

Abstract
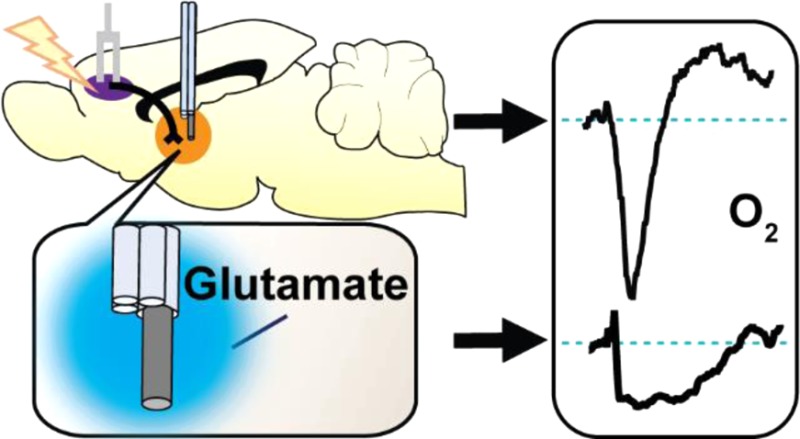
Glutamate is ubiquitous throughout the brain and serves as the primary excitatory neurotransmitter. Neurons require energy to fire, and energetic substrates (i.e., O2, glucose) are renewed via cerebral blood flow (CBF) to maintain metabolic homeostasis. Magnetic resonance brain functionality studies rely on the assumption that CBF and neuronal activity are coupled consistently throughout the brain; however, the origin of neuronal activity does not always coincide with signals indicative of energy consumption (e.g., O2 decreases) at high spatial resolutions. Therefore, relationships between excitatory neurotransmission and energy use must be evaluated at higher resolutions. In this study, we showed that both endogenously released and exogenously ejected glutamate decrease local tissue O2 concentrations, but whether hyperemic O2 restoration followed depended on the stimulus method. Electrically stimulating the glutamatergic corticostriatal pathway evoked biphasic O2 responses at striatal terminals: first O2 decreased, then concentrations increased above baseline. Using iontophoresis to locally eject ionotropic glutamate receptor antagonists revealed that these receptors only influenced the O2 decrease. We compared electrical stimulation to iontophoretic glutamate stimulation, and measured concurrent single-unit activity and O2 to limit both stimulation and recordings to <50 μm radius from our sensor. Similarly, iontophoretic glutamate delivery elicited monophasic O2 decreases without subsequent increases.
Keywords: Glutamate, hyperemia, iontophoresis, ionotropic glutamate receptors, neurovascular coupling, single-unit electrophysiology
Introduction
Despite accounting for a small portion of total body mass, the brain uses a disproportionately large percentage of the body’s resting energy. This discrepancy arises from the energetically expensive demands of neuronal communication and the maintenance of ionic gradients.1 Metabolic substrates (i.e., glucose and O2) are replenished after neuronal activity via increased regional cerebral blood flow (CBF), where they are consumed to generate adenosine triphosphate (ATP) and reestablish ion gradients. To maintain energetic homeostasis, neuronal activity is typically coupled with increased CBF in a process termed functional hyperemia.2 However, dysfunctional decoupling of neuronal activity and CBF can lead to chronic energy deficits where blood flow cannot replenish the energy consumed during neurotransmission. With insufficient energy, ionic gradients collapse, often leading to neuronal death.3 However, CBF delivery is not exclusively controlled by metabolic deficiencies, as the amplitudes of task-evoked CBF increases are maintained when the same task is performed under hypoglycemic or hypoxic conditions.4,5 Further, although CBF delivers excess O2 to overcompensate for energy loss, the O2 concentrations delivered vary depending on stimulus type and intensity.6−8 Together, these suggest that neuronal activity and metabolic deficits play complex roles in the regulation of energy throughout the brain.
Often, CBF is measured indirectly by monitoring extracellular O2 concentrations, where increases in O2 can be attributed to hyperemic increases in blood flow. This is the basis for blood oxygenation level-dependent (BOLD) functional magnetic resonance imaging (fMRI);9 however, positive BOLD fMRI signals (i.e., increases in oxygenated blood) only infer the presence of increased neural activity.10 Fast-scan cyclic voltammetry (FSCV) also measures O2 changes, but the carbon-fiber microelectrodes used with the technique limit analyte detection to a range within a 20 μm radius of the electrode surface.11−13 As with BOLD, these electrochemical measurements also do not provide information about neuronal activity. Previously, we used a multimodal sensor to perform simultaneous FSCV and single-unit electrophysiology.14 Here, we utilize a different voltammetric waveform at our sensor to study colocalized O2 dynamics in conjunction with cell firing. This approach uses a single, minimally invasive carbon-fiber microelectrode and enables simultaneous detection of both neuronal activity and O2 changes in localized environments. Further, we coupled iontophoresis, a local drug delivery method, to FSCV and electrophysiology at the same sensor to form a comprehensive suite of techniques suited for spatially focused functional hyperemia studies.15 Using iontophoresis to eject glutamate, an excitatory neurotransmitter, stimulated local neuronal firing in anesthetized subjects and gave us a high degree of control over the cell activity proximal to our sensor.16−18 Together, we used our multimodal sensor to stimulate and measure neuronal firing and the subsequent O2 changes within a <50 μm radius of brain tissue.
In this study, we compared the effects of electrically stimulated glutamate versus local glutamate iontophoresis on O2 dynamics between the striatum and somatosensory cortex. We first elicited endogenous glutamate release via electrically stimulating the prefrontal cortex (PFC) and found that striatal O2 concentrations both increased and decreased in a manner dependent on stimulation intensity and the availability of ionotropic glutamate receptors (iGluRs). In contrast, glutamate ejected locally within the striatum evoked either O2 decreases exclusively or did not change local O2 concentrations. The ejection method, controlled iontophoresis, uses current to deliver solution aliquots on the order of femto- to picomoles and measures the solution delivery.19 Previously, we showed that iontophoresis ejection currents themselves do not influence cell firing (i.e., ejecting NaCl) but that glutamate iontophoresis has the intended excitatory effect of eliciting cell firing.20 Finally, we compared glutamate iontophoresis evoked changes in O2 and cell firing between cortical and striatal locations. The majority of recording locations responded to glutamate iontophoresis with increased cell firing and subsequent O2 decreases; however, exceptions were found in both cortex and striatum. Together, these data show that global, electrically evoked glutamate release and local, chemically specific glutamatergic excitation produce dissimilar O2 changes and that neuronal firing does not always couple to O2 use throughout the cortex and striatum.
Results
Electrically Stimulating PFC Glutamate Cell Bodies Evokes O2 Responses at Striatal Terminals
Glutamatergic neurons project from the PFC to medium spiny neurons (MSNs) in the striatum.21,22 We recorded O2 changes at locations where glutamate iontophoresis elicited single-unit activity, that is, near the soma of glutamate-sensitive cells (Figure 1A). Unit activity was evoked during the glutamate ejection and ceased upon stimulation termination (Figure 1A). These cells were classified as MSNs, which account for over 90% of the cells in the striatum,23 based on their waveform shapes and low firing rates (Figure 1A). Next, we electrically stimulated the PFC to drive glutamate release in the striatum (Figure 1B) and recorded subsequent O2 changes at glutamate-sensitive neurons. Electrical stimulations interfered with electrophysiological recordings, so we were unable to assess the single-unit activity during PFC stimulations. With FSCV, we observed robust, reproducible O2 responses that consisted of two distinct phases: the first, event 1, was a decrease in O2, followed by an O2 increase to above baseline values, termed event 2 (Figure 1C). These biphasic responses were consistent with poststimulus O2 changes observed both locally using amperometry at microelectrodes8,24 and globally with BOLD fMRI.8,10,24 By comparison, 2 s glutamate iontophoresis ejections in the same locations evoked O2 decreases of a similar magnitude as the electrical stimulations but failed to ever produce an event 2 (Figure 1D).
Figure 1.

Electrically stimulated glutamatergic cell bodies in the PFC elicit biphasic O2 changes in the striatum. (A) (i) The multimodal sensor ejected glutamate iontophoretically to determine proximity to glutamate-sensitive cells (e.g., medium spiny neuron, MSN). Sensor barrel diameters and carbon-fiber length not to scale. (ii) Four replicate perievent rasters, where each black tic represents a recorded single-unit action potential. In anesthetized subjects, glutamate ejections (gray box) evoked single-unit activity bursts that ceased soon after the ejection. (iii) Averaged single-unit waveform of the unit isolated in panel A, part ii. (B) Sagittal view of the chosen stimulated glutamatergic pathway. A bipolar stimulating electrode was placed into the PFC (blue) and a multimodal sensor was inserted into the dorsal striatum (orange). (C) (top) Current taken at the O2 reduction potential (−1.35 V, horizontal dotted line in color plot below) tracked the time course of O2 changes from the representative color plot below. First, O2 decreased during the stimulation (event 1) and subsequently increased above baseline following electrical stimulation (event 2). Cyclic voltammograms from each event (insets, each taken from horizontal white lines in color plot below) confirmed the identity of O2, with background subtraction taken at the color plot black dashed line. Slight hysteresis was attributed to concomitant endogenous ion fluctuations. (bottom) Color plot representation of cyclic voltammetry data recorded around a 100 pulse, 20 Hz, 300 μA electrical stimulation as indicated by the gray bar. Abscissa, applied voltage; ordinate, acquisition time of the cyclic voltammograms. Background-subtracted currents are color coded. (D) Averaged O2 changes evoked from 2 s glutamate iontophoresis ejections at the glutamate-sensitive striatal cells before the first electrical stimulation and 10 min after the last electrical stimulation (n = 6). Shading indicates −SEM. (E) Experimental paradigm for assessing local striatal iGluR influence over O2. PFC cell bodies were electrically stimulated after a cocktail of AP5 (50 mM) and CNQX (10 mM) was ejected at striatal terminals for 60 s. (F) Event 1 was significantly attenuated (**P = 0.001, Student’s t test) under iGluR blockade in the striatum. Error bars indicate +SEM.
To confirm whether glutamatergic neurotransmission was responsible for the biphasic O2 response, we iontophoresed iGluR antagonists for 60 s prior to electrical stimulation (Figure 1E). This was to achieve steady-state ejection conditions and saturate the tissue within ∼100 μm of our sensor.25 After collecting a predrug stimulation response, we delivered the NMDA receptor antagonist AP5 (50 mM) and the AMPA and kainate receptor antagonist CNQX (10 mM) for 60 s from a single iontophoresis barrel attached to the carbon-fiber electrode. After drug delivery, we stimulated the PFC again and compared the striatal O2 changes before and after iGluR antagonism (Figure 1F). We chose a stimulation of 80 pulses delivered at 20 Hz, as this was the shortest stimulation train capable of reliably eliciting biphasic O2 responses (Figure 2A,B). In all cases, the poststimulus O2 decrease (event 1) significantly diminished under local iGluR blockade to 44.8% ± 11.1% of predrug values (t(2,8) = 5.0, P = 0.001, n = 5; Figure 1F), while differences in event 2 amplitudes were negligible. The attenuation of the evoked response with iGluR antagonists confirmed that PFC stimulations released endogenous glutamate into the striatum and indicated that local iGluR activation significantly contributed to O2 decreases but not the overcompensating O2 rebound.
Figure 2.
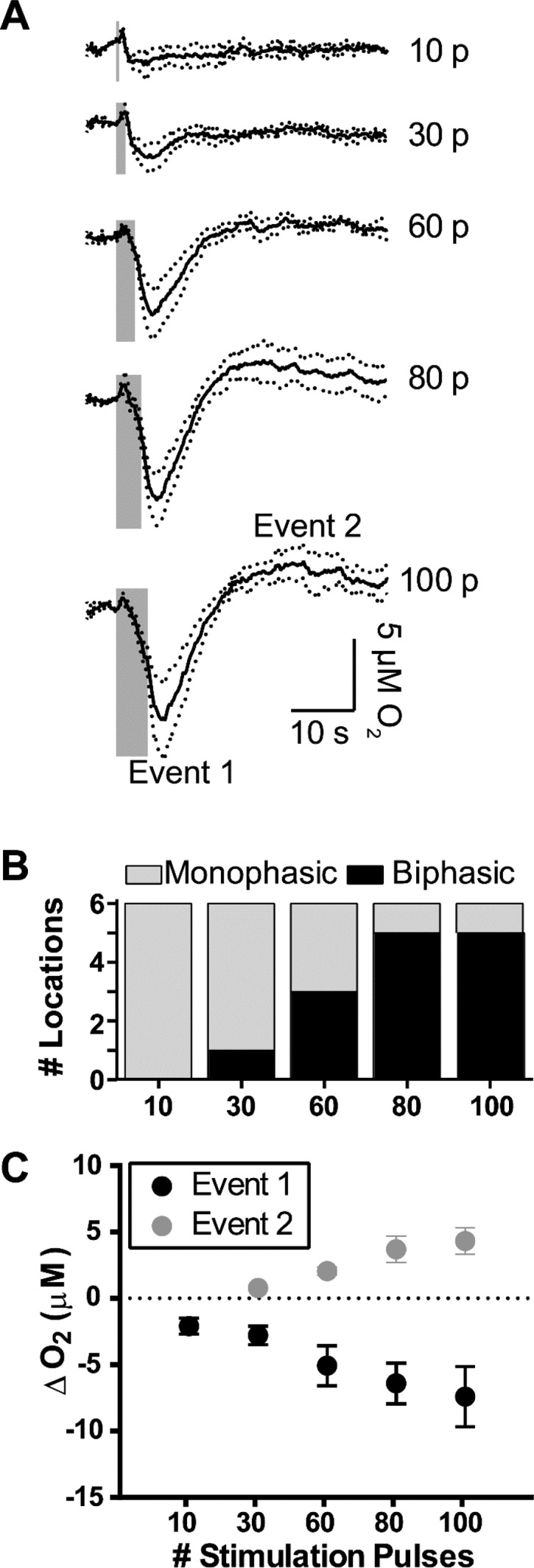
PFC electrical stimulations controlled the biphasic O2 response in the striatum through the number of applied stimulation pulses. (A) Current traces taken at the O2 reduction potential were averaged (n = 6). The number of electrical stimulation pulses used (p) are indicated to the right of the traces. Stimulation frequency and amplitude were held constant at 20 Hz and 300 μA, respectively. Gray bars indicate stimulation durations. Dotted lines indicate ±SEM. (B) The relative occurrence of biphasic O2 responses to electrical stimulation increased with the number of stimulus pulses. (C) The maximum poststimulus O2 decreases (event 1) and increases (event 2) increased linearly (event 1, R2 = 0.990; event 2, R2 = 0.975) with the number of stimulus pulses. Monophasic data (i.e., decreased O2 responses that did not return to baseline within 60 s) were excluded from plotted event 2 points. Data points represent averaged triplicate responses from each location. Error bars indicate ±SEM.
Biphasic O2 Responses Depend on Electrical Stimulation Parameters
We next investigated the dependence of the observed O2 events on the stimulation parameters to explore what stimulus was sufficient to elicit a biphasic, hyperemic response. First, we kept stimulation frequencies and amplitudes constant at 20 Hz and 300 μA, respectively, and changed the number of stimulation pulses from 10–100 pulses (Figure 2A). The number of locations that responded with biphasic O2 changes increased with the number of stimulus pulses (Figure 2B). Furthermore, the relationship was linear between both biphasic event amplitudes and the corresponding number of stimulation pulses (event 1, R2 = 0.990; event 2, R2 = 0.975, Figure 2C). All O2 changes from 10 pulse stimulations were monophasic, so we omitted these data from the linear fit. This confirmed that both mono- and biphasic O2 responses can be elicited at the same location, identified a pulse-dependent stimulus threshold that must be exceeded before biphasic responses occurred, and showed a strong linear relationship between the number of stimulation pulses and the resulting O2 event amplitudes.
To ensure that the pulse-dependent stimulus effects were not caused by an increase in total stimulation length, we kept pulse numbers and amplitudes constant (120 pulses and 300 μA, respectively) and tested 5–60 Hz frequencies. These frequencies did not affect either event 1 or event 2 (when applicable) amplitudes or influence whether responses were mono- or biphasic (Figure 3A,B). Despite consistent amplitude changes across different frequencies, the time interval between stimulations and maximum O2 event changes significantly increased from higher to lower frequency stimulations (one-way ANOVA, event 1, F(6,53) = 6.26, P = 0.0002; event 2, F(6,35) = 5.47, P = 0.003, Figure 3C). Bonferroni post hoc analysis revealed significant differences in event 1 minima times between both 5 and 10 Hz stimulations as compared to higher stimulation frequencies (5 Hz, 13.6 ± 2.5 s; 10 Hz, 13.1 ± 1.0 s; 30 Hz, 6.5 ± 0.5 s; 40 Hz, 6.4 ± 0.8 s; and 60 Hz, 6.2 ± 0.5 s; 5 Hz versus 30, 40, and 60 Hz: P < 0.01 each, n = 6 each; 10 Hz versus 30 and 40 Hz: P < 0.05 each; 10 Hz versus 60 Hz: P < 0.01, n = 6 each, Figure 3C). Time differences between stimulations and event 2 maxima (when present) also increased from 5 and 10 Hz stimulation frequencies to 60 Hz (5 Hz, 33.1 ± 7.5 s, n = 2; 10 Hz, 30.8 ± 2.8 s, n = 4; 60 Hz, 18.6 ± 0.5 s; 5 and 10 Hz versus 60 Hz: P < 0.05 each, Figure 3C). These findings indicated that, without affecting O2 event magnitudes, low frequency stimuli increased the time elapsed between electrical stimulations and both the maximum event 1 and event 2 O2 decreases and increases, respectively.
Figure 3.

PFC stimulation frequencies affected the time to reach maximal O2 decreases (i.e., event 1) and subsequent maximal O2 increases (i.e., event 2) in the dorsal striatum. (A) Current traces taken at the O2 reduction potential were averaged (n = 6), with their respective stimulation frequencies indicated to the right. The number of biphasic pulses applied and their amplitudes were kept constant (80 pulses and 300 μA, respectively). Gray bars indicate stimulation durations. Dotted lines indicate ±SEM. (B) Stimulus frequency did not affect whether or not the responses were biphasic. (C) The stimulus frequency affected the time to reach maximum event 1 O2 decreases and the subsequent event 2 O2 increases. Monophasic O2 responses were excluded from event 2 statistics. Error bars indicate ±SEM. Significance was determined with repeated measures one-way ANOVA and Bonferroni’s post hoc test. *P < 0.05, **P < 0.01.
Glutamate Iontophoresis Provokes Local Single-Unit Firing and O2 Decreases
We next delivered glutamate directly to the recording locations to exclude the confounding effects of nonglutamatergic neurotransmitters that could be released from electrical stimulation. Experiments started in the somatosensory cortex as proof of principle, then extended deeper within the brain through the striatum. We ejected glutamate from an iontophoresis barrel in 2 s pulses, until electrophysiology could resolve single-unit firing from background noise at the carbon fiber (Figure 4A,C). The amount of glutamate ejected was not quantified for each experiment, as the concentration gradient from a 2 s iontophoretic ejection overestimates the quantity ejected when using a standard calibration curve;26 instead, a steady-state glutamate concentration was used to estimate ejected quantities. A 120 s controlled iontophoresis ejection using 100 nA, with an assumed 175 pL s–1 ejection rate based on the ejection current,25 yielded a 3–5 mM glutamate steady-state plateau. Therefore, glutamate iontophoresis for 2 s at 100 nA ejected glutamate on the order of 1–2 pmol. Though steady-state ejections can diffuse solution to a radius of several hundreds of micrometers, a 2 s ejection is unlikely to diffuse much further than the barrel tip,26 especially with the efficiency of glial glutamate transporters.27
Figure 4.

Multimodal sensors characterized a variety of microenvironments based on the effects of local glutamate-elicited excitation via iontophoresis. Recording locations fell into one of two broad categories, those that responded to glutamate with excitatory single-unit activity, that is, glutamate-sensitive cells (A, C), and those that did not (B, D). Within each group, glutamate ejections either were succeeded by an O2 decrease (A, B) or experienced no change in O2 relative to baseline (C, D). Black tics (top) indicate action potentials during each of the four repeated trials shown. Gray boxes indicate the glutamate ejection duration. The averaged O2 traces from the four repeated trials are shown (bottom). Baseline concentrations are shown as a dotted line. Sharp peaks immediately prior to and following ejections reflect an ejection artifact.
We investigated differences between the role of glutamate in superficial and deep brain environments, exploring the somatosensory cortex and the striatum. Cortical cells required >100 nA cathodic current to respond and were most often found at or below 0.9 mm D-V, corresponding to the most densely populated layers IV–VIb. These observations were consistent with published observations,16,17,28 and validated our approach to locally activate cells with iontophoresed glutamate. Meanwhile, in agreement with the literature,29 we observed denser cell responses in the ventral striatum as compared to the dorsal striatum. Striatal cells responded using only 20 to 80 nA ejection currents, which was both less than the >100 nA ejection currents required for cortical cells16 and consistent with previous reports.30,25,26 The difference in ejection currents between brain regions is worth noting, as the amount of glutamate ejected per 2 s scales with ejection current.25 The consistent difference in the ejection currents required to elicit cell firing in the cortex versus the striatum suggested that glutamate sensitivity may differ between these two brain regions. However, cells also required different glutamate ejection currents within the same brain region, which we attribute to either cell proximity to the drug barrel, cell sensitivity to glutamate, or the availability of local excitatory receptors.17 These differences both between and within brain regions may be extremely subtle given that picomole or sub-picomole quantities of glutamate are ejected using currents above and below 100 nA, respectively.
We used FSCV to record O2 changes during glutamate iontophoresis and simultaneous single-unit activity recording. In contrast to electrical stimulations, we observed one of two effects in the recorded environments following glutamate iontophoresis: either an exclusive monophasic O2 decrease (i.e., event 1 not followed by event 2; Figure 4A,B) or negligible O2 changes (Figure 4C,D). The monophasic O2 decreases typically returned to baseline within 40 s (Figure 4A,B). This contradicted a previous study that found a robust O2 increase following a local pressure ejection of 0.5 nmol glutamate aliquots.31 In contrast, our generous estimate of the glutamate delivered from a 2 s iontophoretic ejection was orders of magnitude less, 1–2 pmol. Overall, our sensor recorded one of four responses to glutamate at any given location. Either glutamate evoked cell firing or it did not, and the ejection was either succeeded by O2 decreases or did not perturb local tissue O2 (Figure 4), indicating that sensor responses were sensitive to the natural heterogeneity of the surrounding brain tissue.
Glutamate Inhibits Fast-Spiking Interneuron Activity
While characterizing glutamate-responsive neurons in the striatum, we serendipitously observed fast-spiking interneurons in ventral locations, offering us the unique opportunity to study them in vivo. These cells represent only 5–10% of striatal neurons and were identified as interneurons based on the observed high spontaneous firing rates (Figure 5A). Though we were unable to positively identify the type of fast-spiking interneuron from single-unit recordings alone, the 2 to 20 Hz spontaneous firing rates observed in deeply anesthetized subjects were in line with spontaneous firing rates reported for cholinergic interneurons.23,32 Up to 3 spontaneously active cells could be discerned per fast-spiking interneuron recording location (Figure 5A,B), consistent with interneuron clustering.33 Local glutamate ejections significantly inhibited, rather than augmented, single-unit firing rates in the vast majority of these spontaneously active cells (49.0% ± 8.2% of baseline activity, t(2,36) = 5.91, n = 19, P < 0.0001, Figure 5C); however, glutamate did not affect the single-unit activity of 3 fast-spiking cells that were included in these statistics. One fast-spiking cell responded with augmented single-unit firing and was excluded as a Grubbs statistical outlier (P < 0.05; cell no. 1 in Figure 5A). In some instances, these ejections fully suppressed firing with extended ejection times (Figure 5B). Basal firing rates recovered after glutamate ejections ceased in all cases (examples in Figure 5B). To our knowledge, we are the first to show this explicit chemical relationship between the majority of striatal, fast-spiking interneurons and glutamate in vivo in an intact brain.
Figure 5.
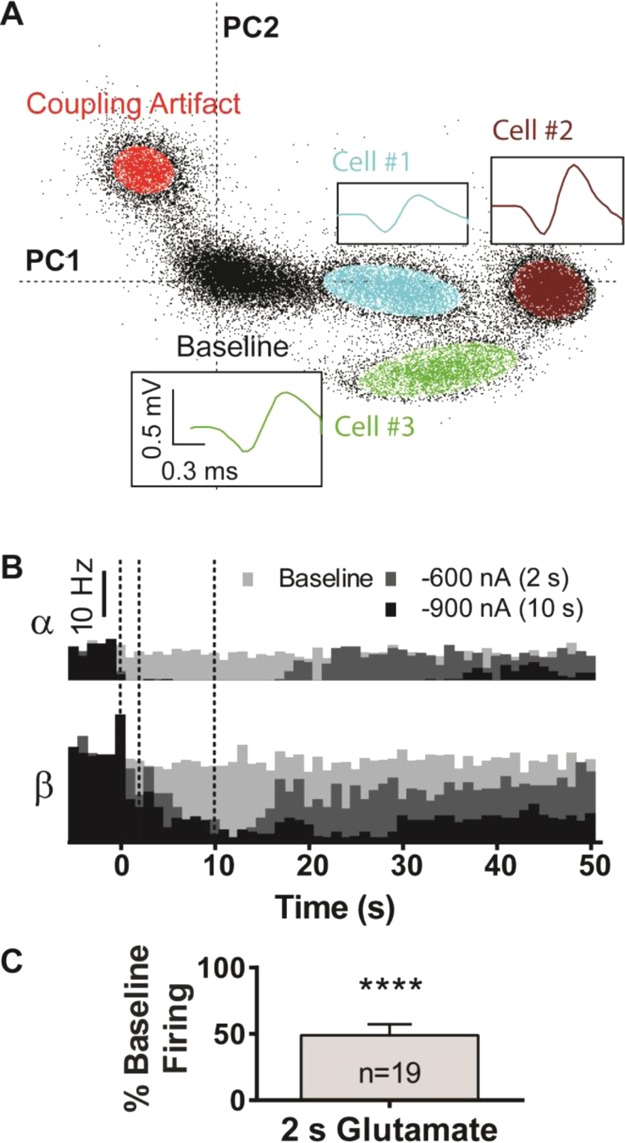
Glutamate inhibited fast spiking interneurons observed in the ventral striatum. (A) A 2D waveform density plot distinguished a cluster of three spontaneously fast-spiking cells at a single carbon fiber at one location, each resolved from the background. High firing rates (>10 Hz) produced large, dense clusters over the time course. Insets: Color-coded average action potential waveforms, with outliers >4σ removed. (B) Firing rate histograms for two interneurons (α and β), recorded at a different location than that of panel A. The firing rate of both cells attenuated during glutamate ejections and recovered after the ejections ended. The dashed lines from left to right represent the start of ejection, end of the 2 s ejection, and end of the 10 s ejection. (C) A 2 s glutamate ejection inhibited single-unit interneuron activity in the ventral striatum (Student’s t-test: t(2,36) = 5.91, P < 0.0001). Firing was normalized to the spontaneous firing during baseline recordings. Error bars indicate SEM.
Transient O2 Events
Finally, while characterizing glutamate responses in both cortical and striatal environments, we also observed spontaneous, discrete O2 changes throughout both regions (Figure 6). These transient O2 events occurred without reliably concurrent cell activity, presented during baseline recordings in absence of glutamate, and persisted throughout data collections unaffected by glutamate ejections. The cyclic voltammograms revealed that the fluctuations were not noise because they only occurred at the O2 reduction potential. The overlapping relationships between these transients and glutamate-stimulated O2 decreases and single-unit activity between cortical and striatal environments are summarized in Figure 7. A total of n = 197 discrete data sets were collected at both glutamate-sensitive and glutamate-insensitive locations in the cortex (n = 59 and 24, respectively) and striatum (n = 79 and 35, respectively, for MSNs). Additionally, we collected O2 data at fast-spiking interneuron locations (n = 16). For categorization purposes, we defined glutamate-sensitive cells as those that responded to glutamate ejections with increased single-unit activity, and transients as baseline-resolved O2 changes that exceeded 3 standard deviations above baseline noise. Interestingly, 90.0% of all recorded locations with glutamate-sensitive cells presented with spontaneous O2 transients. These transients appeared in 93.7% of striatal glutamate-sensitive recording locations (excluding interneurons) and 88.6% of striatal glutamate-insensitive locations (Figure 7). In contrast, spontaneous O2 transients presented at 80.8% of glutamate-sensitive cortical cell locations but only 54.8% of cortical glutamate-insensitive locations (Figure 7). These data revealed the high probability of encountering spontaneous O2 fluctuations in close proximity to glutamate-sensitive neurons, but further studies are needed to determine their significance.
Figure 6.

Locations exhibited spontaneous O2 transients in (A) a layer Vb cortical cell, (B) a medium spiny neuron (MSN) in the striatum, and (C) an interneuron in the ventral striatum. Each black hash (top) indicates a single recorded action potential. Clean and discrete O2 events presented alone, with no other significant electrochemical signals (color plots, middle). Current traces (bottom) were obtained by setting the background current such that all events were O2 decreases/positive current deflections for consistency.
Figure 7.

Majority of glutamate-sensitive cell locations in the striatum and cortex both led to an O2 decrease following glutamatergic excitation and exhibited spontaneous O2 transients. Circles for each cell category (i.e., cortical, striatal, and interneuron cells) are drawn to scale. Numbers indicate the quantity of discrete recording locations within a category. The color of each circle corresponds to a category as defined in the legend.
Discussion
In this study, we extended the use of an existing multimodal sensor to investigate the relationship between glutamatergic neurotransmission and O2 responses in highly localized environments. We assessed the differences between stimulated (global, nonspecific) and iontophoresed (local, specific) glutamate responses. Endogenous glutamate release via electrical PFC stimulation elicited a biphasic O2 pattern in the striatum, first decreasing and then increasing above baseline levels, consistent with the idea that O2 is first locally consumed (event 1) following neurotransmission before being replenished via hyperemic CBF increases (event 2).8,10,24 In previous work, we showed that stimulated O2 increases do not occur in brain slices devoid of CBF, supporting the idea that event 2 is hyperemic in origin.12 Striatal iGluR activation was responsible for a significant portion of the O2 decrease amplitude, but its negligible effect on the O2 increase event (i.e., CBF) suggested that either stimulus strength, the release of other electrically evoked neurotransmitters, or both play this role in hyperemia. Local O2 responses to glutamate iontophoresis resembled those from weak electrical stimulations, that is, monophasic O2 decreases insufficient to provoke CBF-driven O2 increases. Taken together, it is clear that our improved multimodal sensor provides a chemically and spatially selective method of studying exclusive glutamatergic influence on single-unit activity and local O2 changes throughout the intact brain.
Here, we stimulated glutamatergic cell bodies in the PFC to measure O2 changes at terminals in the dorsal striatum. Typical electrical stimulation paradigms use biphasic pulses and optimize stimulation parameters through tuning curves.6,8,34,35 Studies have reported that below a threshold stimulation “strength”, both O2 decreases and cell firing occurred without evoking CBF changes.6,8 We stimulated glutamatergic PFC projections to the striatum and evoked O2 decreases that were only followed by O2 increases past a threshold “strength” related to the number of stimulating pulses. The event amplitudes grew linearly with the number of stimulation pulses, as did the likelihood of the location responding with a biphasic O2 pattern (i.e., first consuming O2, then replenishing O2 to increases above baseline). Together, these results support current functional hyperemia dogma that stimulating neuronal activity leads to increased energy consumption followed by overcompensating amounts of O2 delivered via CBF. Thus, we verified that a stimulation threshold must be exceeded to elicit hyperemic O2 responses in the corticostriatal pathway and identified that this threshold directly depends upon the number of electrical stimulus pulses.
In contrast to previous studies that show both CBF and neuronal activity as being dependent upon stimulation frequency,6,8,34,35 we found no relationship between higher stimulation frequencies and whether the locations expressed biphasic behavior. It is important to note that each of these studies kept the stimulation duration constant while modulating the frequency, which confounds frequency effects with the pulse numbers per stimulus. Further, we found no relationship between the event amplitudes and frequency, which we attribute to using a consistent number of pulses. Lowering the stimulation frequency only increased the amount of time between the stimulation onset and the maximal O2 event responses, which again supported our conclusion that the number of pulses ultimately controls the magnitude of O2 responses. Thus, we concluded that, for a constant number of electrical stimulus pulses, the PFC stimulation frequency has no influence over hyperemic O2 amplitudes in the striatum.
Interestingly, we did not observe dopamine release at the sensor as a result of PFC stimulation, as has been reported in the ventral striatum;21 however, we positioned our electrodes in the dorsal striatum and optimized our sensor location based on robust glutamate-evoked single-unit activity. Additionally, the O2 voltammetric waveform has poor sensitivity for dopamine (2.5 nA/μm at the dopamine oxidation potential [+0.6 V]).36 We observed catechol-like signals in some locations following stimulation, but the absence of a catecholamine reduction peak at −0.3 V and the presence of negative current at −0.1 V suggested that these signals indicated pH shifts.
While manipulating PFC stimulation parameters controlled whether striatal O2 changes were mono- or biphasic, the specific role of glutamate as an excitatory neurotransmitter only influenced local O2 decreases (i.e., event 1). Subsequent O2 overcompensation (i.e., event 2) was insensitive to iGluR blockade at the cell terminals, consistent with studies that related neurovascular coupling to postsynaptic receptor activity.6,37 As a sufficient quantity of stimulation pulses was necessary to evoke O2 overcompensation (i.e., CBF), the process responsible required either a larger recruitment of excited cells or robust, accumulative stimulation in order to release certain neurotransmitters. Meanwhile, glutamate iontophoresed at the same striatal locations as the PFC recordings only evoked O2 decreases, even with high ejection currents (i.e., greater quantities of glutamate released). Extensive electrical PFC stimulations at similar frequencies have been shown to increase glutamate concentrations by several micromolar in the ventral striatum, orders of magnitude greater than what our iontophoretic ejections are estimated to release.38 The picomoles of glutamate iontophoresed with our 2 s ejection are unlikely to diffuse far enough to excite the same number of neurons as a stimulating electrode, but the magnitude of the O2 decreases with iontophoresis were still comparable to “weaker” electrical stimuli (i.e., fewer pulses). Together, this suggests that local O2 overcompensation at striatal terminals depended on PFC neurotransmission. However, the lack of specificity offered by electrical stimulation barred any conclusions about whether this hyperemic relationship was driven by neuronal activity or the stimulated release of vasoactive neurotransmitters, especially since the potential amount of striatal glutamate released versus ejected may differ by orders of magnitude.
It is well-known that electrical stimulations excite spatially extensive cell populations, so we adapted an existing multimodal sensor to excite local neuronal populations, record action potentials, and observe subsequent O2 changes. These techniques have exceptionally high spatial resolutions; steady-state iontophoresis (i.e., ejections 120 s or longer) delivers drug to a radius in the hundreds of micrometers,25 extracellular single-unit recordings detect somatic firing within a ∼50 μm radius,39 and FSCV at a carbon-fiber electrode probes O2 changes within a 20 μm radius.13 A 2 s (i.e., far from steady-state) iontophoretic glutamate ejection, estimated to deliver 1–2 pmol of glutamate, evoked single-unit activity in highly localized cell populations, in agreement with previous studies.16,17,20 Nearly all recording locations at glutamate-sensitive cells (100+) showed either a monophasic O2 decrease that returned to baseline or no O2 change in response to the 2 s glutamate ejection (Figure 7; O2 examples in Figure 4A,C). Glutamate iontophoresis evoked single-unit activity without subsequent O2 changes and vice versa, in agreement with studies showing decoupling at high spatial resolution.7,40 However, the outstanding majority of glutamate-sensitive cells showed a decrease in O2 after glutamate stimulation, similar to a “weak” electrical stimulus. Iontophoresed glutamate could not elicit biphasic responses, even with extended (>5 s) ejections using currents >500 nA (data not shown). Previous work established how to both quantify the drug ejected and track its spatial diffusion,19,25 which we used to confirm that a 2 s ejection released glutamate on the order of picomoles (vide supra). This is unlikely to be excitotoxic41 and establishes glutamate iontophoresis as a safe method of investigating local excitatory dynamics. It is unlikely that glutamate ejections of this magnitude could diffuse to a sufficient number of neurons and match the stimulation strength necessary to elicit a biphasic response, especially with the efficiency of glutamate uptake through neuronal and astrocytic transporters.42
Cell firing is normally inhibited in deeply anaesthetized animals; however, we observed cells in the ventral striatum that fired spontaneously at high rates, a well-known characteristic of interneurons. Glutamate significantly and reversibly inhibited most of the recorded fast-spiking interneurons; however, glutamate had no effect on a small number of fast-spiking cells and increased cell firing only in one single case. While our sensor afforded us the unique advantage of recording interneuron responses to glutamate in an intact brain, technical limitations precluded our ability to definitively categorize the interneurons into their specific subtypes using single-unit activity waveforms alone.23,32 It is possible that the iontophoresed glutamate induces a temporary depolarization block, as has been previously observed with extended glutamate iontophoresis in dopamine neurons and ventral striatum neurons,43,44 which may explain why some neurons required longer ejections at stronger currents to quell firing (Figure 5B). The specific effect of glutamate on interneuron firing rates adds to the growing body of literature investigating the complex interactivity between neurotransmitters through striatal MSNs and fast-spiking interneurons.33,45,46
Spontaneous O2 events, found here in both the cortex and striatum, have been observed in vivo as early as 1957.47 Studies attributed these spontaneous events to factors ranging from the type of anesthetic used48 and to spontaneous astrocytic excitation,49,50 among other possibilities. Studies from Lowry et al. detected baseline current fluctuations when observing O2 via amperometry, a technique that is also capable of determining absolute tissue O2 concentrations.51 FSCV measures changes in O2 rather than absolute concentrations; however, the unique voltammogram shape allowed us to confirm what may be mistaken for noise as O2 (examples in Figure 1C). Though some transients were large (>20 μM), the short (<60 s) durations, continued neuronal sensitivity to glutamate excitation, and lack of concomitant ionic signals precluded these from representing spreading depolarizations.52
When glutamate ejected during a transient event, we observed augmented O2 decreases that returned to baseline on the same time scale as those without transients, suggesting that the transients are controlled by mechanisms that are independent of (or unlikely to compete with) neuronal glutamate receptor-dependent O2 decreases. Indeed, spontaneous O2 events did not always correspond with single-unit firing (Figure 6 and 7) despite the spatial resolution of O2 detection being more confined than that of extracellular single-unit detection (∼20 and 50 μm radii, respectively).13,39 Nearly 30% of oxidative metabolism in the brain originates from astrocytes,53 undetectable with single-unit electrophysiology, making them attractive potential sources of these transients. That a larger percentage of transients were observed in deeper brain regions also indicated a possible environmental factor to their presence. The large overlap between locations with spontaneous transients and glutamate-sensitive neurons would make multiunit cellular recording an interesting future topic of exploration. Though we did not investigate any underlying chemical or physical source(s) of these transients, research from the Venton group has been investigating the relationship between adenosine and O2 changes, both as stimulated events and as spontaneous transients.54,55
As technology progresses to stimulate the brain more selectively and specifically, increasing evidence suggests that disconnects between functional hyperemia relationships exist in healthy subjects. Here, we elicited a hyperemic response from electrically stimulated glutamate release in the striatum, but targeted, exogenous glutamate excitation produced only decreases in recorded O2. Modifying an existing multimodal sensor that paired iontophoresis with electrophysiology and FSCV permitted the simultaneous detection of highly localized O2 and cell firing responses to selective glutamatergic stimuli in an intact, anesthetized animal. This technique serves as a starting point for investigating spatially targeted cerebrovascular coupling as it relates to glutamate elicited neurotransmission and O2 responses.
Methods
Animal Care
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill (UNC) in accordance with the Guide for Care and Use of Laboratory Animals (eighth edition). Male Sprague–Dawley rats (300–450 g, Charles River, Wilmington, MA, USA) were pair-housed at UNC animal facilities, given food and water ad libitum, and kept on a 12 h light/dark cycle. Care was taken to reduce the number of animals used. A total of 6 animals were used for stimulation experiments, and glutamate iontophoresis data were pooled and analyzed from 70 animals originally used for other protocols. A single rat expired during a stimulation experiment, and was excluded from this study.
Surgery
Animals were anesthetized with urethane (1.5 g/kg ip) and placed in a stereotaxic frame (Kopf, Tujunga, CA, USA). Coordinates are relative to bregma from the Paxinos and Watson (2007) atlas. Depths were measured relative to dura mater. Holes were drilled in the dorsal striatum (+0.7 or +2.0 mm A-P, +3.2 mm M-L, −2.5 to −3.5 mm D-V) for the carbon-fiber electrode and the PFC (+3.0 mm A-P, +0.8 mm M-L, −3.0 mm D-V) for the stimulating electrode. A Ag/AgCl reference electrode wire was placed in the contralateral hemisphere and served also as an iontophoresis ground. Holes were drilled for glutamate iontophoresis studies in the striatum (+2.2 mm A-P, +1.7 mm M-L, −6.4 to −7.8 mm D-V) and somatosensory cortex (+0.6 mm A-P, +2.8 mm M-L and +2.0 mm A-P, +2.6 mm M-L, −0.9 to −2.0 mm D-V for each).
Drugs and Solutions
dl-2-Amino-5-phosphonopentanoic acid sodium salt (AP5, NMDA receptor antagonist), and 1,2,3,4-tetrahydro-7-nitro-2,3-dioxoquinoxaline-6-carbonitrile disodium salt (CNQX, AMPA receptor/kainate antagonist) were obtained from Abcam (Cambridge, MA, USA). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA). Iontophoresis solutions of glutamate, AP5, and CNQX were 200, 50, and 10 mM, respectively, with the latter two combined into a single cocktail solution. AP5 and CNQX concentrations were chosen based on existing literature,56 with a higher CNQX concentration to compensate for experimentally observed difficulties ejecting the drug at higher currents. Drugs were dissolved in 5 mM NaCl and ejected iontophoretically using cathodic currents (−25 to −400 nA).
Voltammetric O2 Measurements and Iontophoresis
Multimodal sensors were fabricated from a four-barrelled glass capillary as described previously.15 Briefly, a ∼5 μm diameter carbon fiber, pulled into a glass capillary, was inserted into a 4-barrel capillary and cut under a light microscope to an exposed length between 80–120 μm. The remaining iontophoresis barrels were filled with drug solutions.
FSCV measurements were obtained and analyzed with the high-definition cyclic voltammetry (HDCV) computer program.14 The voltammetric waveform was 11 ms in duration, first scanned from a holding potential of 0.0 V to +0.8 V, then to −1.4 V, and finally back to the holding potential (vs Ag/AgCl reference electrode) at a scan rate of 400 V s–1. Carbon-fiber surfaces were conditioned in the brain by scanning the waveform at 60 Hz, followed by 10 Hz, each for 15 min. Simultaneous electrochemical and electrophysiological measurements were acquired with a 5 Hz repetition rate as described previously.14 Briefly, each 200 ms data acquisition period consisted of single-unit activity recordings for 179 ms, a voltammetric waveform lasting 11 ms, and a 0 V potential held both prior to and following each waveform for 5 ms each. O2 current versus time traces were exported at the O2 reduction potential on the forward scan (−1.35 V) to exclude the nonfaradaic current that immediately follows switching potentials from adsorbing species that disrupt the electrical double-layer capacitance (e.g., calcium).12 O2 concentrations were estimated from the current at the O2 reduction potential (−1.35 V) using a −0.35 nA μM–1 calibration factor, such that negative currents indicate O2 increases and positive currents indicate O2 decreases. The calibration methods used have been described elsewhere,12 where the O2 reduction current is measured from solutions of known O2 content passing through an airtight flow cell. Custom instrumentation controlled the alternating connection of the carbon fiber between a current transducer (cyclic voltammetry) and a voltage follower (electrophysiology measurements).14
Iontophoretic ejections were performed by applying constant current (NeuroPhore BH-2 System, Harvard Apparatus, Holliston, MA, USA). Ejection timing was controlled by HDCV. Iontophoretic barrels were primed at least 400 μm dorsal to the measurement region, outside the brain regions of interest, to test barrel functionality without affecting receptors at the actual recording locations.
Electrically-Stimulated Endogenous Glutamate Release
Stimulus isolators (NL800A, Neurolog, Digitimer, Hertfordshire, UK) generated electrical stimulations at a bipolar stainless steel electrode (Plastics One, West Lafayette, IN, USA) in the PFC glutamatergic cell bodies. A multimodal sensor was placed in the ipsilateral dorsal striatum. One drug barrel contained glutamate (200 mM) while another contained an AP5 (50 mM) and CNQX (10 mM) cocktail to antagonize iGluRs. Recordings were taken at locations where glutamate reponsive cells were present, defined as locations where single-unit activity accompanied a 2 s glutamate ejection using cathodic currents <100 nA. The electrode depth was optimized for the strongest biphasic response while maintaining sensitivity to iontophoresed glutamate. One location was used per animal. Dorsal striatum locations were verified with histology as described below.
Stimulations were optimized to maximize biphasic O2 responses while maintaining physiologically relevant stimulus lengths. Stimulation frequencies from 5 to 60 Hz were tested; all stimulations were comprised of 120 biphasic pulses of 0.2 ms width and 300 μA amplitude. These frequencies were chosen with the knowledge that high frequency corticostriatal stimulations raise striatal glutamate levels57 and are within the range of stimulations used in other studies.58,59 To measure the effect of stimulation duration, we held stimulation frequency and current amplitude constant (20 Hz and 300 μA, respectively) and varied pulse numbers from 10 to 100. A random number generator controlled the order in which different frequencies or pulse numbers were used.
Pharmacological studies investigated the role of local iGluRs on O2 events in the striatum following PFC stimulation. To antagonize NMDA, AMPA, and kainate receptors, a drug cocktail (50 mM AP5 and 10 mM CNQX) was iontophoretically ejected into the dorsal striatum recording location for 60 s using cathodic current. The ejection duration was chosen to reach near-steady state diffusion, such that local (∼100 μm radius)25 receptors would become saturated with antagonists. After the drug ejections ended, 10–30 s elapsed before the postdrug PFC stimulation (20 Hz, 80 pulses) to measure O2 changes under local iGluR antagonism. Glutamate was iontophoresed before the first electrical stimulation in each subject and again 10 min after the end of the final drug cocktail stimulation to confirm drug clearance.
Local Glutamatergic Excitation with Iontophoresis
A multimodal sensor was placed in either the cortex or the striatum, with one barrel containing a glutamate solution (200 mM glutamate dissolved in 5 mM NaCl). Each probe was used for multiple locations, if background currents remained stable, to both avoid tissue damage and minimize the number of animals. Recording locations were at least 300 μm apart.15
Both the electrophysiological software and HDCV continuously display data in real time, which allowed us to locate glutamate-sensitive cells quickly. The sensor was lowered into the brain region of interest and glutamate was ejected (2 s). Locations were designated as cell locations if they produced reproducible single-unit firing that time-locked to the glutamate ejection. If no time-locked response occurred, the sensor was lowered further and the glutamate ejection repeated until a cell was located. We optimized the depth of cell recording locations for maximal single-unit activity responses to glutamate. Because multiple data sets could be collected per animal, data sets were also taken at locations that were insensitive to glutamate to characterize other metrics (i.e., O2 responses and spontaneous O2 transients, vide infra). Each recording location data set consisted of 3–6 replicates each of baseline O2 activity (nothing ejected) and 2 s glutamate ejections. Time between subsequent glutamate ejections was 120 s.
Single-Unit Activity Electrophysiology
Single-unit activity was measured at the carbon-fiber for 179 ms between each voltammetric scan. The data were amplified (×5000), fed through a bandpass filter (300–3000 Hz, Krohn-Hite Corp., Brockton, MA), and digitized using commercially available software (Digitizer, Plexon, Dallas, TX, USA). Single units were analyzed using Offline Sorter (Plexon, Dallas, TX, USA). Individual waveforms coinciding with a coupling artifact were excluded. After cell sorting, perievent rasters depicting the firing rate of cells over time were compiled using commercially available software (NeuroExplorer, NexTechologies, Madison, AL) and exported in 0.18 s bins for statistical analysis.
Histology
Following data collection, the probe location was lesioned by cyclically applying a ramp of 0–10 V DC potential three times over 20 s. Animals were euthanized with urethane cardiac puncture. Brains were removed and fixed in 10% formalin for >7 days. Brains were then cryoprotected in 30% sucrose for >48 h, before coronal sections (50 μm) were taken with a freezing microtome (Leica, Germany). Slices were mounted on microscope slides and viewed under a light microscope to confirm electrode placements.
Statistical Analyses
Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). A repeated measures one-way analysis of variance (ANOVA) with post hoc Bonferroni’s test was used to evaluate significance between different electrical stimulation parameters. A Student’s t-test was used to determine significance between pre- and postdrug O2 events elicited by electrical stimulation. Differences were significant when *P < 0.05. Venn diagrams were generated using Venn Diagram Plotter software (Pacific Northwest National Laboratory, Richland, WA, USA; https://omics.pnl.gov/).
Acknowledgments
The authors acknowledge Collin McKinney from the UNC Electronics Shop for assistance in assembling the instrumentation used in this research, and Meg Fox for her helpful comments.
Glossary
Abbreviations
- ANOVA
analysis of variance
- AP5
dl-2-amino-5-phosphonopentanoic acid
- ATP
adenosine triphosphate
- BOLD
blood oxygenation level-dependent
- CBF
cerebral blood flow
- CNQX
1,2,3,4-tetrahydro-7-nitro-2,3-dioxoquinoxaline-6-carbonitrile
- fMRI
functional magnetic resonance imaging
- FSCV
fast-scan cyclic voltammetry
- HDCV
high-definition cyclic voltammetry
- iGluRs
ionotropic glutamate receptors
- MSN
medium spiny neuron
- PFC
prefrontal cortex
- UNC
University of North Carolina at Chapel Hill
Author Contributions
L.R.W. drafted the manuscript, designed experiments, and carried out the acquisition, analysis, and interpretation of data and statistics. N.G.B. acquired and analyzed data. S.C. acquired data and designed instrumental setup. R.M.W. germinated the scope of research and designed experiments. All authors contributed to the editing of this manuscript.
This research received financial support from the NIH (Grant DA 032530).
The authors declare no competing financial interest.
References
- Sokoloff L. (1989) Circulation and energy metabolism of the brain. Basic Neurochemistry 2, 338–413. [Google Scholar]
- Roy C. S.; Sherrington C. S. (1890) On the regulation of the blood-supply of the brain. J. Physiol. 11, 85. 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake C. T.; Iadecola C. (2007) The role of neuronal signaling in controlling cerebral blood flow. Brain and Language 102, 141–152. 10.1016/j.bandl.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Powers W. J.; Hirsch I. B.; Cryer P. E. (1996) Effect of stepped hypoglycemia on regional cerebral blood flow response to physiological brain activation. AJP: Heart and Circulatory Physiology 270, H554–H559. [DOI] [PubMed] [Google Scholar]
- Mintun M.; Shulman G.; Snyder A.; Raichle M. (2000) Cerebral blood flow during regional activation is not regulated to maintain oxygen delivery. J. Neurosci. 26, 645–611. [Google Scholar]
- Nielsen A. N.; Lauritzen M. (2001) Coupling and uncoupling of activity-dependent increases of neuronal activity and blood flow in rat somatosensory cortex. J. Physiol. 533, 773–785. 10.1111/j.1469-7793.2001.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. K.; Peterson M. R.; Freeman R. D. (2003) Single-neuron activity and tissue oxygenation in the cerebral cortex. Science 299, 1070–1072. 10.1126/science.1079220. [DOI] [PubMed] [Google Scholar]
- Offenhauser N.; Thomsen K.; Caesar K.; Lauritzen M. (2005) Activity-induced tissue oxygenation changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. J. Physiol. 565, 279–294. 10.1113/jphysiol.2005.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S.; Lee T.-M.; Kay A. R.; Tank D. W. (1990) Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc. Natl. Acad. Sci. U. S. A. 87, 9868–9872. 10.1073/pnas.87.24.9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman E. M. (2014) Coupling mechanism and significance of the BOLD signal: a status report. Annu. Rev. Neurosci. 37, 161. 10.1146/annurev-neuro-071013-014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman J. B.; Wightman R. M. (1991) Simultaneous electrochemical measurements of oxygen and dopamine in vivo. Anal. Chem. 63, 24–28. 10.1021/ac00001a005. [DOI] [PubMed] [Google Scholar]
- Bucher E. S.; Fox M. E.; Kim L.; Kirkpatrick D. C.; Rodeberg N. T.; Belle A. M.; Wightman R. M. (2014) Medullary norepinephrine neurons modulate local oxygen concentrations in the bed nucleus of the stria terminalis. J. Cereb. Blood Flow Metab. 34, 1128–1137. 10.1038/jcbfm.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom R. C.; Meaney T.; Tople R.; Wightman R. M. (1987) Spatiotemporal description of the diffusion layer with a microelectrode probe. Anal. Chem. 59, 2005–2010. 10.1021/ac00142a024. [DOI] [Google Scholar]
- Takmakov P.; McKinney C. J.; Carelli R. M.; Wightman R. M. (2011) Instrumentation for fast-scan cyclic voltammetry combined with electrophysiology for behavioral experiments in freely moving animals. Rev. Sci. Instrum. 82, 074302. 10.1063/1.3610651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle A. M.; Owesson-White C.; Herr N. R.; Carelli R. M.; Wightman R. M. (2013) Controlled iontophoresis coupled with fast-scan cyclic voltammetry/electrophysiology in awake, freely moving animals. ACS Chem. Neurosci. 4, 761–771. 10.1021/cn400031v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnjević K.; Phillis J. (1963) Iontophoretic studies of neurones in the mammalian cerebral cortex. J. Physiol. 165, 274. 10.1113/jphysiol.1963.sp007057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour Y.; Dutar P.; Jobert A.; Dykes R. (1988) An iontophoretic study of single somatosensory neurons in rat granular cortex serving the limbs: a laminar analysis of glutamate and acetylcholine effects on receptive-field properties. Journal of Neurophysiology 60, 725–750. [DOI] [PubMed] [Google Scholar]
- Nicola S. M.; Surmeier D. J.; Malenka R. C. (2000) Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu. Rev. Neurosci. 23, 185–215. 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick D. C.; Wightman R. M. (2016) Evaluation of Drug Concentrations Delivered by Microiontophoresis. Anal. Chem. 88, 6492. 10.1021/acs.analchem.6b01211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick D.; McKinney C.; Manis P. B.; Wightman R. (2016) Expanding neurochemical investigations with multi-modal recording: simultaneous fast-scan cyclic voltammetry, iontophoresis, and patch clamp measurements. Analyst 141, 4902–4911. 10.1039/C6AN00933F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S.; Grenhoff J.; Chouvet G.; Gonon F. G.; Svensson T. H. (1993) Prefrontal cortex regulates burst firing and transmitter release in rat mesolimbic dopamine neurons studied in vivo. Neurosci. Lett. 157, 53–56. 10.1016/0304-3940(93)90641-W. [DOI] [PubMed] [Google Scholar]
- Parent A.; Hazrati L.-N. (1995) Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res. Rev. 20, 91–127. 10.1016/0165-0173(94)00007-C. [DOI] [PubMed] [Google Scholar]
- Zhou F. M.; Wilson C. J.; Dani J. A. (2002) Cholinergic interneuron characteristics and nicotinic properties in the striatum. J. Neurobiol. 53, 590–605. 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]
- Ances B. M.; Buerk D. G.; Greenberg J. H.; Detre J. A. (2001) Temporal dynamics of the partial pressure of brain tissue oxygen during functional forepaw stimulation in rats. Neurosci. Lett. 306, 106–110. 10.1016/S0304-3940(01)01868-7. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick D. C.; Edwards M. A.; Flowers P. A.; Wightman R. M. (2014) Characterization of solute distribution following iontophoresis from a micropipet. Anal. Chem. 86, 9909–9916. 10.1021/ac5026072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick D.; Walton L.; Edwards M.; Wightman R. (2016) Quantitative analysis of iontophoretic drug delivery from micropipettes. Analyst 141, 1930–1938. 10.1039/C5AN02530C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt N. C. (2001) Glutamate uptake. Prog. Neurobiol. 65, 1–105. 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Dykes R.; Lamour Y. (1988) An electrophysiological study of single somatosensory neurons in rat granular cortex serving the limbs: a laminar analysis. Journal of Neurophysiology 60, 703–724. [DOI] [PubMed] [Google Scholar]
- Meitzen J.; Pflepsen K. R.; Stern C. M.; Meisel R. L.; Mermelstein P. G. (2011) Measurements of neuron soma size and density in rat dorsal striatum, nucleus accumbens core and nucleus accumbens shell: differences between striatal region and brain hemisphere, but not sex. Neurosci. Lett. 487, 177–181. 10.1016/j.neulet.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin E. A.; Rebec G. V. (1996) Dopaminergic modulation of glutamate-induced excitations of neurons in the neostriatum and nucleus accumbens of awake, unrestrained rats. Journal of Neurophysiology 75, 142–153. [DOI] [PubMed] [Google Scholar]
- Lourenço C. F.; Santos R. M.; Barbosa R. M.; Cadenas E.; Radi R.; Laranjinha J. (2014) Neurovascular coupling in hippocampus is mediated via diffusion by neuronal-derived nitric oxide. Free Radical Biol. Med. 73, 421–429. 10.1016/j.freeradbiomed.2014.05.021. [DOI] [PubMed] [Google Scholar]
- Witten I. B.; Lin S.-C.; Brodsky M.; Prakash R.; Diester I.; Anikeeva P.; Gradinaru V.; Ramakrishnan C.; Deisseroth K. (2010) Cholinergic interneurons control local circuit activity and cocaine conditioning. Science 330, 1677–1681. 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara A. A.; Chen V.; Herring B. E.; Mendenhall J. M.; Berlanga M. L. (2003) Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Res. 986, 22–29. 10.1016/S0006-8993(03)03165-2. [DOI] [PubMed] [Google Scholar]
- Mathiesen C.; Caesar K.; Akgören N.; Lauritzen M. (1998) Modification of activity-dependent increases of cerebral blood flow by excitatory synaptic activity and spikes in rat cerebellar cortex. J. Physiol. 512, 555–566. 10.1111/j.1469-7793.1998.555be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeyer H. W.; Enager P.; Thomsen K. J.; Lauritzen M. J. (2007) Nonlinear neurovascular coupling in rat sensory cortex by activation of transcallosal fibers. J. Cereb. Blood Flow Metab. 27, 575–587. 10.1038/sj.jcbfm.9600372. [DOI] [PubMed] [Google Scholar]
- Venton B. J.; Michael D. J.; Wightman R. M. (2003) Correlation of local changes in extracellular oxygen and pH that accompany dopaminergic terminal activity in the rat caudate–putamen. J. Neurochem. 84, 373–381. 10.1046/j.1471-4159.2003.01527.x. [DOI] [PubMed] [Google Scholar]
- Iordanova B.; Vazquez A. L.; Poplawsky A. J.; Fukuda M.; Kim S.-G. (2015) Neural and hemodynamic responses to optogenetic and sensory stimulation in the rat somatosensory cortex. J. Cereb. Blood Flow Metab. 35, 922–932. 10.1038/jcbfm.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z.-B.; Tzschentke T. M.; Brodin E.; Wise R. A. (1998) Electrical stimulation of the prefrontal cortex increases cholecystokinin, glutamate, and dopamine release in the nucleus accumbens: an in vivo microdialysis study in freely moving rats. J. Neurosci. 18, 6492–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen K. H.; Einevoll G. T. (2008) Amplitude variability and extracellular low-pass filtering of neuronal spikes. Biophys. J. 94, 784–802. 10.1529/biophysj.107.111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Herron P.; Chhatbar P. Y.; Levy M.; Shen Z.; Schramm A. E.; Lu Z.; Kara P. (2016) Neural correlates of single-vessel haemodynamic responses in vivo. Nature 534, 378. 10.1038/nature17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W. (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Rothstein J. D.; Dykes-Hoberg M.; Pardo C. A.; Bristol L. A.; Jin L.; Kuncl R. W.; Kanai Y.; Hediger M. A.; Wang Y.; Schielke J. P.; Welty D. F. (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. 10.1016/S0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- White F. J.; Hu X.-T.; Zhang X.-F.; Wolf M. E. (1995) Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J. Pharmacol. Exp. Ther. 273, 445–454. [PubMed] [Google Scholar]
- Grace A.; Bunney B. (1986) Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: analysis using in vivo intracellular recording. J. Pharmacol. Exp. Ther. 238, 1092–1100. [PubMed] [Google Scholar]
- Gras C.; Herzog E.; Bellenchi G. C.; Bernard V.; Ravassard P.; Pohl M.; Gasnier B.; Giros B.; El Mestikawy S. (2002) A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J. Neurosci. 22, 5442–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier D. J.; Ding J.; Day M.; Wang Z.; Shen W. (2007) D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235. 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Davies P.; Bronk D. (1957) Oxygen tension in mammalian brain. Federation Proceedings 689. [PubMed] [Google Scholar]
- Hudetz A., Biswal B., Shen H., Lauer K., and Kampine J. (1998) Spontaneous fluctuations in cerebral oxygen supply, Oxygen Transport to Tissue XX, pp 551–559, Springer. [DOI] [PubMed] [Google Scholar]
- Volterra A.; Meldolesi J. (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640. 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Khakh B. S.; Sofroniew M. V. (2015) Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952. 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry J. P.; Griffin K.; McHugh S. B.; Lowe A. S.; Tricklebank M.; Sibson N. R. (2010) Real-time electrochemical monitoring of brain tissue oxygen: a surrogate for functional magnetic resonance imaging in rodents. NeuroImage 52, 549–555. 10.1016/j.neuroimage.2010.04.187. [DOI] [PubMed] [Google Scholar]
- Ayata C.; Lauritzen M. (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol. Rev. 95, 953–993. 10.1152/physrev.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L.; Peng L.; Dienel G. A. (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 27, 219–249. 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- Cechova S.; Venton B. J. (2008) Transient adenosine efflux in the rat caudate–putamen. J. Neurochem. 105, 1253–1263. 10.1111/j.1471-4159.2008.05223.x. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Venton B. J. (2017) Correlation of transient adenosine release and oxygen changes in the caudate-putamen. J. Neurochem. 140, 13–23. 10.1111/jnc.13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Lou N.; Xu Q.; Tian G.-F.; Peng W. G.; Han X.; Kang J.; Takano T.; Nedergaard M. (2006) Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 9, 816–823. 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Perschak H.; Cuenod M. (1990) In vivo release of endogenous glutamate and aspartate in the rat striatum during stimulation of the cortex. Neuroscience 35, 283–287. 10.1016/0306-4522(90)90082-F. [DOI] [PubMed] [Google Scholar]
- Lada M. W.; Vickroy T. W.; Kennedy R. T. (1998) Evidence for Neuronal Origin and Metabotropic Receptor-Mediated Regulation of Extracellular Glutamate and Aspartate in Rat Striatum In Vivo Following Electrical Stimulation of the Prefrontal Cortex. J. Neurochem. 70, 617–625. 10.1046/j.1471-4159.1998.70020617.x. [DOI] [PubMed] [Google Scholar]
- Taber M. T.; Fibiger H. C. (1995) Electrical stimulation of the prefrontal cortex increases dopamine release in the nucleus accumbens of the rat: modulation by metabotropic glutamate receptors. J. Neurosci. 15, 3896–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]