

Abstract
Botulinum toxins (BoNT) A and E block neurotransmitter release by specifically cleaving the C- terminal ends of SNAP-25, a plasma membrane SNARE protein. Here, we find that SNAP-25A and E, the cleavage products of BoNT A and E respectively, terminate membrane fusion via completely different mechanisms. Combined studies of single molecule FRET and single vesicle fusion assays reveal that SNAP-25E is incapable of supporting SNARE pairing and thus, vesicle docking. In contrast, SNAP-25A facilitates robust SNARE pairing and vesicle docking with somewhat reduced SNARE zippering, which leads to severe impairment of fusion pore opening. The EPR results show that the discrepancy between SNAP-25A and E might stem from the extent of the dynamic destabilization of the t-SNARE core at the N-terminal half which plays a pivotal role in nucleating SNARE complex formation. Thus, the results provide insights into the structure/dynamics-based mechanism by which BoNT A and E impair membrane fusion.
Keywords: SNARE, EPR, smFRET, botulinum toxin (BoNT), membrane fusion
eTOC Blurb
In this work, Khounlo et al show that cleavage of SNAP-25 by BoNT A and E terminates SNARE-mediated membrane fusion at different stages. EPR shows that this discrepancy stems from structural destabilization of the N-terminal half of t-SNARE.
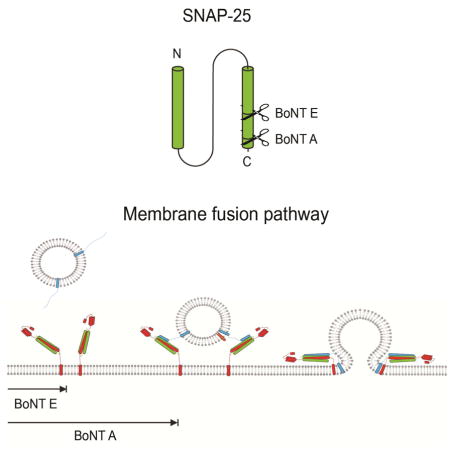
Introduction
Synaptic communication involves neurotransmitter release from the neuron to the synaptic cleft. The release of neurotransmitters requires synaptic vesicle docking onto the target plasma membrane, formation of a fusion pore, and complete fusion of two membranes. It is widely believed that this membrane fusion process is mediated by soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs). The SNARE proteins consist of VAMP2 on the synaptic vesicle (v-SNARE) and syntaxin 1A and SNAP-25 on the target membrane (t-SNAREs). The cognate v- and t-SNAREs, when brought into proximity, form a highly stable ternary SNARE complex that is thought to drive fusion of two membranes (Jahn and Scheller, 2006; Rizo and Rosenmund, 2008; Sudhof and Rothman, 2009; Wickner and Schekman, 2008).
More specifically, the highly conserved SNARE motifs, one from syntaxin 1A, two from SNAP- 25 and one from VAMP2, assemble into a parallel four helix bundle (Fasshauer et al., 1998; Poirier et al., 1998; Stein et al., 2009; Sutton et al., 1998). It has been proposed that SNARE complex formation is a multi-step process where zippering starts from the membrane-distal N-terminal region and progresses towards the membrane-proximal C-terminal region (Gao et al., 2012; Min et al., 2013; Shin et al., 2014; Sorensen et al., 2006). However, the coupling mechanism between zippering steps and membrane remodeling steps has been elusive (Lou and Shin, 2016).
Prior to their interaction with VAMP2, syntaxin 1A and SNAP-25 form a binary 1:1 t-SNARE complex on the plasma membrane (Fasshauer and Margittai, 2004; Fiebig et al., 1999; Rizo and Rosenmund, 2008). It has been shown that VAMP2 has a significantly higher affinity to the t- SNARE complex compared to the individual t-SNAREs (Calakos et al., 1994). Moreover, only with the t-SNARE complex, not with individual t-SNAREs, does VAMP2 assemble into the SNARE complex and elicit synaptic exocytosis. The importance of the t-SNARE complex may be further emphasized with the fact that some botulinum toxins (BoNT) inhibit synaptic exocytosis by enzymatically cleaving individual t-SNAREs (Gerona et al., 2000; Rossetto et al., 2014).
BoNTs are a class of protein toxins with eight distinct serotypes produced from clostridia. BoNT consists of four distinct domains that function to bind to the nerve terminals, translocate into the cytosol, and cleave SNAREs via the metalloprotease activity (Lacy et al., 1998). While all BoNT serotypes induce flaccid paralysis by inhibiting neurotransmitter release at the neuromuscular junction, individual isoforms target different SNAREs and cleave them at different positions (Rossetto et al., 2014).
Both BoNT A and E site-specifically cleave SNAP-25 at the C-terminal SNARE motif leaving 9 and 26 residue shortened versions SNAP-25A and SNAP-25E, respectively (Rossetto et al., 2014; Schiavo et al., 1993). This cleavage is sufficient to reduce or abolish membrane fusion. While BoNT E completely abolishes neurotransmitter release, BoNT A seems to have a milder effect considering some membrane fusion is rescued with elevated levels of Ca2+ (Gerona et al., 2000; Lundh et al., 1977). This may imply that BoNT E and A impair membrane fusion at different steps. Thus, an understanding of the impact of the cleavage on the structure and dynamics of the SNARE complexes could provide valuable insights into the mechanism by which SNAREs mediate membrane fusion.
In order to cohesively investigate the effect of BoNT A and E cleavage on SNARE complex formation and membrane fusion, we probed the structure and dynamics of t-SNAREs using electron paramagnetic resonance (EPR), followed by observing SNARE zippering with single molecule fluorescent resonance energy transfer (smFRET). We finally used single vesicle fusion assays to dissect the individual membrane fusion steps. Our results show that the BoNT A and E, although otherwise similar except that BoNT E cuts 17 residues more from SNAP-25 than BoNT A, impairs membrane fusion through entirely different mechanisms. While SNAP-25E blocks SNARE complex formation and vesicle docking, SNAP-25A allows robust vesicle docking, but reduces SNARE zippering and significantly impairs membrane fusion. The EPR results show that such big differences might stem from the extent of the dynamic destabilization of the t-SNARE core at the N-terminal half which plays a pivotal role in nucleating SNARE complex formation.
Results
Dynamic structure of the 1:1 binary t-SNARE complex of syntaxin 1A and SNAP-25
Although the structures of the ternary SNARE complex have been thoroughly investigated in both solution and membrane mimetics (Poirier et al., 1998; Shin et al., 2014; Stein et al., 2009), the t- SNARE complex is less well defined. Considering that the t-SNARE complex serves as a precursor to the ternary SNARE complex, further insights into the structure could shed light on the mechanistic steps in the ternary SNARE complex formation.
There are two forms of the t-SNARE complex: the non-productive 2:1 (syntaxin 1A: SNAP-25) complex and the productive, on-pathway 1:1 complex (Fasshauer and Margittai, 2004; Xiao et al., 2001). The 2:1 t-SNARE complex has been previously investigated with EPR (Xiao et al., 2001; Zhang et al., 2002). The structure is a parallel four-helix bundle, basically identical to the ternary SNARE complex, but with the second syntaxin 1A SNARE motif replacing VAMP2. In contrast, the structure of the 1:1 t-SNARE complex has been elusive, most likely due to the dynamic nature of the structure.
Based on their smFRET experiments in live cells, An and Almers have previously proposed that the N-terminal SNARE motif of SNAP-25 (SN), within the 1:1 t-SNARE complex, forms a robust helical complex with syntaxin 1A while the C-terminal SNARE motif of SNAP-25 (SC) is detached from the complex and freely diffuses in solution (An and Almers, 2004). To verify this model, we first probed the dynamics of SN in the 1:1 complex using site-directed spin labeling EPR. This technique was chosen because the lineshape is highly sensitive to the motion of the nitroxide spin label, which is a reflection of the local structural environment. In one case, we attached the nitroxide to an engineered single cysteine at position 42 in the N-terminal region of SN and in another case, the nitroxide was attached at position 74 in the C-terminal region of SN (Figure 1A). Prior to complex formation, EPR spectra from both spin labeled mutants showed narrow lineshapes, prototypical of a freely diffusing random coil. However, when SNAP-25 was complexed with syntaxin 1A in the 1:1 stoichiometry, we observed extensive line-broadening in both cases (Figure 1B). This suggests that SN underwent a conformational change from a random coil to a helical structure at both N-terminal and C-terminal regions upon formation of the t- SNARE complex.
Figure 1. EPR spectra and analysis of spin-labeled SNAP-25 as monomers and part of the 1:1 binary t-SNARE complex.

(A) Schematic representation of the site-directed spin labeling EPR of the t-SNARE complex with syntaxin 1A (red) and SNAP-25 (green). SNAP-25 is denoted with spin labeled positions and BoNT A and E cleavage sites. The conserved zero layer is represented by a dashed line. The inset depicts chemical structure of the spin label (MTSSL) attached to the cysteine side chain. (B) Room temperature EPR spectra for A42C and A74C in the SN domain in monomeric SNAP-25 or in the t-SNARE complex. (C) Representative EPR spectral subtraction analysis. The composite binary EPR spectrum (black) was subtracted by the monomer spectra (red) to obtain the broad, interacting spectral component (blue). (D) Bound fraction of the labeled positions (SN domain) in the t-SNARE complex obtained from the spectral subtraction analysis. The data are shown as means ± SD. (E) Room temperature EPR spectra for the labeled positions in the SC domain in monomeric SNAP-25 or in the t-SNARE complex. The red arrow indicates areas of EPR lineshape broadening. (F) Bound fraction of the labeled positions (SC domain) in the t-SNARE complex obtained from the spectral subtraction analysis. The average bound fraction (red) of spin labeled positions G168C, T173C, N175C, and N196C is 0.61. Position 203 was excluded when calculating the mean due to its position being near the end the SC domain which is known to be frayed and unstructured. The data are shown as means ± SD. (G) Model of dynamic equilibrium of the SNAP-25.
We still observed some narrow components in the EPR spectra, which represent signals from an unstructured polypeptide. The percentage of the narrow spectral components was quantitatively determined with the spectral subtraction method (Figure 1C) (Thorgeirsson et al., 1996). We found that the narrow components were approximately 5–10% of the composite spectra (Figure 1D). These numbers are consistent with the previously reported dissociation equilibrium of the 1:1 binary t-SNARE complex, where Kd ~0.4 μM (Weninger et al., 2008). One could argue that the narrow component may have been due to the predominant existence of the 2:1 complex in the 1:1 mixture. However, if this were the case, a much larger narrow component would be observed due to the significant fraction of SNAP-25 remaining as monomers.
We then investigated the structure and dynamics of SC in the 1:1 complex using EPR. We generated five single cysteine mutants: G168C, T173C, N175C, N196C, and L203C of SC. These mutants were specifically selected to be around the central conserved residue Q174 (zero layer); two were positioned on the N-terminal half and three were positioned on the C-terminal half (Figure 1A). As monomers, all spin labeled SNAP-25 mutants displayed a narrow EPR spectra similar to what was observed with the SN, indicative of a freely diffusing polypeptide chain with little secondary or tertiary structure. However, when bound to syntaxin 1A all positions except 203C exhibited a composite two component spectra with one broad component reflecting the structured species and another narrow component reflecting the unstructured species (Figure 1E and F). We did not observe much spectral change with 203C, consistent with the previous finding that the C-terminal end is frayed for the 1:1 complex (Zhang et al., 2016).
Quantitative spectral subtraction analysis revealed that approximately 40% of SC was unstructured (Figure 1F), which was 4 times more than what was expected from the dissociation equilibrium between syntaxin 1A and SNAP-25. Taking into account the global association-dissociation equilibrium, we estimate that approximately 30% of SC remains unstructured when SNAP-25 is complexed with syntaxin 1A (Figure 1F). Thus, the results show that a significant fraction of SC (~30%) is unstructured while SN is complexed with syntaxin 1A. The results are partially consistent with the dynamic structure proposed by An and Almers in that SC has the tendency to dissociate from the complex (An and Almers, 2004). However, our observations suggest that the majority of SC (~70%) is still bound to syntaxin 1A and SN and together they form a three helix bundle (Figure 1G).
Cleavage of SC by botulinum toxins increases the dynamics of SC
Having characterized the 1:1 t-SNARE complex with EPR, we investigated the impact of the proteolytic cleavage of SC by BoNT A and E. To this end, we prepared recombinant SNAP-25 mutants of reduced lengths, SNAP-25A (aa. 1-197) and SNAP-25E (aa. 1-180). For SNAP-25A and SNAP-25E, we attached nitroxide spin labels at the same positions as those of the wild-type described in the previous section (Figure 2A).
Figure 2. EPR spectra and analysis of spin-labeled SNAP-25, SNAP-25A, and SNAP-25E as monomers or part of the 1:1 binary t-SNARE complex.

(A) Schematic representation of the site-directed spin labeling positions of SNAP-25, SNAP-25A and SNAP-25E. The zero layer is denoted by a dashed line. (B) Room temperature EPR spectra of SNAP-25, SNAP-25A and SNAP- 25E spin labeled variants on the SN motif as monomers or part of the t-SNARE complex. (C) EPR spectra of SNAP-25, SNAP-25A, and SNAP-25E spin labeled variants on the SC motif as monomers or part of the t-SNARE complex. The red arrows point to the broad component of the EPR spectra. (D) Bound fraction of the labeled positions in the t-SNARE complex obtained from spectral subtraction analysis for SC and SN motifs are shown in the left and right graph, respectively. The data for SNAP-25 (black circle), SNAP-25A (orange triangle), and SNAP-25E (blue rectangle) are shown as means ± SD.
For spin labeled positions 42 and 74 on SN, not much difference in the spectral line shape between wild-type SNAP-25 and the shortened mutants was observed. Further quantitative spectral subtraction analysis confirmed that the amounts of the narrow spectral components reflecting the global dissociation of the t-SNARE complex remained within ± 5% (Figure 2B). This suggests that BoNT A or E-induced cleavage of SNAP-25 does not alter the stability of SN in the t-SNARE complex.
However, when we examined the EPR spectra from SNAP-25E, the broad spectral components reflecting the structured conformation was reduced significantly (Figure 2C). Quantitative spectral subtraction analysis showed that the bound fraction was decreased for SNAP-25E as much as ~55% for the spin labeled positions in SC (Figure 2D). Thus, our results show that the cleavage of 26 residues of SC causes the dynamic destabilization of the already dynamic t-SNARE complex.
In contrast, the dynamic destabilization that was brought about by the cleavage of SC by BoNT A appeared to be milder than what was observed with the cleavage by BoNT E (Neal et al., 1999). The effect is pronounced on positions 175 and 196, which are located in the C-terminal half of SC. In contrast, the change is not visible in the N-terminal half. Thus, the EPR analysis shows that while BoNT E affects the dynamics of the entire SC motif, the effect of BoNT A cleavage is confined within the C-terminal half of SC.
Cleavage of SC by BoNT E impairs ternary SNARE complex formation
The EPR results suggest that although the SN in the t-SNARE complex was bound robustly to the syntaxin 1A, the SC was partially bound and destabilized. This destabilization was significantly increased for SNAP-25E, while the change was confined only within the C-terminal half for SNAP- 25A. We then asked how the increased dynamics of the t-SNARE complexes due to the BoNT cleavage affects the formation of the ternary SNARE complex or SNARE zippering.
To answer this question, we observed SNARE zippering at the membrane proximal region of a single trans-SNARE complex assembled between two nanodiscs (Figure 3A) with smFRET. Experimentally, we site-specifically labeled VAMP2 A72C and syntaxin 1A V241C with the fluorescence donor Cy3 and the fluorescence acceptor Cy5, respectively. We then prepared two populations of nanodiscs with the labeled proteins, one reconstituted with Cy3-labeled VAMP2 (v- nanodisc) and the other with the t-SNARE complex (t-nanodisc). The t-SNARE complex was prepared by premixing Cy5-labeled syntaxin 1A with either SNAP-25, SNAP-25A, or SNAP-25E. The t-nanodiscs were mixed with v-nanodiscs to allow the formation of the trans-SNARE complex and then immobilized onto the polyethylene glycol (PEG)-covered quartz imaging surface via streptavidin and biotin-PEG-DSPE conjunction. After washing out free nanodiscs with sufficient buffer, the smFRET efficiencies of the single nanodisc pairs were analyzed from the fluorescence image. We found, using photobleaching counting, that most nanodiscs (~90%) have single fluorescent dyes (Figure S1). The results show that more than 90% of t-nanodiscs had the 1:1 t-SNARE complex. We only analyzed the nanodisc pairs that have single acceptor and donor dyes, which was verified with photobleaching after the FRET measurements.
Figure 3. smFRET analysis of the ternary trans- and cis-SNARE complex using nanodiscs.

(A) Schematic of a nanodisc sandwich harboring a single trans-SNARE complex with the FRET pair at the C-terminal region (CC). VAMP2 A72C-Cy3 and syntaxin 1A V241C-Cy5 were used for CC. (B) Relative docked nanodisc sandwiches for SNAP-25A and SNAP-25E normalized to SNAP-25 are shown as means ± SD. Docking was significantly reduced with SNAP-25E. (C) Histogram of the FRET efficiency distribution for SNAP-25 (top), SNAP-25A (middle) and SNAP-25E (bottom). The distribution showed two distinct populations in the high and in the low FRET regions for SNAP-25 and SNAP-25A. The distribution for SNAP-25E is not well organized. Total of 307, 380 and 69 traces were analyzed for SNAP-25, SNAP-25A and SNAP-25E respectively. (D) The fraction of the high FRET population from the trans-SNARE complex. Approximately half of the population is distributed in the high FRET region for SNAP-25 and SNAP-25A. (E) Schematic a single cis-SNARE complex (CC) anchored to a single nanodisc by the syntaxin 1A transmembrane domain. (F) Relative co-localized Cy3-Cy5 spots for SNAP-25A and SNAP-25E normalized to SNAP-25. Co-localized spots are significantly reduced with SNAP- 25E. Data are shown as means ± SD. (G) Histogram of the FRET efficiency distribution for SNAP- 25 (top), SNAP-25A (middle) and SNAP-25E (bottom). The distribution showed one distinct population in the high FRET regions for SNAP-25 and SNAP-25A. The distribution for SNAP- 25E was not well organized. Total of 422, 411 and 47 traces were analyzed for SNAP-25, SNAP- 25A and SNAP-25E respectively. (H) The fraction of the high FRET population from the cis- SNARE complex. In (B), (D), (F) and (H), the data are shown as means ± SD (*p < 0.05, **p < 0.01, and ***p < 0.005 by Student’s t test; n = 3 independent experiments).
When we counted the number of co-localized donor and acceptor signals, which represented nanodisc-to-nanodisc docking, there was no apparent difference between the wild-type SNAP-25 and SNAP-25A. However, we observed a dramatic decrease of docking, as much as a factor of 1/5, with SNAP-25E (Figure 3B).
We took a closer look into the docked nanodisc sandwich and further examined FRET efficiencies coming from the nanodisc pairs. For wild-type SNAP-25, the histogram showed both a low and a high FRET population, which was consistent with the results from our previous study (Shin et al., 2014; Yin et al., 2016). For SNAP-25E, the high FRET population completely disappeared with some small remaining populations at low FRET (Figure 3C). Our observations were quantitatively confirmed by analyzing the high FRET fraction of the total docked nanodisc pairs (Figure 3D). Although the overall low and high FRET distribution is similar to that of the wild-type, SNAP-25A had ~20% of the high FRET population shifted towards the low FRET region, which indicates that SNARE zippering was mildly hampered due to the cleavage. Thus, the results show that for SNAP-25E, both t- and v-SNARE pairing and SNARE zippering are severely hampered. However, for SNAP-25A the changes are rather mild.
For comparison, we examined the post-fusion state by preparing the cis-SNARE complex in a nanodisc. The cis-SNARE complex was prepared using VAMP2 without the transmembrane domain such that the complex is anchored to the nanodisc via the transmembrane helix from syntaxin 1A (Figure 3E). Consistent with the previous results, we observed severely diminished v- and t-SNARE pairing for SNAP-25E, while no apparent difference between SNAP-25A and wild-type (Figure 3F and G). FRET histograms displayed a dominant high FRET population which peaked at the FRET efficiency E = 0.90 for wild-type SNAP-25 and SNAP-25A, but SNAP-25E did not show any dominant population (Figure 3G and H). Thus, the results suggest that the t-SNARE complex prepared with SNAP-25E is unable to form a well-structured ternary SNARE complex even in the cis conformation.
Cleavage of SC by botulinum toxins decreases/abolishes Ca2+-triggered vesicle fusion
The results from the smFRET experiments show that the t-SNARE complex composed of SNAP-25E and syntaxin loses its ability to bind to VAMP2. However with SNAP-25A, we observed only a mild decrease in SNARE zippering. This is intriguing because previous in vivo studies have shown that both BoNT A and E both elicit the effective inhibition of synaptic exocytosis (Gerona et al., 2000). This raises the possibility that cleavage by BoNT E inhibits the synaptic vesicles at the vesicle docking step, while BoNT A inhibits synaptic exocytosis at a later membrane fusion step.
To test this possibility, we examined Ca2+-triggered SNARE-mediated membrane fusion in the presence of auxiliary factors synaptotagmin 1 and complexin. Synaptotagmin 1 is a major Ca2+ sensor that triggers SNARE-mediated membrane fusion with the Ca2+ signal (Brose et al., 1992; Fernandez-Chacon et al., 2001), while complexin is believed to finely regulate membrane fusion (Tang et al., 2006; Xue et al., 2007). Using the in vitro single-vesicle content-mixing assay, we examined how the SNAP-25A and E affect docking and membrane fusion at the single vesicle level.
Experimentally, we prepared two populations of vesicles representing the synaptic vesicles and the target membrane. The t-vesicles, reconstituted with syntaxin 1A and SNAP-25, were injected into the flow chamber and tethered on the PEGylated imaging surface through streptavidin-biotin conjugation. The v-vesicles, encapsulating 20 mM sulforhodamine B (SRB) and reconstituted with both VAMP2 and synaptotagmin 1, were injected into the flow chamber to allow vesicle docking. After docking, a buffer wash was used to remove any unbound v-vesicles. Throughout the process, the concentration of complexin was maintained at 100 nM, which has been recently shown to confer physiologically relevant Ca2+ sensitivity (Kim et al., 2016). After the docked vesicle-vesicle pairs were prepared, Ca2+ was injected into the flow chamber to trigger membrane fusion (Figure 4A). Fluorescence dequenching of SRB caused a stepwise increase in the fluorescence intensity of the v-vesicles, which was used to identify content-mixing from individual vesicle pairs (Figure 4A and B).
Figure 4. SNAP-25A is unable to trigger SNARE-mediated membrane fusion with 10 μM Ca2+.

(A) Schematic of the in vitro single-vesicle content-mixing assay. After the t-vesicles are immobilized on the imaging surface, unbound t-vesicles were washed out and subsequent docking and washing of unbound v-vesicles were also performed. Once the v-vesicles and t-vesicles are docked, we injected Ca2+ into the flow chamber to evoke content-mixing which are detected by a sudden step-wise increase of fluorescent intensity. The flow chamber maintained constant 100 nM Cpx concentration throughout the experiment. (B) A representative fluorescent intensity time trace is shown in blue. The stepwise increase indicates content-mixing. The red arrow indicates the time Ca2+ was added to the flow chamber. (C) Bar graph of the average number of docked vesicles-vesicle pairs from 3 recordings. Bar graph of the average fusion percentage from 3 recordings are shown for (D) 10 μM Ca2+ and for (E) 500 μM Ca2+. (F) Schematics of the in vitro single-vesicle lipid-mixing assay. The single-vesicle lipid-mixing assay was identical to the content-mixing assay prior to Ca2+ injection with the exception of using DiI and DiD instead of SRB. (G) The FRET distribution between the docked vesicle-vesicle pairs for prepared with SNAP-25 and SNAP-25A are shown left and right, respectively.
As expected, while the number of docked vesicles was similar for t-vesicles prepared with the wild-type and those with SNAP-25A, we observed almost no docking with SNAP-25E (Figure 4C). This agreed well with the aforementioned smFRET experiments. When we flew in 10 μM Ca2+, approximately 45% of the docked vesicles-vesicle pairs showed fusion with the wild-type, similar to our previous results (Kim et al., 2016). The first-order time constant of the content mixing kinetics was 5 seconds in the kinetic measurement that was carried out on the timeframe of 1 minute. In sharp contrast, we observed no fusion with both SNAP-25A and SNAP-25E (Figure 4D). Taken together, the results suggest that BoNT E blocks synaptic membrane fusion by prohibiting docking, while BoNT A stops fusion after docking, but prior to the fusion pore opening.
A previous in vivo study reported that the inhibition of the release by BoNT A can be rescued via treatment with high Ca2+ concentrations (~200 μM) (Gerona et al., 2000; Lundh et al., 1977). To test whether our in vitro system faithfully recapitulates the in vivo results, we performed the single vesicle content-mixing assay using 500 μM Ca2+. Indeed, the fusion activity with t-vesicles prepared with SNAP-25A was rescued up to ~60% of the wild-type. However, membrane fusion was still completely abolished with SNAP-25E even at 500 μM Ca2+ (Figure 4E).
To further dissect steps where membrane fusion was inhibited, we performed the single vesicle- to-vesicle lipid mixing assay (Figure 4F). The experiment was prepared identically to the content- mixing assay except for incorporating lipid dyes instead of content dyes. By doing so, we are able to characterize the physical state of the docked vesicle pairs just prior to Ca2+ injection. We observed a homogenous FRET histogram centered at E = ~0.4 with the wild-type SNAP-25, indicating that hemifusion may be the dominant species prior to the Ca2+ injection. However, with SNAP-25A the FRET histogram is spread over a wider range and is skewed towards low FRET values (Figure 4G). Meanwhile, the docked vesicle pairs were extremely rare with SNAP-25E to the extent that we were unable to obtain an accurate FRET histogram. Thus, the results suggest that for the wild-type, the vesicles are prepared in the hemifused state ready to fuse upon Ca2+ injection (Kweon et al., 2017). In contrast, SNAP-25A is incapable of priming the fusion complex, while SNAP- 25E is unable to even mediate vesicle docking.
Discussion
It is well established that BoNTs inhibit neurotransmitter release from the neuron by site- specifically cleaving SNAREs (Rossetto et al., 2014; Neale et al., 1999). However, the underlying molecular mechanisms by which the BoNT-shortened SNAREs fail to elicit neurotransmitter release are not clearly understood.
In this work, we investigated the effects of SNAP-25 cleavage by BoNT A and E on the initial interactions with syntaxin 1A with EPR, on subsequent interactions and zippering with the vesicle v-SNARE VAMP2 using single molecule FRET, and on specific membrane fusion steps with the single vesicle-vesicle docking and fusion assay.
By employing a combination of these techniques, we were able to comprehensively dissect how cleavage of SNAP-25, by BoNT A and E, impacts SNARE-mediated membrane fusion from the very early steps of SNARE assembly through the final steps of membrane fusion. In comparison to the wild type SNAP-25, the EPR results show that the cleavage of 26 residues at the C-terminal end by BoNT E significantly destabilizes the C-terminal SNARE motif (SC). Importantly, the destabilization infiltrates into the N-terminal half which serves as the nucleating core for the interaction with VAMP2. Consequently, we observed the dramatic decrease of SNARE complex formation and vesicle docking, which resulted in almost no membrane fusion even at high Ca2+ concentrations.
While the EPR spectra show that SNAP-25E destabilizes the entire SC motif, SNAP-25A appears to have a milder effect and the destabilization was confined within the C-terminal half of SC. Our results suggest that the structural integrity of the N-terminal core of the t-SNARE complex is still preserved despite the deletion of the 9 residues at the C-terminus. In addition, the smFRET results with SNAP-25A show that the docking probability remains unchanged with the FRET distribution shifted mildly towards low FRET values, indicative of somewhat impaired SNARE zippering. However, the single vesicle fusion assay shows complete inhibition of fusion with physiologically relevant 10 μM Ca2+. Taken together, our results suggest that BoNT A tampers with membrane fusion just prior to the fusion pore formation step, in sharp contrast to BoNT E which inhibits at the very early step of SNARE complex formation.
It is remarkable that, despite only being a 17 residue difference in length, SNAP-25A and E terminate membrane fusion at completely different steps along the fusion pathway. While SNAP- 25A is able to support robust docking, SNAP-25E loses its ability to interact with VAMP2 almost completely, seriously impairing vesicle docking. Our results are in line with the previous observation in the presynapse that the size of the readily releasable vesicle pool for the BoNT A-treated neuron, determined by the high K+ treatment, is similar to the control, while that for the BoNT E-treated neuron is significantly reduced (Neal et al., 1999). Furthermore, our results show that the C-terminal part of SC plays an important role in maintaining the stability of the N-terminal core of the t-SNARE complex, which is necessary for the interaction with VAMP2. For SNAP-25A, the t-SNARE core is able to tolerate the loss of C-terminal 9 residues. However, the loss of 26 residues in SNAP-25E is sufficiently large to disrupt the stability of the N-terminal core of the t-SNARE complex.
Now one might ask why SNAP-25A is able to lead the membrane fusion process up to docking, but utterly fail thereafter. We observed some decrease of the high FRET population with SNAP- 25A, indicating some impaired SNARE zippering for SNAP-25A. However, it appeared that the shift was only mild and one might wonder if this is sufficient to explain the major blockage of membrane fusion. One possible scenario could be that, although not significant individually, this effect could be amplified due to the expected cooperativity among the multiple SNARE complexes that are believed to participate at the active zone (Montecucco et al., 2005). We also note that the membrane proximal C-terminal region of the SNARE complex plays an important role when interacting with synaptotagmin 1 and complexin (Chen et al., 2002; Gerona et al., 2000; Kim et al., 2012). Thus, we cannot rule out the possibility that the loss of 9 residues in SNAP-25A hampers the necessary interaction with the accessory proteins.
In the past, clostridial neurotoxins, including BoNT A and E, have played a crucial role in revealing SNAREs as the core fusion machinery for neurotransmitter release (Gerona et al., 2000; Lundh et al., 1977; Pirazzini M et al., 2017). In this study, we performed a comprehensive analysis of their impact on the structure and dynamics of SNARE complex and consequential effects on membrane fusion steps. The results from this study reveal new insights into the mechanism by which SNARE complex formation is coupled to individual fusion steps.
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Yeon-Kyun Shin (colishin@iastate.edu).
Experimental Model and Subject Details
All the experiments were performed in vitro.
Method Details
Plasmid construction and mutagenesis
DNA sequence encoding: syntaxin 1A (amino acids 1-288 with three cysteines replaced by alanines), SNAP-25 (amino acids 1-206 with four native cysteines replaced by alanines), SNAP- 25A (amino acids 1-198), SNAP-25E (amino acids 1-180), VAMP2 (amino acids 1-116 with 1 cysteine replaced by alanines), soluble VAMP2 (amino acids 1-96), complexin (Cpx, amino acids 1-134) were inserted into the pGEX-KG vector as GST fusion proteins. Synaptotagmin 1 (amino acids 50-421 with four cysteines replaced by alanines) was inserted into pET-28b vector as C- terminal His-tagged proteins. Modified apoA1 (amino acids, 1-258) was inserted into pNFXeX vector as a N-terminal His-tagged protein. All cysteine mutants, including SNAP-25 A42C, SNAP- 25 A74C, SNAP-25 G168C, SNAP-25 T173C, SNAP-25 N175C, SNAP-25 N196C, and SNAP-25 L203C were generated by the QuickChange site-directed mutagenesis kit and confirmed by DNA sequencing (Iowa State University DNA Sequencing Facility).
Cell Culture
All recombinant proteins were expressed in Escherichia coli BL21 (DE3) bacterial strain cells. Cells were first grown in LB medium at 37°C with shaking at 200 rpm until optical density reached 0.6–0.8 (600 nm). Isopropyl-β-D-thiogalactopyranoside (IPTG) (0.4 mM final concentration) was added to induce protein expression. After induction cells were further incubated at 16°C, shaking at 100 rpm for another 14–16 h.
Protein Purification
For GST-tagged proteins, cells were pelleted and resuspended in 15 mL of PBST (PBS, pH 7.4, containing 0.2 Triton X-100) for membrane proteins and 15 ml of PBS (pH 7.4) for the soluble proteins with final concentrations of 1 mM 4-(2- aminoethyl)- benzenesulfonyl fluoride (AEBSF) and 4 mM DTT. Cells were lysed by sonication on ice and centrifuged at 25,000 g for 30 min at 4°C. The cleared supernatant was nutated with 2 ml of GST beads for 1 hour at 4°C. His-tagged synaptotagmin 1 and ApoA1 were purified using the same protocol except for using the Ni-NTA column.
After intense washing, the bound proteins were then eluted by 0.02 unit/μL thrombin in cleavage buffer (PBS, pH 8.0, containing 2 mM DTT) with/without 0.8 noctyl-D-glucopyranoside (OG) for GST-tagged membrane and soluble proteins, respectively. For His-tagged proteins, elution was carried out with buffer of 25 mM HEPES, 400 mM KCl, 500 mM imidazole and 0.8% OG. Purified proteins were examined by SDS-PAGE (15%, w/v) and the purity was at least 85% for all proteins. The protein concentration was determined using RC DC kit.
Site-directed spin and fluorophore labeling
For site-directed spin labeling of single cysteine mutants for EPR, the protein were incubated with 5 mM dithiothreitol (DTT) for 1 hour at 4°C. The protein was then subjected to the PD-10 size exclusion column and incubated for 16 hours at 4°C with 10-fold molar excess of 1-Oxyl-2,2,5,5- tetramethylpyrroline-3-methyl methanethiosulfonate (MTSSL). The proteins were then subjected to another PD-10 size exclusion column to remove any excess spin labels.
For site-directed fluorophore labeling of single cysteine mutants, the proteins were desalted with a PD MiniTrap G-25 column to eliminate free DTT and then incubated with a 10-fold molar excess of maleimide-derivative fluorophore indodicarbocyanine (Cy5) or indocarbocyanine (Cy3), respectively, overnight at 4 °C. The labelled protein was purified using the PD MiniTrap G- 25 column and free dye was further separated from the protein sample using centrifugal filters (3kDa, Amicon Millipore Corporation, Bedford, MA). The labeling efficiency of each SNARE protein was measured spectrophotometrically (NanoDrop 2000c Spectrophotometer, Thermo Fisher Scientific, Waltham, MA). Further details can be found in our recent paper (Yin et al., 2016).
EPR data collection
EPR spectra were obtained using a Bruker ESP 300 spectrometer (Bruker, Germany) equipped with a low-noise microwave amplifier (Miteq, Hauppauge, NY) and loop gap resonator (Medical Advances, Milwaukee, WI). The modulation amplitude was set at no greater than one-fourth of the line width. Spectra were collect at room temperature in the first-derivative mode.
SNARE-incorporated nanodiscs preparation
The following lipids were used to generate nanodiscs. t-nanodiscs were prepared with 1,2- dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 1-palmitoyl-2-oleoyl-snglycero-3- phosphocholine (POPC), phosphatidylinositol-4,5-bisphosphate (PIP2, from porcine brain), cholesterol, and biotin-PEG-DSPE with a molar ratio of 62.9:15:20:2:0.1. The v-nanodiscs are composed of POPC, DOPS, and cholesterol with molar ratios of 75:5:20. Lipid film was made by drying the lipid mixtures with air and further incubation in house vacuum for 16–18 hours. The lipid film was then resuspended in HEPES buffer (25 mM HEPES, pH 7.4, and 150 mM KCl) to a final concentration of 50 mM. We then took 5 μL of the resuspended lipid mixture and dissolved it in sodium cholate such that the final concentration of sodium cholate would become 50 mM after the addition of apoA1 and SNARE proteins. Then, the binary t-SNARE complex (syntaxin 1A and SNAP proteins with a molar ratio of 1:2, pre-incubated at room temperature for 1 h) for t-nanodisc and VAMP2 for v-nanodiscs were added along with apoA1 into their respective detergent-solubilized lipid mixture. The final molar ratio of lipids, apoA1, and SNARE(s) was 300:5:1. After a 20 min incubation period, the reconstitution of protein into nanodiscs was initiated by rapid removal of detergent with 50% (w/v) SM-2 Bio-Beads. Homogenously sized t- and v-nanodiscs were purified by gel filtration using a SuperdexTM 200 GL 10/300 column (GE Healthcare Life Sciences, Piscataway, NJ).
Preparation of trans-/cis-SNAREpin nanodiscs
The PEGylated imaging surface (25 × 75 × 1.0 mm) was prepared as previously described (Yin et al., 2016). The flow chambers were assembled with strips of double sided tape placed between the quartz slide and coverslip. Streptavidin (0.2 mg/mL) was incubated in the flow chamber for 10 min and washed out prior to subsequent nanodisc immobilization.
The experiments using trans-SNAREpin nanodiscs, t-nanodiscs, and v-nanodiscs were pre-incubated at room temperature for 45 minutes with a molar ratio of 1:5 to allow the formation of a trans- SNAREpin between two nanodiscs. The unbound nanodiscs were then washed out. The experiments with cis-SNAREpin nanodiscs, t-nanodiscs, and soluble VAMP2 were pre-incubated at room temperature for 45 minutes with a molar ratio of 1:5 to allow the formation of a cis-SNAREpin in one nanodisc. The unbound t-nanodiscs and soluble VAMP2 were then washed out.
smFRET collection
The imaging of the samples was performed at room temperature with the oxygen scavenger system [0.4% glucose (Sigma Aldrich, St. Louis, MO), 4 mM Trolox, 1 mg/mL glucose oxidase, 0.04 mg/ml catalase in HEPES buffer.
Proteoliposome Reconstitution
For the single vesicle content mixing assay, molar ratios of lipids used for vesicle preparation were 15:63:20:2:0.1 (DOPS: POPC: Cholesterol: PIP2: biotin-PEG-DSPE) for the t-vesicles, and 5:75:20 (DOPS: POPC: Cholesterol) for the v-vesicles, respectively. We added 1% 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate (DiI) and 1,1′-Dioctadecyl- 3,3,3′,3′-Tetramethylindodicarbocyanine, 4-Chlorobenzenesulfonate (DiD) to the v- and t- vesicles, respectively, for the single vesicle lipid mixing assay (Thermo Fisher Scientific, Waltham, MA). The lipids were dried in a glass tube with nitrogen gas and stored overnight in a vacuum desiccator. The lipid film was resuspended with HEPES buffer (25mM HEPES, 100 mM KCl, pH 7.4) whereas, in the single vesicle content mixing assay, v-vesicle lipid film was resuspended with HEPES buffer containing 20 mM SRB (Thermo Fisher Scientific, Waltham, MA). After 10 times freeze-thaw cycles between hot water and liquid nitrogen, we used an extruder to make unilamellar vesicles with polycarbonate filter (100 nm pore size, Avanti Polar Lipids, Alabaster, AL). The binary t-SNARE complex, pre-mixed at room temperature for 30 min, were mixed with liposomes (10 mM in total lipid concentration) while VAMP2 and Syt1 were reconstituted with SRB (20 mM)-containing liposomes for ~10 min. We used a 200:1 lipid/protein molar ratio for both t- and v-vesicles. The mixture was diluted with HEPES buffer (3 times the lipid/protein mixture volume) and then dialyzed in 2 L dialysis buffer at 4°C overnight. For the v- vesicles, free SRB was removed using the PD-10 desalting column (GE Healthcare Life Sciences, Piscataway, NJ) after dialysis.
Single vesicle content-mixing data collection
The imaging surface was prepared identical to the nanodisc smFRET experiments. We introduced a mixture containing 125 μM t-vesicles with 100 nM Cpx in HEPES buffer into the flow chamber. Once t-vesicles were immobilized on the PEGylated surface, the unbound t-vesicles were washed using HEPES buffer containing 100 μM Cpx. Then mixture of 100 nM Cpx and v-vesicles (20 mM SRB) in HEPES buffer was injected and incubated for 10 min for vesicle-vesicle docking. The unbound v-vesicles were washed out using HEPES buffer containing 100 nM Cpx. We imaged the vesicle-vesicle pairs as we injected Ca2+ into the flow chamber.
Single vesicle lipid-mixing data collection
The single vesicle lipid mixing experiment was performed identical to the content-mixing assay with exception of Ca2+. Data from docked vesicle-vesicle pairs were obtained taking multiple images from randomly selected areas in the flow chamber. The immobilized spots were analyzed and we measured the FRET efficiencies of individual vesicle pairs and plotted onto a histogram.
Quantification and Statistical Analysis
EPR data analysis
The Bruker Xepr software was used to collect all EPR spectra (Figure 1b, 1e, 2b, and 2c). The spectral subtraction method involves obtaining the EPR spectra of the monomer and the composite at the same position. The monomer was subtracted from the composite in order to determine the amount of monomer present (Figure 1c). This was used to quantitatively determine the population of mobile and immobile spin labeled proteins (Figure 1d, 1f, and 2d).
smFRET data analysis
Images were recorded using smFRET Package software that was a gift from the Taekjip Ha lab. While recording, the first 10–20 frames were excited by a red laser (635 nm) so the t- nanodisc could be identified. We then switched to the green laser (532 nm) to identify v- and t- pairing from the nanodiscs. The co-localized spots, which have both the acceptor and donor signal channel, were then selected and analyzed.
From the selected spots, the acceptor and donor time traces were analyzed to obtain the FRET histograms (Figure 3c and 3g). As mentioned in our previous work (Yin et al., 2016), we seldom observed any significant transitions in the FRET value using 100 msec time resolution. Thus, we assigned a single FRET value (mean) for each co-localized spot. The assigned FRET value was obtained from the period in which both dyes were photon emitting. Significant FRET values were quantified using the student’s t-test. The t-test was performed using the GraphPad software (Figure 3b, 3d, 3f, and 3h).
Single vesicle content/lipid mixing analysis
Images were obtained using the same software as the smFRET data analysis. However, the data was analyzed using a home built software developed in MATLAB R2014b. For the single vesicle content mixing assay, a stepwise jump in the fluorescence intensity was monitored as an indication of content-mixing (Figure 4b). The fusion percentage was the average number of fused vesicles over the total number of docked vesicles in 3 separate recordings (Figure 4d and 4e). For the single vesicle lipid mixing assay, the FRET distribution was calculated in the same way as the smFRET data analysis (Figure 4g).
Data and Software Availability
Data resources
The following DNA and protein sequences have been deposited in UniProt under the corresponding ID codes.
Software resources
The following software can be found at the corresponding links.
Bruker Xepr used to collect and analyze EPR spectra: https://www.bruker.com/products/mr/epr/epr-software/xepr/overview.html
smFRET Package software used to image and analyze FRET distributions was a gift from the Taekjip Ha lab. Information on how to obtain the software can be directed to our lead author.
GraphPad Software used to perform the student t-test: https://www.graphpad.com/quickcalcs/ttest1.cfm
The MATLAB R2014b software was built in house and analyze content mixing assays. Information on how to obtain the software can be directed to our lead author.
Supplementary Material
Highlights.
EPR shows that the 1:1 binary t-SNARE complex is partially unstructured
Cleavage of SNAP-25 by BoNT A and E elicits uncoiling at the C-terminus
BoNT E impairs ternary SNARE complex formation and inhibits vesicle docking
BoNT A does not affect SNARE nucleation but impairs fusion pore formation
Acknowledgments
This work was supported by the grant from the National Institute of Health R01 GM051290.
There are no conflicts of interest with this papers.
Footnotes
Author Contributions
R.K., J.K., and L.Y. contributed equally to the data collection, analysis, and interpretation of the experiments along with the drafting and revising the manuscript. R.K. contributed to the EPR, J.K. to the single vesicle fusion, and L.Y. to the smFRET experiments. Y.-K.S. was responsible for the design of the experiments and writing the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An SJ, Almers W. Tracking SNARE complex formation in live endocrine cells. Science. 2004;306:1042–1046. doi: 10.1126/science.1102559. [DOI] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Calakos N, Bennett MK, Peterson KE, Scheller RH. Protein-protein interactions contributing to the specificity of intracellular vesicular trafficking. Science. 1994;263:1146–1149. doi: 10.1126/science.8108733. [DOI] [PubMed] [Google Scholar]
- Chen X, Tomchick DR, Kovrigin E, Arac D, Machius M, Sudhof TC, Rizo J. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/s0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Eliason WK, Brunger AT, Jahn R. Identification of a minimal core of the synaptic SNARE complex sufficient for reversible assembly and disassembly. Biochemistry. 1998;37:10354–10362. doi: 10.1021/bi980542h. [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Margittai M. A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J Biol Chem. 2004;279:7613–7621. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Fiebig KM, Rice LM, Pollock E, Brunger AT. Folding intermediates of SNARE complex assembly. Nature Structural Biology. 1999;6:117–123. doi: 10.1038/5803. [DOI] [PubMed] [Google Scholar]
- Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y. Single Reconstituted Neuronal SNARE Complexes Zipper in Three Distinct Stages. Science. 2012;337:1340–1343. doi: 10.1126/science.1224492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerona RR, Larsen EC, Kowalchyk JA, Martin TF. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem. 2000;275:6328–6336. doi: 10.1074/jbc.275.9.6328. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Kim J, Zhu YC, Shin YK. Preincubation of t-SNAREs with Complexin I Increases Content-Mixing Efficiency. Biochemistry. 2016;55:3667–3673. doi: 10.1021/acs.biochem.6b00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Choi BK, Choi MG, Kim SA, Lai Y, Shin YK, Lee NK. Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. Embo J. 2012;31:2144–2155. doi: 10.1038/emboj.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweon D, Kong B, Shin YK. Hemifusion in Synaptic Vesicle Cycle. Front Mol Neurosci. 2017;10:65. doi: 10.3389/fnmol.2017.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat Struct Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- Lou XC, Shin YK. SNARE zippering. Biosci Rep. 2016;36:7. doi: 10.1042/BSR20160004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundh H, Leander S, Thesleff S. Antagonism of the paralysis produced by botulinum toxin in the rat. The effects of tetraethylammonium, guanidine and 4-aminopyridine. J Neurol Sci. 1977;32:29–43. doi: 10.1016/0022-510x(77)90037-5. [DOI] [PubMed] [Google Scholar]
- Min D, Kim K, Hyeon C, Cho YH, Shin YK, Yoon TY. Mechanical unzipping and rezipping of a single SNARE complex reveals hysteresis as a force-generating mechanism. Nature Communications. 2013;4:10. doi: 10.1038/ncomms2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco C, Schiavo G, Pantano S. SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem Sci. 2005;30:367–372. doi: 10.1016/j.tibs.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Neale EA, et al. Botulinum neurotoxin A blocks synaptic vesicle exocytosis but not endocytosis at the nerve terminal. J Cell Biol. 1999;147(6):1249–60. doi: 10.1083/jcb.147.6.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirazzini M, et al. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol Rev. 2017;69(2):200–235. doi: 10.1124/pr.116.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier MA, Xiao WZ, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nature Structural Biology. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto O, Pirazzini M, Montecucco C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nat Rev Microbiol. 2014;12:535–549. doi: 10.1038/nrmicro3295. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Santucci A, Dasgupta BR, et al. Botulinum neurotoxins serotypes A and E cleave SNAP-25 at distinct COOH-terminal peptide bonds. FEBS Lett. 1993;335(1):99–103. doi: 10.1016/0014-5793(93)80448-4. [DOI] [PubMed] [Google Scholar]
- Shin J, Lou X, Kweon DH, Shin YK. Multiple conformations of a single SNAREpin between two nanodisc membranes reveal diverse pre-fusion states. Biochemical Journal. 2014;459:95–102. doi: 10.1042/BJ20131668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–U105. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane Fusion: Grappling with SNARE and SM Proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 angstrom resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson TE, Russell CJ, King DS, Shin YK. Direct determination of the membrane affinities of individual amino acids. Biochemistry. 1996;35:1803–1809. doi: 10.1021/bi952300c. [DOI] [PubMed] [Google Scholar]
- Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT. Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP-25 complex. Structure. 2008;16:308–320. doi: 10.1016/j.str.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–664. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Poirier MA, Bennett MK, Shin YK. The neuronal t-SNARE complex is a parallel four-helix bundle. Nat Struct Biol. 2001;8:308–311. doi: 10.1038/86174. [DOI] [PubMed] [Google Scholar]
- Xue M, Reim K, Chen X, Chao HT, Deng H, Rizo J, Brose N, Rosenmund C. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol. 2007;14:949–958. doi: 10.1038/nsmb1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Kim J, Shin YK. Complexin splits the membrane-proximal region of a single SNAREpin. Biochem J. 2016;473:2219–2224. doi: 10.1042/BCJ20160339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Chen Y, Kweon DH, Kim CS, Shin YK. The four-helix bundle of the neuronal target membrane SNARE complex is neither disordered in the middle nor uncoiled at the C-terminal region. J Biol Chem. 2002;277:24294–24298. doi: 10.1074/jbc.M201200200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Rebane AA, Ma L, et al. Stability, folding dynamics, and long-range conformational transition of the synaptic t-SNARE complex. Proc Natl Acad Sci USA. 2016;113(50):E8031–E8040. doi: 10.1073/pnas.1605748113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.