decreased. These results suggest that MR1704 improves glucose homeostasis through glucose‐dependent insulin secretion with a low risk of hypoglycemia and pancreatic toxicity. MR1704 shows promise as a new, glucose‐lowering drug to treat type 2 diabetes mellitus.
Keywords: FFAR1/GPR40, glucose‐stimulated insulin secretion, MR1704, type 2 diabetes mellitus
Abbreviations
- [Ca2+]i
intracellular calcium
- ALT
alanine aminotransferase
- AUC
area under the curve
- CHO
Chinese hamster ovary
- CLtot
total clearance
- FFA
free fatty acid
- GK
Goto‐Kakizaki
- GPR40/FFAR1
G protein‐coupled receptor 40/Free fatty acid receptor 1
- HbA1c
hemoglobin A1C
- OGTT
oral glucose tolerance test
- PPARγ
peroxisome proliferator‐activated receptor gamma
- SU
sulfonylurea
- T2DM
type 2 diabetes mellitus
- Vdss
steady‐state distribution volume
Introduction
Type 2 diabetes mellitus (T2DM) is a chronic disease characterized by hyperglycemia due to a relative deficiency of insulin secretion and/or insufficient insulin action in target organs such as the liver, muscle, and adipose tissue. T2DM is a worldwide epidemic disease involving 382 million estimated people in 2013. The number of people with T2DM is increasing in every country, resulting in an increase in the number of people with T2DM to an estimated 592 million by 2035 (Guariguata et al. 2014). Global expenditure due to diabetes mellitus and its complications represents a massive burden to healthcare budgets worldwide. In 2013, the International Diabetes Federation estimated that at least USD 548 billion was spent on diabetes treatment, 11% of total expenditure on adults.
Management of T2DM is complex, time consuming, and expensive. Lifestyle intervention, such as dietary changes, increased exercise, and education, are the first choices of T2DM treatment programs. Regarding first‐line medication, metformin, which improves hyperglycemia by suppressing hepatic gluconeogenesis, is the most cost‐effective agent, in particular for obese patients. If lifestyle intervention alone has not achieved hemoglobin A1C (HbA1c) goals, pharmaceutical treatment should be started immediately (Inzucchi et al. 2012). Since T2DM is a progressive disorder and is characterized by several different causes, metformin monotherapy may not be sufficient, and guidelines recommend combining glucose‐lowering agents with different drug classes (Inzucchi et al. 2015).
Sulfonylureas (SU) are insulin secretagogues, which bind to an ATP‐sensitive potassium channel on pancreatic β‐cells and stimulate insulin secretion; they are widely used second‐line medications because of their effectiveness in controlling glucose levels (Bryan et al. 2005). However, SUs continuously stimulate insulin secretion, irrespective of glucose levels, and they can cause hypoglycemia, which is a frequent undesirable effect of pharmacological treatment in T2DM. In addition, studies have demonstrated the existence of secondary failure of SU efficacy (Kahn et al. 2006), and it is hypothesized that SUs may promote the loss of β‐cell function. Second‐ and third‐line glucose‐lowering agents include dipeptidyl peptidase (DPP)‐4 inhibitors, thiazolidinediones (TZDs) glucagon‐like peptide 1 (GLP‐1) agonists, sodium–glucose cotransporter 2 (SGLT2) inhibitors, insulin, and alpha‐glucosidase inhibitors (Inzucchi et al. 2015). Although these glucose‐lowering agents have less risk of hypoglycemia compared with that of SUs, each has a unique safety and efficacy profile. There is still a need for novel glucose‐lowering agents to meet the unmet medical need to treat diabetes patients.
G protein‐coupled receptor 40/Free fatty acid receptor 1 (GPR40/FFAR1) is highly expressed in pancreatic β‐cells and can be activated by medium‐ to long‐chain free fatty acids (FFAs) (Briscoe et al. 2003; Itoh et al. 2003; Kotarsky et al. 2003). Activation of GPR40/FFAR1 by FFAs or synthetic agonists enhances insulin secretion, at least partly, through the amplification of intracellular calcium signaling in a glucose‐dependent manner, without hypoglycemia, which is a problem with SUs, glinides, and insulin. In addition, data on GPR40/FFAR1 agonists (Tan et al. 2008; Lin et al. 2011; Tsujihata et al. 2011; Ito et al. 2013; Tanaka et al. 2013; Sunil et al. 2014), transgenic mice overexpressing hGPR40/FFAR1 (Nagasumi et al. 2009), and GPR40/FFAR1 null mice (Latour et al. 2007; Kebede et al. 2008; Lan et al. 2008; Alquier et al. 2009; Matsuda‐Nagasumi et al. 2013) suggest that activation of GPR40/FFAR1 improves glucose homeostasis and does not cause pancreatic toxicity.
Takeda Pharmaceutical Company reported that fasiglifam (TAK‐875), a GPR40/FFAR1 selective agonist, was safe and well tolerated in healthy volunteers (Naik et al. 2012). Takeda also found a glucose‐lowering effect of fasiglifam in T2DM patients, with less risk of hypoglycemia compared with that of glimepiride, demonstrating proof‐of‐concept that fasiglifam is a novel class of glucose‐lowering drug (Burant et al. 2012; Kaku et al. 2013). Taken together, GPR40/FFAR1 is highlighted as a novel drug target for the treatment of T2DM patients.
In 2013, Takeda announced the voluntary termination of the fasiglifam development program due to concerns about liver safety. Fasiglifam's hepatotoxicity was revealed by increased liver enzyme levels (Kaku et al. 2015). Whether fasiglifam's hepatotoxicity is mechanistically derived via activation of liver GPR40 or not is unknown.
In this study, MR1704, a structurally different GPR40/FFAR1 agonist from fasiglifam, exhibited favorable pharmacokinetic profiles and pharmacological effects in various in vitro and in vivo models. Our results indicate that MR1704 is a novel glucose‐lowering GPR40 agonist with an improved toxicity profile.
Materials and Methods
Compounds
MR1704 ((R)‐5‐[4‐[3‐[2,6‐Dimethyl‐4‐[3‐(methylsulphonyl)propoxy]‐phenyl]benzyloxy]phenyl]‐isothiazol‐3‐ol 1‐oxide), GW1100 (1‐(4‐Ethoxycarbonylphenyl)‐2‐(4‐fluo robenzylthio)‐5‐(2‐ethoxy‐5‐pyrimidinylmethyl)‐4‐pyrimidinone;Benzoic acid, 4‐[5‐[(2‐ethoxy‐5‐pyrimidinyl)methyl]‐2‐[[(4‐fluorophenyl)methyl]thio]‐4‐oxo‐1(4H)‐pyrimidinyl]‐, ethyl ester), AR231453 (N‐[2‐Fluoro‐4‐(methylsulphonyl)phenyl]‐5‐nitro‐6‐[4‐[3‐(1‐methylethyl)‐1,2,4‐oxadiazol‐5‐yl]‐1‐piperidinyl]‐5‐nitro‐4‐pyrimidinamine) and fasiglifam were synthesized in the medicinal chemistry laboratory at Mochida Pharmaceutical Company Limited. These compounds were dissolved in dimethyl sulphoxide (DMSO) for in vitro experiments.
Animals
Male Sprague‐Dawley rats were purchased from Japan SLC and male Goto‐Kakizaki (GK) rats were purchased from CLEA Japan. Rats were fed ad libitum with chow (CE‐2, CLEA Japan). They were maintained in an air‐conditioned room with a temperature of 23°C and humidity of 55% on a regulated 12 h light/dark cycle (lights on from 7:30 a.m. to 7:30 p.m.) with free access to food and tap water. Blood samples were collected from the tail vein, jugular vein, or retro‐orbital plexus under isoflurane anesthesia. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Mochida Pharmaceutical Research Center. All surgery was performed under isoflurane anesthesia, and all efforts were made to minimize suffering.
Intracellular calcium assays
CHO‐DXB11 cells were kindly donated by Dr. L Chasin (Columbia University) and were grown in Ham's F‐12 medium containing 10% heat‐inactivated fatal bovine serum (FBS), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin (Thermo Fisher Scientific, Waltham, MA). The CHO‐DXB11 cells stably expressing human, mouse, or rat GPR40/FFAR1 (NP_005294.1, NP_918946.2, or NP_695216.1), and human GPR120/FFAR4 (NP_001182684) were generated by transfecting CHO‐DXB11 cells with each corresponding expression vector. These cells were cultured in 96‐well plates. Effects of the compounds were determined by measuring the changes in duplicate in intracellular Ca2+ with the FLIPR® Calcium Assay Kit (Molecular Devices, Sunnyvale, CA), using a fluorescence imaging plate reader (FDSS; Hamamatsu Photonics K.K., Shizuoka, Japan). In GPR120/FFAR4 FLIPR® assays, alpha‐linolenic acid (Wako, Osaka, Japan) was used as a control. The 50% effective concentration (EC50) was calculated, using a sigmoidal curve‐fitting program (Symyx Assay Explorer 3.1 SP2; Symyx Technologies Inc., Santa Clara, CA).
cAMP reporter assays
CHO‐K1 cells were purchased from DS Pharma Biomedical (Osaka, Japan) and were grown in Ham's F‐12 medium containing 10% heat‐inactivated FBS, 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin. The CHO‐K1 cells stably expressing human GPR119 (NP_848566) were co‐transfected with a Cre‐luciferase reporter vector (pGL4.29; Promega, Madison, WI) and a renilla luciferase control vector (pGL4.73; Promega).These transfectants were cultured in 96‐well plates, incubated with the drug for 4 h, and then lysed. The firefly and renilla luciferase activities in the lysates were measured in duplicate using a dual‐luciferase assay system (Promega) and a chemiluminescence detection plate reader (ARVO SX 1420 multilabel counter; Perkin‐Elmer). In these assays, AR231453 was used as a control. The EC50 was also calculated, using Symyx Assay Explorer 3.1 SP2.
Pharmacokinetic study in rats
MR1704 was dissolved in N, N‐dimethylacetamide (Wako)/polyethylene glycol 400 (Wako)/water (4/4/2, v/v/v) for intravenous administration, and suspended in 1% Tween® 80 (MP Biochemicals, Santa Ana, CA) containing 0.5% carboxymethyl cellulose (Nacalai, Kyoto, Japan) for oral administration. MR1704 was administered to male Sprague‐Dawley rats (7 weeks old) at a dose of 3 mg kg−1 in 1 mL intravenously or at doses of 0.3, 1, 3, and 10 mg kg−1 in 10 mL orally. The rats used for oral administration were fasted overnight before the experiment. Pharmacokinetic parameters were calculated, using the analysis program (WinNonlin noncompartmental analysis program, version 6.1; Pharsight, Cary, NC).
Glucose‐dependent insulin secretion in rat islets
Pancreatic islets of Langerhans were isolated from male Sprague‐Dawley rats (7 or 8 weeks old) by collagenase digestion and Ficoll–Conray gradient centrifugation. These islets were cultured overnight in RPMI 1640 medium containing 10% heat‐inactivated FBS. Each group of 10 islets was handpicked, transferred to a 24‐well plate, and incubated for 1 h in Krebs‐Ringer bicarbonate HEPES buffer (KRBH)/0.1% BSA with 2 mmol·L−1 glucose. The islets were treated with MR1704 or KCl (Wako) in KRBH/0.1% BSA with 2 mmol·L−1 or 16 mmol·L−1 glucose for 1 h. To determine the mode of action of MR1704, a selective GPR40/FFAR1 antagonist, GW1100, was added to the assay. The culture supernatants were collected, and the amount of insulin was assayed, using an ultrasensitive rat insulin ELISA kit (Morinaga, Yokohama, Japan).
Measurement of caspase 3/7 activity
FLIPR® assay and induction of caspase 3/7 activity were evaluated in CHO‐DXB11 cells stably expressing GPR40/FFAR1 and mouse insulinoma MIN6 cells in the presence of 10% FBS and 0.5% BSA. MIN6 cells were kindly donated by Dr. Miyazaki (Osaka University) and were grown in Dulbecco's Modified Eagle Medium (DMEM) containing 10% heat‐inactivated FBS, 100 U mL−1 penicillin, 100 mg mL−1 streptomycin and 0.0005% 2‐mercaptoethanol (Thermo Fisher Scientific). These cells were cultured in 96‐well plates with palmitic acid (Sigma‐Aldrich, St. Louis, MO) or MR1704 for 72 h. Caspase 3/7 activity was then measured, using an Apo‐ONE® Homogeneous Caspase‐3/7 Assay kit (Promega).
Oral glucose tolerance test and effects on fasting glucose levels in Sprague‐Dawley rats
Male Sprague‐Dawley rats (7 or 8 weeks old) were fasted overnight and divided into groups (n=10 per group) based on body weight. The rats were orally administered the vehicle control or MR1704 60 min before oral glucose challenge (1 g kg−1). Blood samples were collected before compound administration (pre), 5 min before glucose challenge, and 5, 15, 30, 60, and 120 min after glucose challenge. The plasma glucose and plasma insulin levels were measured using Glucose CII test WAKO (Wako) and an ultrasensitive rat insulin ELISA kit (Morinaga), respectively. To see the effects on fasting glucose levels, male Sprague‐Dawley rats (7 weeks old) were fasted overnight and divided into five groups (n=8 per group) based on their fasting blood glucose levels and body weight. The rats were orally administered the vehicle control, MR1704, or glibenclamide (Wako). Blood samples were collected before compound administration (pre) and 1, 2, 4, 8, and 12 h after dosing.
Acute and chronic treatment with MR1704 in GK rats
Male GK rats (9–10 weeks old) were fasted overnight and divided into groups (n = 9–10 per group) based on their fasting glucose levels and body weight. The rats were orally administered the vehicle control or MR1704 and then subjected to the oral glucose challenge (1 g kg−1) after 60 min. Blood samples were collected before compound administration (pre), 5 min before glucose challenge, and 5, 15, 30, 60, 120, and 180 min after glucose challenge. For evaluation of efficacy of chronic treatment with MR1704, male GK rats (8 weeks old) were divided into four groups based on their glucose levels and body weight (n = 8–10 per group). GK rats were orally administered either the vehicle control or MR1704 once daily from 5:00 p.m. to 7:00 p.m. Glucose levels were measured once weekly between 8:30 p.m. and 10:00 p.m. under a red light. Glycated hemoglobin (HbA1c) levels were measured after the 5‐week treatment period, using a DCA2000 analyzer (SIEMENS, Munich, Germany). Fat‐free pancreas was dissected from the rats after the treatment period. Insulin was extracted from the pancreas using acid‐ethanol (0.23 mol·L−1 HCl in ethanol).
Statistical analyses
Data were expressed as means ± SEM or means ± SD. Statistical comparisons between two groups were performed with Student's t‐test. For multiple comparisons, statistical analyses between groups were performed with Dunnett's multiple comparison tests, using SAS version 9.1 (SAS Institute Inc., Cary, NC).
Results
Characteristics of MR1704 in vitro
MR1704 increased intracellular [Ca2+] ([Ca2+]i) in CHO‐DXB11 cells that stably expressed human, mouse, or rat GPR40/FFAR1 (Fig. 1). The EC50 values for human, mouse, and rat GPR40/FFAR1 were 35 ± 6, 25 ± 4, and 27 ± 3 nmol·L−1, respectively. In contrast, MR1704 did not show any agonistic activities in PPAR gamma‐, GPR120/FFAR4‐ or GPR119‐expressing cells at 3 μmol·L−1 (Table 1).
Figure 1.
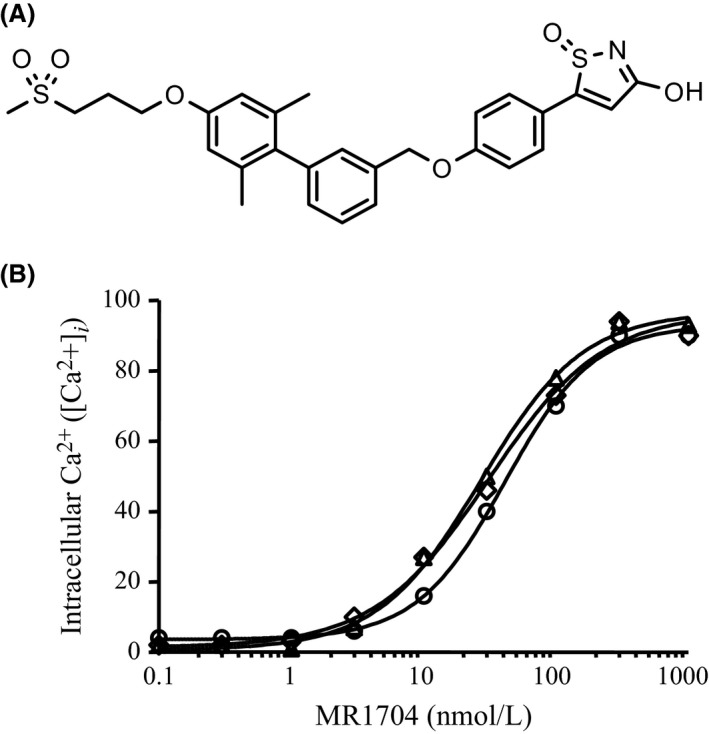
(A) Chemical structure of MR1704. (B) Agonistic activities of MR1704, intracellular Ca2+ ([Ca2+]i) was monitored in CHO‐DXB11 cells expressing human (○), mouse (▵) or rat (◇) GPR40. Increases in [Ca2+]i (%) are expressed as the percentage of the maximum activity (Emax) of fasiglifam. Each data point represents mean of duplicate determinations.
Table 1.
Summary of the effects of MR1704
| MR1704 | |
|---|---|
| hGPR40/FFAR1 EC50 | 35 ± 6 nmol·L−1 |
| mGPR40/FFAR1 EC50 | 25 ± 4 nmol·L−1 |
| rGPR40/FFAR1 EC50 | 27 ± 3 nmol·L−1 |
| hGPR119 activation (at 3 nmol·L−1) | 1% |
| hGPR120 activation (at 3 nmol·L−1) | 4% |
| PPAR gamma activation (at 3 nmol·L−1) | 1% |
EC50 values are means ± SD of 6 assays: GPR/FFAR, G protein‐coupled receptor/free fatty acid receptor; PPAR, peroxisome proliferator‐activated receptor.
Pharmacokinetic study of MR1704 in rats
Plasma concentrations of MR1704 in rats after intravenous or oral dosing and pharmacokinetic parameters are summarized in Figure 2 and Table 2. After intravenous administration, plasma levels of MR1704 declined biphasically with a half‐life (t 1/2) of 1.92 h for the final phase. The total clearance (CLtot) and steady‐state distribution volume (V dss) were low (0.337 L·h−1·kg−1) and moderate (0.584 L·kg−1), respectively. After oral administration, MR1704 was absorbed efficiently, and plasma concentrations reached maximum values at 0.83–5.33 h at all tested doses. The maximum plasma concentrations (C max) were 0.102, 0.264, 0.755, and 1.13 μg mL−1, and the bioavailability estimated from the area under the plasma concentration‐time curve (AUC) was 57, 49, 53, and 28% at doses of 0.3, 1, 3, and 10 mg·kg−1, respectively. In the oral dose ranges of 0.3–3 mg·kg−1, these parameters demonstrated linear kinetics.
Figure 2.
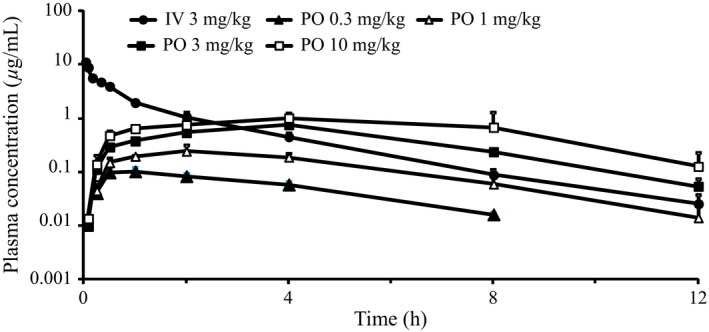
Plasma concentrations of MR1704 following a single administration to male Sprague‐Dawley rats. Each symbol represents the mean and SD of 3 animals.
Table 2.
Pharmacokinetic properties of MR1704
| Route | Dose (mg∙kg−1) | T max (h) | C max (μg·mL−1) | t 1/2 (h) | AUC0‐∞ (μg·h·mL−1) | CLtot (L·h−1·kg−1) | Vdss (L·kg−1) | F (%) |
|---|---|---|---|---|---|---|---|---|
| i.v. | 3 | — | — | 1.92 (0.24) | 8.96 (0.80) | 0.337 (0.031) | 0.584 (0.035) | |
| p.o. | 0.3 | 0.83 (0.29) | 0.102 (0.022) | 2.48 (0.12) | 0.511 (0.071) | — | — | 57 |
| 1 | 2.67 (1.15) | 0.264 (0.035) | 2.10 (0.44) | 1.45 (0.19) | — | — | 49 | |
| 3 | 4.00 (0.00) | 0.755 (0.124) | 2.13 (0.48) | 4.71 (0.14) | — | — | 53 | |
| 10 | 5.33 (2.31) | 1.13 (0.33) | 2.08 (0.20) | 8.43 (3.77) | — | — | 28 |
Data are expressed as the means (SD) of 3 animals.
i.v., intravenous; p.o., oral; F, bioavailability; CLtot, total clearance; Vdss, steady state distribution volume.
Augmentation of glucose‐dependent insulin secretion by MR1704 in rat islets
Insulin secretion in primary rat islets was significantly elevated in high (16 mmol·L−1) glucose conditions (Fig. 3A). KCl, a depolarizing stimulus, was used as a positive control. KCl significantly potentiated insulin secretion with high (16 mmol·L−1) and low (2 mmol·L−1) glucose. MR1704 (3 μmol·L−1) also markedly potentiated insulin secretion in high glucose, but not low glucose conditions. A selective GPR40/FFAR1 antagonist, GW1100 (1 μmol·L−1), did not have effects on insulin secretion in high glucose conditions (Fig. 3B). However, MR1704‐potentiated insulin secretion with high glucose was completely abolished by adding GW1100.
Figure 3.
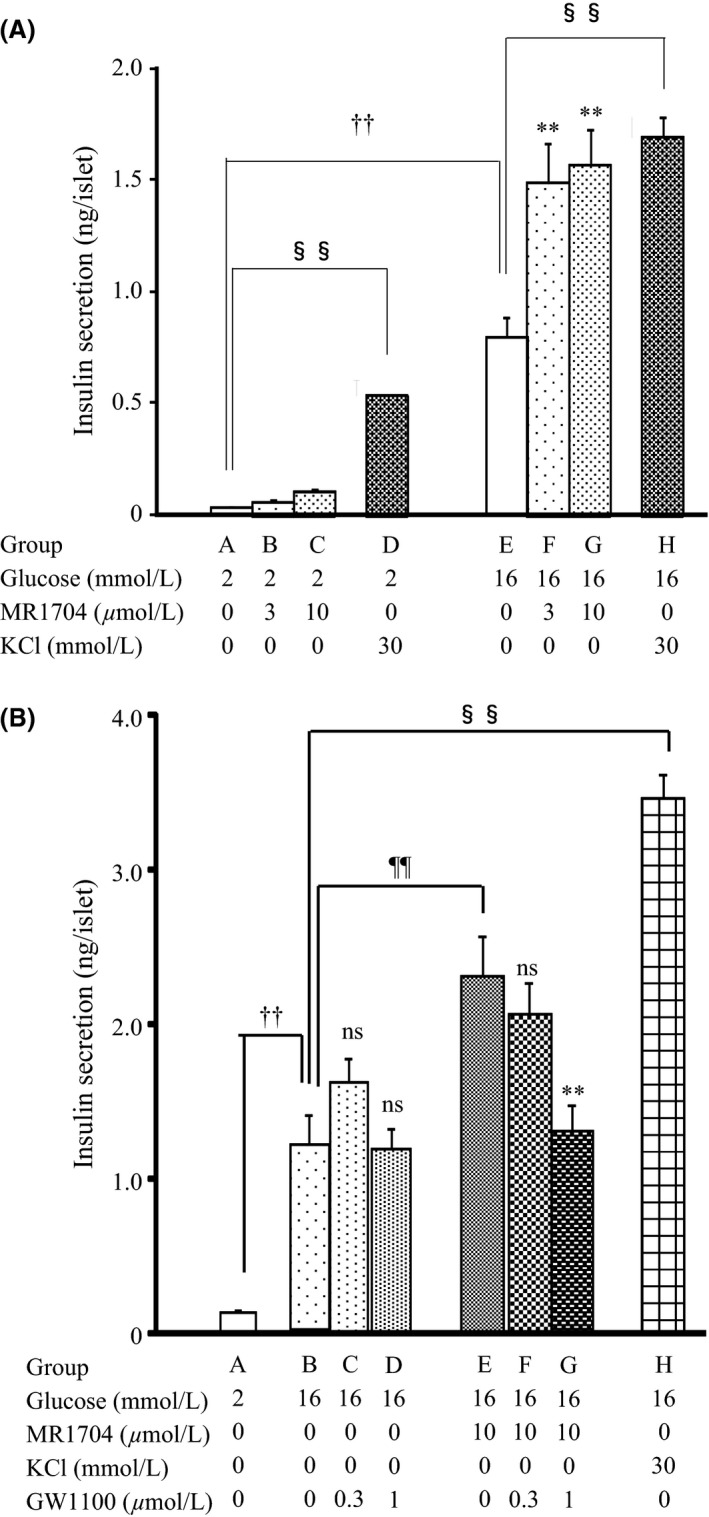
(A) The effects of MR1704 on glucose‐stimulated insulin secretion in rat primary islets. Each column represents the mean + SEM of eight wells. †† P < 0.01. Comparisons between group A and group E were analyzed via Student's t‐test. §§ P < 0.01. Comparisons between group A and group D and between group E and group H were analyzed via Student's t‐test. **P < 0.01. Comparisons between group A and group B or C and between group E and group F or G were analyzed via Dunnett's multiple comparison tests. (B) The effects of GW1100 on MR1704‐potentiated insulin secretion. Each column represents the mean + SEM of eight wells. †† P < 0.01, §§ P < 0.01, and ¶¶ P < 0.01. Comparisons between group A and group B, group B and group H, and group B and group E, respectively, were analyzed via Student's t‐test. **P < 0.01 and ns, not significant. Comparisons between group B and group C or D and between group E and group F or G were analyzed via Dunnett's multiple comparison tests.
Effects of MR1704 on apoptosis in MIN6 Cells
In the presence of 10% FBS and 0.5% BSA, palmitic acid and MR1704 showed agonist activity in CHO‐DXB11 cells stably expressing GPR40/FFAR1 (Fig. 4A and C). EC50 values of palmitic acid and MR1704 against GPR40/FFAR1 were 29 and 0.21 μmol·L−1, respectively. These results indicate that palmitic acid and MR1704 sufficiently stimulate GPR40/FFAR1 within these concentration range in the presence of 10% FBS and 0.5% BSA. In MIN6 cells, 300 μmol·L−1 palmitic acid with 0.5% BSA induced caspase 3/7 activity after a 72‐h exposure. In contrast, MR1704 with 0.5% BSA, even at 10 μmol·L−1, did not induce caspase 3/7 activity (Fig. 4B and D).
Figure 4.
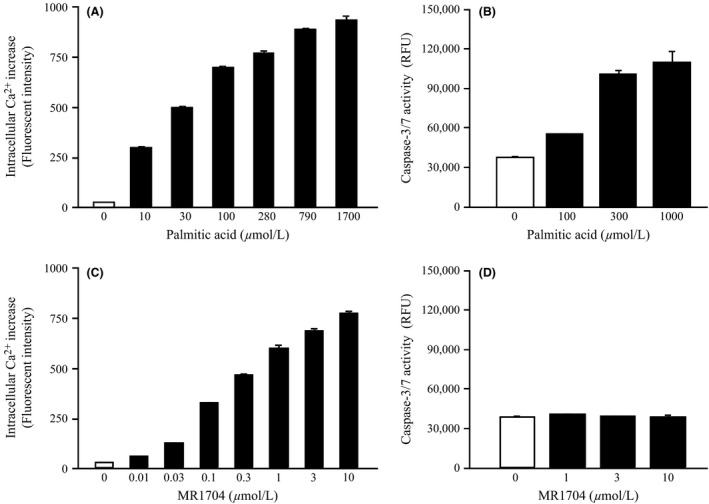
Effects of palmitic acid, and MR1704 on caspase‐3/7 activities in mouse insulinoma MIN6 cells. Intracellular Ca2+ concentration increases in CHO‐DXB11 cells stably expressing mouse GPR40 (A, C) and caspase‐3/7 activities in MIN6 cells (B, D) in the presence of 10% fatal bovine serum and 0.5% BSA are indicated. Data are expressed as the means + SD of triplicate determinations.
Glucose‐lowering effects of MR1704 during oral glucose tolerance test in normal rats
MR1704 reduced plasma glucose excursion compared with that of vehicle treatment in Sprague‐Dawley rats during oral glucose tolerance tests (OGTT). A significant reduction in the AUC of glucose levels was observed at 1 mg·kg−1. Fifteen minutes after glucose challenge, plasma insulin secretion was significantly enhanced at 1 mg·kg−1 (Fig. 5).
Figure 5.
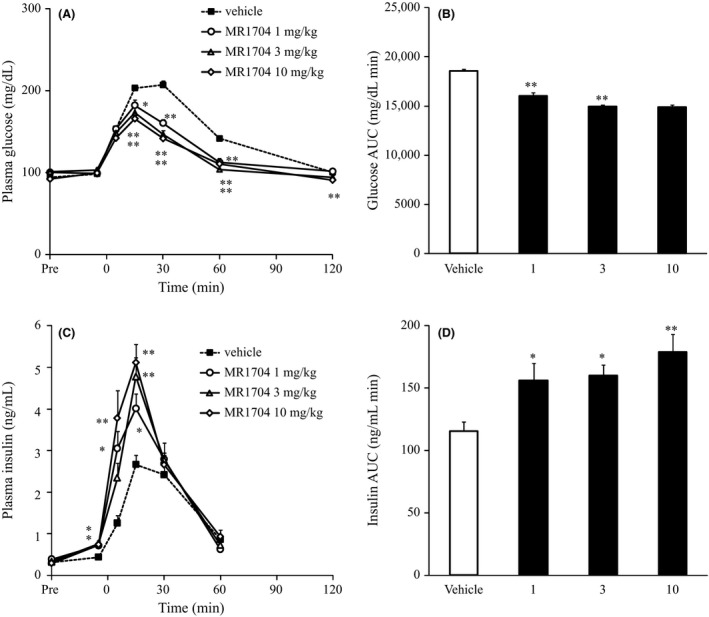
Effects of MR1704 on plasma glucose and plasma insulin levels during oral glucose tolerance tests in Sprague‐Dawley rats. The changes in plasma glucose levels (A), AUC of plasma glucose (−5–120 min) (B), changes in plasma insulin levels (C), and AUC of plasma insulin (−5–60 min) (D) are indicated. Data are expressed as the means + SEM of 10 animals. *P < 0.05 and **P < 0.01. Comparisons between the vehicle‐treated group and MR1704‐treated group were analyzed via Dunnett's multiple comparison tests. AUC, area under the curve.
Glucose‐lowering effects of MR1704 on fasting glucose levels in normal rats
MR1704 did not reduce the fasting plasma glucose levels, even at 300 mg·kg−1, in Sprague‐Dawley rats from 1 to 12 h after dosing. On the contrary, glibenclamide significantly reduced the fasting plasma glucose levels (Fig. 6).
Figure 6.
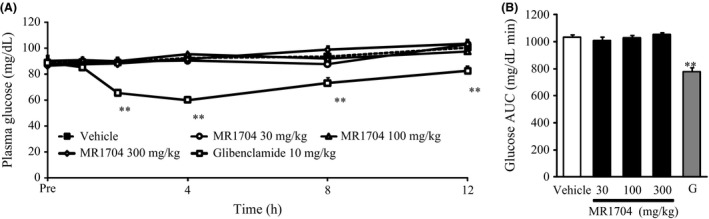
Effects of MR1704 and glibenclamide on the fasting plasma glucose levels in Sprague‐Dawley rats. The changes in plasma glucose levels (A) and AUC of plasma glucose (1–12 h) (B) are indicated. Data are expressed as the means + SEM of 8 animals. G: Glibenclamide 10 mg·kg−1. **P < 0.01. Comparisons between the vehicle‐treated group and compound‐treated groups were analyzed via Dunnett's multiple comparison tests.
Glucose‐lowering effects of MR1704 in diabetic Goto‐Kakizaki rats
In OGTT, MR1704 reduced plasma glucose excursion compared with that of vehicle treatment in GK rats. There was a significant reduction in the AUC of glucose levels at 1 mg·kg−1. Fifteen minutes after glucose challenge, 1 mg·kg−1 MR1704 had significantly enhanced plasma insulin secretion (Fig. 7).
Figure 7.
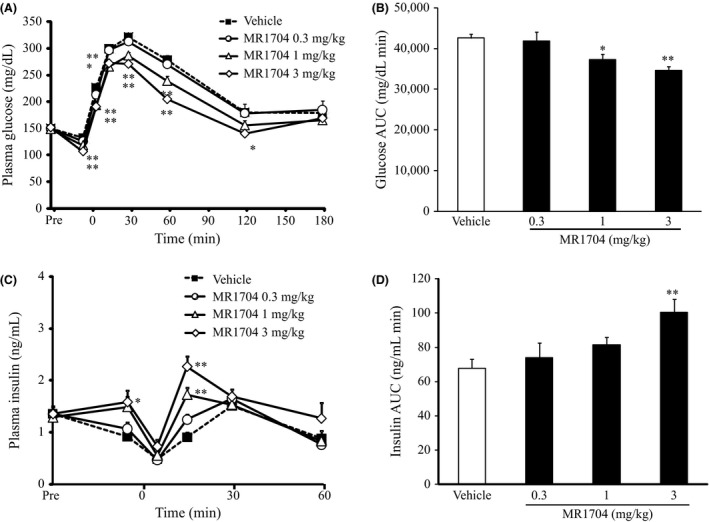
Effects of MR1704 on plasma glucose and plasma insulin levels during oral glucose tolerance tests in Goto‐Kakizaki (GK) rats. The changes in plasma glucose levels (A), AUC of plasma glucose (−5–180 min) (B), changes in plasma insulin levels (C), and AUC of plasma insulin (−5 to 60 min) (D) are indicated. Data are expressed as the means + SEM of 9–10 animals. *P < 0.05 and **P < 0.01. Comparisons between the vehicle‐treated group and MR1704‐treated group were analyzed via Dunnett's multiple comparison tests.
As acute treatment with MR1704 improved postprandial hyperglycemia, potential anti‐diabetic effects of MR1704 were examined chronically in GK rats. GK rats were treated with MR1704 and monitored for changes in their non‐fasting glucose levels and body weight during the 5‐week treatment period. Oral administration of at least 1 mg·kg−1 MR1704 once daily reduced nonfasting glucose levels without attenuation of drug efficacy during the study period (Fig. 8A). In addition, the MR1704‐treated groups and vehicle‐treated group had comparable body weight gain (Fig. 8B). Moreover, 3 mg·kg−1 MR1704 significantly reduced glycated hemoglobin (HbA1c) levels after the 5‐week treatment period (Table 3) without reducing pancreatic insulin content.
Figure 8.
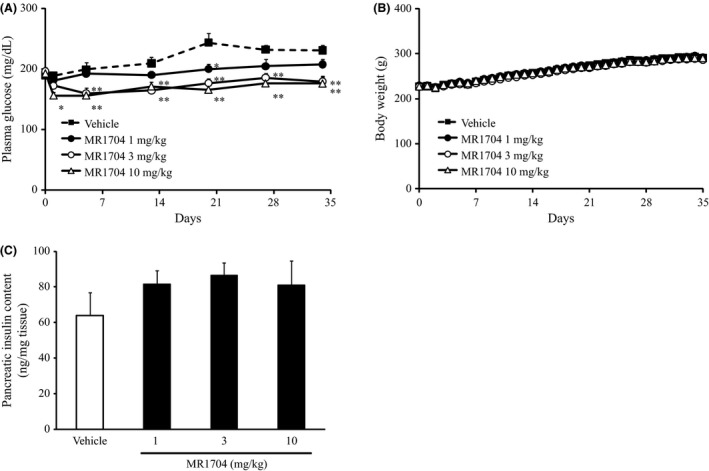
Effects of MR1704 on plasma glucose levels (A) and body weight (B). Data are expressed as the means + SEM of 8–10 animals. *P < 0.05 and **P < 0.01. Comparisons between the vehicle‐treated group and MR1704‐treated groups were analyzed via Dunnett's multiple comparison tests. Effects on the pancreatic insulin content after the treatment period (C). Data are expressed as the means + SEM of 5–10 pancreases. No significant difference was observed between the vehicle‐treated group and MR1704‐treated groups via Dunnett's multiple comparison test.
Table 3.
Long‐term effects of MR1704 on HbA1c levels
| Treatment | HbA1c levels (%) |
|---|---|
| Vehicle | 4.21 ± 0.06 |
| MR1704 1 mg·kg−1 | 4.09 ± 0.06 |
| MR1704 3 mg·kg−1 | 3.93 ± 0.08* |
| MR1704 10 mg·kg−1 | 3.91 ± 0.04** |
Data are expressed as the means ± SEM of 8–10 animals.
*P<0.05 and **P<0.01 versus the vehicle‐treated group by Dunnett's multiple comparison test. HbA1c, Hemoglobin A1C.
Discussion
GPR40/FFAR1 is a Gq‐coupled, family A G‐protein coupled receptor (GPCR) highly expressed on β‐cells of human and rat islets. Activation of GPR40/FFAR1 by FFAs or synthetic compounds has been reported to enhance insulin secretion in a glucose‐dependent manner, and is considered to have potential as a novel target for the treatment of T2DM (Mancini and Poitout 2015). In this study, we focused on effects of MR1704, a novel GPR40/FFAR1 agonist, on glucose homeostasis and pancreatic toxicity both in vitro and in vivo. Consequently, we found that MR1704 exhibited favorable pharmacokinetic profiles and pharmacological effects in various models without detectable risk of hypoglycemia and pancreatic toxicity.
The present results showed that MR1704 dose‐dependently increased [Ca2+]i in CHO cells stably expressing human, mouse, or rat GPR40/FFAR1, with similar EC50. No influence of MR1704 was observed in cells expressing GPR119 or GPR120/FFAR4. Despite activation of GPR40/FFAR1 by some TZD peroxisome proliferator‐activated receptor gamma (PPARγ) agonists (Kotarsky et al. 2003), MR1704 was found to have no effect on PPARγ in a nuclear receptor reporter activation assay. Collectively these data clarify that MR1704 is a potent, selective GPR40/FFAR1 agonist. Moreover, MR1704 also markedly potentiated insulin secretion in high glucose, but not low glucose conditions in primary rat islets. It is known that increased [Ca2+]i is related to potentiated insulin secretion (Prentki et al. 1997); this result may explain the glucose‐dependent insulinotropic effects of GPR40/FFAR1. It is noteworthy that MR1704‐potentiated insulin secretion in high glucose conditions was completely abolished by adding a selective GPR40/FFAR1 antagonist, GW1100.
Long‐chain FFAs acutely enhance insulin secretion, while chronic exposure to FFAs causes toxicity, such as death or dysfunction of β‐cells. Since long‐chain FFAs such as palmitic acid activate GPR40/FFAR1, it has been postulated that GPR40/FFAR1 activation mediates toxicity caused by FFAs. However, there have been discrepancies in the phenotypes of FFAR1/GPR40 genetically modified mice. One group reported that loss of GPR40/FFAR1 protects mice from obesity‐induced hyperinsulinemia, hepatic steatosis, hypertriglyceridemia, increased hepatic glucose output, hyperglycemia, and glucose intolerance (Steneberg et al. 2005). Conversely, overexpression of GPR40/FFAR1 in β‐cells of mice leads to impaired β‐cell function, hypoinsulinemia, and diabetes (Steneberg et al. 2005). In contrast, other studies of GPR40/FFAR1 null mice did not reproduce these metabolic abnormalities (Latour et al. 2007; Kebede et al. 2008; Lan et al. 2008; Alquier et al. 2009; Matsuda‐Nagasumi et al. 2013). In the present study, chronic exposure to MR1704 did not induce caspase 3/7 activity in MIN6 cells, in contrast to palmitic acid, a long‐chain FFA. It should be noted that both palmitic acid and MR1704 sufficiently stimulate GPR40/FFAR1 in these experiments. Since it has been reported that fasiglifam, a structurally different GPR40/FFAR1 agonist, also does not cause pancreatic toxicity (Tsujihata et al. 2011; Ito et al. 2013), our results provide additional support that activation of GPR40/FFAR1 is independent of the toxicity caused by FFAs.
As MR1704 exhibited favorable pharmacokinetics with rapid absorption, high Cmax, and high bioavailability in rats, the glucose‐lowering effects of MR1704 were evaluated during OGTT in healthy Sprague‐Dawley rats. A single oral administration of MR1704 enhanced insulin secretion and reduced plasma glucose levels. This is consistent with previous reports of other GPR40/FFAR1 agonists (Tan et al. 2008; Lin et al. 2011; Tsujihata et al. 2011; Ito et al. 2013; Tanaka et al. 2013; Sunil et al. 2014). In contrast, orally administered MR1704 did not exhibit glucose‐lowering effects in fasted Sprague‐Dawley rats, even at 300 mg·kg−1. A previous study demonstrated that fasiglifam, a GPR40 agonist, did not affect clinical parameters of glucose homeostasis in healthy human adults, and was not associated with increased risk of hypoglycemia in T2DM patients (Leifke et al. 2012). These results confirm that administration of GPR40/FFAR1 agonists is not associated with unexpected hypoglycemia and enhances insulin secretion only when blood glucose levels are elevated.
We also found that a single oral administration of MR1704 enhanced insulin secretion and reduced plasma glucose levels during OGTT in diabetic GK rats. Postprandial hyperglycemia caused by insufficient insulin secretion in response to blood glucose levels is observed in T2DM patients. The GK rat is one of the best‐characterized animal models of insufficient insulin secretion in response to blood glucose levels (Portha et al. 2009).
Chronic treatment with MR1704 for 5 weeks significantly decreased glycated hemoglobin (HbA1c) levels. Although one risk of long‐term exposure to a GPCR agonist is receptor desensitisation, the glucose‐lowering effects persisted during the study period in GK rats. In addition, improvement of glucose homeostasis was not associated with reducing the pancreatic insulin content. It is known that insulin secretagogues, such as SUs have risks of pancreatic β‐cell exhaustion. Although further direct evidence should be provided, MR1704 may have a low risk of pancreatic β‐cell exhaustion.
The clinical data on fasiglifam indicate that hepatotoxicity, such as alanine aminotransferase (ALT), increases with treatment (Kaku et al. 2015). Although the reason for hepatotoxicity due to fasiglifam is unknown, a GPR40/FFAR1 agonist as an oral therapeutic agent should overcome those adverse events. Although the precise mechanism of hepatotoxicity is unclear, it has not been reported that GPR40/FFAR1 is expressed in the human liver. Further clinical studies on GPR40/FFAR1 agonists with chemical and structural diversity would clarify the therapeutic potential of this target.
In conclusion, this study has shown that MR1704, a GPR40/FFAR1 that is a structurally different agonist from fasiglifam, exhibited favorable pharmacokinetic profiles and improved glucose homeostasis without risk of hypoglycemia, pancreatic toxicity, and pancreatic β‐cell exhaustion. The improvements in metabolic abnormalities observed in the present study are in agreement with other reported results of studies using GPR40/FFAR1 agonists (Tan et al. 2008; Lin et al. 2011; Tsujihata et al. 2011; Ito et al. 2013; Tanaka et al. 2013; Sunil et al. 2014). Collectively, our results indicate that MR1704 has potential to be a novel glucose‐lowering agent, overcoming unmet needs in T2DM patients.
Disclosures
None declared.
Acknowledgements
We thank the members of Mochida Pharmaceutical Company Limited, including Nao Yoshida and Kayoko Nagaya, for their technical assistant and general support. All authors are employees of Mochida Pharmaceutical Co. Ltd, and may own stock or hold stock options in Mochida Pharmaceutical Co. Ltd.
Tsuda N., Kawaji A., Sato T., Takagi M., Higashi C., Kato Y., Ogawa K., Naba H., Ohkouchi M., Nakamura M., Hosaka Y., Sakaki J.. A novel free fatty acid receptor 1 (GPR40/FFAR1) agonist, MR1704, enhances glucose‐dependent insulin secretion and improves glucose homeostasis in rats. Pharma Res Per, 5(4), 2017, e00340, https://doi.org/10.1002/prp2.340
References
- Alquier T, Peyot ML, Latour MG, Kebede M, Sorensen CM, Gesta S, et al. (2009). Deletion of GPR40 impairs glucose‐induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes 58: 2607–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. (2003). The orphan G protein‐coupled receptor GPR40 is activated by medium and long chain fatty acids. The Journal of Biological Chemistry 278: 11303–11311. [DOI] [PubMed] [Google Scholar]
- Bryan J, Crane A, Vila‐Carriles WH, Babenko AP, Aguilar‐Bryan L (2005). Insulin secretagogues, sulfonylurea receptors and K(ATP) channels. Curr Pharm Des 11: 2699–2716. [DOI] [PubMed] [Google Scholar]
- Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, et al. (2012). TAK‐875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet 379: 1403–1411. [DOI] [PubMed] [Google Scholar]
- Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE (2014). Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract 103: 137–149. [DOI] [PubMed] [Google Scholar]
- Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. (2012). Management of hyperglycemia in type 2 diabetes: a patient‐centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 35: 1364–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. (2015). Management of hyperglycemia in type 2 diabetes, 2015: a patient‐centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 38: 140–149. [DOI] [PubMed] [Google Scholar]
- Ito R, Tsujihata Y, Matsuda‐Nagasumi K, Mori I, Negoro N, Takeuchi K (2013). TAK‐875, a GPR40/FFAR1 agonist, in combination with metformin prevents progression of diabetes and β‐cell dysfunction in Zucker diabetic fatty rats. Br J Pharmacol 170: 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. (2003). Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 422: 173–176. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. (2006). Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 355: 2427–2443. [DOI] [PubMed] [Google Scholar]
- Kaku K, Araki T, Yoshinaka R (2013). Randomized, double‐blind, dose‐ranging study of TAK‐875, a novel GPR40 agonist, in Japanese patients with inadequately controlled type 2 diabetes. Diabetes Care 36: 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku K, Enya K, Nakaya R, Ohira T, Matsuno R (2015). Efficacy and safety of fasiglifam (TAK‐875), a G protein‐coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double‐blind, placebo‐controlled, phase III trial. Diabetes Obes Metab 17: 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebede M, Alquier T, Latour MG, Semache M, Tremblay C, Poitout V (2008). The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high‐fat feeding. Diabetes 257: 2432–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B (2003). A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Comm 301: 406–410. [DOI] [PubMed] [Google Scholar]
- Lan H, Hoos LM, Liu L, Tetzloff G, Hu W, Abbondanzo SJ, et al. (2008). Lack of FFAR1/GPR40 does not protect mice from high‐fat diet‐induced metabolic disease. Diabetes 57: 2999–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour MG, Alquier T, Oseid E, Tremblay C, Jetton TL, Luo J, et al. (2007). GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 56: 1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifke E, Naik H, Wu J, Viswanathan P, Demanno D, Kipnes M, et al. (2012). A multiple‐ascending‐dose study to evaluate safety, pharmacokinetics, and pharmacodynamics of a novel GPR40 agonist, TAK‐875, in subjects with type 2 diabetes. Clin Pharmacol Ther 92: 29–39. [DOI] [PubMed] [Google Scholar]
- Lin DC, Zhang J, Zhuang R, Li F, Nguyen K, Chen M, et al. (2011). AMG 837: a novel GPR40/FFA1 agonist that enhances insulin secretion and lowers glucose levels in rodents. PLoS ONE 6: e27270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini AD, Poitout V (2015). GPR40 agonists for the treatment of type 2 diabetes: life after ‘TAKing’ a hit. Diabetes Obes Metab 17: 622–629. [DOI] [PubMed] [Google Scholar]
- Matsuda‐Nagasumi K, Takami‐Esaki R, Iwachidow K, Yasuhara Y, Tanaka H, Ogi K, et al. (2013). Lack of GPR40/FFAR1 does not induce diabetes even under insulin resistance condition. Diabetes Obes Metab 15: 538–545. [DOI] [PubMed] [Google Scholar]
- Nagasumi K, Esaki R, Iwachidow K, Yasuhara Y, Ogi K, Tanaka H, et al. (2009). Overexpression of GPR40 in pancreatic beta‐cells augments glucose‐stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes 58: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik H, Vakilynejad M, Wu J, Viswanathan P, Dote N, Higuchi T, et al. (2012). Safety, tolerability, pharmacokinetics, and pharmacodynamic properties of the GPR40 agonist TAK‐875: results from a double‐blind, placebo‐controlled single oral dose rising study in healthy volunteers. J Clin Pharmacol 52: 1007–1016. [DOI] [PubMed] [Google Scholar]
- Portha B, Lacraz G, Kergoat M, Homo‐Delarche F, Giroix MH, Bailbé D, et al. (2009). The GK rat beta‐cell: a prototype for the diseased human beta‐cell in type 2 diabetes? Mol Cell Endocrinol 297: 73–85. [DOI] [PubMed] [Google Scholar]
- Prentki M, Tornheim K, Corkey BE (1997). Signal transduction mechanisms in nutrient‐induced insulin secretion. Diabetologia 40: S32–S41. [DOI] [PubMed] [Google Scholar]
- Steneberg P, Rubins N, Bartoov‐Shifman R, Walker MD, Edlund H (2005). The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab 1: 245–258. [DOI] [PubMed] [Google Scholar]
- Sunil V, Verma MK, Oommen AM, Sadasivuni M, Singh J, Vijayraghav DN, et al. (2014). CNX‐011‐67, a novel GPR40 agonist, enhances glucose responsiveness, insulin secretion and islet insulin content in n‐STZ rats and in islets from type 2 diabetic patients. BMC Pharmacology & Toxicology. https://doi.org/10.1186/2050-6511-15-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CP, Feng Y, Zhou YP, Eiermann GJ, Petrov A, Zhou C, et al. (2008). Selective small‐molecule agonists of G protein‐coupled receptor 40 promote glucose‐dependent insulin secretion and reduce blood glucose in mice. Diabetes 57: 2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Yoshida S, Oshima H, Minoura H, Negoro K, Yamazaki T, et al. (2013). Chronic treatment with novel GPR40 agonists improve whole‐body glucose metabolism based on the glucose‐dependent insulin secretion. The Journal of Pharmacology and Experimental Therapeutics 346: 443–452. [DOI] [PubMed] [Google Scholar]
- Tsujihata Y, Ito R, Suzuki M, Harada A, Negoro N, Yasuma T, et al. (2011). TAK‐875, an orally available G protein‐coupled receptor 40/free fatty acid receptor 1 agonist, enhances glucose‐dependent insulin secretion and improves both postprandial and fasting hyperglycemia in type 2 diabetic rats. The Journal of Pharmacology and Experimental Therapeutics 339: 228–237. [DOI] [PubMed] [Google Scholar]