

Abstract
Despite the known importance of androgen receptor (AR) signaling in prostate cancer (PCa), the processes downstream of AR that drive disease development and progression remain poorly understood. This knowledge gap has thus limited the ability to treat cancer. Here, it is demonstrated that androgens increase the metabolism of glutamine in PCa cells. This metabolism was required for maximal cell growth under conditions of serum starvation. Mechanistically, AR signaling promoted glutamine metabolism by increasing the expression of the glutamine transporters SLC1A4 and SLC1A5, genes commonly overexpressed in PCa. Correspondingly, gene expression signatures of AR activity correlated with SLC1A4 and SLC1A5 mRNA levels in clinical cohorts. Interestingly, MYC, a canonical oncogene in PCa and previously described master regulator of glutamine metabolism, was only a context-dependent regulator of SLC1A4 and SLC1A5 levels, being unable to regulate either transporter in phosphatase and tensin homolog (PTEN) wild-type cells. In contrast, rapamycin was able to decrease the androgen-mediated expression of SLC1A4 and SLC1A5 independent of PTEN status, indicating that mechanistic target of rapamycin complex 1 (mTORC1) was needed for maximal AR-mediated glutamine uptake and PCa cell growth. Taken together, these data indicate that three well-established oncogenic drivers (AR, MYC and mTOR) function by converging to collectively increase the expression of glutamine transporters, thereby promoting glutamine uptake and subsequent prostate cancer cell growth.
Implications: AR, MYC and mTOR converge to increase glutamine uptake and metabolism in prostate cancer through increasing the levels of glutamine transporters.
Keywords: androgen receptor (AR), glutamine, SLC1A4, SLC1A5, prostate cancer
Introduction
Prostate cancer is the second most commonly diagnosed malignancy amongst men in Western countries (1). Since the 1940s, it has been known that the development and progression of prostate cancer relies heavily on androgens (2). Androgens function by binding to and activating a ligand-inducible transcription factor called the androgen receptor (AR). In the context of prostate cancer, AR then, in combination with additional oncogenic signals, promotes prostate cancer cell proliferation and survival (2). Despite AR's established role in prostate cancer, it is still not completely understood which AR-mediated downstream processes, either alone or in combination with other oncogenic cascades, drive the disease.
Altered cellular metabolism is now recognized as one of the hallmarks of cancer (3). Although the majority of metabolic cancer research focuses on glucose metabolism, it has become clear that cancer cells also readily metabolize glutamine to fulfill their metabolic needs (4, 5). In this context, glutamine catabolism (glutaminolysis) can be used to balance the influx and efflux of carbon and nitrogen through the tricarboxylic acid (TCA) cycle. Glutaminolysis can promote anaplerosis (x2212) the replenishment of intermediates of the TCA cycle in part for biosynthetic purposes (x2212) by converting glutamine to α-ketoglutarate, a key intermediate of the TCA cycle (6).
Glutamine-mediated anaplerosis/glutaminolysis begins with the initial uptake of glutamine via cell surface transporters such as SLC1A4 (also called ASCT1) and SLC1A5 (commonly referred to as ASCT2) (6). Once inside the cell, glutamine is committed to glutaminolysis by the enzyme glutaminase (GLS), which converts glutamine to glutamate. The only way this metabolism can be reversed is through the action of glutamine synthetase (GLUL), which converts glutamate back into glutamine (7). Thus, in the absence of appreciable glutamine synthetase activity, glutamate can then be converted to α-ketoglutarate where it enters the TCA cycle.
The oncogene MYC is a known regulator of the initial steps of glutaminolysis, during which MYC up-regulates mitochondrial glutaminase as well as glutamine transporters, promoting influx of the amino acid and its subsequent metabolism (8). In prostate cancer, MYC can function as a transformative factor. In the mouse prostate, Myc overexpression promotes prostatic intraepithelial neoplasia (PIN) followed by invasive adenocarcinoma in a dose-dependent manner (9). Interestingly, recent work has demonstrated that AR signaling can increase glutamine metabolism in prostate cancer cells (10). Additionally, AR has been demonstrated to modulate MYC expression in a context-dependent manner (11-13). Given MYC's previously described role in glutamine metabolism, we hypothesized that androgens promoted prostate cancer cell growth in part through augmenting MYC-mediated glutamine metabolism.
Materials and Methods
Cell culture, plasmids, and reagents
LNCaP and VCaP human prostate cancer cell lines were obtained from ATCC (Manassas, VA) and maintained and tested for androgen responsiveness just prior to experiments as previously described (14). PTEN-P8 and PTEN-CaP8 were obtained from ATCC and maintained in Dulbecco's Modified Eagle's Medium supplemented with 8% fetal bovine serum (FBS), 25 μg/ml bovine pituitary extract, 5 μg/ml human recombinant insulin and 6 ng/ml human recombinant epidermal growth factor (15). PrEC-LHS, PrEC-LHSR and PrEC-LHMK human prostate cancer cells were kindly provided by Dr. William Hahn (Dana-Farber Cancer Institute, Boston, MA, USA) and previously described (16). Cell lines were validated biannually by genotyping and mycoplasma-free confirmation through the use of a PCR-based assay. For all experiments, cells were first plated in phenol red-free medium containing charcoal-stripped FBS (CS-FBS) for 72 hours to minimize endogenous hormone signaling. Cells were then switched for the remainder of the assay to a customized experimental medium (Sigma, St. Louis, MO) that lacked serum, non-essential amino acids, sodium pyruvate, additional glucose and HEPES buffer. This experimental medium was supplemented with 2 mM L-glutamine unless otherwise noted (ex. Fig. 1A).
Figure 1.
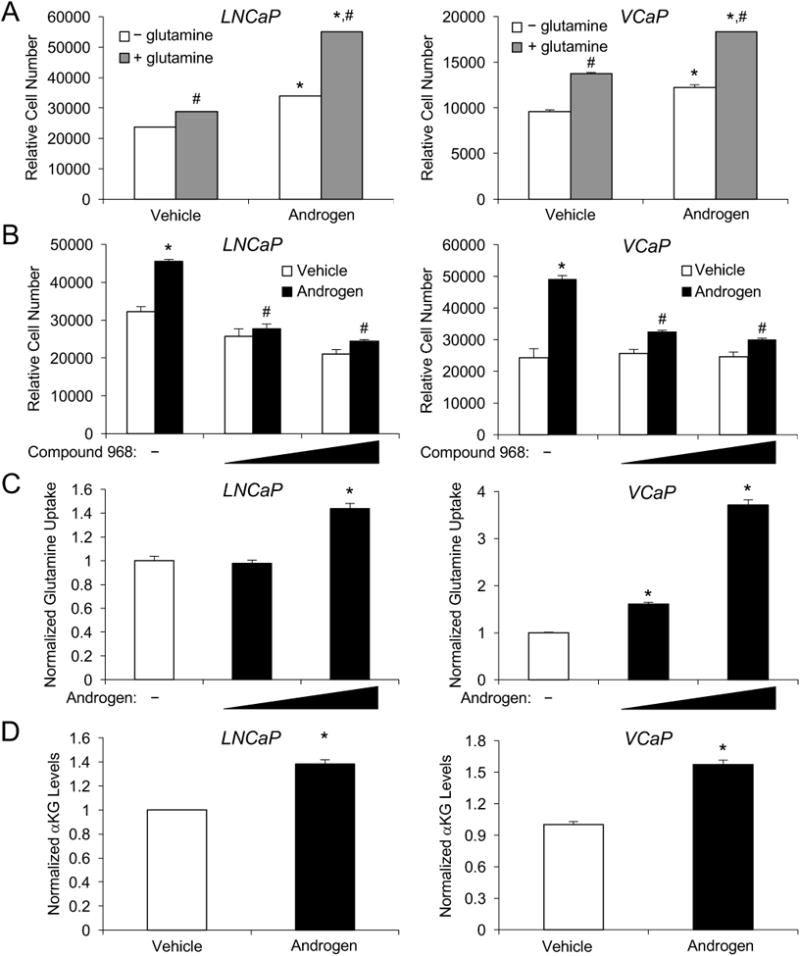
Androgens and glutamine increase prostate cancer cell growth. A, indicated cells were treated with vehicle (ethanol) or androgen (100 pM R1881) for 7 days in serum-free medium ± 2 mM glutamine. Cells were lysed and relative cell number was measured using a fluorescent DNA dye. *, significant (P<0.05) changes from vehicle. #, significant (P<0.05) changes from no glutamine. B-D, cells were grown in serum-free medium supplemented with 2 mM glutamine. B, cells were treated with vehicle (DMSO), 10 μM or 20 μM of Compound 968, a glutaminase inhibitor, followed by treatment ± androgen (100 pM R1881) for 7 days. Relative cell numbers were then quantitated as in A. *, significant (P<0.05) changes from vehicle (no androgen). #, significant (P<0.05) changes from vehicle (no Compound 968). C, cells were treated with vehicle, 10 pM or 100 pM androgen (R1881). Spent medium was then collected and analyzed for glutamine levels using a bioanalyzer and normalized to cellular DNA content. *, significant (P<0.05) changes from vehicle. D, cells were treated for 3 days with vehicle or androgen (100 pM R1881). Intracellular levels of α-ketoglutarate were then quantitated using an enzymatic assay and values were normalized to cellular DNA content. *, significant (P<0.05) changes from vehicle.
Stable cell lines were created using a pINDUCER10 construct that enabled the expression of a short hairpin RNA targeting MYC in the presence of doxycycline (Supplementary Fig. 4A). Additional lentiviral vectors have previously been described (17). Stealth siRNAs were from Life Technologies (Grand Island, NY). The sequences for all shRNAs and siRNAs used in this study are listed in Supplementary Table 1. Antibodies recognizing MYC (cat#: 5605), SLC1A4 (cat#: 8442), SLC1A5 (cat#: 8057), phospho-S6 (cat#: 4856), total S6 (cat#: 2317) and cleaved poly (ADP-ribose) polymerase (PARP) (cat#: 5625) were obtained from Cell Signaling (Beverly, MA). The antibody recognizing glutaminase (cat#: ab156876) was from Abcam (Cambridge, MA). Antibody recognizing AR (cat#: sc-7305) and GAPDH (cat#: G9545) were from Santa Cruz (Dallas, TX) and Sigma, respectively. Compound 968, a glutaminase inhibitor, was obtained from EMD Millipore (Billerica, MA). Methyltrienolone (R1881, a synthetic androgen) was from PerkinElmer (Waltham, MA). Rapamycin was from Cell Signaling and used at concentrations previously shown to block mTORC1 signaling and androgen-mediated proliferation (18, 19). Docetaxel was from Sigma.
Proliferation assays
Cells were plated in phenol red-free, CS-FBS-containing medium at a density of 5x103 cells per well in 96-well plates for 3 days. After this, the medium was switched to a serum-free medium that contained a final concentration of ± 2 mM glutamine without sodium pyruvate, non-essential amino acids, HEPES or additional glucose (experimental medium). Cells were then treated and incubated for 3 or 7 days as indicated. At the end, cell numbers were quantitated using a fluorescent DNA dye as previously described (18).
For experiments using siRNAs, all cell lines were plated as stated above. Cells were then transfected with 100 nM final concentration siRNAs for 3 days. Afterwards, the cells were transfected a second time and treated as indicated and allowed to incubate an additional 4 days. Cell proliferation was then quantified as described above.
Glutamine uptake assays
LNCaP cells were plated at a density of 3×104 cells per well, while VCaP cells were plated at 1.2×105 cells per well in 24-well plates. After 3 days, the cells were switched to 2 mM glutamine-containing experimental medium and transfected and/or treated as indicated. Afterwards, the medium was collected and glutamine levels were analyzed using a YSI 2700 Bioanalyzer (YSI Life Sciences, Yellow Springs, OH). Glutamine uptake levels were normalized to cellular DNA content.
RNA isolation, cDNA synthesis, and qPCR
RNA isolation, cDNA synthesis and qPCR were performed as previously described using 36B4 as a control (14). All primers used in this study are listed in Supplementary Table 1.
Immunoblot analysis
Immunoblot analysis was performed as previously described (20). All antibodies were used at the manufacturers’ recommended concentrations. Results shown are representative blots. Densitometry was performed using ImageJ software (National Institutes of Health (NIH), Bethesda, MD) and normalized to indicated controls. Results are presented as normalized mean values + SEM from experimental repeats (n ≥ 3).
Creation of LNCaP-shMYC cell lines
Stable cell lines were created using the pINDUCER10 system and puromycin selection as previously described (17, 21). The sequence for the MYC-targeting shRNA is presented in Supplementary Table 1.
α-Ketoglutarate assays
Cells were plated at a density of 5×10 cells/well and treated as described. The assay was performed using the coupled enzymatic assay according to the manufacturer's instructions (Sigma; cat#: MAK054). In brief, a-ketoglutarate concentration is determined by a coupled enzyme assay that results in a colorimetric (570 nm) product that is proportional to the amount of a-ketoglutarate present. Total α-ketoglutarate levels were normalized to cellular DNA content.
Fluorescence-activated cell sorting (FACS) analysis
The percentage of cells in the sub-G1 phase of the cell cycle was determined based on relative DNA content as assessed by FACS analysis. After 72 hours treatment, cells were detached by incubating with 0.25% trypsin-EDTA, washed with phosphate-buffered saline (PBS) and fixed overnight in 70% ethanol at 4°C. Fixed cells were then centrifuged (100 × g, 5 min), washed 1× in PBS, resuspended in PBS containing RNase A and propidium iodide (PI) (50 μg/ml each; ThermoFisher (Waltham, MA): cat#s: EN0531 and P1304MP) and analyzed on a Gallios Flow Cytometer (Beckman Coulter, Inc. (Brea, CA)). The percentage of sub-G1 population was determined using the MULTICYCLE software program (Phoenix Flow Systems, San Diego, CA).
Bioinformatic analyses of gene expression in clinical datasets
For the gene expression signature comparisons, transcriptomic profiles of human prostate cancer cohorts were downloaded from The Cancer Genome Atlas (TCGA). Androgen-induced signatures (Hieronymus AR and Nelson AR) were generated from previously defined data (22, 23). For each of the signatures, an activity score for each sample in each cohort was generated as previously described (24). Briefly, the gene expression values of prostate cancer cohorts were converted to z-scores with respect to normal samples. The activity score for each sample for a signature was evaluated by adding the z-scores of upregulated genes and subtracting the z-scores of downregulated genes. Correlation between pairs of gene signature activity scores were evaluated using the Pearson Correlation Coefficient as implemented in the Python statistical library SciPy; significance was assessed at P<0.05.
Statistical analysis
Multiple comparisons were performed by using a one-way analysis of variance (ANOVA), followed by post hoc Tukey's test. Analyses were done using GraphPad Prism, Version 5 (GraphPad Software, La Jolla, CA). All experiments were repeated at least three times unless otherwise noted.
Results
Androgens promote glutamine-mediated prostate cancer cell growth
The majority of cancers depend on increased glucose uptake and glycolysis as first described by Otto Warburg in the 1920s (25). It is now recognized that many cancers additionally exhibit an increased affinity for the amino acid glutamine, a metabolic shift that is likely a result of altered oncogenic and/or tumor suppressive signaling events that are to date not completely defined. Given AR's predominant role in prostate cancer, we tested whether androgens could augment prostate cancer cell growth in part through increasing glutamine consumption. We hypothesized that this intersection of hormone signaling and glutamine metabolism might be most pronounced under conditions of limited nutrient availability. To test this, we first assessed the effects of androgen treatment on prostate cancer cell growth in the presence or absence of glutamine under conditions with no additional non-essential amino acids, sodium pyruvate or serum. The concentration of androgen selected (100 pM R1881) was chosen because it represents the concentration at which peak androgen-mediated proliferation occurs in these cells ((19, 26, 27) and Supplementary Fig. S1A). Glucose was still required for cell seeding and survival. In both AR-positive, hormone-responsive LNCaP and VCaP cells, glutamine was consistently required for maximal androgen-mediated prostate cancer cell growth (Fig. 1A). To confirm a requirement for glutamine metabolism in androgen-mediated prostate cancer cell growth, we next treated cells with or without androgen and with increasing concentrations of compound 968, an inhibitor of glutaminase, a rate-limiting step of glutamine metabolism. Addition of the glutaminase inhibitor significantly decreased androgen-mediated prostate cancer cell growth in both LNCaP and VCaP cells (Fig. 1B). Interestingly, compound 968 had limited effect, particularly in VCaP cells, on basal prostate cancer cell growth, suggesting some specificity to androgen-mediated signaling. Given that androgens appeared to increase glutamine utilization, we then tested whether androgens increased cellular glutamine uptake. As shown in Fig. 1C, androgens significantly increased glutamine uptake in both LNCaP and VCaP cells at the same concentrations that stimulated cell growth. Similar to cell growth, androgens exhibited a biphasic dose response on glutamine uptake (Supplementary Fig. S1B) suggesting prostate cancer cell growth correlates with glutamine uptake. Consistent with these findings, androgens also increased the intracellular levels of the TCA cycle metabolite α-ketoglutarate, a key intermediate of glutamine-mediated anaplerosis/glutaminolysis (Fig. 1D). These results are consistent with our previous mass spectrometry findings that androgen treatment increased intracellular levels of all the TCA intermediates including α-ketoglutarate (10, 20). Taken together, these results suggest that AR signaling increases glutamine uptake and metabolism to increase prostate cancer cell growth.
AR signaling increases the expression of the glutamine transporters SLC1A4 and SLC1A5
Since androgens increased glutamine uptake, we next tested whether AR signaling increased the expression of glutamine transporters. We focused on the major glutamine transporters SLC1A4 and SLC1A5 because they were commonly upregulated in prostate cancer in multiple clinical datasets (Table 1) while other reported transporters were not 1) expressed in our prostate cancer models, 2) upregulated in prostate cancer clinical datasets or 3) regulated by androgens (ex. SLC7A5 and SLC38A5)(24, 28-34). In LNCaP cells, androgens increased SLC1A5 mRNA and protein levels (Fig. 2A). While SLC1A4 was expressed at a high basal level in LNCaP cells, its expression was not further changed following androgen treatment (Fig. 2A). Conversely, both SLC1A4 and SLC1A5 were significantly increased by androgens in VCaP cells (Fig. 2B). To assess whether AR could also regulate these genes in patients, we leveraged two different previously published, curated AR gene signatures of identified AR target genes (genes that were regulated in response to androgens and modulated by AR antagonists)(22, 23). Using a bioinformatics approach, we determined that these AR gene signatures positively correlated with increased mRNA transcript levels of SLC1A4 and SLC1A5 in the TCGA clinical dataset (Figs. 2C and D, R>0, P<0.05), suggesting AR may also regulate the expression of these genes in patients. Of note, while other groups have observed dramatic regulation of glutaminase (GLS) by additional oncogenic cascades such as MYC (8), we did not detect a robust, androgen-mediated change in GLS protein levels in either cell model despite the apparent androgen-mediated increase in GLS mRNA levels in VCaP cells. In addition, the AR gene signatures described above did not correlate with GLS expression in patients (P>0.05) nor was GLS overexpressed in clinical datasets (data not shown). However, it is important to note that while GLS protein levels did not change significantly in response to androgens, its basal expression was high unlike the expression for GLUL, the gene encoding glutamine synthetase (Figs. 2A and B). This is important because glutamine synthetase carries out the reverse reaction of glutaminase. The combined presence of high glutaminase levels and undetectable levels of glutamine synthetase indicated that any increase in glutamine uptake would subsequently lead to the rapid forward movement through glutaminolysis, consistent with our observed increase in α-ketoglutarate levels (Fig. 1D).
Table 1.
Fold increased expression of the glutamine transporters SLC1A4 and SLC1A5 in prostate cancer samples compared to benign controls in clinical datasets.
| Transporter | Dataset | Fold Change | P value | # of samples |
|---|---|---|---|---|
| SLC1A4 | Vanaja et al | 1.687 | 7.37E-4 | 40 |
| Holzbeierlein et al | 1.175 | .011 | 54 | |
| Taylor et al | 1.123 | .003 | 185 | |
| Welsh et al | 1.405 | .004 | 34 | |
| Wallace et al | 1.486 | .027 | 89 | |
| Singh et al | 1.476 | .034 | 102 | |
| Arredouani et al | 1.513 | .012 | 21 | |
| SLC1A5 | Magee et al | 1.518 | .018 | 15 |
| Singh et al | 2.106 | 3.24E-4 | 102 | |
| Wallace et al | 1.745 | 5.11E-4 | 89 | |
| Welsh et al | 1.399 | .007 | 34 |
Figure 2.

AR signaling increases the expression of the glutamine transporters SLC1A4 and SLC1A5. LNCaP (A) and VCaP (B) cells were treated for 3 days with either vehicle or androgen (100 pM R1881) in serum-free medium containing 2 mM glutamine. Left, qRT-PCR was used to quantify gene expression and normalized to 36B4 mRNA levels and vehicle control. Note, the expression of GLUL (the gene encoding glutamine synthetase – the enzyme that regulates the metabolism of glutamate back to glutamine) was not detected. *, significant (P<0.05) changes from vehicle. Right, Western blot analysis was done on whole cell lysates. GAPDH was used as a loading control. C and D, expression of SLC1A4 or SLC1A5 correlated significantly with two, distinct, previously described AR gene signatures (C, Hieronymus et al(22) and D, Nelson et al(23)) in transcriptomic profiles of prostate cancer patients from TCGA. Similar results were obtained using these AR activity signatures across multiple clinical cohorts.
Mechanistically, SLC1A4 and SLC1A5 appeared to be secondary targets of AR. In support of this, treatment of LNCaP cells for shorter time periods (16 hours compared to the 72 hour treatment shown in Figs. 2A and B), while sufficient to increase the expression of known primary AR target genes such as FKBP5, was not sufficient to increase SLC1A4 or SLC1A5 expression (Supplementary Fig. S2A). Likewise, 16-hour androgen treatment did not increase SLC1A5 expression in VCaP cells, but did increase FKBP5 mRNA levels (Supplementary Fig. S2B). Although androgens increased SLC1A4 expression at 16 hours posttreatment, this induction was blocked by an inhibitor of protein translation, cycloheximide. In contrast, cycloheximide had no effect on androgen-mediated FKBP5 expression (Supplementary Fig. S2B). Collectively, these results indicate that AR signaling increased the expression of the glutamine transporters SLC1A4 and SLC1A5 via an indirect mechanism.
Functional role of SLC1A4 and SLC1A5 in hormone-sensitive prostate cancer cells
Given the AR-mediated regulation of SLC1A4 and SLC1A5 (Fig. 2) and the requirement for glutamine for maximal androgen-mediated prostate cancer cell growth (Fig. 1), we next wanted to test the functional roles of these glutamine transporters. To do this, we assessed the impact of silencing SLC1A4 or SLC1A5 expression in prostate cancer cells (Fig. 3A and Supplementary Fig. S3) on glutamine uptake (Fig. 3B) and cell growth (Fig. 3C). Knockdown of SLC1A5 consistently decreased androgen-mediated glutamine uptake (Fig. 3B) and cell growth (Fig. 3C) in both LNCaP and VCaP cells. Again, there were modest effects on basal cell growth, indicating some specificity for androgen-mediated signaling. Knockdown of SLC1A4 with siRNA #1 also decreased both androgen-mediated glutamine uptake (Fig. 3B) and cell growth (Fig. 3C) in VCaP cells. Unfortunately, despite multiple attempts, we were unable to achieve effective knockdown of SLC1A4 with siRNA #2 in VCaP cells at either the mRNA (Supplementary Fig. S3B) or protein level (Fig. 3A). Correspondingly, this siRNA then functioned as an additional negative control as no effect was observed on either glutamine uptake or cell growth as would be expected. Surprisingly, knockdown of SLC1A4 (Fig. 3A and Supplementary Fig. S3A) decreased glutamine uptake (Fig. 3B) and cell growth (Fig. 3C) in LNCaP cells. This was unexpected because androgens did not increase SLC1A4 expression in LNCaP cells (Fig. 2A and Supplementary Fig. S3A). Thus, it appears that in a cell-type dependent manner AR signaling may potentiate SLC1A4 activity through additional mechanisms, unknown at this time, beyond gene expression (ex. posttranslational modifications, etc).
Figure 3.
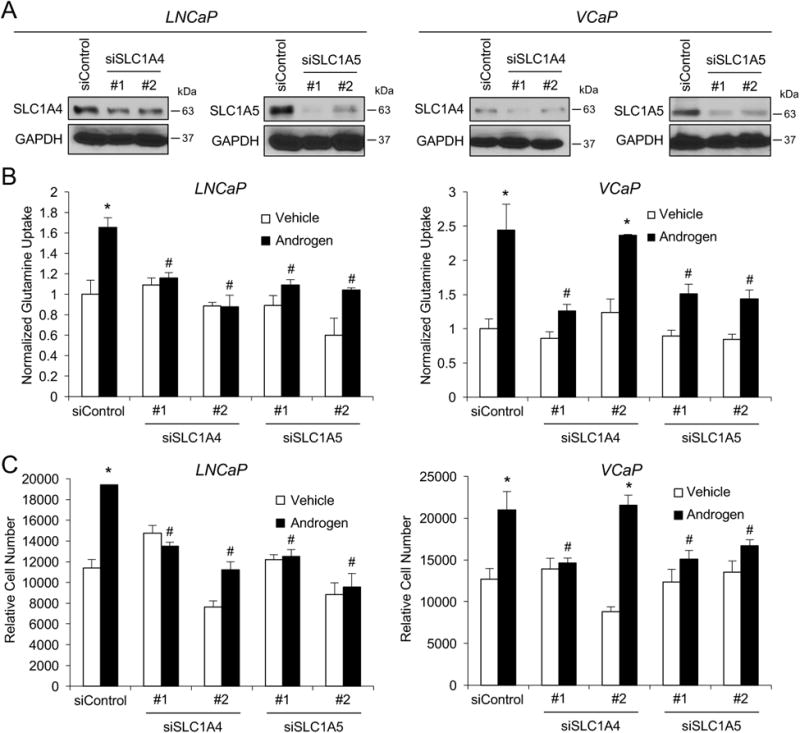
SLC1A4 and SLC1A5 are required for maximal androgen-mediated prostate cancer cell growth. A, prostate cancer cells were transfected for 3 days with indicated siRNAs. Cells were then harvested and lysates were subjected to Western blot analysis. B and C, prostate cancer cells were transfected with indicated siRNAs and treated for 7 days with vehicle or 100 pM R1881 (androgen). Then, glutamine uptake (B) or cell numbers (C) were assessed as described in Figure 1. *, significant (P<0.05) changes from vehicle. #, significant (P<0.05) changes from siControl.
MYC is a contextual regulator of SLC1A5 in prostate cancer cell models
A master regulator of glutamine metabolism is MYC (4, 6, 8, 35), a canonical oncogene in prostate cancer (9, 36, 37). Previous work has suggested that AR signaling could modulate MYC (c-MYC) expression (38-40). As such, we hypothesized that androgens promoted prostate cancer cell growth through MYC-dependent glutaminolysis. Specifically, we sought to determine what role MYC played, if any, in the regulation of SLC1A4 and SLC1A5 expression and function under our conditions of serum starvation. To facilitate these studies, we created stable derivatives of LNCaP cells that could inducibly express an shRNA targeting MYC in the presence of doxycycline (LNCaP-shMYC)(Supplementary Fig. S4). Here, androgens increased the protein levels of MYC and SLC1A5 but not SLC1A4 (Fig. 4A), consistent with our earlier results (Fig. 2A). Doxycycline-mediated knockdown of MYC decreased androgen-mediated SLC1A5 protein levels but had no effect on SLC1A4 or basal SLC1A5 levels (Fig. 4A). In contrast to previous work done in PC-3 prostate cancer cells (8), silencing of MYC also had no impact on GLS protein levels. Regardless, MYC knockdown decreased both glutamine uptake (Fig. 4B) and prostate cancer cell growth (Fig. 4C). The significant decrease in basal glutamine uptake and trend towards decreased baseline cell growth following MYC knockdown indicate that MYC, independent of AR signaling, likely has additional functions in LNCaP cells besides the regulation of SLC1A5 that contribute to glutamine uptake and, perhaps not surprisingly given MYC's known role in proliferation, cell growth.
Figure 4.
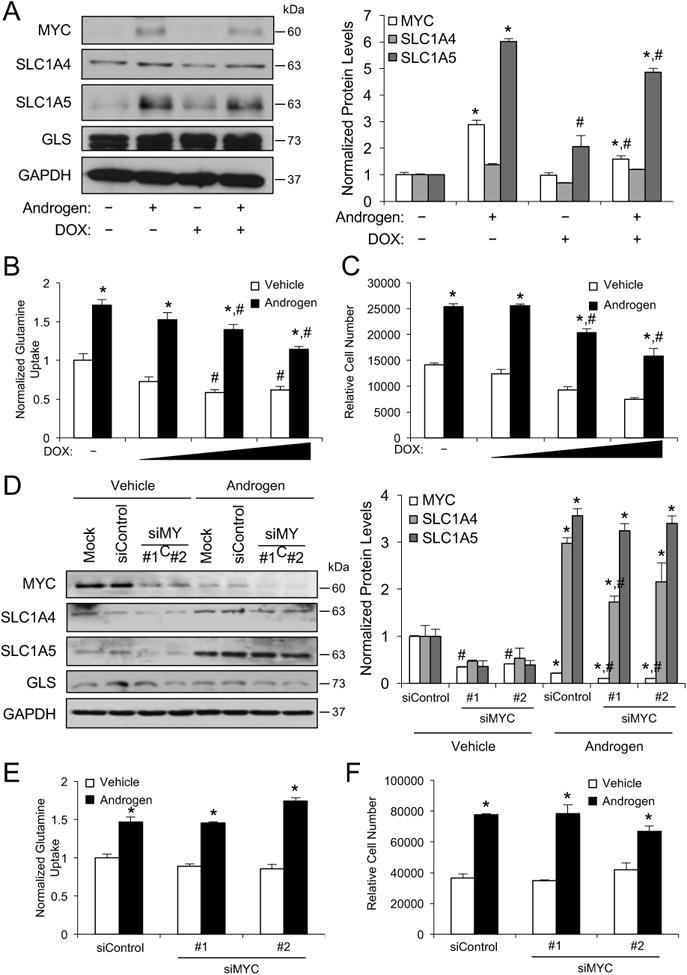
Regulation of MYC levels by AR and glutamine transporter levels by MYC are cell-type dependent. A, LNCaP stable cells that inducibly express an shRNA targeting MYC (LNCaP-shMYC) following doxycycline (DOX) treatment were treated for 3 days ± 700 ng/ml DOX with vehicle or 100 pM R1881 (androgen). Cells were then lysed and subjected to Western blot analysis. Left, representative blots. Right, densitometry summary of Western blot repeats (n = 3). Data are normalized to experimental GAPDH loading control. B-C, LNCaP-shMYC cells were treated with a dose response of DOX (0, 300, 700, 1500 ng/ml) ± androgen (100 pM R1881) for 3 days and then assayed for glutamine uptake (B) or proliferation (C) as described in Figure 1. A-C, *, significant (P<0.05) changes from vehicle (no androgen). #, significant (P<0.05) changes from no DOX. D-F, VCaP cells were transfected with mock or siRNAs targeting scramble control or MYC (#1 and #2) and then treated ± androgen (100 pM R1881) and subjected to Western blot analysis (D) or assessed for glutamine uptake (E) or proliferation (F). D-F, *, significant (P<0.05) changes from vehicle (no androgen). #, significant (P<0.05) changes from siControl.
Unfortunately, we were unable to create stable derivatives of VCaP cells using the same lentiviral approach as we have found that these cells are particularly resistant to lentiviral modulation. As an alternative, we silenced MYC expression using two different siRNAs and assessed the effect of MYC knockdown on SLC1A4 and SLC1A5 expression and androgen-mediated glutamine uptake and cell growth. As previously reported (41), androgen treatment reduced MYC protein levels in VCaP cells (Fig. 4D). Similar to LNCaP cells, MYC knockdown had no consistent effect on SLC1A4 or GLS protein levels (Fig. 4D). In direct contrast to the regulation we observed in LNCaP cells (Fig. 4A), knockdown of MYC had no effect on androgen-mediated SLC1A5 levels in VCaP cells (Fig. 4D). Consistent with these findings, depletion of MYC in VCaP cells did not change basal or androgen-mediated glutamine uptake (Fig. 4E) or cell growth (Fig. 4F). Thus, MYC appears dispensable for glutamine uptake and cell growth in VCaP cells but was required for maximal androgen-mediated SLC1A5 expression, glutamine uptake and cell growth in LNCaP cells under our conditions of limited nutrient availability. Together, these data indicate that MYC acts as contextual regulator of glutamine metabolism in prostate cancer cells.
mTOR stimulates expression of the glutamine transporters SLC1A4 and SLC1A5
Given MYC's previously described role as a master regulator of glutamine metabolism, it was surprising to us that MYC did not have a more pronounced role in our prostate cancer cell models. Hence, we suspected additional pathways that are 1) hyperactivated in prostate cancer and 2) known to be influenced by AR signaling could regulate SLC1A4 and SLC1A5 and therefore glutamine metabolism. The mechanistic target of rapamycin (mTOR), formerly known as the mammalian target of rapamycin, is one of the most commonly activated proteins in prostate cancer and has previously been shown to be regulated by AR signaling (18, 19, 42). Its role as a sensor for amino acid levels made it an ideal candidate to test. As shown in Figs. 5A and B, treatment with androgens increased the expression of SLC1A5 in LNCaP cells and SLC1A4 and SLC1A5 in VCaP cells, consistent with our results described in Fig. 2. As previously reported, androgens also increased mTOR signaling in prostate cancer cells as assessed by the phosphorylation of S6, a well-characterized downstream target of mTOR signaling (18, 19). Co-treatment with rapamycin, a selective inhibitor of the mTORC1 complex, decreased both basal and androgen-mediated SLC1A5 expression in LNCaP cells and suppressed the androgen-mediated induction of SLC1A4 and SLC1A5 in VCaP cells (Figs. 5A and B). This effect appeared to not be due to any changes in MYC (Supplementary Fig. S5) or effects on cell death (Supplementary Fig. S6). The effects of rapamycin on basal SLC1A5 expression are likely due to the fact that LNCaP cells have high basal mTOR signaling as a result of a mutation in phosphatase and tensin homolog (PTEN) that renders this upstream tumor suppressor inactive (43). Conversely, VCaPs express wild-type PTEN and do not have constitutively active phosphoinositide 3-kinase (PI3K)/Akt signaling (44). To our knowledge, this is the first description of mTOR regulation of SLC1A4 or SLC1A5 expression in prostate cancer. Consistent with this regulation and with the described roles for SLC1A4 and SLC1A5 above, rapamycin also blocked both androgen-mediated glutamine uptake (Fig. 5C) and cell growth (Fig. 5D).
Figure 5.
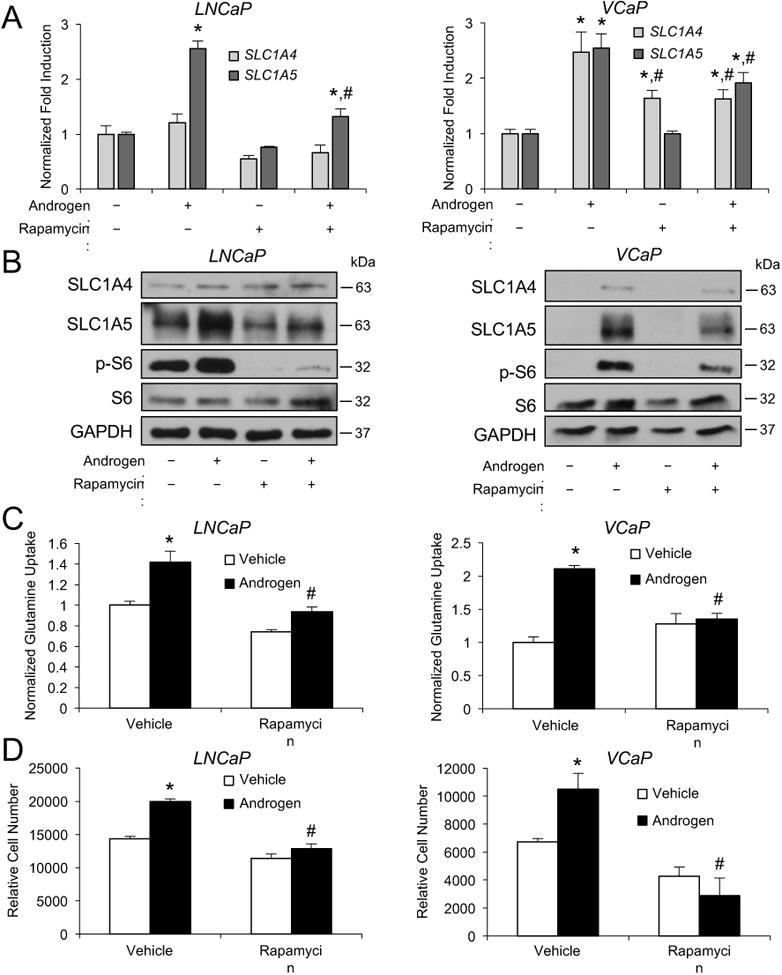
mTOR activity increases SLC1A4 and SLC1A5 expression, glutamine uptake and cell growth. A and B, prostate cancer cells cells were treated with vehicle or 10 nM rapamycin in addition to vehicle or androgen (100 pM R1881) for 3 days in serum-free medium containing 2 mM glutamine. Cells were then lysed and subjected to qRT-PCR (A) or Western blot (B) analysis. *, significant (P<0.05) changes from vehicle (no androgen). #, significant (P<0.05) changes from vehicle (no rapamycin). C and D, cells were treated as in A and B. C, glutamine uptake was then quantitated and normalized as described in Fig. 1. D, cell numbers were then also quantitated as described in Fig. 1. *, significant (P<0.05) changes from vehicle (no androgen). #, significant (P<0.05) changes from vehicle (no rapamycin).
Because of the differences in transporter regulation between LNCaP and VCaP cells, we next wanted to mechanistically determine whether these differences were due to variations in PTEN status. We focused on PTEN-regulated signaling because PTEN is a commonly altered tumor suppressor in prostate cancer (45) and its status is different in LNCaP and VCaP cells with PTEN being wild type in VCaP cells but inactivated in LNCaP cells (43). One of the difficulties with directly comparing LNCaP and VCaP cells is that these two popular models are genetically unrelated. To begin to address this issue, we leveraged two genetically defined sets of prostate cell models to compare the impact of specific cancer signaling pathways on SLC1A4 and SLC1A5 expression. First, we used a series of cell models derived from normal human prostate epithelial cells (PrECs) that were altered in a stepwise manner through the introduction of retroviruses encoding various oncogenes (16). Here, PrEC LHS (PrEC cells engineered to express the SV40 large T antigen (LT), small t antigen (ST) and hTERT, causing the cells to become immortalized but nontransformed), LHSR (LHS cells engineered to also express H-ras, causing the cells to become transformed) and LHMK (PrEC cells engineering to express SV40 LT, hTERT, MYC and PI3K, causing the cells to become transformed) cells were treated for 72 hours with vehicle or 10 nM rapamycin (Supplementary Fig. S7A). In all three PrEC-derived cell lines, SLC1A4 expression was unchanged regardless of treatment suggesting that in this system, H-ras, MYC, PI3K and mTOR were all unable to regulate SLC1A4 levels. Conversely, LHMK cells exhibited a moderately higher level of SLC1A5 expression that was more dramatically decreased by treatment with rapamycin relative to LHS and LHSR cells. This indicated that the combination of MYC and PI3K overexpression, perhaps also with the loss of ST, was sufficient to increase SLC1A5 expression and make these cells more susceptible to mTORC1 inhibition (with regards to SLC1A5 regulation). Interestingly, rapamycin increased MYC levels in LHS and LHSR cells, indicating that mTORC1 inhibited MYC in these cells. Conversely, mTORC1 augmented MYC expression in the LHMK cells as rapamycin decreased MYC levels. Taken together, PrEC LHMK cells more closely resembled LNCaP cells in that MYC, PI3K signaling (a byproduct of PTEN inactivation) and mTOR all coordinated to promote SLC1A5 expression.
There are two important drawbacks to using the PrEC models with respect to our study. First, PrEC cells do not express endogenous AR, making it difficult to assess the impact of androgen signaling. Second, LHMK cells have three genetic alterations (expression of MYC and PI3K, but no ST) compared to the LHS and LHSR cells, which complicated drawing conclusions about a single alteration. To circumvent these drawbacks, we next utilized two isogenic mouse cell lines that differed only in their Pten status (15). The PTEN-P8 cell line is heterozygous for Pten deletion. Its isogenic partner, PTEN-CaP8, is homozygous for Pten deletion. To test the role of Pten status in SLC1A4 and SLC1A5 expression, cells were cotreated for 72 hours with increasing concentrations of androgen (R1881: 0, 0.1 and 10 nM) and vehicle or 10 nM rapamycin. Unfortunately, while these cells were first reported to express AR, we did not detect any androgen regulation of SLC1A4, SLC1A5, MYC or mTOR signaling (Supplementary Fig. S7B). However, PTEN-CaP8 cells expressed higher levels of p-S6 and SLC1A4 compared to PTEN-P8 cells, an effect that was blocked by rapamycin. This suggested that in this model loss of Pten increased mTOR-mediated SLC1A4 expression. While SLC1A5 levels were unaffected, it should be noted that basal levels of SLC1A5 were high in both cell lines. It is still unclear whether this was due to inactivation of the first Pten allele. Interestingly, MYC levels were higher in the PTEN-CaP8 cells compared to PTEN-P8 cells. However, rapamycin only decreased MYC levels in the PTEN-P8 cells, indicating that Pten homozygous deletion rendered MYC expression insensitive to changes in mTORC1 activity. Regardless, SLC1A4 and SLC1A5 levels did not correlate with MYC expression, implying that MYC is not a major regulator of these two transporters in this system. This comes with the caveat that this is a mouse model system. Collectively, the data from these mouse cell lines, combined with the human PrEC models described above, further support a model of SLC1A4 and SLC1A5 levels being contextually regulated by MYC and mTOR. Importantly, in the models in which PTEN and/or PI3K were unaltered (VCaP, PrEC LHS, PrEC LHSR and PTEN-P8), MYC was unable to increase expression of either of the two transporters, suggesting that in PTEN/PI3K wild type prostate cancer cells, glutamine uptake via SLC1A4 and SLC1A5 are MYC-independent (Fig. 6).
Figure 6.
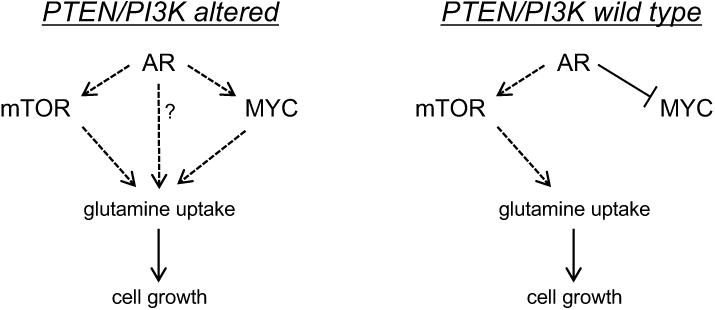
Working model of the regulation of the glutamine transporters by AR, mTOR and MYC signaling in prostate cancer cells. Prostate cancer cells can augment cell growth by increasing glutamine metabolism. This metabolism can be initiated by various oncogenic signaling cascades that, in a cell type-dependent manner, increase the expression of SLC1A4 and SLC1A5, two of the primary glutamine transporters. Of note, MYC's role in glutamine uptake may be dependent on PTEN/PI3K status. In addition, AR may increase SLC1A4 function through an unknown mechanism.
Discussion
Prostate cancer has an atypical metabolism. Benign prostate is characterized by the existence of a truncated TCA cycle that occurs as a result of high zinc levels in prostatic epithelial cells (46, 47). Zinc inhibits mitochondrial aconitase, shunting carbons that entered the TCA cycle out in the form of secreted citrate (46). One of the first transformation events that occurs during the evolution of prostate cancer is a drop in intracellular zinc levels due to the decreased expression of zinc transporters (46, 47). This decreased zinc leads to a derepression of aconitase that ultimately increases forward flux through the TCA cycle and augments oxidative phosphorylation (OXPHOS). To date, the majority of attention has focused on glucose's contribution to cancer metabolism. However, it is now recognized that glutamine metabolism may also contribute to oncogenesis under certain circumstances (4, 48, 49). Here, we demonstrate that under conditions of serum starvation, multiple oncogenic signaling pathways can increase the uptake and metabolism of glutamine, which is required for maximal prostate cancer cell growth (Fig. 1).
While many of the oncogenic pathways that govern sugar metabolism have been elucidated (ex. PI3K-Akt), those controlling glutamine metabolism are still emerging. Previous work has demonstrated that AR increases glutaminolysis in prostate cancer cells (10). Here, we demonstrated that AR-mediated glutamine metabolism is also augmented by the increased uptake of the amino acid through indirectly increasing the expression of two transporters, SLC1A4 and SLC1A5 (Figs. 1-3). Interestingly, AR promoted SLC1A4 and SLC1A5 expression in a cell-type specific manner through several mechanisms including MYC- and mTOR-dependent as well as -independent pathways. Further, both MYC and mTOR signaling are prevalent oncogenic cascades in prostate cancer that can be stimulated through AR-independent mechanisms (42, 50). Hence, SLC1A4 and SLC1A5 appear to serve as functional, downstream conduits for AR, MYC and mTOR.
Analyses of several cancer types indicated that the oncogene MYC could function as a master regulator of glutamine metabolism through directly increasing the expression of SLC1A5 and indirectly increasing the levels of GLS (4, 6, 8). The MYC-mediated modulation of GLS occurs through the supression of miR-23a/b (8). Although we also observed MYC-mediated expression of SLC1A5 in LNCaP cells, we did not detect significant changes in GLS protein levels in either LNCaP or VCaP cells (Fig. 4). This data contrasts previous work in PC-3 prostate cancer cells that demonstrated that MYC was required for stabilizing GLS protein levels (8). These variances may be due to the differences in the cell types as PC-3 cells more closely resemble small cell-like or neuroendocrine-like prostate cancer cells whereas LNCaP and VCaP cells are phenotypically similar to the adenocarcinoma cells that are more prevalently observed in the clinic (51). Previous studies imply a complex relationship between AR and MYC in the prostate (12, 13, 38-40). Evidence suggests that in the normal/benign prostate, AR inhibits MYC expression (12, 13). Conversely, as prostatic epithelial cells become transformed, the AR-mediated downregulation of MYC is either lost or reversed (13). In this regard, the AR/MYC relationship in VCaP cells appears to still resemble what is observed in the benign prostate while the connection appears to have already switched in LNCaP cells where AR increases MYC (Figs. 4 and 6). What exactly causes this regulatory switch is still poorly understood.
Because of mTOR's 1) established role in amino acid metabolism (52) and 2) known regulation by AR (18, 19), we postulated that AR may also influence glutamine uptake through mTOR. Consistent with this idea, we found that rapamycin decreased androgen-mediated SLC1A5 mRNA and protein levels (Fig. 5 and Supplementary Fig. S6A). In addition, rapamycin impaired the androgen-mediated SLC1A4 expression in VCaP cells (Fig. 5) and Pten null-mediated SLC1A4 expression in PTEN-CaP8 cells (Supplementary Fig. S7B). These data indicated that mTOR, and more specifically the mTORC1 complex, could also potentiate glutaminolysis. Interestingly, others have shown that glutamine flux through the SLC1A5 transporter activates mTOR signaling in breast cancer (53). Taken together, mTOR signaling and glutamine uptake may form a positive feedback loop.
We suspect that our findings may have translational significance. There is current interest in blocking glutamine metabolism in cancer (4). To that end, inhibitors of glutaminase such as CB-839 are in early phase clinical trials (NCT02071927, NCT02944435, NCT02071888, NCT02861300, NCT02771626, NCT02071862). Targeting glutamine transporters may offer an alternative therapeutic approach. This approach would be advantageous because it targets the potential pathological meeting point of three driver cascades (AR, MYC and mTOR). Further, as cell surface molecules, these transporters may be more readily druggable. Accordingly, novel inhibitors of SLC1A5 have recently been described (54). In addition, several groups are evaluating glutamine analogs for their value in positron emission tomography (PET) imaging of cancer (55). Our data here could inform radiologists regarding specific cellular signaling events that may influence results. In May 2016, the U.S. Food and Drug Administration approved Axumin™, also known as fluciclovine or anti-1-amino-3-18F-fluorocyclobutane-1-carboxylic acid (FACBC), for PET imaging of men with suspected prostate cancer recurrence. Fluciclovine is an amino acid analog that has been reported to be taken up into cells in part by SLC1A5-mediated transport (56). The uptake of fluciclovine appears to correlate with the levels of PSA/KLK3, an AR-regulated biomarker. Our results shown here would strongly suggest that the mechanistic explanation for this phenomenon is due in part to the AR-mediated expression of SLC1A5 and possibly SLC1A4. In future, it would be of interest to determine whether other regulators of these transporters such as mTOR signaling also track with increased fluciclovine PET imaging sensitivity.
Our study examined the regulation and role of two transporters, SLC1A4 and SLC1A5, in the earliest steps of glutaminolysis, namely glutamine uptake. It still remains to be determined how glutamine is subsequently metabolized by the cancer cell. Glutamine can be used in anaplerotic reactions to refill TCA cycle intermediates (4, 55). Accordingly, proliferating cells often metabolize glutamine to restore components of the TCA cycle in part for biosynthetic purposes (6). Carbons and nitrogens are syphoned off throughout this process to contribute to the synthesis of nucleic acids, other amino acids and hexosamines, the latter of which can contribute to posttranslational modifications. Additionally, glutamine, via its metabolism through glutamate, can be used for the biosynthesis of glutathione and therefore help modulate oxidative stress. Alternatively, nitrogens can also be released in the form of ammonia. Certainly, future studies using stable isotope tracing will help delineated how glutamine is further metabolized and for what it is being used.
Supplementary Material
Acknowledgments
We thank members of the Frigo Laboratory for their technical support, critical reading of the manuscript and their suggestions. We also thank Zhang Weihua (University of Houston) for use of his bioanalyzer and Thomas Westbrook (Baylor College of Medicine) for the pINDUCER constructs.
Notes: This study was supported by NIH grants R21CA191009 and R01CA184208 (D.E.F.). This work was also partially supported by a CPRIT Proteomics and Metabolomics Core Facility Support Award RP120092 (C.C. and K.R.) and CPRIT award RP170295 (C.C. and J.D.).
References
- 1.Society AC. Cancer Facts & Figures 2015. Atlanta: American Cancer Society; 2015. [Google Scholar]
- 2.Schmidt LJ, Tindall DJ. Androgen receptor: past, present and future. Curr Drug Targets. 2013;14:401–7. doi: 10.2174/1389450111314040002. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–33. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol. 2012;23:362–9. doi: 10.1016/j.semcdb.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–7. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–34. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellwood-Yen K. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 10.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget. 2015;6:31997–2012. doi: 10.18632/oncotarget.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni M, Chen Y, Fei T, Li D, Lim E, Liu XS, et al. Amplitude modulation of androgen signaling by c-MYC. Genes Dev. 2013;27:734–48. doi: 10.1101/gad.209569.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antony L, van der Schoor F, Dalrymple SL, Isaacs JT. Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/beta-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate. 2014;74:1118–31. doi: 10.1002/pros.22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci. 2014;10:627–42. doi: 10.7150/ijbs.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, et al. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011;71:528–37. doi: 10.1158/0008-5472.CAN-10-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, Sawyers CL, et al. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007;67:6083–91. doi: 10.1158/0008-5472.CAN-06-4202. [DOI] [PubMed] [Google Scholar]
- 16.Berger R, Febbo PG, Majumder PK, Zhao JJ, Mukherjee S, Signoretti S, et al. Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer Res. 2004;64:8867–75. doi: 10.1158/0008-5472.CAN-04-2938. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y, Han JJ, Tennakoon JB, Mehta FF, Merchant FA, Burns AR, et al. Androgens promote prostate cancer cell growth through induction of autophagy. Mol Endocrinol. 2013;27:280–95. doi: 10.1210/me.2012-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsouko E, Khan AS, White MA, Han JJ, Shi Y, Merchant FA, et al. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis. 2014;3:e103. doi: 10.1038/oncsis.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–92. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 20.Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene. 2014;33:5251–61. doi: 10.1038/onc.2013.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108:3665–70. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–30. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002;99:11890–5. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofman K, Swinnen JV, Verhoeven G, Heyns W. E2F activity is biphasically regulated by androgens in LNCaP cells. Biochem Biophys Res Commun. 2001;283:97–101. doi: 10.1006/bbrc.2001.4738. [DOI] [PubMed] [Google Scholar]
- 27.Sonnenschein C, Olea N, Pasanen ME, Soto AM. Negative controls of cell proliferation: human prostate cancer cells and androgens. Cancer Res. 1989;49:3474–81. [PubMed] [Google Scholar]
- 28.Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–82. [PubMed] [Google Scholar]
- 29.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–27. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welsh JB, Sapinoso LM, Su AI, Kern SG, Wang-Rodriguez J, Moskaluk CA, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–8. [PubMed] [Google Scholar]
- 31.Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–36. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 32.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–9. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 33.Arredouani MS, Lu B, Bhasin M, Eljanne M, Yue W, Mosquera JM, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin Cancer Res. 2009;15:5794–802. doi: 10.1158/1078-0432.CCR-09-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, et al. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 2001;61:5692–6. [PubMed] [Google Scholar]
- 35.Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 2010;70:859–62. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, McLeod DG, et al. Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate Cancer Prostatic Dis. 2010;13:311–5. doi: 10.1038/pcan.2010.31. [DOI] [PubMed] [Google Scholar]
- 37.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–67. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao L, Schwartzman J, Gibbs A, Lisac R, Kleinschmidt R, Wilmot B, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS One. 2013;8:e63563. doi: 10.1371/journal.pone.0063563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 2013;73:5277–87. doi: 10.1158/0008-5472.CAN-13-0549. [DOI] [PubMed] [Google Scholar]
- 40.Kokontis J, Takakura K, Hay N, Liao S. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res. 1994;54:1566–73. [PubMed] [Google Scholar]
- 41.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rai JS, Henley MJ, Ratan HL. Mammalian target of rapamycin: a new target in prostate cancer. Urol Oncol. 2010;28:134–8. doi: 10.1016/j.urolonc.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 43.Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998;58:2720–3. [PubMed] [Google Scholar]
- 44.Liu L, Dong X. Complex impacts of PI3K/AKT inhibitors to androgen receptor gene expression in prostate cancer cells. PLoS One. 2014;9:e108780. doi: 10.1371/journal.pone.0108780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research. N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costello LC, Franklin RB, Feng P. Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion. 2005;5:143–53. doi: 10.1016/j.mito.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franz MC, Anderle P, Burzle M, Suzuki Y, Freeman MR, Hediger MA, et al. Zinc transporters in prostate cancer. Mol Aspects Med. 2013;34:735–41. doi: 10.1016/j.mam.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2016;35:3201–8. doi: 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q, Hardie RA, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol. 2015;236:278–89. doi: 10.1002/path.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and Prostate Cancer. Genes Cancer. 2010;1:617–28. doi: 10.1177/1947601910379132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gong Y, Chippada-Venkata UD, Galsky MD, Huang J, Oh WK. Elevated circulating tissue inhibitor of metalloproteinase 1 (TIMP-1) levels are associated with neuroendocrine differentiation in castration resistant prostate cancer. Prostate. 2015;75:616–27. doi: 10.1002/pros.22945. [DOI] [PubMed] [Google Scholar]
- 52.Tsun ZY, Possemato R. Amino acid management in cancer. Semin Cell Dev Biol. 2015;43:22–32. doi: 10.1016/j.semcdb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–34. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schulte ML, Khodadadi AB, Cuthbertson ML, Smith JA, Manning HC. 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: A novel scaffold for inhibition of ASCT2-mediated glutamine transport. Bioorg Med Chem Lett. 2016;26:1044–7. doi: 10.1016/j.bmcl.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rajagopalan KN, DeBerardinis RJ. Role of glutamine in cancer: therapeutic and imaging implications. J Nucl Med. 2011;52:1005–8. doi: 10.2967/jnumed.110.084244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okudaira H, Shikano N, Nishii R, Miyagi T, Yoshimoto M, Kobayashi M, et al. Putative transport mechanism and intracellular fate of trans-1-amino-3-18F-fluorocyclobutanecarboxylic acid in human prostate cancer. J Nucl Med. 2011;52:822–9. doi: 10.2967/jnumed.110.086074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.