

Abstract
Small-molecule drug discovery has traditionally focused on occupancy of a binding site that directly affects protein function, which typically precludes targeting proteins that lack such amenable sites. Furthermore, high systemic drug exposures may be needed to maintain sufficient target inhibition in vivo, increasing the risk of undesirable off-target effects. Induced protein degradation is an alternative approach that is ‘event-driven’: upon drug binding, the target protein is tagged for elimination. Emerging technologies based on proteolysis-targeting chimeras (PROTACs) that exploit cellular quality control machinery to selectively degrade target proteins are attracting considerable attention in the pharmaceutical industry owing to the advantages they could offer over traditional small-molecule strategies. These advantages include the potential to reduce systemic drug exposure, the ability to counteract increased target protein expression that often accompanies inhibition of protein function and the potential ability to target proteins that are not currently therapeutically tractable, such as transcription factors, scaffolding and regulatory proteins.
1) Introduction
Although the genomic revolution has allowed a plethora of novel protein targets to be linked to disease states1–4, standard small-molecule drug discovery strategies are unable to exploit all of these opportunities because many of these potential targets do not have suitable binding pockets that directly modulate protein function5. Due to their intracellular location, such proteins are often not tractable targets for antibody therapeutics either. The challenge that this presents for drug discovery is highlighted by the fact that there are very few marketed drugs that target scaffolding proteins, transcription factors, and other non-enzymatic proteins inside the cell6,7. Furthermore, small-molecule therapeutic strategies that focus on occupation of a binding site on a protein may require high systemic drug exposures to achieve sufficient site-occupancy in vivo8, potentially increasing the risk of off-target adverse effects.
One method for modulating intracellular protein concentrations is through nucleic acid-based agents such as antisense oligonucleotides (ASOs) or agents that exploit RNA interference (RNAi), such as small interfering RNAs (siRNAs). More recently, the advent of CRISPR/Cas9 technology9,10 offers the potential of modifying the genome itself to achieve gene knockout; however, the clinical potential of this technology is yet to be explored (see recent reviews11–13). While nucleic acid-based tools have proven useful in research, their development as drug candidates has faced many challenges: unmodified nucleotides are highly unstable in serum14, whereas modified nucleotides tend to accumulate in the kidney15,16 and can be immunogenic17,18. Nucleic acid-based agents encapsulated by nanoparticles to improve their delivery properties are still captured in the liver19–22. Additionally, RNAi can engage off-target mRNA, leading to undesired effects23–25. Finally, efficacy is ultimately dependent on target protein half-life; thus, long-lived proteins are less affected by these approaches. For a more detailed analysis, the reader is referred to these excellent reviews26–28. Although significant progress has been made, as indicated by the many ongoing clinical trials, only two ASO therapeutics (fomirisen29 and mipomersen30) that reduce the production of a particular protein have been approved in the United States31.
Small-molecule induced protein degradation is emerging as a strategy that also has the potential to target a broader range of proteins than standard small-molecule strategies that focus on binding site occupancy, yet maintain the pharmaceutical advantages of that such compounds typically have compared with nucleic acid-based agents. This shift from inhibition of the target protein to its removal permits the targeting of proteins that have generally been considered “undruggable”32,33. Furthermore, removing “druggable” targets from the system can be synergistic with current inhibitor-based regimens. For example, inhibition of certain cellular pathways promotes feedback-mediated increased expression of the target protein, leading to pharmacological insufficiency34,35. Induced protein degradation not only reduces the number of active proteins needed to be inhibited but also counteracts compensatory protein overexpression often observed upon loss of protein function. Additionally, many “druggable” targets are recognized to have “undruggable” scaffolding roles that contribute to resistance mechanisms36–42.
Induced protein degradation is not a recent concept. Inhibitors of the molecular chaperone Heat Shock Protein 90 (HSP90), which helps other proteins fold properly, first entered clinical trials in 1999. The initial excitement generated by HSP90 inhibitors was founded on the discovery that cancer cells upregulate HSP90 to drive proliferation and survival pathways43 and are more sensitive to HSP90 inhibition due to an overactive conformation of HSP90 found in cancer cells44. All reported HSP90 inhibitors target the ATP-binding domain45–47, affecting HSP90 chaperone cycling and leading to degradation of HSP90 client proteins48,49. There are more than 30 ongoing clinical trials associated with HSP90 inhibitors, yet none has gained FDA-approval (Box 1). HSP90 inhibitors have been hindered by poor in vivo pharmacological properties and severe hepatotoxicity attributed to their benzoquinone moiety50–52. Furthermore, due to the diversity of HSP90 client proteins, which includes more than half the human kinome53–55, it has been difficult to determine the exact cellular context in which an oncological vulnerability can be exploited by a broad inducer of protein degradation, although attempts have been made to bias the degradation56,57. Several excellent reviews offer a more thorough survey of the HSP90 inhibitor literature58–60.
Box 1. HSP90 Inhibitors as Broad Inducers of Protein Degradation.
Geldanamycin184,185, the first HSP90 inhibitor identified, exhibits low nanomolar activity against the NIH panel of 60 cancer cells lines186. However, as a clinical agent, geldanamycin has poor solubility, is readily metabolized and is hepatotoxic186. Second-generation compounds tanespimycin (17-AAG, KOS-953, CNF-101; Kosan Biosciences/Bristol-Myers Squibb) and alvespimycin (17-DMAG, KOS1022; Kosan Biosciences/Bristol-Myer Squibb) have improved solubility but remain hepatotoxic50–52. Tanespimycin entered clinical trials in 1999; surprisingly, despite broad protein degradation, tanespimycin demonstrated good tolerability187. More recently in 2008, tanespimycin reported promising results from Phase II trials for breast carcinoma (NCT00773344)188. However, Phase III trials were not pursued by Bristol-Myer Squibb, likely due to production and patent issues58. A third natural HSP90 inhibitor, retaspimycin (IPI-504; Infinity Pharmaceuticals) entered Phase III trials for gastrointestinal stromal tumor (GIST) in 2008 (NCT00688766), which were terminated due to drug-related deaths189.
To address the solubility and hepatotoxicity issues of natural inhibitors, synthetic inhibitors were pursued. CNF2024/BIIB021 (Biogen Idec/Conforma Therapeutics)190 was the first purine-based inhibitor to enter the clinic (NCT00345189) and was found to be relatively effective against GIST in Phase II trials (NCT00618319)191; however, Biogen halted its development after discontinuing its oncology pipeline192. One of the most potent HSP90 inhibitors, NVP-AUY922 (Vernalis/Norvatis)193 entered multiple Phase II trials for non-small cell lung cancer (NSCLC), pancreatic cancer, GIST and breast cancer; however, Novartis recently halted all development of the compound. Ganetespib (STA-9090; Synta Pharmaceuticals)194 also demonstrated some promise in a Phase II for NSCLC195; however, the following Phase III trial (NCT01798485) was recently terminated due to futility196. Identified through fragment-based drug discovery, onalespib (AT13387; Astex Pharmaceuticals)197 has entered Phase I/II trials for NSCLC and breast cancer (NCT02535338) after achieving limited efficacy for GIST198. Although the development of synthetic inhibitors appears to have solved many of the pharmacological limitations that plagued the natural inhibitors, a clear clinical indication for HSP90 inhibitors has yet to be identified given the diversity of HSP90 client proteins.
In contrast to the general client protein knockdown achieved with HSP90 inhibitors, selective estrogen receptor degraders (SERDs) and selective androgen receptor degraders (SARDs) exhibit high specificity in target protein degradation. Interestingly, these compounds were originally developed as modulators of protein function but were subsequently serendipitously discovered to be protein degraders61. Though the efficacy of these compounds has been impressive, this strategy is not generalizable to other targets: the compounds can only induce the degradation of estrogen and androgen receptors.
More recently, we and others have developed platform technologies that combine the modularity of nucleic acid-based strategies with the in vivo pharmacology of small-molecule therapeutics. Based on the concept of ‘event-driven’ pharmacology, proteolysis-targeting chimera (PROTAC) and hydrophobic tagging (HyT) technologies utilize bifunctional molecules, whereby one end binds to the protein of interest while the other hijacks cellular quality control mechanisms to induce the degradation of the target protein (Figure 1). Only a transient binding event is required for activity and thus, in contrast to the stoichiometric site-occupancy of the ‘occupancy-driven’ model, these ‘event-driven’ molecules can cycle through multiple rounds of activity, removing super-stoichiometric quantities of proteins. Additionally, compared to the dissociation kinetics of an inhibitor from an active site, restoration of protein function requires re-synthesis of the protein, providing a kinetic advantage. Furthermore, the protein ligand does not need to occupy a binding site that affects protein function (such as an enzyme active site) in order to function as a degrader — binding at any “nook” or “cranny” on a target could potentially induce degradation. This advantage could be exploited by utilizing thermal shift-, surface plasmon resonance (SPR)- or nuclear magnetic resonance (NMR)-based screens that focus on the identification of ligands that simply bind to a target, rather than screens that focus on identifying ligands that affect the function of the protein62–66. Moreover, by turning protein ligands into degraders, it might be possible to resurrect high-throughput screening (HTS) hits that bind to the desired targets but were set aside because they could not adequately block protein function67–69. Finally, several studies have determined that the degradation selectivity of PROTACs not only retains, but can also exceed, the binding selectivity of the protein ligand, such that PROTAC binding to off-target proteins does not necessarily lead to the degradation of the off-target protein; thus, PROTACs offer an added layer of specificity that can be modulated by which E3 ligase is recruited to the targets70–72.
Figure 1. Mechanism of induced protein degradation technologies.
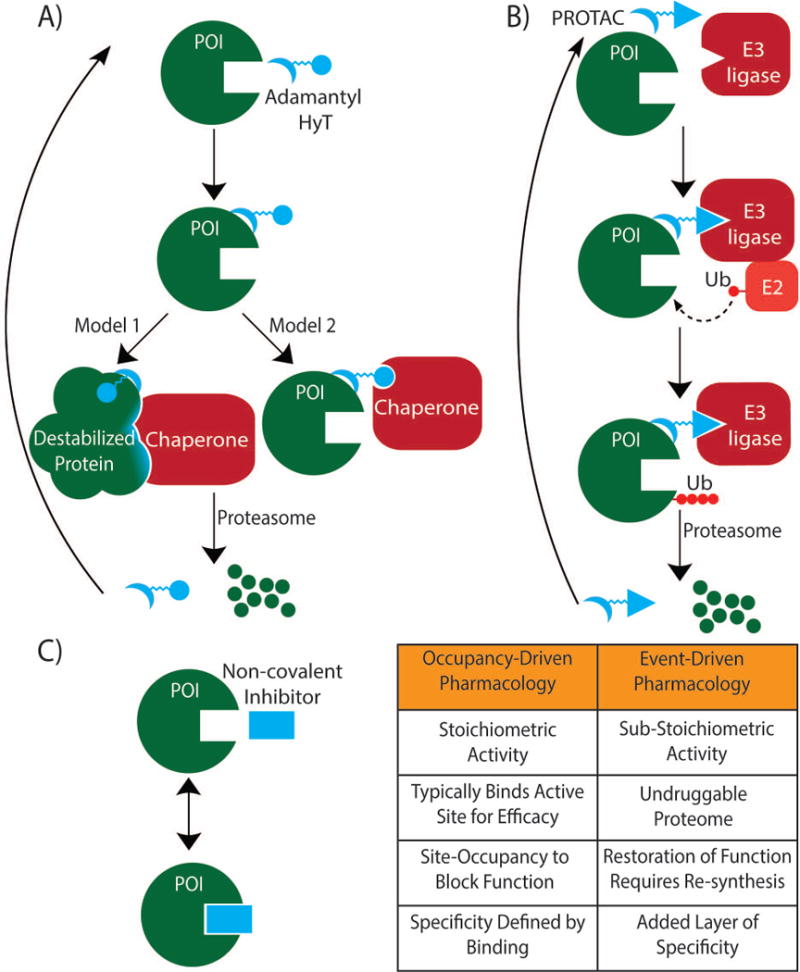
A) Hydrophobic tagging (HyT) using bifunctional adamantane-based molecules. One proposal for the mode of action is that the hydrophobic tag destabilizes the protein of interest (POI), thereby recruiting endogenous chaperones to the unfolded protein. The protein is then shuttled for degradation to the proteasome. An alternative proposal is that chaperones recognize the hydrophobic tag directly and mediate the proteasomal degradation of the tagged protein. B) PROTAC technology works through active recruitment of an E3 ligase in order to tag proteins for disposal. Bifunctional PROTAC molecules bind to the POI with one end while the other end binds an E3 ligase to form a ternary complex. The recruited E3 ligase then mediates the transfer of ubiquitin from an E2 enzyme to the POI. The ternary complex dissociates, the ubiquitinated POI is removed by the proteasome and the PROTAC can bind another POI (assuming the interaction with the POI is non-covalent). C) The traditional ‘occupancy-driven pharmacology’ model is predicated upon stoichiometric drug binding to the POI in order to modulate protein function. With non-covalent inhibitors, the drug is capable of dissociating from the POI, leading to restoration of protein function. Furthermore, inhibitors based on ‘occupancy-driven’ pharmacology typically bind to an active site on the POI (although allosteric inhibitors and inhibitors targeting protein–protein interactions are an exception). By contrast, induced protein degradation technologies are ‘event-driven’, and can potentially exploit binding anywhere on the POI in order to achieve degradation.
Substantial progress has recently been made, in particular with the development of PROTAC compounds demonstrating that PROTACs are not just useful chemical tools, but also viable therapeutic candidates. This progress also supports the idea that induced protein degradation is a novel drug discovery paradigm with the potential to target a broad range of proteins currently considered therapeutically intractable. In this review, we summarize the main approaches that have been investigated so far to induce degradation of targeted proteins, highlighting the progress that has been made and discussing the opportunities and challenges in establishing drug discovery platforms based on these approaches.
2) Selective hormone receptor degraders
The estrogen receptor (ER), a nuclear hormone receptor, is one of the few transcription factors amenable to modulation with small-molecule drugs. Upon binding to estrogens (17β-estradiol, estrone, and estriol), ER binds DNA to exert a cell-specific transcriptional response that is dependent on co-regulatory proteins present73; thus, estrogens have a diverse effect on the cardiovascular, musculoskeletal, immune and reproductive systems in both males and females74. As ER signaling is crucial for breast cancer progression, ER-positive breast carcinomas are responsive to hormonal therapy such as the selective estrogen receptor modulator (SERM) tamoxifen75. However, depending on the cellular context, SERMs can act as either an antagonist or agonist: in breast tissue, tamoxifen was found to act as an antagonist, whereas in the uterus, it was characterized as an agonist and led to an increased risk of endometrial cancers76. Furthermore, long-term estrogen deprivation by SERMs can lead to compensatory feedback signaling, resulting in pharmacological insufficiency77.
Addition of an alkylsulphinyl moiety to the endogenous ER ligand 17β-estradiol led to the development of fulvestrant (ICI82780, Faslodex; AstraZeneca)78,79 that destabilizes the ER (Table 1)61. Fulvestrant binding to ERα induces a structural change that leads to increased surface hydrophobicity and subsequent degradation80,81. Fulvestrant-induced degradation affects the ligand-independent functions of ER82 that are not readily addressed by tamoxifen83,84 and likely has less risk of endometrial cancer than tamoxifen due to its pure anti-estrogen antagonism85. Thus, fulvestrant is the only FDA-approved SERD for ER-positive metastatic breast carcinomas that have progressed after tamoxifen therapy86. However, due to fulvestrant’s lack of oral bioavailability and poor systemic exposure87,88, newer generation SERDs are in development89–92.
Table 1.
Proteins Successfully Targeted for Degradation
| Target | Mechanism | Year |
|---|---|---|
| Akt | VHL (polypeptidic)129 | 2016 |
| Androgen Receptor | β-TrCP (polypeptidic)122 | 2003 |
| VHL (polypeptidic)125 | 2004 | |
| MDM2 (nutlin-3)134 | 2008 | |
| cIAP (small-molecule)145 | 2011 | |
| HyT (adamantane)116 | 2015 | |
| Aryl Hydrocarbon Receptor | VHL (polypeptidic)214 | 2007 |
| BCR-ABL | VHL (small-molecule)71 | 2016 |
| CRBN (small-molecule)71 | 2016 | |
| BET (BRD2/3/4) | VHL (small-molecule)162,163 | 2015 |
| CRBN (small-molecule)72,160 | 2015 | |
| CRABP-I and -II | cIAP (small-molecule)138 | 2010 |
| DHFR | HyT (Arg-Boc3)107 | 2012 |
| ERRα | VHL (small-molecule)70 | 2015 |
| ErbB3 | HyT (adamantane)114 | 2014 |
| Estrogen Receptor | Fulvestrant78 | 1991 |
| β-TrCP (polypeptidic)122 | 2003 | |
| VHL (polypeptidic)127 | 2008 | |
| cIAP (small-molecule)145 | 2011 | |
| FKBP12 | CRBN (small-molecule)72 | 2015 |
| FRS2α | VHL (polypeptidic)131 | 2013 |
| GST | HyT (Arg-Boc3)107 | 2012 |
| HaloTag | HyT (adamantane)110 | 2011 |
| VHL (small-molecule)177 | 2015 | |
| cIAP (small-molecule)215 | 2015 | |
| MetAP-2 | β-TrCP (polypeptidic)118 | 2001 |
| PI3K | VHL (polypeptidic)131 | 2013 |
| RAR | cIAP (small-molecule) 138 | 2010 |
| RIPK2 | VHL (small-molecule)70 | 2015 |
| TACC3 | cIAP (small-molecule)146 | 2014 |
| Tau | VHL (polypeptidic)130 | 2016 |
BCR-ABL, breakpoint cluster region-Abelson tyrosine kinase fusion protein; BET, bromodomain and extra-terminal; BRD4, bromodomain containing protein 4; CRABP, Cellular retinoic acid-binding protein; DHFR, dihydrofolate reductase; ErbB3, erythroblastosis oncogene B3; ERRα, estrogen-related receptor α; FKBP12, FK506 binding protein 12; FRS2α, fibroblast growth factor receptor substrate 2α; GST, glutathione-S-transferase; MetAP-2, methionine aminopeptidase-2; PI3K, phosphatidylinositol-3-kinase; RAR, retinoic acid receptor; RIPK2, receptor-interacting serine-threonine kinase 2; TACC3, transforming, acidic coiled-coil containing protein 3.
Despite its issues, fulvestrant’s limited clinical success prompted the pursuit of an analogous approach to target the androgen receptor (AR) in prostate cancer, i.e., the development of selective androgen receptor degraders (SARDs). The AR is also a ligand-dependent transcription factor amenable to a traditional inhibitor approach. As a nuclear hormone receptor, AR responds to endogenous androgens including 5α-dihydrotestosterone (5α-DHT) and testosterone in order to mediate the development and maintenance of male reproductive organs93. For this reason, prostate cancer is also initially dependent on AR signaling; thus, antiandrogen therapy with flutamide or bicalutamide has demonstrated good clinical efficacy for prostate cancer94,95; however, resistance eventually develops, commonly through overexpression or amplification of the androgen receptor35,96, leading to castration-resistant prostate cancer (CRPC)97,98. A more potent androgen antagonist, enzalutamide (MDV3100, Xtandi; Medivation)99, established that a subset of CRPC is still dependent on androgen signaling. To address these resistance mechanisms, the first-in-class nonsteroidal SARD AZD3514100 was developed and was shown to downregulate AR101–103. Despite promising phase I (NCT01162395) efficacy data103, the development of AZD3514 was discontinued due to tolerability issues104. Though much work105 has been done in the field of SARDs, there are currently no known SARDs on the market.
The field of induced ER and AR degradation remains active, with multiple compounds in clinical trials. Targeted degradation not only combats compensatory upregulation associated with inhibition of function but also affects functions not amenable to traditional inhibitors (i.e. ligand-independent function of ERα).However, SARDs and SERDs are only capable of inducing the degradation of androgen and estrogen receptors; they cannot be repurposed to target other proteins for degradation.
3) Hydrophobic tagging (HyT)
To extend the concept of induced protein instability to a larger spectrum of protein targets, a new platform technology was recently developed by us and others: hydrophobic tagging (HyT). Similar to fulvestrant-induced surface hydrophobicity, this approach appends hydrophobic moieties (i.e., adamantane or Boc3-Arg) to the surface of target proteins in an attempt to mimic a partially unfolded protein state and co-opt the cytosolic unfolded protein response to degrade the target106. Compared to SARDs and SERDs, this approach is modular with respect to which proteins can be targeted as long as a ligand to the target can be identified.
The ability of Boc3-Arg to induce protein degradation was demonstrated in 2012, with the targeting of glutathione-S-transferase α1 (GST-α1) through the covalent inhibitor etacrynic acid linked to Boc3-Arg at 80μM (Figure 2)107. Similarly, dihydrofolate reductase was degraded using micromolar concentrations of the non-covalent inhibitor trimethoprim coupled to Boc3-Arg. This induced target protein degradation pathway occurs via the proteasome in an ubiquitin- and ATP-independent manner, although the roles of chaperones and other quality control pathways remain unknown. Interestingly, it was recently found that the Boc3-Arg moiety can inhibit the translational machinery as mediated by the mammalian Target of Rapamycin Complex 1 (mTORC1) pathway108. This off-target effect of Boc3-Arg could potentially limit the clinical use of Boc3-Arg as a hydrophobic tag.
Figure 2. Structures of compounds used for induced protein degradation technologies.
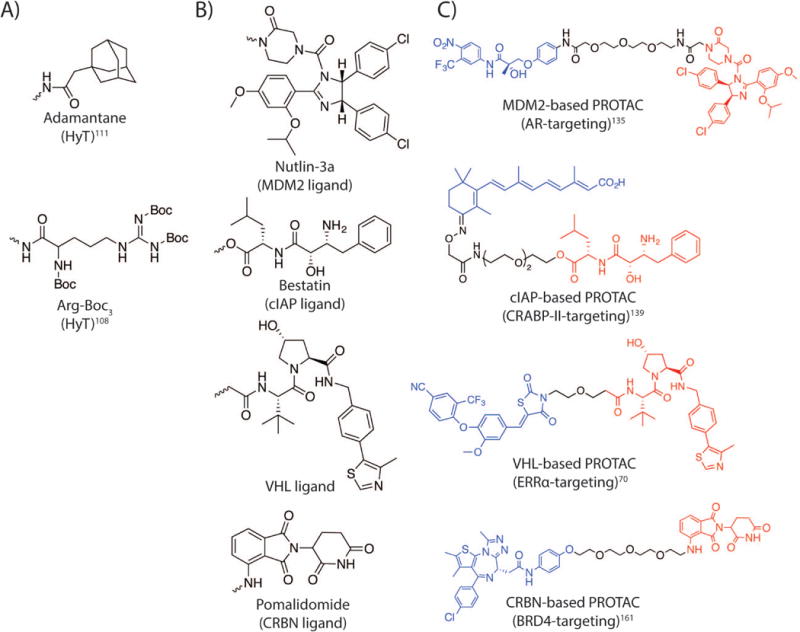
A) Hydrophobic tags (HyTs) utilized to recruit chaperones to a protein of interest (POI): adamantane and Arg-Boc3. B) E3 ligase recruiting ligands employed to hijack E3 ligases to ubiquitinate and degrade POIs. The wavy line illustrates the attachment point utilized in the studies discussed. C) The chemical structures of proteolysis-targeting chimeras (PROTACs) utilizing different E3 ligases to target the POIs for degradation. The POI ligands are shown in blue; the linkers are shown in black; and the E3 ligase recruiting ligands are shown in red.
An earlier HyT strategy, the adamantane-based HyT strategy, has been applied to a broader range of targets. This technology also consists of a bifunctional molecule, with one end binding to a protein of interest and the other end engaging quality control mechanisms through a hydrophobic adamantyl moiety109. First reported in 2011, ectopically expressed cytosolic and transmembrane HaloTag fusion proteins110–112 were shown to be susceptible to HyT-mediated degradation. Pharmacokinetic studies with adamantane-based HyT in mice demonstrated that 75% of the hydrophobic tag remained after 24h and the HyT compound was sufficient to suppress xenografted HaloTag-HRasG12V-driven tumor growth in mice by 80%110. Immunoprecipitation of HaloTag fusion protein tagged with the adamantyl group demonstrated increased association with HSP70, suggesting that HSP70 mediates adamantyl-mediated degradation 113. Furthermore, higher molecular weight bands corresponding to ubiquitination were observed in the immunoprecipitates, suggesting that the ubiquitin-proteasome system (UPS) plays a role in adamantane-based HyT. In the same study, a second small-molecule ligand (HALTS1) was discovered to stabilize HaloTag fusion proteins, allowing for bidirectional control of protein levels (Box 2)113. Despite the promising introduction of HyT technology as a tool for modulation of intracellular protein levels, degradation of targets with HyT was only demonstrated via covalent interaction with the artificial HaloTag system at the time.
Box 2. Induced Protein Instability for Discovery Biology.
Several excellent reviews discuss the development of small-molecule induced protein instability as chemical biology tools172,176,199. However, two recent publications highlight the application of these chemical tools for discovery biology. In 2014, the Hydrophobic Tagging (HyT) technology was utilized to uncover an acute resolvable endoplasmic reticulum (ER) unfolded protein response (UPR) pathway that previously evaded analysis using earlier chemical and genetic tools200. Cells expressing ER-localized HaloTag fusion proteins were treated with adamantane-based HyT, leading to protein unfolding in the ER lumen. The ER stress induced by the HyT was transient, resolvable and non-apoptotic in nature in comparison to the pro-apoptotic chemical tools, thapsigargin201 and tunicamycin202. This resolvable stress identified the estrogen signaling as a component of a late-stage ER stress resolution pathway.
In 2015, a similar small-molecule induced protein instability technology elucidated a specific nuclear UPR distinct from the cytoplasmic heat shock response203. Furthermore, unfolding of a specific protein in the cytoplasm engaged a p53 transcriptional program that was not found in the general heat shock response. Cells primed with the inducible stress were more capable of surviving subsequent unfolded protein stress. These two studies highlight the advantages of chemical tools over genetic methods in terms of specificity, temporal control and dose-response for discovery biology.
In 2014, the HyT technology was further extended to endogenously expressed proteins with the degradation of erythroblastosis oncogene B3 (ErbB3/HER3), a pseudokinase considered to be “undruggable” by an ‘occupancy-driven’ approach114. Gray and co-workers discovered a selective ErbB3 covalent ligand from a HTS with an apparent IC50 of 23 nM115; however, given that ErbB3 has minimal kinase activity, a small-molecule binder would have little consequence on the scaffolding role of ErbB3. Coupling adamantane to the ErbB3 ligand, Gray and co-workers converted the ligand into a HyT degrader termed TX2-121-1. Covalent binding of TX2-121-1 to ErbB3 led to the degradation of ErbB3 at 500nM, inhibiting downstream signaling and exhibiting an antiproliferative effect on ErbB3-dependent cell lines. Elimination of the covalent interaction reduced the efficacy of the adamantane-based HyT. The extension of the technology to the previously “undruggable” ErbB3 was a significant step in the development of HyT as a therapeutic strategy; however, the degradation of ErbB3 was still dependent on a covalent interaction, and so the degradation would be stoichiometric, rather than sub-stoichiometric.
Recently, our group has expanded the clinical potential of the HyT technology by targeting the androgen receptor (AR) with a non-covalent binder116. A high-affinity AR agonist, RU59063117, was coupled with an adamantyl moiety, thus converting an agonist into an antagonistic degrader. Upon incubation with LNCaP prostate tumor cells, AR degradation was observed in the sub-micromolar range, reducing the expression of AR target genes and leading to decreased cell proliferation. As the first demonstration of a successful adamantane-based HyT strategy with a non-covalent interaction, this study was a step towards expanding the target spectrum for this approach. Despite this success, more examples of targets being degraded through a non-covalent interaction need to be demonstrated before HyT becomes a potential therapeutic strategy. Indeed, the in vivo potential of adamantane-based and Boc3-Arg HyTs in a non-artificial system remains untested.
The HyT platform illustrates the potential of a modular post-translational protein knockdown strategy by targeting traditionally intractable targets (i.e. ErbB3) or by utilizing non-covalent protein ligands to induce protein degradation; however, there is some concern that hydrophobic moieties of such compounds can remain bound by serum proteins resulting in low free fraction of the compound available to induce degradation of target proteins. Moreover, it is unclear whether HyT-mediated degradation is dependent on recognition of the adamantane/Arg-Boc3 moiety or on the induced destabilization of the protein. If the latter, HyT would be less effective against more stable proteins, limiting the targets that could be accessed for degradation. Additionally, the degradation pathways for adamantane- and Arg-Boc3-based HyT may be different. Thus, as the mechanism of HyT is unresolved, HyT technology remains a chemical tool for discovery biology rather than a viable therapeutic strategy. In contrast, PROTACs, which utilize small-molecule E3 ligase ligands to induce proximity-induced ubiquitination of a target protein, are much closer to clinical translation.
4) Proteolysis-targeting chimeras (PROTACs)
Peptide-based PROTACs
In several proof-of-concept experiments published in 2001, our lab, in collaboration with Ray Deshaies, reported the first PROTACs —bifunctional molecules that harness the ubiquitin–proteasome system (UPS) by recruiting an E3 ligase to a protein of interest, leading to the proximity-induced ubiquitination and subsequent degradation of the protein (Box 3)118. These early PROTACs were composed of a phosphopeptide portion of IκBα to recruit the F-box protein β-TrCP, a component of the Skp1-Cullin1-F box E3 ligase complex (SCFβ–TrCP)119, and the angiogenesis inhibitor ovalicin to covalently bind the target protein, methionine aminopeptidase-2 (MetAP-2)120,121. Addition of this PROTAC to Xenopus egg extracts led to MetAP-2 degradation. The technology was further demonstrated in a cellular system to induce the degradation of AR by microinjecting a β-TrCP-based PROTAC with dihydroxytesterone (DHT) as the protein ligand into HEK293 cells122. Though these were the first demonstrations of the PROTAC technology, there was much room for improvement: the potencies of these PROTACs were in the micromolar range and cellular permeability was very low; furthermore, the dependency on IκBα phosphorylation for E3 ligase recruitment was limiting given the action of cellular phosphatases.
Box 3. The Ubiquitin-Proteasome System (UPS).
The ubiquitin-proteasome system (UPS) is the primary mechanism for removing misfolded proteins from the cytosol and nucleus. The UPS consists of a coordinated enzymatic cascade that results in the covalent attachment of ubiquitin (a 76-amino-acid protein) to a lysine residue of a targeted protein, eventually leading to the removal of this protein substrate by the proteasome. This cascade begins with an ubiquitin-activating enzyme (E1)204 creating a reactive thioester on ubiquitin in an ATP-dependent manner. The ubiquitin is then transferred to the ubiquitin-conjugating enzyme (E2)205. The last enzyme, ubiquitin ligase enzyme (E3)183, specifically binds the target protein and catalyzes the proximity-induced transfer of the activated ubiquitin from the E2 protein to the target protein.
In order to regulate intracellular protein concentrations, E3 ligases recognize a number of degradation signals. For example, the E3 ligase von Hippel-Lindau (VHL) recognizes a core hydroxylated proline residue on hypoxia-inducible factor 1α (HIF1α)124 while β-TrCP binds specifically to phosphorylated residues on IκBα and β-catenin206. A variety of E3 ligases are utilized in PROTAC technology: CRBN, VHL and β-TrCP act as substrate recognition subunits in multi-subunit Cullin-Ring E3 ligase (CRL) complexes170,183 while MDM2 forms homo- or hetero-oligomeric E3 ligase complexes for productive ubiquitination207,208. Since ubiquitin has seven lysines, substrate-linked ubiquitin can be further modified to produce polyubiquitin chains with unique topographies. Ubiquitin chains linked through Lys-48 have been classically associated with proteasomal degradation209,210 while other chain types, such as Lys-63 and Lys-11, have been associated with subcellular localization, endocytosis and transcriptional regulation211,212. In contrast, a single ubiquitination event (monoubiquitination) has been recognized to play a part in transmembrane protein endocytosis and signaling213. PROTAC technology tethers an E3 ligase to a target protein with the goal of inducing polyubiquitination and subsequent removal of the target in the proteasome.
The first improvement in peptidic PROTACs was a switch to a smaller E3 ligase recruiting peptide fragment that did not require phosphorylation for activity. The E3 ligase von Hippel-Lindau (VHL) recognizes a core hydroxylated proline in a seven-amino-acid recognition sequence of hypoxia-inducible factor 1α (HIF1α)123,124. An AR-targeting PROTAC was developed using DHT conjugated to the HIF1α peptide fragment with a poly-D-arginine tag appended for cell penetrance125. Addition of the PROTAC at 25μM led to significant AR depletion in HEK293 cells. Analogous PROTACs using estradiol demonstrated ERα degradation, leading to reduced proliferation of ERα-dependent breast cancer cell lines126–128. More recently, VHL-recruiting peptidic PROTACs were utilized to induce the degradation of Akt129 and Tau130. The latter’s ability to induce degradation of a traditionally “undruggable” protein highlights the advantages of ‘event-driven’ over ‘occupancy-driven’ pharmacology.
The advantage of ‘event-driven’ pharmacology is further emphasized with the application of the PROTAC technology to the conditional removal of proteins. As the PROTAC technology works post-translationally (in contrast to nucleic acid-based approaches), there is a pharmacological opportunity to remove a select subset of proteins. Our group introduced a phospho-dependent PROTAC, whereby activation of the NGF receptor (TrkA) or ErbB3/HER3 was coupled to the post-translational degradation of downstream signaling components, i.e., fibroblast growth factor receptor substrate 2α (FRS2α) and phosphatidylinositol-3-kinase (PI3K), respectively131. Upon growth factor stimulation, TrkA or ErbB3 phosphorylates a peptidic PROTAC, which is then recognized by either FRS2α or PI3K.FRS2α or PI3K, bound to their respective phosphorylated peptidic PROTAC, is then ubiquitinated by VHL. In a similar conditional knockdown study, the β-catenin binding motif of E-cadherin was fused to β-TrCP. The resulting fusion protein was capable of inducing the degradation of only the cytosolic population of β-catenin, sparing the membrane-bound population132. However, the potencies of these peptidic PROTACs remain in the micromolar range, likely due to the poor cell penetration associated with peptide-based therapeutics. Fortunately, incremental improvements have led to the development of potent small-molecule PROTACs with in vivo stability.
Small-molecule PROTACs
The current pharmacopeia that targets intracellular proteins remains largely dominated by small-molecules, as such targets are not accessible to monoclonal antibodies and so far, few nucleotide-based therapies have been approved. Given the clinical limitations of peptide-based PROTACs, efforts have increasingly focused on the generation of small-molecule PROTACs, beginning in 2008 with the creation of a PROTAC utilizing nutlin-3a133 to recruit the E3 ligase mouse double minute 2 homolog (MDM2; nutlin-3a IC50 against MDM2 = 0.09μM) to the AR134. PROTAC-induced AR degradation was observed at 10μM in cells; however, the maximum knockdown of the AR was not as dramatic as with the peptidic VHL ligand. Despite the success of this initial foray into all-small-molecule PROTACs, subsequent medicinal chemistry efforts were unsuccessful at achieving more effective degradation of the targets using the E3 ligase MDM2. However, newer generations of nutlins, RG7112 and idasanutlin (RG7388; Genentech), have been developed with increased potency135,136 and it remains to be determined if these can be successfully incorporated into PROTACs with improved potency and knockdown efficiency. Moreover, dose-limiting cytopenias and gastrointestinal effects reported in phase I trials with idasanutlin are possible concerns for any nutlin-based PROTAC drug candidates137.
Another small-molecule-based PROTAC approach for targeted protein degradation, termed Specific and Non-genetic Inhibitor-of-apoptosis proteins (IAPs)-dependent Protein Eraser (SNIPER), was reported in 2010 by the Hashimoto group138. Utilizing bestatin (ubenimex; Nippon Kayaku) esters139 to repurpose the E3 ligase cellular inhibitor of apoptosis protein 1 (cIAP1), Hashimoto and co-workers demonstrated the degradation of cellular retinoic acid-binding proteins (CRABP-I and -II) with all-trans retinoic acid (ATRA) as the target ligand. Unfortunately, bestatin is not specific for cIAP1 as it inhibits both arginyl aminopeptidases and leukotriene A4 hydrolase139,140. Additionally, these bestatin esters also induced the autoubiquitination and degradation of the recruited E3 ligase cIAP1, limiting the full potential of the technology141. However, to overcome some of these limitations, medicinal chemistry efforts have reduced the autoubiquitination observed with bestatin142 or have recruited related E3 ligases for SNIPER143. In addition to the CRABP family, cIAP-based PROTACs were also used to degrade estrogen receptor144, androgen receptor145, retinoic acid receptors145 and transforming acidic coiled-coil-3146. Despite the range of targets degraded, SNIPER PROTACs are still maximally effective only in the micromolar range. Given the micromolar potencies of cIAP-based PROTACs and the fact that bestatin is not specific for cIAP1, SNIPER PROTACs have yet to be tested in mouse models.
Recent efforts have focused on identifying an appropriate small-molecule replacement for the HIF1α peptide fragment to provide an alternative E3 ligase for the all small-molecule PROTAC approach. The first small-molecule ligand for the VHL E3 ligase was identified through in silico and fragment-based screens in 2012 with a reported Kd of 5.4μM147–149. This affinity is more than 20-fold weaker than a 10-mer HIF1α fragment interaction with VHL (Kd = 0.2μM). Subsequent structure-activity relationship (SAR) efforts yielded a VHL ligand with a Kd of 185nM150. Utilizing this improved ligand, a VHL-based small-molecule PROTAC was described in 2015 with nanomolar potency for nearly complete degradation of estrogen-related receptor α (ERRα) and receptor-interacting serine-threonine kinase 2 (RIPK2) without stabilization of the endogenous target HIF1α at concentrations of up to 30µM of the ERRα-targeted PROTAC70. In vivo degradation of ERRα was achieved with approximately 40% knockdown of protein in heart, kidney and tumor xenograft. Moreover, PROTACs demonstrated sub-stoichiometric activity where one PROTAC molecule was capable of inducing multiple rounds of target protein ubiquitination. Finally, through comparative proteomic studies, PROTACs were not only specific for the target protein but also had less off-target degradation than initially suggested by the ligand binding specificity. This study was the first demonstration of a small-molecule VHL-based PROTAC with sub-stoichiometric activity and activity in mouse models.
Concurrent with the establishment of VHL-based PROTACs, another PROTAC system based on a fourth small-molecule E3 ligase recruiting ligand was developed. In 2010, work from Handa and colleagues identified the E3 ligase Cereblon (CRBN) as the primary target of thalidomide151. In 2014, the Kaelin and Ebert groups discovered that phthalimides (thalidomide, lenalidomide, and pomalidomide) induced the degradation of the lymphoid transcription factors Ikaros Family Zinc Finger 1 and 3 (IKZF1 and IKZF3)152–154 by binding to CRBN (Kd = 150–250nM)155 and repurposing the E3 ligase to ubiquitinate the transcription factors. Further studies have found that not all of the phthalimides have the same substrate degradation spectrum and thus are utilized in different clinical situations: lenalidomide can induce the degradation of casein kinase Iα (CKIα); in contrast, thalidomide and pomalidomide are incapable of inducing CKIα degradation156–158. Recently, screening for CRBN ligands that degraded other targets led to the identification of CC-885, a phthalimide derivative that targets G1 to S phase transition protein 1 homologue (GSPT1; also known as ERF3A)159.
Considering the ability of phthalimide to bind CRBN, our group and others sought to utilize phthalimide as an E3 ligase recruiting ligand to hijack CRBN to degrade proteins of interest. Conjugation of a small-molecule BRD4 binding moiety (OTX015) to pomalidomide yielded a PROTAC capable of degrading the epigenetic regulator BRD4 at picomolar potencies160. In comparison to the BRD4 inhibitors JQ1 and OTX015, the CRBN-based PROTAC provided a more sustained suppression of c-myc levels by counteracting known increased BRD4 expression associated with BRD4 inhibition161. This sustained suppression translated into superior antiproliferative and apoptotic effects against Burkitt’s lymphoma cell lines compared to the inhibitors. In a separate but parallel study, the BRD4 inhibitor JQ1 was conjugated to a thalidomide derivative72. The resulting PROTAC, termed dBET1, was capable of inducing BRD4 degradation at low nanomolar concentrations. Proteomic studies with this PROTAC determined that the compound significantly reduced c-myc, BRD2, BRD3, and BRD4 levels with minimal off-target degradation. This bifunctional compound was also found to be superior to JQ1 against human AML and lymphoma cell lines. Finally, dBET1 was investigated in a xenograft model of AML and a disseminated leukemia mouse model; the PROTAC was found to be active in the AML model and outperformed JQ1 in the disseminated model. The superiority of ‘event-driven’ versus ‘occupancy-driven’ pharmacology in preclinical models is encouraging and sets the stage for clinical applications in the near future.
In addition to CRBN-based PROTACs, a picomolar VHL-based pan-BET-targeting PROTAC was also developed, which demonstrated superior anti-proliferative activity compared to BET inhibitors162. The pan-BET-targeting PROTAC also exhibited activity against a 22RV-1 CRPC mouse xenograft, thus expanding the application of PROTAC technology to solid tumor malignancies. Similarly, Ciulli and co-workers coupled the VHL recruiting ligand to JQ1 to create a PROTAC capable of differentially degrading BRD4 over BRD2 and BRD3163. Maximum degradation of BRD4 occurred at nanomolar concentrations while effects on BRD3 and BRD2 required low micromolar PROTAC concentrations. This selective degradation of BRD4 over BRD3 and BRD2 leads to a differential gene expression pattern between the parent inhibitor JQ1 and the BRD4-targeting PROTAC. This observation suggests that PROTACs can provide an added layer of specificity relative to an inhibitor: off-target proteins can be spared from PROTAC-induced degradation in spite of target binding.
This idea was further studied by exploring VHL-based PROTACs and CRBN-based PROTACs in a head-to-head comparison71. Utilizing the protein tyrosine kinase inhibitors bosutinib (SK-606, Bosulif; Pfizer)164 and dasatinib (BMS-354825, SPRYCEL; Bristol-Myer Squibb)165, ABL- and BCR-ABL-targeting PROTACs were generated via conjugation to either a VHL recruiting ligand or a CRBN recruiting ligand. Surprisingly, the degradation profile of ABL-targeting and BCR-ABL-targeting PROTACs depended on the identity of the inhibitor warhead (bosutinib or dasatinib) and on the E3 ligase recruited (VHL or CBN) but was independent of target engagement by the PROTACs. In this case, CRBN E3 ligase appeared more promiscuous in its degradation profile as compared to VHL, consistent with previous studies targeting BRD4. One explanation could lie within the unique flexibility of the CRBN E3 ligase complex containing Cullin 4A157,166–168 compared to the VHL E3 ligase complex169,170, thus, allowing for a larger ubiquitination zone. These two studies into E3 ligase selectivity in the PROTAC system open up the possibility of converting a promiscuous inhibitor into a selective degrader.
With the progress made from the 2001 report of PROTAC-induced degradation in a Xenopus extract to the more recent targeted degradation achieved in cells and mouse models, interest in PROTACs has grown 171. As a platform technology, PROTACs offer a modular ‘plug-and-play’ aspect that allows for general applicability: once a target protein ligand is identified, the ligand can be linked to an E3 ligase recruiting ligand for PROTAC development172. The removal of target proteins allows PROTACs to counteract compensatory accumulation that is often reported with inhibitors and provide a kinetic advantage similar to covalent inhibitors (i.e., restoration of protein function requires target protein re-synthesis). However, in contrast to the stoichiometric property of covalent ‘occupancy-driven’ inhibitors, the ‘event-driven’ catalytic nature of non-covalent PROTACs has the potential to require less total drug exposure. In addition, the possibility of conditional knockdowns and an increased layer of specificity hold the promise of minimizing off-target effects. Finally, proof-of-concept experiments in mouse tumor xenografts demonstrate that the in vivo efficacy of ‘event-driven’ targeted protein degradation can be superior to the ‘occupancy-driven’ pharmacology of the parent inhibitor compounds161,163.
The promise of induced protein degradation as a novel therapeutic modality has attracted the attention of the pharmaceutical industry173 and led to the creation of companies focused on developing such technologies. Arvinas (based in New Haven, Connecticut, USA) was launched in 2013174 and recently announced plans to advance a PROTAC-based drug candidate into Phase I clinical trials in 2017175, and C4 therapeutics (based in Cambridge, Massachusetts, USA) was launched in 2016. Either as tool compounds176,177 or as a future drug discovery paradigm, the myriad of potential PROTAC applications has not yet been fully explored. However, from the few innovative reports to date, it is clear that induced protein degradation as a novel pharmaceutical paradigm holds much promise.
5) Challenges and Opportunities Ahead
With the progress made in the field of ASO and the more recent publications in the field of small-molecule induced degradation, a complementary approach to the traditional therapeutic strategies inhibiting protein function — the modulation of protein levels — has emerged (Figure 3). Nucleic acid-based therapeutics initially held much promise with their ease of synthesis and modularity; however, dependency on protein half-life and challenging in vivo pharmacological properties have hindered the broad development of nucleic acid-based clinical drug candidates. General inducers of degradation, such as HSP90 inhibitors, have suffered from challenging pharmacological properties, tolerability and the lack of a clear clinical indication; however, given the volume and range of ongoing clinical trials, HSP90 inhibitors may still find their way to the market. With the approval of fulvestrant, SERDs and SARDs remain another promising class of drugs that can address mechanisms of resistance arising from a traditional inhibitor approach. However, bioavailability and systemic exposure remain a challenge for this class of drugs. Additionally, these small-molecule approaches are not broadly applicable to other targets.
Figure 3. Timeline of the induced protein degradation field.
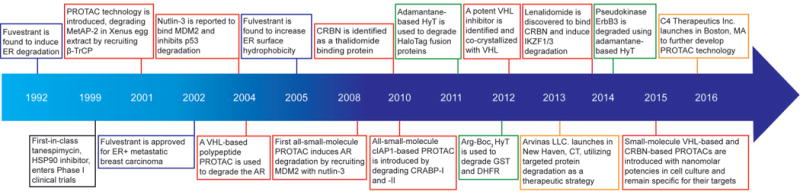
Selected events related to selective estrogen receptor degraders are highlighted in blue; proteolysis-targeting chimeras (PROTAC) technology in red,; hydrophobic tagging (HyT) in green,; and biotechnology company launches in yellow. MetAP-2, methionine aminopeptidase-2; ER, estrogen receptor; MDM2, mouse double minute 2 homolog; AR, androgen receptor; CRBN; Cereblon; cIAP1, cellular inhibitor of apoptosis protein 1; CRABP, cellular retinoic acid-binding protein; VHL, Von Hippel-Lindau; GST, glutathione-S-transferase; DHFR, dihydrofolate reductase; IKZF, Ikaros family zinc finger; ErbB3, erythroblastosis oncogene B3.
In theory, HyT technology can be broadly applied to targets yet the unknown mechanism of HyT might limit the adoption of the technology. By contrast, the PROTAC technology offers a modular approach to sub-stoichiometric targeted protein degradation, which only requires accessible surface lysines to be present on the target protein in order to harness the UPS (Box 3). However, PROTACs are not considered traditional “small” molecules since they do not follow Lipinski’s rule of five178 owing to their relatively large molecular mass. Unfortunately, this larger size of PROTACs provides more opportunity for metabolic modification of the compounds. First-pass metabolism could limit the oral delivery of PROTACs yet a lower bioavailability might not significantly hinder PROTAC efficacy given the sub-stoichiometric nature of the technology when using non-covalent ligands. In regards to future clinical application, the potencies achievable in cell culture as well as the efficacies observed in tumor xenografts are encouraging.
As the field of PROTAC-mediated target protein degradation matures, the idea of additional specificity could be explored further: is it possible to achieve tissue-specific or disease-specific induced protein degradation by utilizing tissue/disease-specific E3 ligases? Small-molecule screens have already identified a number of candidate E3 ligase inhibitors that can potentially be utilized as recruiting ligands179–182 and we have already found that different E3 ligases provide different degradation profile71. A more thorough understanding of E3 ligase tissue expression could potentially offer an increased therapeutic window with an appropriate E3 ligase-recruiting PROTAC. Similarly, subcellular localization and regulation of E3 ligases could offer opportunities for conditional removal of target proteins183: perhaps a nuclear localized E3 ligase would permit conditional degradation of activated nuclear receptors while sparing the cytoplasmic population. Additionally, as suggested by studies with peptidic PhosphoPROTACs, perhaps developing ‘state-specific’ protein ligands will permit conditional protein knockdown; on the other end of the PROTAC, we can also imagine a conditional unmasking of the E3 ligase recruiting ligand either by chemical or optical means to permit temporal and spatial control of target degradation analogous to the endogenous system of cryptic degrons. The concept of increased specificity is only the tip of the iceberg for this emerging technology.
Finally, PROTAC technology could have its biggest impact via the development of degrader molecules capable of targeting the “undruggable” proteome. Despite the recent success of small-molecule PROTACs, this technology has to date only been applied to classically “druggable” proteins. However, the ability to sub-stoichiometrically degrade transcription factors, scaffolding and other non-enzymatic proteins would greatly increase the current drug target space. One could envision a process in which appropriate protein binders are identified via screens and then conjugated to pre-made linkers attached to E3 ligase ligands to generate potent degrader compounds. In this way, PROTAC generation could become part of the routine strategy in drug discovery, thereby making the “undruggable” proteome finally pharmaceutically vulnerable.
Acknowledgments
The authors would like to thank Saul Jamie-Figueroa, Taavi Neklesa, Kanak Raina and the rest of the Crews laboratory for their helpful comments. C.M.C. gratefully acknowledges the NIH for its support (R35-CA197589). A.C.L acknowledges support from the NIH (MSTP NIH/NIGMS T32GM007205). C.M.C. is founder, consultant and shareholder of Arvinas, LLC.
Glossary
- ‘Occupancy-driven’ Pharmacology
A drug discovery paradigm based on identifying compounds that modulate protein function through stoichiometric drug binding and occupation of the binding site to modulate protein function
- ‘Event-driven’ Pharmacology
A drug discovery paradigm based on identifying compounds that lead to the removal of the target protein from the system following drug binding
- Sub-stoichiometric
pertains to molecular compounds that have a greater effect than the 1:1 activity seen with a stoichiometric agent
- HaloTag
a bacterial dehalogenase engineered by the Promega Corporation to covalently bind chloroalkanes and has been used primarily for protein purification and imaging purposes
- Erythroblastosis oncogene B3 (ErbB3/HER3)
A member of the epidermal growth factor receptor (EGFR) family that has minimal kinase activity and acts primarily as a scaffold for signal transduction
- IκBα
Inhibitor of NF-κB that contains a destruction motif that, when phosphorylated, is recognized by the E3 ligase substrate recognition component β-TrCP
- von Hippel-Lindau (VHL)
the substrate recognition portion of the E3 ligase complex VHL-elongin B/C-cullin 2 (VHL-ELOBC-CUL2) that mediates the transfer of ubiquitin onto hypoxia-inducible factor 1α (HIF 1α)
- Poly-D-arginine tag
An arginine-containing peptide fragment based on the HIV Tat peptide that facilitates cellular uptake of proteins
- BRD4
A member of the Bromodomain and Extra-Terminal (BET) family that recognizes acetylated lysines of histones to modulate the epigenetic code
- BCR-ABL
an oncogenic fusion of the breakpoint cluster region protein (BCR) and the Abelson (ABL) tyrosine kinase leading to chronic myelogenous leukemia
- Rule of five
the rule of five is a widely used guideline to predict the likelihood of poor absorption or permeability characteristics for small molecules. It is based on upper limits for physiochemical properties of the compounds, including molecular mass (for which 500 Da is the cut-off)
- Degron
a peptide segment recognized by the cellular quality control system that influences the degradation rate of the full-length protein
References
- 1.Campbell J, et al. Large-Scale Profiling of Kinase Dependencies in Cancer Cell Lines. Cell Reports. 2016;14:2490–2501. doi: 10.1016/j.celrep.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cowley GS, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Scientific Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang T, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 6.Lazo JS, Sharlow ER. Drugging Undruggable Molecular Cancer Targets. Annual Review of Pharmacology and Toxicology. 2016;56:23–40. doi: 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- 7.Jin L, Wang W, Fang G. Targeting Protein-Protein Interaction by Small Molecules. Annual Review of Pharmacology and Toxicology. 2014;54:435–456. doi: 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- 8.Adjei AA. What Is the Right Dose? The Elusive Optimal Biologic Dose in Phase I Clinical Trials. Journal of Clinical Oncology. 2006;24:4054–4055. doi: 10.1200/jco.2006.07.4658. [DOI] [PubMed] [Google Scholar]
- 9.Cong L, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jinek M, et al. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346 doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 12.Hsu Patrick D, Lander Eric S, Zhang F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotech. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Smidt PC, Le Doan T, de Falco S, van Berkel TJ. Association of antisense oligonucleotides with lipoproteins prolongs the plasma half-life and modifies the tissue distribution. Nucleic Acids Res. 1991;19:4695–4700. doi: 10.1093/nar/19.17.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geary RS, et al. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J Pharmacol Exp Ther. 2001;296:890–897. [PubMed] [Google Scholar]
- 16.McMahon BM, et al. Pharmacokinetics and tissue distribution of a peptide nucleic acid after intravenous administration. Antisense Nucleic Acid Drug Dev. 2002;12:65–70. doi: 10.1089/108729002760070803. [DOI] [PubMed] [Google Scholar]
- 17.Marques JT, Williams BR. Activation of the mammalian immune system by siRNAs. Nat Biotechnol. 2005;23:1399–1405. doi: 10.1038/nbt1161. [DOI] [PubMed] [Google Scholar]
- 18.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annual review of immunology. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 19.Dahlman JE, Kauffman KJ, Langer R, Anderson DG. Nanotechnology for in vivo targeted siRNA delivery. Adv Genet. 2014;88:37–69. doi: 10.1016/b978-0-12-800148-6.00003-1. [DOI] [PubMed] [Google Scholar]
- 20.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilhelm S, et al. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials. 2016;1:16014. doi: 10.1038/natrevmats.2016.14. [DOI] [Google Scholar]
- 22.Peer D, Lieberman J. Special delivery: targeted therapy with small RNAs. Gene Ther. 2011;18:1127–1133. doi: 10.1038/gt.2011.56. [DOI] [PubMed] [Google Scholar]
- 23.Fedorov Y, et al. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12:1188–1196. doi: 10.1261/rna.28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson AL, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotech. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 25.Qiu S, Adema CM, Lane T. A computational study of off-target effects of RNA interference. Nucleic Acids Research. 2005;33:1834–1847. doi: 10.1093/nar/gki324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melnikova I. RNA-based therapies. Nat Rev Drug Discov. 2007;6:863–864. doi: 10.1038/nrd2314. [DOI] [PubMed] [Google Scholar]
- 28.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 29.Perry CM, Balfour JAB. Fomivirsen. Drugs. 1999;57:375–380. doi: 10.2165/00003495-199957030-00010. [DOI] [PubMed] [Google Scholar]
- 30.Wong E, Goldberg T. Mipomersen (Kynamro): A Novel Antisense Oligonucleotide Inhibitor for the Management of Homozygous Familial Hypercholesterolemia. Pharmacy and Therapeutics. 2014;39:119–122. [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha G. Antisense battles small molecule for slice of rare lipid disorder market. Nat Biotech. 2013;31:179–180. doi: 10.1038/nbt0313-179. [DOI] [PubMed] [Google Scholar]
- 32.Crews CM. Targeting the undruggable proteome: the small molecules of my dreams. Chem Biol. 2010;17:551–555. doi: 10.1016/j.chembiol.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 34.Duncan James S, et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Visakorpi T, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 36.Hatzivassiliou G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 37.Heidorn SJ, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rauch J, Volinsky N, Romano D, Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun Signal. 2011;9:23. doi: 10.1186/1478-811x-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan X, Thapa N, Sun Y, Anderson RA. A kinase-independent role for EGF receptor in autophagy initiation. Cell. 2015;160:145–160. doi: 10.1016/j.cell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vivanco I, et al. A kinase-independent function of AKT promotes cancer cell survival. eLife. 2014;3:e03751. doi: 10.7554/eLife.03751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weihua Z, et al. Survival of Cancer Cells Is Maintained by EGFR Independent of Its Kinase Activity. Cancer cell. 2008;13:385–393. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moulick K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamal A, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 45.Grenert JP, et al. The Amino-terminal Domain of Heat Shock Protein 90 (hsp90) That Binds Geldanamycin Is an ATP/ADP Switch Domain That Regulates hsp90 Conformation. Journal of Biological Chemistry. 1997;272:23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- 46.Stebbins CE, et al. Crystal Structure of an Hsp90–Geldanamycin Complex: Targeting of a Protein Chaperone by an Antitumor Agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 47.Prodromou C, et al. Identification and Structural Characterization of the ATP/ADP-Binding Site in the Hsp90 Molecular Chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 48.Schneider C, et al. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proceedings of the National Academy of Sciences. 1996;93:14536–14541. doi: 10.1073/pnas.93.25.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and Proteasomal Degradation of the p185c-erbB-2 Receptor Protein-tyrosine Kinase Induced by Geldanamycin. Journal of Biological Chemistry. 1996;271:22796–22801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 50.Samuni Y, et al. Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radical Biology and Medicine. 2010;48:1559–1563. doi: 10.1016/j.freeradbiomed.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banerji U, et al. Phase I Pharmacokinetic and Pharmacodynamic Study of 17-Allylamino, 17-Demethoxygeldanamycin in Patients With Advanced Malignancies. Journal of Clinical Oncology. 2005;23:4152–4161. doi: 10.1200/jco.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 52.Taldone T, Gozman A, Maharaj R, Chiosis G. Targeting Hsp90: small-molecule inhibitors and their clinical development. Current opinion in pharmacology. 2008;8:370–374. doi: 10.1016/j.coph.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McClellan AJ, et al. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 54.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 55.Taipale M, et al. Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuduk SD, et al. Synthesis and evaluation of geldanamycin-testosterone hybrids. Bioorg Med Chem Lett. 2000;10:1303–1306. doi: 10.1016/s0960-894x(00)00208-0. [DOI] [PubMed] [Google Scholar]
- 57.Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ. Synthesis and evaluation of geldanamycin-estradiol hybrids. Bioorg Med Chem Lett. 1999;9:1233–1238. doi: 10.1016/s0960-894x(99)00185-7. [DOI] [PubMed] [Google Scholar]
- 58.Katerina S, Evangelia P. HSP90 Inhibitors: Current Development and Potential in Cancer Therapy. Recent Patents on Anti-Cancer Drug Discovery. 2014;9:1–20. [PubMed] [Google Scholar]
- 59.Jego G, Hazoume A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett. 2013;332:275–285. doi: 10.1016/j.canlet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 60.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dauvois S, Danielian PS, White R, Parker MG. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci U S A. 1992;89:4037–4041. doi: 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giannetti AM. From experimental design to validated hits a comprehensive walk-through of fragment lead identification using surface plasmon resonance. Methods in enzymology. 2011;493:169–218. doi: 10.1016/b978-0-12-381274-2.00008-x. [DOI] [PubMed] [Google Scholar]
- 63.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 64.Mashalidis EH, Sledz P, Lang S, Abell C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nature protocols. 2013;8:2309–2324. doi: 10.1038/nprot.2013.130. [DOI] [PubMed] [Google Scholar]
- 65.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2016.109. advance online publication. [DOI] [PubMed] [Google Scholar]
- 66.Renaud JP, et al. Biophysics in drug discovery: impact, challenges and opportunities. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2016.123. advance online publication. [DOI] [PubMed] [Google Scholar]
- 67.Bleicher KH, Bohm HJ, Muller K, Alanine AI. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discov. 2003;2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 68.Macarron R, et al. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 69.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–716. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 70.Bondeson DP, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lai AC, et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Winter GE, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heldring N, et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiological Reviews. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 74.Nilsson S, et al. Mechanisms of Estrogen Action. Physiological Reviews. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 75.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 76.Group, E. B. C. T. C. Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet (London, England) 1998;351:1451–1467. [PubMed] [Google Scholar]
- 77.Martin LA, et al. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. 2003;278:30458–30468. doi: 10.1074/jbc.M305226200. [DOI] [PubMed] [Google Scholar]
- 78.Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- 79.Buzdar AU, Robertson JF. Fulvestrant: Pharmacologic Profile versus Existing Endocrine Agents for the Treatment of Breast Cancer. Annals of Pharmacotherapy. 2006;40:1572–1583. doi: 10.1345/aph.1G401. [DOI] [PubMed] [Google Scholar]
- 80.Wu YL, et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 81.Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007;67:9549–9560. doi: 10.1158/0008-5472.can-07-1590. [DOI] [PubMed] [Google Scholar]
- 82.Kato S, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 83.Connor CE, et al. Circumventing Tamoxifen Resistance in Breast Cancers Using Antiestrogens That Induce Unique Conformational Changes in the Estrogen Receptor. Cancer Research. 2001;61:2917–2922. [PubMed] [Google Scholar]
- 84.Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. British Journal of Cancer. 2004;90:S2–S6. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vergote I, Abram P. Fulvestrant, a new treatment option for advanced breast cancer: tolerability versus existing agents. Annals of Oncology. 2006;17:200–204. doi: 10.1093/annonc/mdj047. [DOI] [PubMed] [Google Scholar]
- 86.Di Leo A, et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol. 2010;28:4594–4600. doi: 10.1200/jco.2010.28.8415. [DOI] [PubMed] [Google Scholar]
- 87.van Kruchten M, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 2015;5:72–81. doi: 10.1158/2159-8290.cd-14-0697. [DOI] [PubMed] [Google Scholar]
- 88.Bross PF, et al. Fulvestrant in Postmenopausal Women with Advanced Breast Cancer. Clinical Cancer Research. 2003;9:4309–4317. [PubMed] [Google Scholar]
- 89.Govek SP, et al. Optimization of an indazole series of selective estrogen receptor degraders: Tumor regression in a tamoxifen-resistant breast cancer xenograft. Bioorg Med Chem Lett. 2015;25:5163–5167. doi: 10.1016/j.bmcl.2015.09.074. [DOI] [PubMed] [Google Scholar]
- 90.Lai A, et al. Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts. J Med Chem. 2015;58:4888–4904. doi: 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- 91.Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs. 2015;26:948–956. doi: 10.1097/cad.0000000000000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weir HM, et al. AZD9496: An oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1 mutant breast tumours in preclinical models. Cancer Res. 2016 doi: 10.1158/0008-5472.can-15-2357. [DOI] [PubMed] [Google Scholar]
- 93.Matsumoto T, et al. The Androgen Receptor in Health and Disease. Annual Review of Physiology. 2013;75:201–224. doi: 10.1146/annurev-physiol-030212-183656. [DOI] [PubMed] [Google Scholar]
- 94.Crawford ED, et al. A Controlled Trial of Leuprolide with and without Flutamide in Prostatic Carcinoma. New England Journal of Medicine. 1989;321:419–424. doi: 10.1056/NEJM198908173210702. [DOI] [PubMed] [Google Scholar]
- 95.Kolvenbag GJ, Blackledge GR, Gotting-Smith K. Bicalutamide (Casodex) in the treatment of prostate cancer: history of clinical development. The Prostate. 1998;34:61–72. doi: 10.1002/(sici)1097-0045(19980101)34:1<61::aid-pros8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 96.Linja MJ, et al. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 97.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 98.Yap TA, et al. Drug discovery in advanced prostate cancer: translating biology into therapy. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2016.120. advance online publication. [DOI] [PubMed] [Google Scholar]
- 99.Scher HI, et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. New England Journal of Medicine. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 100.Bradbury RH, et al. Small-molecule androgen receptor downregulators as an approach to treatment of advanced prostate cancer. Bioorg Med Chem Lett. 2011;21:5442–5445. doi: 10.1016/j.bmcl.2011.06.122. [DOI] [PubMed] [Google Scholar]
- 101.Bradbury RH, et al. Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer. Bioorg Med Chem Lett. 2013;23:1945–1948. doi: 10.1016/j.bmcl.2013.02.056. [DOI] [PubMed] [Google Scholar]
- 102.Loddick SA, et al. AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol Cancer Ther. 2013;12:1715–1727. doi: 10.1158/1535-7163.mct-12-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Omlin A, et al. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer – results of two parallel first-in-human phase I studies. Investigational New Drugs. 2015;33:679–690. doi: 10.1007/s10637-015-0235-5. [DOI] [PubMed] [Google Scholar]
- 104.Williams R. Discontinued drugs in 2012: oncology drugs. Expert opinion on investigational drugs. 2013;22:1627–1644. doi: 10.1517/13543784.2013.847088. [DOI] [PubMed] [Google Scholar]
- 105.Li H, et al. Characterization of a New Class of Androgen Receptor Antagonists with Potential Therapeutic Application in Advanced Prostate Cancer. Molecular Cancer Therapeutics. 2013;12:2425–2435. doi: 10.1158/1535-7163.mct-13-0267. [DOI] [PubMed] [Google Scholar]
- 106.Neklesa TK, Crews CM. Chemical biology: Greasy tags for protein removal. Nature. 2012;487:308–309. doi: 10.1038/487308a. [DOI] [PubMed] [Google Scholar]
- 107.Long MJ, Gollapalli DR, Hedstrom L. Inhibitor mediated protein degradation. Chem Biol. 2012;19:629–637. doi: 10.1016/j.chembiol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Coffey RT, Shi Y, Long MJ, Marr MT, 2nd, Hedstrom L. Ubiquilin-mediated Small Molecule Inhibition of Mammalian Target of Rapamycin Complex 1 (mTORC1) Signaling. J Biol Chem. 2016;291:5221–5233. doi: 10.1074/jbc.M115.691584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kubota H. Quality control against misfolded proteins in the cytosol: a network for cell survival. J Biochem. 2009;146:609–616. doi: 10.1093/jb/mvp139. [DOI] [PubMed] [Google Scholar]
- 110.Neklesa TK, et al. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat Chem Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis. Methods Mol Biol. 2007;356:195–208. doi: 10.1385/1-59745-217-3:195. [DOI] [PubMed] [Google Scholar]
- 112.Tae HS, et al. Identification of hydrophobic tags for the degradation of stabilized proteins. Chembiochem. 2012;13:538–541. doi: 10.1002/cbic.201100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neklesa TK, et al. A bidirectional system for the dynamic small molecule control of intracellular fusion proteins. ACS Chem Biol. 2013;8:2293–2300. doi: 10.1021/cb400569k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xie T, et al. Pharmacological targeting of the pseudokinase Her3. Nat Chem Biol. 2014;10:1006–1012. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lim SM, et al. Development of small molecules targeting the pseudokinase Her3. Bioorg Med Chem Lett. 2015;25:3382–3389. doi: 10.1016/j.bmcl.2015.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gustafson JL, et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew Chem Int Ed Engl. 2015;54:9659–9662. doi: 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Teutsch G, et al. Non-steroidal antiandrogens: synthesis and biological profile of high-affinity ligands for the androgen receptor. J Steroid Biochem Mol Biol. 1994;48:111–119. doi: 10.1016/0960-0760(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 118.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yaron A, et al. Identification of the receptor component of the I kappa B alpha-ubiquitin ligase. Nature. 1998;396:590–594. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 120.Griffith EC, et al. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chemistry & Biology. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]
- 121.Sin N, et al. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proceedings of the National Academy of Sciences. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sakamoto KM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 123.Hon WC, et al. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 124.Min JH, et al. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 125.Schneekloth JS, Jr, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 126.Bargagna-Mohan P, Baek SH, Lee H, Kim K, Mohan R. Use of PROTACS as molecular probes of angiogenesis. Bioorganic & Medicinal Chemistry Letters. 2005;15:2724–2727. doi: 10.1016/j.bmcl.2005.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rodriguez-Gonzalez A, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang D, Baek SH, Ho A, Kim K. Degradation of target protein in living cells by small-molecule proteolysis inducer. Bioorganic and Medicinal Chemistry Letters. 2004;14:645–648. doi: 10.1016/j.bmcl.2003.11.042. [DOI] [PubMed] [Google Scholar]
- 129.Henning RK, et al. Degradation of Akt using protein-catalyzed capture agents. J Pept Sci. 2016 doi: 10.1002/psc.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chu TT, et al. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chemical Biology. 2016;23:453–461. doi: 10.1016/j.chembiol.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 131.Hines J, Gough JD, Corson TW, Crews CM. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proceedings of the National Academy of Sciences. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cong F, Zhang J, Pao W, Zhou P, Varmus H. A protein knockdown strategy to study the function of β-catenin in tumorigenesis. BMC Molecular Biology. 2003;4:1–11. doi: 10.1186/1471-2199-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vassilev LT, et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 134.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ding Q, et al. Discovery of RG7388, a Potent and Selective p53-MDM2 Inhibitor in Clinical Development. Journal of Medicinal Chemistry. 2013;56:5979–5983. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- 136.Vu B, et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Medicinal Chemistry Letters. 2013;4:466–469. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Andreeff M, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22:868–876. doi: 10.1158/1078-0432.ccr-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. Journal of the American Chemical Society. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 139.Umezawa H, Aoyagi T, Suda H, Hamada M, Takeuchi T. Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. The Journal of antibiotics. 1976;29:97–99. doi: 10.7164/antibiotics.29.97. [DOI] [PubMed] [Google Scholar]
- 140.Orning L, Krivi G, Fitzpatrick FA. Leukotriene A4 hydrolase. Inhibition by bestatin and intrinsic aminopeptidase activity establish its functional resemblance to metallohydrolase enzymes. Journal of Biological Chemistry. 1991;266:1375–1378. [PubMed] [Google Scholar]
- 141.Sekine K, et al. Small Molecules Destabilize cIAP1 by Activating Auto-ubiquitylation. Journal of Biological Chemistry. 2008;283:8961–8968. doi: 10.1074/jbc.M709525200. [DOI] [PubMed] [Google Scholar]
- 142.Itoh Y, et al. Development of target protein-selective degradation inducer for protein knockdown. Bioorganic & Medicinal Chemistry. 2011;19:3229–3241. doi: 10.1016/j.bmc.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 143.Itoh Y, et al. Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorganic & Medicinal Chemistry Letters. 2012;22:4453–4457. doi: 10.1016/j.bmcl.2012.04.134. [DOI] [PubMed] [Google Scholar]
- 144.Demizu Y, et al. Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorganic & Medicinal Chemistry Letters. 2012;22:1793–1796. doi: 10.1016/j.bmcl.2011.11.086. [DOI] [PubMed] [Google Scholar]
- 145.Itoh Y, Kitaguchi R, Ishikawa M, Naito M, Hashimoto Y. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg Med Chem. 2011;19:6768–6778. doi: 10.1016/j.bmc.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 146.Ohoka N, et al. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis. 2014;5:e1513. doi: 10.1038/cddis.2014.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Buckley DL, et al. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. Journal of the American Chemical Society. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Buckley DL, et al. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angewandte Chemie International Edition. 2012;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Van Molle I, et al. Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1alpha protein-protein interface. Chem Biol. 2012;19:1300–1312. doi: 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Galdeano C, et al. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J Med Chem. 2014;57:8657–8663. doi: 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ito T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 152.Kronke J, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lu G, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gandhi AK, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. British Journal of Haematology. 2014;164:811–821. doi: 10.1111/bjh.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fischer ES, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kronke J, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Petzold G, Fischer ES, Thoma NH. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4 ubiquitin ligase. Nature. 2016 doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 158.Adès L, Fenaux P. Immunomodulating Drugs in Myelodysplastic Syndromes. ASH Education Program Book. 2011:556–560. doi: 10.1182/asheducation-2011.1.556. (2011) [DOI] [PubMed] [Google Scholar]
- 159.Matyskiela ME, et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature. 2016 doi: 10.1038/nature18611. advance online publication. [DOI] [PubMed] [Google Scholar]
- 160.Lu J, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shimamura T, et al. Efficacy of BET Bromodomain Inhibition in Kras-Mutant Non–Small Cell Lung Cancer. Clinical Cancer Research. 2013;19:6183–6192. doi: 10.1158/1078-0432.ccr-12-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Raina K, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of the National Academy of Sciences. 2016 doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chemical Biology. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Golas JM, et al. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63:375–381. [PubMed] [Google Scholar]
- 165.O’Hare T, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–4505. doi: 10.1158/0008-5472.can-05-0259. [DOI] [PubMed] [Google Scholar]
- 166.Duda DM, et al. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fischer ES, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 168.Liu J, Nussinov R. Flexible Cullins in Cullin-RING E3 Ligases Allosterically Regulate Ubiquitination. The Journal of Biological Chemistry. 2011;286:40934–40942. doi: 10.1074/jbc.M111.277236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liu J, Nussinov R. The Mechanism of Ubiquitination in the Cullin-RING E3 Ligase Machinery: Conformational Control of Substrate Orientation. PLoS Computational Biology. 2009;5:e1000527. doi: 10.1371/journal.pcbi.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 171.Deshaies RJ. Protein degradation: Prime time for PROTACs. Nat Chem Biol. 2015;11:634–635. doi: 10.1038/nchembio.1887. [DOI] [PubMed] [Google Scholar]
- 172.Toure M, Crews CM. Small-Molecule PROTACS. New Approaches to Protein Degradation. Angew Chem Int Ed Engl. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 173.Lou KJ. PROTAC the protein. SciBx. 2012;5 doi: 10.1038/scibx.2012.514. [DOI] [Google Scholar]
- 174.Bouchie A, Allison M, Webb S, DeFrancesco L. Nature Biotechnology’s academic spinouts of 2013. Nat Biotech. 2014;32:229–238. doi: 10.1038/nbt.2846. [DOI] [PubMed] [Google Scholar]
- 175.Chi KR. Drug developers delve into the cell’s trash-disposal machinery. Nat Rev Drug Discov. 2016;15:295–297. doi: 10.1038/nrd.2016.86. [DOI] [PubMed] [Google Scholar]
- 176.Buckley DL, Crews CM. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew Chem Int Ed Engl. 2014;53:2312–2330. doi: 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Buckley DL, et al. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chemical Biology. 2015;10:1831–1837. doi: 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 179.Maculins T, et al. A Generic Platform for Cellular Screening Against Ubiquitin Ligases. Scientific Reports. 2016;6:18940. doi: 10.1038/srep18940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chan CH, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Orlicky S, et al. An allosteric inhibitor of substrate recognition by the SCFCdc4 ubiquitin ligase. Nat Biotech. 2010;28:733–737. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Aghajan M, et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat Biotech. 2010;28:738–742. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Deshaies RJ, Joazeiro CAP. RING Domain E3 Ubiquitin Ligases. Annual Review of Biochemistry. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 184.DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. The Journal of antibiotics. 1970;23:442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- 185.Rinehart KL, Sasaki K, Slomp G, Grostic MF, Olson EC. Geldanamycin. I. Structure assignment. Journal of the American Chemical Society. 1970;92:7591–7593. doi: 10.1021/ja00729a018. [DOI] [PubMed] [Google Scholar]
- 186.Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer chemotherapy and pharmacology. 1995;36:305–315. doi: 10.1007/bf00689048. [DOI] [PubMed] [Google Scholar]
- 187.Ramanathan RK, et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:3385–3391. doi: 10.1158/1078-0432.ccr-04-2322. [DOI] [PubMed] [Google Scholar]
- 188.Modi S, et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:5132–5139. doi: 10.1158/1078-0432.ccr-11-0072. [DOI] [PubMed] [Google Scholar]
- 189.Demetri GD, A. L. C. Von Mehren M, Chmielowski B, Bauer S, Chow WA, Rodenas E, McKee K, Grayzel DS, Kang Y. 2010 Gastrointestinal Cancers Symposium. 2010 [Google Scholar]
- 190.Lundgren K, et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol Cancer Ther. 2009;8:921–929. doi: 10.1158/1535-7163.mct-08-0758. [DOI] [PubMed] [Google Scholar]
- 191.Dickson MA, et al. Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Annals of Oncology. 2013;24:252–257. doi: 10.1093/annonc/mds275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mitchell P. Biogen Idec restructures, sharpens neurology focus. Nat Biotech. 2011;29:7–8. doi: 10.1038/nbt0111-7. [DOI] [PubMed] [Google Scholar]
- 193.Eccles SA, et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–2860. doi: 10.1158/0008-5472.can-07-5256. [DOI] [PubMed] [Google Scholar]
- 194.Lin TY, et al. The novel HSP90 inhibitor STA-9090 exhibits activity against Kit-dependent and -independent malignant mast cell tumors. Experimental hematology. 2008;36:1266–1277. doi: 10.1016/j.exphem.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ramalingam S, et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1) Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2015;26:1741–1748. doi: 10.1093/annonc/mdv220. [DOI] [PubMed] [Google Scholar]
- 196.Suman Chatterjee SB, Socinski Mark A, Burns Timothy F. HSP90 Inhibitors in Lung Cancer: Promise Still Unfulfilled. Clinical Advances in Hematology & Oncology. 2016;14:346–356. [PubMed] [Google Scholar]
- 197.Woodhead AJ, et al. Discovery of (2,4-Dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl]methanone (AT13387), a Novel Inhibitor of the Molecular Chaperone Hsp90 by Fragment Based Drug Design. Journal of Medicinal Chemistry. 2010;53:5956–5969. doi: 10.1021/jm100060b. [DOI] [PubMed] [Google Scholar]
- 198.Wagner AJ, et al. Dose-escalation study of a second-generation non-ansamycin HSP90 inhibitor, onalespib (AT13387), in combination with imatinib in patients with metastatic gastrointestinal stromal tumour. European Journal of Cancer. 2016;61:94–101. doi: 10.1016/j.ejca.2016.03.076. [DOI] [PubMed] [Google Scholar]
- 199.Rakhit R, Navarro R, Wandless TJ. Chemical biology strategies for posttranslational control of protein function. Chem Biol. 2014;21:1238–1252. doi: 10.1016/j.chembiol.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Raina K, et al. Targeted protein destabilization reveals an estrogen-mediated ER stress response. Nat Chem Biol. 2014;10:957–962. doi: 10.1038/nchembio.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- 202.Takatsuki A, Arima K, Tamura G. Tunicamycin, a new antibiotic. I. Isolation and characterization of tunicamycin. The Journal of antibiotics. 1971;24:215–223. doi: 10.7164/antibiotics.24.215. [DOI] [PubMed] [Google Scholar]
- 203.Miyazaki Y, Chen LC, Chu BW, Swigut T, Wandless TJ. Distinct transcriptional responses elicited by unfolded nuclear or cytoplasmic protein in mammalian cells. Elife. 2015;4 doi: 10.7554/eLife.07687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nature reviews Molecular cell biology. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Winston JT, et al. The SCF(β-TRCP)–ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes & Development. 1999;13:270–283. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 Is a RING Finger-dependent Ubiquitin Protein Ligase for Itself and p53. Journal of Biological Chemistry. 2000;275:8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 208.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chau V, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 210.Lu Y, Lee BH, King RW, Finley D, Kirschner MW. Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science. 2015;348:1250834. doi: 10.1126/science.1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Komander D, Rape M. The Ubiquitin Code. Annual Review of Biochemistry. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 212.Husnjak K, Dikic I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annual Review of Biochemistry. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- 213.MacGurn JA, Hsu PC, Emr SD. Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev Biochem. 2012;81:231–259. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 214.Lee H, Puppala D, Choi EY, Swanson H, Kim KB. Targeteddegradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. Chembiochem. 2007;8:2058–2062. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- 215.Tomoshige S, Naito M, Hashimoto Y, Ishikawa M. Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules. Organic & biomolecular chemistry. 2015;13:9746–9750. doi: 10.1039/c5ob01395j. [DOI] [PubMed] [Google Scholar]