

Abstract
Recently, surface-enhanced Raman scattering (SERS) nanoprobes (NPs) have shown promise in the field of cancer imaging due to their unparalleled signal specificity and high sensitivity. Here we report the development of a DNA aptamer targeted SERS NP. Recently, aptamers are being investigated as a viable alternative to more traditional antibody targeting due to their low immunogenicity and low cost of production. We developed a strategy to functionalize SERS NPs with DNA aptamers, which target Mucin1 (MUC1) in human breast cancer (BC). Thorough in vitro characterization studies demonstrated excellent serum stability and specific binding of the targeted NPs to MUC1. In order to test their in vivo targeting capability, we co-injected MUC1-targeted SERS NPs, and as controls non-targeted and blocked MUC1-targeted SERS NPs in BC xenograft mouse models. A two-tumor mouse model with differential expression of MUC1 (MDA-MB-468 and MDA-MB-453) was used to control for active versus passive targeting in the same animals. The results showed that the targeted SERS NPs home to the tumors via active targeting of MUC1, with low levels of passive targeting. We expect this strategy to be an advantageous alternative to antibody-based targeting and useful for targeted imaging of tumor extent, progression, and therapeutic response.
Keywords: SERS, Raman, nanoprobe, cancer imaging, targeting, aptamer, MUC1
Graphical Abstract
A MUC1 DNA aptamer targeted SERS nanoparticle is developed by surface functionalization of SERS nanoparticles. These nanoparticles are optically and structurally stable in biological fluid and target MUC1 overexpressing breast cancer cells in vitro. By co-injection with a nontargeted nanoparticle, we show that the MUC1 aptamer targeted nanoparticles home specifically to MUC1 overexpressing tumor tissue in vivo.
1. Introduction
In recent years, Raman imaging has emerged as a promising modality in the field of bioimaging.[1]Raman reporter coated gold nanoparticles (AuNPs) with an encapsulant layer of silica have emerged as the preferred architecture for the fabrication of bright SERS NPs (with enhancement factors in the order of 107–1014).[2] Owing to the narrow, ‘fingerprint’-like Raman spectral signatures, SERS NPs have shown very high specificity of detection and multiplexing capabilities.[3]Additionally, SERS NPs exhibit little to no susceptibility to photobleaching and have been shown to be non-toxic.[4] The ultrahigh sensitivity and specificity of detection, the multiplexing capabilities, and the photostability are key advantages of the newest generations of SERS NPs over imaging agents based on fluorescence.[1c] Due to these perennial advantages, SERS NPs have gained a lot of attention as a highly sensitive tool for cancer imaging.[5] SERS NPs functionalized with polyethylene glycol (PEG) chains exhibit relatively long blood circulation time and accumulate in cancer tissues by the virtue of the so-called enhanced permeability and retention (EPR) effect.[6] This uptake mechanism is also known as “passive targeting”. However, if one desires to detect the expression of a specific marker of interest in a tumor, then “active targeting” via specific interaction of the NP with a surface ligand with the tumor cells or cells in the tumor microenvironment is required. Several studies have shown promising results in achieving specific targeting of cancer biomarkers in vitro and in vivo using antibodies as the targeting moiety.[7]Nevertheless, major disadvantages of using antibodies for active SERS NP targeting are considerable immunogenicity and high cost of production of humanized monoclonal antibodies, both of which could hinder clinical translation.[8]
Recently, aptamers, also termed chemical antibodies, have emerged as a promising candidate for targeting cancer biomarkers with high selectivity and specificity.[9] Aptamers possess several advantages over monoclonal antibodies, such as: (i) low molecular weight, (ii) low or completely absent immunogenicity, (iii) reproducible, large scale and inexpensive synthesis via solid phase chemistry, and (iv) available chemical modifications which allow different imaging agents to be attached using straight forward conjugation techniques.[10]Therefore, efficient conjugation of aptamers with radioisotopes, fluorophores and nanoparticles have produced several promising aptamer-targeted imaging modalities to date.[11]More encouragingly, in recent clinical trials aptamers have shown no significant immunogenicity advocating the use of aptamers as a targeting moiety.[12] Recently aptamer functionalized nanoparticles and SERS NPs gained attention for in vitro cancer targeting while in vivo targeted cancer detection still remains challenging.[13]
In this work, we have developed a novel DNA aptamer targeted SERS NPs for in vivo imaging of MUC1 overexpression in BC. MUC1 is a transmembrane glycoprotein overexpressed in 90% of BC, and other cancer types as well. [14] MUC1 has been shown to have an important role in cancer progression and metastasis and is overexpressed in early stages of triple negative breast cancer.[15] The overexpression of MUC1 also alters the MUC1 glycosylation pattern and exposes certain epitopes in the core protein, which act as an active site for specific binding. Recently, several monoclonal antibodies have been developed for MUC1 specific tumor imaging and therapy in preclinical studies.[16] However, antibody targeting inherits several limitations such as long plasma half-life, immunogenicity, and relatively large molecular weight.[17] To alleviate these shortcomings, nucleic acid-based aptamers are being pursued as a potential alternative to more traditional antibody targeting. In this work, we chose a well-studied DNA aptamer that was shown to bind to MUC1 with subnanomolar dissociation constant[18] and demonstrated the active targeting capabilities of MUC1 DNA aptamer targeted SERS NPs in vivo as depicted in Figure 1. We postulated that DNA aptamer functionalized SERS NPs, a new member of the spherical nucleic acid (SNA) family, will inherit low nuclease degradation compared to the free aptamer due to unique spatial distribution and produce minimal immune response.[19]
Figure 1.

Schematic representation of the proposed concept: (a) SERS NPs, consisting of AuNP core, IR dye coating, and silica shell, were functionalized with MUC1 DNA aptamers. (b) The MUC1-NPs were administered in a human BC xenograft mouse model via intravenous injection. The MUC1-NPs selectively homed to MUC1 overexpressing BC tissue and were detected by identifying the fingerprint-like spectral signature of the SERS NPs with a confocal Raman Microscope.
2. Results and discussion
2.1. Synthesis and characterization of MUC1 aptamer grafted SERS NPs
The synthesis of aptamer grafted SERS NPs was carried out by stepwise surface functionalization of the previously reported SERS NPs (Figure 2a, step 1, 2).[20] The synthesis of SERS NPs started with the formation of 70 nm AuNP cores using a modified protocol using a seed-mediated growth method described by Perrault et.al. [21] (See experimental section for details). We used hydroxylamine (HA) instead of hydroquinone (HQ) as a reducing agent because AuNPs derived from the HQ synthesis did not produce bright SERS particles, likely due to high surface binding and subsequent passivation by HQ molecules. In a typical synthesis procedure, first 15 nm AuNP seeds were synthesized and subsequently grown into 70 nm AuNPs using HAuCl4 as a gold precursor and HA as a reducing agent in the presence of 15 nm AuNP seeds. Within a few seconds of the reaction, the color of the solution turned deep red confirming the synthesis of 70 nm AuNP cores. Further, synthesized core AuNPs were silica coated in the presence of IR dyes (IR780 perchlorate or IR792 perchlorate) by a modified Stöber process to produce the SERS NPs. The silica surface of the synthesized SERS NPs was stepwise modified in order to attach DNA as explained in Figure 2b. In the first step, surface hydroxyl groups were thiolated using 10% (3-marcaptopropyl)trimethoxysilane (MPTMS) in 85% ethanol and 5% water. Next, the thiol groups were reacted with maleimide-PEG2-biotin to attach biotin moieties onto the silica surface. In the last step, biotinylated SERS NPs were incubated with 1000 molar excess of premixed neutravidin and biotinylated DNA (1:1 ratio) at room temperature in 10 mM MES buffer. The DNA functionalized NPs were purified by centrifugation and dispersion in 10 mM MES buffer. We did not observe any change in Raman signal intensities of SERS NPs during the functionalization procedure (data not shown). This strategy allowed us to functionalize the SERS NPs essentially with any DNA sequence with a biotin modification, typically purchased from a commercial source (IDT DNA Inc.).
Figure 2.

(a) Synthesis and DNA functionalization of SERS NPs. First, 70 nm spherical AuNP cores were silica coated in the presence of IR dye (IR 780 perchlorate or IR 792 perchlorate) to produce SERS NPs. The SERS NPs were functionalized with DNA molecules by sequential modification of the silica shell surface. To verify the presence of the DNA sequence, a 10 nm AuNP functionalized with the complementary DNA strand was hybridized to produce SERS NP core - 10 nm AuNP satellite hybrid nanostructures. (b) Illustration of DNA functionalization: first, the surface hydroxyl groups were converted to sulfhydryl groups using (3-mercaptopropyl)trimethoxysilane (MPTMS) in a water-ethanol mixture. Next, sulfhydryl groups were reacted with a maleimide-PEG2-biotin linker to attach biotin molecules to the surface. Subsequently, biotin labeled DNA molecules were attached to the biotinylated surface of the SERS NPs using neutravidin, a biotin-binding protein as a crosslinker. (c) Representative TEM images of the DNA functionalized SERS NPs. (d) The DLS hydrodynamic diameter measurements show an increase in size from ~80 nm to ~137 nm after silica coating and ~170 nm after DNA functionalization. Error bars represent standard deviations of three measurements. (e) Representative TEM images of the SERS NP core – 10nm AuNP satellite hybrid nanostructures. The inset shows 10 nm AuNP satellites being attached to the SERS NP surface. All the scale bars are 100 nm.
We characterized the SERS NPs using a transmission electron microscope (TEM) and dynamic light scattering (DLS). A typical TEM image of the SERS NPs is presented in Figure 2c (see Figure S1 for additional TEM images), where the electron dense AuNP cores and the silica shell are clearly visible. From the TEM images, average AuNP core and the SERS NP diameters was found to be 69±7 nm and 132±9 nm respectively, demonstrating a high degree of monodispersity of sizes and shapes. The dynamic light scattering (DLS) measurements yielded hydrodynamic diameters of ~80 nm, ~140 nm and ~168 nm of AuNPs, SERS NPs and DNA functionalized SERS NPs, respectively as shown in Figure 2d. The DLS diameter was found to be ~10 nm larger than the average diameter in TEM measurements due to the added hydration layer on the NP surface.
The presence of DNA molecules on the SERS NP surface was verified by two independent methods. In the first method, we hybridized a complementary strand modified with IR 700 dye at the 5′ end of the DNA-functionalized SERS NPs. After removal of excess dye modified strands by centrifugation and redispersion of the NPs, we measured the fluorescence in two emission channels, 700 nm, and 800 nm. We found a higher fluorescence signal in the 700 nm channel compared to before hybridization, due to the specific DNA hybridization of IR700 DNA strands to the particles (Figure S2). The signal in the 800 nm channel was emitted by the IR780 dye present in the SERS NPs itself. In the second method, we hybridized AuNPs with a diameter of 10 nm, functionalized with the complementary DNA to the SERS NPs. We acquired TEM images of the SERS NPs after hybridization, showing SERS-NP-core +10 nm-AuNP-satellite structures (Figure 2e and Figure S3). These results clearly suggested that SERS NPs are successfully functionalized with DNA. We estimated the number of DNA strands to be ~265 ± 30 per SERS NP (See methods section for details).
2.2. In vitro stability of the DNA functionalized SERS NPs
In order to serve as an effective targeted imaging agent in vivo, the SERS NPs, as well as the DNA corona, should exhibit considerable serum stability in vitro within the measurement time window. Therefore, we first investigated the in vitro serum stability of SERS NPs by measuring the Raman spectrum of the SERS NPs after 0, 2, 4 and 16 hours of incubation in 50 % mouse serum at 37 °C, as shown in Figure 3a. We observed Raman spectra to be very consistent in peak positions and intensities before and after incubation. The measured peak intensities did not alter with time; the 960 cm−1 peak intensity measured around 10,000 counts using the same acquisition conditions (Figure 3b). We also examined the TEM images of the same SERS NPs before and after 16 hours of serum incubation shown in Figure 3c and 3d, respectively. From the visual inspection of the TEM images, we observed minimal degradation of silica shells after serum exposure. These results suggested that the SERS NPs were optically and structurally stable in an in vivo milieu.
Figure 3.

(a) Raman spectra of the MUC1-NPs after 0 (black), 2 (red), 4 (blue) and 16 (purple) hours post incubation in 50 % mouse serum at 37 °C temperature. The Raman spectra were acquired at 0.05% laser power, 1-second exposure time and using a 5× objective. (b) The intensity of 960 cm−1 peak was plotted against incubation time showing an excellent signal stability of the SERS NPs. The error bars are the standard deviation of three independent measurements. (c), (d) The TEM images of the SERS NPs before and after 16 hours of serum exposure showing integrity of silica shell. Scale bars for inset images are 100 nm.
We further investigated the stability of the DNA corona upon exposure to serum. We first hybridized an IR700 dye labeled complementary DNA with SERS NPs and incubated for 16 hours in 50 % mouse serum at 37 °C. Subsequently, we washed, re-dispersed in 10 nM MES buffer and measured the fluorescence of the SERS NPs as shown in Figure S2. We observed a decrease in the intensity of the 700 nm emission channel by only 10%, suggesting that the DNA corona was fairly stable in biological conditions. This stability can be attributed to the spherical configuration of the DNA chains which are resistant to nuclease degradation, consistent with previous reports.[22] These results suggested that both Raman signal and DNA corona of the SERS NPs should be stable in an in vivo environment.
2.3. Design of MUC1 targeted NPs and nontargeted NPs
Since tumor xenograft mouse models can exhibit passive uptake of SERS NPs due to the EPR effect, we decided to include a nontargeted SERS NP control to decouple the active and passive targeting. The multiplexing capabilities of SERS NPs allowed us to use two different flavors of SERS NPs with distinct Raman signatures for targeted NPs and nontargeted NPs without changing the overall NP architecture as shown in Figure 4a. Both the MUC1 targeted NPs (MUC1-NPs) and nontargeted NPs (NT-NPs) consisted of a similar internal structure of a 70 nm AuNP core, IR dye (IR 780 for MUC1-NPs and IR 792 for NT-NPs) and a silica shell. However, the structure of the DNA corona of these particles were significantly different. The MUC1-NP corona consisted of a 25 base double-stranded stem and a 25 base MUC1 DNA aptamer sequence separated by a T5 sequence. The double stranded region acted as a rigid handle to spatially separate the MUC1 aptamer sequence from the NP surface. The NT-NPs were also functionalized with the double-stranded stem but lacked the MUC1 aptamer region (see SI Figure S4 for design details). We observed a slightly larger (10 nm) hydrodynamic diameter of MUC1-NPs than NT-NPs in DLS measurements, due to the presence of the aptamer region of the size approximately 5 nm (Figure S5). These NPs have distinct Raman spectra due to the presence of different Raman reporter molecules absorbed onto the AuNP core (Figure 4b). We further determined the limit of detection of these NPs in tissue phantoms using the same imaging conditions as used for in vivo imaging. The direct classical least square (DCLS) algorithm in the WiRE 3.4 software (Renishaw) was used to generate the 2D Raman maps of both SERS NPs. From the Raman map, the limit of detection for both NPs was found to be 5–10 fM, consistent with our previous reports (Figure S4).[3, 23] We further tested whether the DCLS algorithm could differentiate the signals from SERS NPs mixed at different ratios. We mixed MUC1-NPs (IR 780) and NT-NPs (IR 792) in different ratios and used the DCLS algorithm to deconvolute the composite spectra. The algorithm was found to be sensitive up to a ratio of 20:1 showing the high sensitivity of the multiplexed measurements using the DCLS algorithm (Figure 4c).
Figure 4.

(a) A schematic representation of the NT-NP and MUC1-NP. IR 792 and IR 780 dyes are used as Raman reporters for NT-NPs and MUC1-NPs, respectively. MUC1-NPs are functionalized with MUC1 aptamers on a ds-DNA corona, while the NT-NPs lack the MUC1 aptamer region. (b) The Raman spectra of NT-NP (green) and MUC1-NP (red) showing distinct spectral signatures. (c) Determination of limit of detection of NT-NPs and MUC1-NPs in tissue phantoms (100% laser power, 1.5 s integration time, 5× objective). Different wells have different concentrations and ratio of MUC1-NPs and NT-NPs as described in the table above. The limit of detection of individual NPs was found to be ~10 fM and ratios up to 1:20 ratio of either particle can be distinguished using the DCLS algorithm inbuilt in the WiRE 3.4 software.
2.4. In vitro targeting of MUC1 overexpressing cell lines
Inspired by the excellent stability profile in serum and highly sensitive multiplexed imaging capability in tissue phantoms, we explored the targeting capabilities of the SERS NPs in MUC1 overexpressing BC cells in vitro. First, we tested the binding of the MUC1 aptamer to the MUC1 overexpressing MDA-MB-468 and MUC1 negative MDA-MB-453 cell lines as verified by western blot (SI Figure S6c). We performed a flow cytometry-based assay for the binding of a fluorescently labeled aptamer and a random DNA sequence as a control with the cell lines (Figure S6 a, b). Upon quantification of cellular fluorescence intensity, we found that the random sequence exhibited a weak binding to both MDA-MB-468 and MDA-MB-453 cells, presumably due to nonspecific binding. However, the MUC1 aptamer demonstrated a higher and specific binding with MDA-MB-468 cells while showing the same fluorescence intensity in MDA-MB-453 cells as the non-aptamer control. This result indicated specific targeting by the MUC1 aptamers in a MUC1 overexpressing cell line. In order to verify that the selective binding of the MUC1 aptamer to MUC1 overexpressing BC cells was unaltered when grafted onto the SERS NP surface, we incubated MDA-MB-468 cells adherent on a plate with an equal concentration of MUC1-NPs and NT-NPs (both with IR780 dye in order to compare the peak intensity). After subsequent washing steps, we mapped the Raman intensity of the plates at 960 cm−1. We found that the MUC1-NPs adhered to the MDA-MB-468 cells more than NT-NPs (Figure S6 d, e), demonstrating in vitro targeting of MUC1-NPs. These findings motivated us to further test the targeting capabilities of MUC1-NPs in vivo.
2.5. In vivo targeting in a one-tumor xenograft mouse model
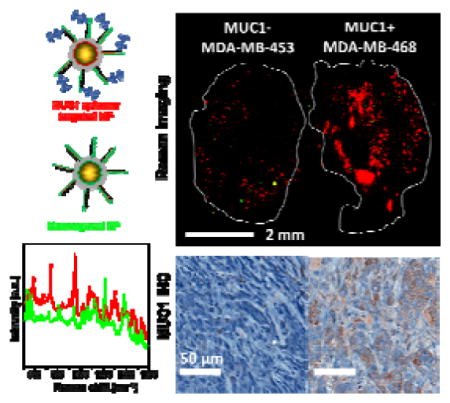
In order to validate the targeting capabilities of MUC1-NPs in vivo, an equimolar cocktail of MUC1-NPs and NT-NPs was injected via tail vein into athymic nude mice (n=4) that were inoculated with MUC1 overexpressing MDA-MB-468 cells. This strategy allowed us to evaluate the targeting capabilities of SERS NPs in the same animal, considering the EPR effect can be variable between different animals even if the tumor type and size are identical.[23]The Raman spectrum of the injected SERS NP cocktail contained the major peaks from MUC1-NPs (740 and 960 cm−1) and NT-NPs (1210 cm−1) (Figure 5a). After 16–18 hours post-injection, tumors and reticuloendothelial system (RES) rich organs such as liver were excised and imaged with a confocal Raman microscope. We observed that the injected SERS NPs primarily accumulated in RES organs like liver and spleen as shown in the Figure S7, consistent with previous reports.[3, 6] We also found a commensurate accumulation of both the NPs in the RES organs and observed the major peak heights to be equal to the injected dose shown in Figure S8. Interestingly, when we imaged the tumor tissue in a Raman microscope, we observed preferential accumulation of MUC1-NPs (Figure 5c, d). We observed foci of MUC1-NPs to be scattered all over the tumor volume, shown as red dots. Interestingly, the NT-NPs did not have significant accumulation, with very few observed foci (green color) in the tumor. These results suggested the targeting capabilities of NPs were operational even after systemic delivery in the mouse model and the EPR mediated targeting was minimal. Histological evaluation of the resected tumors using hematoxylin and eosin (H&E) staining confirmed the presence of tumor tissue in the imaged specimen (Figure 5e). Furthermore, we performed immunohistochemical (IHC) staining of MUC1 expression levels of the tumor tissue (Figure 5f) validating the MUC1 overexpression was scattered throughout the whole tumor volume. These findings further led us to further investigate whether the MUC1 negative tumors would exhibit any uptake of the targeted and the non-targeted SERS NPs in vivo.
Figure 5.

(a) The Raman spectra of NT-NP (green) and MUC1-NP (red) showing distinct spectral signatures. The spectra of the injected NP cocktail (black) contain the major peaks from NT-NP (1210 cm−1) and MUC1-NPs (740 and 960 cm−1) (b) Photograph of a nude mouse with an MDA-MB-468 tumor xenograft in the left flank. (c) Bright field image of the excised tumor. (d) Ex vivo Raman image of the tumor (100% laser power, 1.5 s integration time, 5× objective). The predominantly red signal corresponds to the prevalence of MUC1-NPs throughout the tumor volume. Very few foci of NT-NPs accumulation represented by the green signal. (e) Hematoxylin and eosin (H&E) staining and (f) Immunohistochemical staining of MUC1 in the fixed tumor tissue demonstrated overexpression of MUC1 in the sectioned and imaged specimen.
2.6. In vivo targeting in a two-tumor xenograft mouse model
In order to verify the specific uptake of MUC1-NPs in MUC1 positive tumors, we developed bilateral human BC xenograft model for such a study by staggered inoculation of MDA-MB-453 (MUC1 negative) and MDA-MB-468 (MUC1 positive) cell lines into both flanks of athymic nude mice (n=3). After the tumors had grown to similar sizes, we injected the cocktail of MUC1-NPs and NT-NPs and excised the tumors 16–18 hours post injection. A representative ex vivo Raman image of both tumors acquired in the same scan is shown in Figure 6b, c. We detected abundant signal from the MUC1-NPs shown in red throughout the MUC1 positive tumor derived from the MDA-MB-468 cells, and in contrast very few, sparsely distributed foci from NT-NP (green dots). More interestingly, we observed a 10-fold lower uptake of the targeted MUC1-NP in the tumor derived from the MDA-MB-453 cell line. The signal from NT-NPs was scarce, similar to the contralateral MUC1 positive tumors and consistent with the results of the one-tumor mouse model. The Raman mapping of these tumors clearly suggested selective targeting of MUC1-NPs to MDA-MB-468 tumors, while non-specific EPR-based uptake remained negligible. We further corroborated the Raman imaging results by histological correlation of the fixed tissue sections. IHC staining confirmed high MUC1 overexpression in tumors derived from the MDA-MB-468 cell line overexpressed MUC1, in contrast to the tumors derived from MDA-MB-453 cells line, in contrast to the tumors derived from MDA-MB-453 cells (Figure 6d, e). These findings strongly supported the selective targeting of MUC1 overexpressing BC tumors by MUC1-NPs.
Figure 6.

(a) Photograph of a nude athymic mouse with MDA-MB-468 tumor (R) and MDA-MB-453 (L) xenograft. (b) Bright field image of the excised tumors. (c) Ex vivo Raman image of the tumors (100% laser power, 1.5 s integration time, 5× objective). The predominantly red signal corresponds to the prevalence of MUC1-NPs throughout the tumor volume. On the other hand, the MDA-MB-453 (L) tumor showed minimal uptake of both, MUC1-NPs and NT-NPs. (d) Hematoxylin and eosin (H&E) staining of the cancer tissue. (e) Immunohistochemical (IHC) staining of MUC1 in the fixed tumor tissue demonstrating overexpression of MUC1 in the MDA-MB-468 tumor in contrast to the MDA-MB-453 tumor.
2.7. Blocking targeting moieties suppresses targeted NP homing
We further verified the MUC1 aptamer-mediated targeting, by attaching a blocking module in the same manner as we used to verify successful DNA functionalization. The 10 nm AuNPs functionalized with a complementary DNA sequence were attached to the MUC1-NP to shield the aptamer from being exposed and able to bind its target. The schematic representation and the TEM images of the construct are shown in Figure 7a and Figure 7b respectively. We incubated the blocked MUC1-NPs in serum at 37 °C to assess the stability in vitro. We did not observe significant dissociation of the 10 nm AuNP shields from the blocked MUC1-NPs after the serum exposure, suggesting sufficient serum stability (Figure S3). These blocked NPs when injected in the two tumor xenograft mouse model, did not exhibit considerable uptake in either the MUC1 negative or MUC1 positive tumors as shown in Figure 7c, d. This result strongly supported the fact that targeting was dependent on the DNA aptamers on the SERS NP surface.
Figure 7.

(a) Schematic representation of the blocked MUC1-NPs. The aptamer moiety is sterically shielded by 10 nm AuNPs. (b) TEM images of the blocked MUC1-NPs after 16 hours of serum incubation at 37 °C showing excellent structural integrity of the hybrid structures. (c) Bright field image of the excised tumors. (d) Ex vivo Raman image of the tumor (100% laser power, 1.5 s integration time, 5× objective). Both tumors exhibited very low uptake of the blocked NPs, suggesting that NP homing is due to the active targeting of the aptamers grafted on the MUC1-NPs.
3. Conclusion
In conclusion, we have developed a strategy to functionalize SERS NPs with a MUC1 aptamer sequence. These targeted SERS NPs were found to be stable in biological conditions and could be detected even at low fM concentrations. Further, we showed that MUC1 aptamers and MUC1-NPs exhibited selective binding to MUC-1 overexpressing breast cancer cells in vitro. The multiplexing capability inherent to the methodology of Raman Imaging allowed us to inject differently functionalized NPs into the same animal, thus comparing directly passive and targeted homing of tumor lesions in vivo. We were able to demonstrate that the major uptake of the NPs was indebted to active rather than passive / EPR driven targeting. The lack of passive uptake could be attributed to the fact that the NP surface is different from the traditional PEG-based polymers, as supported by previous findings.[11]
These results advocate the use of DNA aptamer sequences as targeting agents for SERS or other imaging modalities considering the existing library of aptamer sequences for cancer biomarkers and the ease of discovering one for suitable targets by sytematic evolution of ligands by exponential enrichment (SELEX).[24] Due to the high multiplexing potential of different SERS probes, we envision multiplexed detection of cancer in vivo using different DNA aptamers as targeting moieties. We also believe stimuli-responsive SERS imaging agents that will selectively light up tumors overexpressing a specific antigen is not far from reality.
4. Experimental Section
Materials
All reagents were purchased of the highest purity from Sigma-Aldrich (St. Louis, MO) and were used as received. The maleimide-PEG2-biotin linker and NeutrAvidin were obtained from Thermo Fisher Scientific. Dialysis cassettes (MWCO 3.5 kDa; slide-a-lyzer G2) were purchased from Thermo-Fisher Scientific (Waltham, MA). All DNA sequences were purchased from IDT DNA Inc. as HPLC purified grade and used without further purifications.
Methods
Synthesis of 70 nm AuNP core
The AuNP core was synthesized using a modified protocol using a seed mediated growth method described by Perrault et al.[24] We used hydroxylamine instead of hydroquinone as a reducing agent. In a typical synthesis procedure, we first synthesized ~ 15 nm AuNP cores. To 99 ml deionized water, 1 ml 25 mM HAuCl4 was added and the solution was heated on a heating plate to boil. To the boiling solution, 1 ml of 3.3% sodium citrate solution was added. After 15 minutes the color of the solution had changed to red confirming the formation of 15 nm AuNP cores. In the next step, to 100 ml deionized water 125 μL of 200 mM HAuCl4, 30 μL 500 mM trisodium citrate and 700 μL 15 nm AuNP cores were added Under stirring. Further, 250 μL of 1M hydroxylamine hydrochloride solution was mixed. Within few seconds the color of the solution turned deep red confirming the synthesis of 70 nm AuNP.
Synthesis of SERS NPs
Synthesis of SERS NPs was carried out using a modified version of the previously described protocol.[23] In brief, in a 50 ml falcon tube, 10 ml 2-Propanol, 500 μL tetraethylorthosilicate (TEOS), and 20 μL 25 mM IR dye (IR780 or IR792 perchlorate) dissolved in anhydrous N,N-dimethylformamide (DMF) were mixed. In another tube, 3 ml ethanol, 1.2 ml of 4 nM 70 nm AuNP solution and 200 μL ammonia solution were combined. Then the contents of two tubes were mixed under vigorous stirring and kept gently shaking for 15 minutes at room temperature. After the reaction was completed, the particles were washed three times with pure ethanol.
DNA functionalization of SERS NPs
As synthesized SERS NPs were functionalized with DNA in several steps. In step one, 1 nM SERS NPs were thiolated in 1 mL 85 % ethanol, 5% DI water and 10% (3-marcaptopropyl)trimethoxy silane (MPTMS) at 70 °C temperature. After 2 hours of reaction, the particles were washed 3 times with ethanol and 1 time with DI water. In the next step, the thiolated particles were biotinylated using a maleimide-PEG2-biotin linker. To 1 nM thiolated SERS NP solution in 10 mM MES buffer (pH 7.5), 100 μL of 10 mM maleimide-PEG2-biotin solution was added and kept for 2 hours at room temperature. Next, the particles were washed with water 3 times and redispersed in 10 mM MES buffer (pH 7.4). Biotinylated SERS NPs were then added to 1000 times excess neutravidin and biotinylated DNA (premixed 1:1 ratio in 10 mM MES buffer). The SERS NPs were incubated for 3–4 hours and centrifuged 2 times to remove excess neutravidin and DNA, and redispersed in 10 mM MES buffer for injection.
Characterizations of SERS NPs
The SERS NPs were characterized by transmission electron microscopy (JEOL 1200, 80 kV, 80,000×–120,000× magnification) to study the SERS NP structures. The size and concentration of the SERS NPs were measured on a Nanoparticle Tracking Analyzer (Malvern Instruments, Malvern, UK). DLS data was measured using Zetasizer Nano ZS (Malvern Instruments). a Raman spectra were obtained in an InVia system equipped with a 785-nm laser (Renishaw Inc., Hoffman Estates, IL).
Estimation of the grafting density of DNA
The number of DNA per SERS NP was measured using a fluorophore-modified biotinylated DNA strand. The fluorescence intensity was measured before and after the incubation with the SERS NPs (biotinylated). The difference between the fluorescence intensity is proportional to the number of the biotinylated DNA attached to the particle. We determined the numbers of DNA by comparing the difference with the fluorescence intensity of known concentration of the same DNA.
SERS nanoprobe limit of detection
To determine the limit of detection of the MUC1-NPs and NT-NPs, the NPs were mixed in desired concentration ratios in 1% agarose and casted in a 96 well plate. The phantom was scanned using the exact setup used for the actual ex vivo tumor imaging (100% laser power, 1.5 second integration time, 5× objective). The direct classical least square (DCLS) algorithm in the WiRE 3.4 software (Renishaw) was used to generate the 2D Raman maps of two SERS NPs. The Raman maps were analyzed in ImageJ (https://imagej.nih.gov/ij/) software to determine the limit of detection.
Western blot assay of MUC1 expression
Cells were lysed 15 minutes on ice in 10% RIPA buffer (9806, Cell Signaling Technology), 0.5% PMSF (8553s, Cell Signaling Technology), 1% protease/phosphatase inhibitor cocktail (5872, Cell Signaling Technology). After protein quantification by using BCA assay kit (23227, ThermoFisher scientific), 20 μg protein was mixed with reducing agent and LDS sample buffer (NP0004+NP0007, ThermoFisher scientific). Samples were resolved by 10% SDS-polyacrylamide gel electrophoresis, and electrotransfer to a PVDF membrane. Immunoblotting was done with the MUC-1 antibody (4538, Cell Signaling Technology) or β-actin antibody in 4 °C for overnight staining. The staining was completed with horseradish peroxidase-conjugated secondary antibody (diluted at 1:10000), and the protein band was detected by chemiluminescence (ECL) system (32109, ThermoFisher scientific) on autoradiography film.
Cell binding of aptamer
The cellular binding of aptamers was determined by flow cytometry analysis of cells after incubation with an AF488 labeled aptamer, or a control DNA (Figure S5 a and b). The cells MDA-MB-468 or MDA-MB-453 cells were washed two times in FACS buffer (1×PBS, 0.5% BSA) and suspended in the binding buffer (1×PBS, 5 mM MgCl2, pH 7.2) and incubated with MUC1 aptamer or control DNA, at the concentration of 100 nM for 30 min. Then the cells were washed with FACS buffer three times and run in a flow cytometer.
Animal studies
All animal experiments were approved by the Institutional Animal Care and Use Committees of Memorial Sloan Kettering Cancer Center.
Tumor models
Four to six week old female outbred homozygous nude mice (Foxn-1nu / Foxn-1nu, Jackson Laboratory) were subcutaneously injected with 1–2 × 106 MDA-MB-468 and/or MDA-MB-453 cells (HTB-132 & HTB-131D, ATCC) mixed with 0.04 mL of Matrigel (354248, Corning) into each lower ventral side of the mammary fat pad. Since MDA-MB-453 grows less rapidly, we injected MDA-MB-453 2 weeks before the injection of MDA-MB-468 to create the 2-tumor mouse model. We waited for four weeks post inoculation or until tumor sizes reached 0.5 × 0.5 cm.
Ex vivo Raman imaging
Mice were administered 150 μL of 2 nM SERS NP (corresponding to a dose of 35 mg/kg) in 10 mM MES buffer (pH 7.1–7.3) via tail vein injection 18–20 hours prior to imaging. Mice were euthanized by carbon dioxide asphyxiation and organs were imaged ex vivo.
All Raman scans were performed on an InVia Raman microscope (Renishaw) equipped with a piezo-controlled stage for spatial mapping, a 300-mW,785-nm diode laser and a 1-inch CCD detector with a spectral resolution of 1.07 cm−1. The SERS spectra were acquired through a 5× objective lens (Leica). Typically, Raman scans were performed at 100 mW laser power, with 1.5 second acquisition time, using the StreamLine high-speed acquisition mode. All Raman images were acquired and analyzed under the same conditions, with the same laser power, Raman integration times, focal plane (same objective lens), and a threshold setting of 0.1. The focal plane was found by focusing on the region of interest with a white light camera. The Raman scans have a resolution of 14 μm in the x-direction, and 80–200 μm in the y-direction. A typical scan of tumors took around 50–70 minutes to complete depending on the scan area.
Supplementary Material
Acknowledgments
We thank the MSKCC electron microscopy and molecular cytology core facilities for technical support and Dr. Chrysafis Andreou for helpful discussions. The following funding sources (to M.F.K.) are acknowledged: NIH R01 EB017748 and K08 CA16396; M.F.K. is a Damon Runyon-Rachleff Innovator supported (in part) by the Damon Runyon Cancer Research Foundation (DRR-29-14); Pershing Square Sohn Prize by the Pershing Square Sohn Cancer Research Alliance; MSKCC Center for Molecular Imaging & Nanotechnology (CMINT) Grant; MSKCC Technology Development Grant; Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. Acknowledgments are also extended to the grant-funding support provided by the MSKCC NIH Core Grant (P30-CA008748).
Footnotes
Supplementary Figures S1–S8 are provided. Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Suchetan Pal, Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
Dr. Stefan Harmsen, Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
Dr. Anton Oseledchyk, Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
Hsiao-Ting Hsu, Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Prof. Moritz F. Kircher, Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. Center for Molecular Imaging and Nanotechnology (CMINT), Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. Molecular Pharmacology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. Weill Cornell Medical College, Cornell University, New York, NY 10065, USA.
References
- 1.a) Lane LA, Qian X, Nie S. Chemical Reviews. 2015;115:10489. doi: 10.1021/acs.chemrev.5b00265. [DOI] [PubMed] [Google Scholar]; b) Zavaleta CL, Kircher MF, Gambhir SS. Journal of Nuclear Medicine. 2011;52:1839. doi: 10.2967/jnumed.111.087775. [DOI] [PubMed] [Google Scholar]; c) Andreou C, Kishore SA, Kircher MF. Journal of Nuclear Medicine. 2015;56:1295. doi: 10.2967/jnumed.115.158196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Nie S, Emory SR. Science. 1997;275:1102. doi: 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]; b) Harmsen S, Huang R, Wall MA, Karabeber H, Samii JM, Spaliviero M, White JR, Monette S, O’Connor R, Pitter KL. Science translational medicine. 2015;7:271ra7. doi: 10.1126/scitranslmed.3010633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zavaleta CL, Smith BR, Walton I, Doering W, Davis G, Shojaei B, Natan MJ, Gambhir SS. Proceedings of the National Academy of Sciences. 2009;106:13511. doi: 10.1073/pnas.0813327106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Thakor AS, Luong R, Paulmurugan R, Lin FI, Kempen P, Zavaleta C, Chu P, Massoud TF, Sinclair R, Gambhir SS. Science Translational Medicine. 2011;3:79ra33. doi: 10.1126/scitranslmed.3001963. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Andreou C, Neuschmelting V, Tschaharganeh DF, Huang CH, Oseledchyk A, Iacono P, Karabeber H, Colen RR, Mannelli L, Lowe SW, Kircher MF. ACS Nano. 2016;10:5015. doi: 10.1021/acsnano.5b07200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Andreou C, Neuschmelting V, Tschaharganeh DF, Huang CH, Oseledchyk A, Iacono P, Karabeber H, Colen RR, Mannelli L, Lowe SW, Kircher MF. ACS Nano. 2016;10:5015. doi: 10.1021/acsnano.5b07200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Harmsen S, Bedics MA, Wall MA, Huang R, Detty MR, Kircher MF. Nat Commun. 2015;6:6570. doi: 10.1038/ncomms7570. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Harmsen S, Huang R, Wall MA, Karabeber H, Samii JM, Spaliviero M, White JR, Monette S, O’Connor R, Pitter KL, Sastra SA, Saborowski M, Holland EC, Singer S, Olive KP, Lowe SW, Blasberg RG, Kircher MF. Science translational medicine. 2015;7:271ra7. doi: 10.1126/scitranslmed.3010633. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Huang R, Harmsen S, Samii JM, Karabeber H, Pitter K, Holland EC, Kircher MF. Theranostics. 2016;6:1075. doi: 10.7150/thno.13842. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kircher MF, de la Zerda A, Jokerst JV, Zavaleta CL, Kempen PJ, Mittra E, Pitter K, Huang R, Campos C, Habte F, Sinclair R, Brennan CW, Mellinghoff IK, Holland EC, Gambhir SS. Nature medicine. 2012;18:829. doi: 10.1038/nm.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Nayak TR, Andreou C, Oseledchyk A, Marcus WD, Wong HC, Massague J, Kircher MF. Nanoscale. 2017;9:1110. doi: 10.1039/c6nr08217c. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Oseledchyk A, Andreou C, Wall MA, Kircher MF. ACS Nano. 2017;11:1488. doi: 10.1021/acsnano.6b06796. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Spaliviero M, Harmsen S, Huang R, Wall MA, Andreou C, Eastham JA, Touijer KA, Scardino PT, Kircher MF. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. 2016 doi: 10.1007/s11307-016-0932-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maeda H. Journal of Controlled Release. 2012;164:138. doi: 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 7.a) Dinish US, Balasundaram G, Chang YT, Olivo M. Scientific Reports. 2014;4:4075. doi: 10.1038/srep04075. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang R, Harmsen S, Samii JM, Karabeber H, Pitter KL, Holland EC, Kircher MF. Theranostics. 2016;6:1075. doi: 10.7150/thno.13842. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pirollo KF, Chang EH. Trends in Biotechnology. 26:552. doi: 10.1016/j.tibtech.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 8.a) Harding FA, Stickler MM, Razo J, DuBridge RB. mAbs. 2010;2:256. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jeyakumar A, Younis T. Clinical Medicine Insights Oncology. 2012;6:179. doi: 10.4137/CMO.S6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Cerchia L, de Franciscis V. Trends Biotechnol. 2010;28:517. doi: 10.1016/j.tibtech.2010.07.005. [DOI] [PubMed] [Google Scholar]; b) Keefe AD, Pai S, Ellington A. Nat Rev Drug Discov. 2010;9:537. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Famulok M, Hartig JS, Mayer G. Chemical Reviews. 2007;107:3715. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 10.Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Mol Ther Nucleic Acids. 2014;3:e182. doi: 10.1038/mtna.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Casey AD, Weibo C, Hao H. Current Topics in Medicinal Chemistry. 2015;15:1138. [Google Scholar]; b) Zhu G, Niu G, Chen X. Bioconjugate Chemistry. 2015;26:2186. doi: 10.1021/acs.bioconjchem.5b00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lao Y-H, Phua KKL, Leong KW. ACS Nano. 2015;9:2235. doi: 10.1021/nn507494p. [DOI] [PubMed] [Google Scholar]
- 13.a) Li LL, Yin Q, Cheng J, Lu Y. Advanced Healthcare Materials. 2012;1:567. doi: 10.1002/adhm.201200116. [DOI] [PubMed] [Google Scholar]; b) Wu P, Gao Y, Zhang H, Cai C. Analytical chemistry. 2012;84:7692. doi: 10.1021/ac3015164. [DOI] [PubMed] [Google Scholar]; c) Chang M, Yang CS, Huang DM. ACS Nano. 2011;5:6156. doi: 10.1021/nn200693a. [DOI] [PubMed] [Google Scholar]; d) Shahdordizadeh M, Yazdian-Robati R, Ramezani M, Abnous K, Taghdisi SM. Journal of Materials Chemistry B. 2016;4:7766. doi: 10.1039/c6tb02564a. [DOI] [PubMed] [Google Scholar]
- 14.Hollingsworth MA, Swanson BJ. Nat Rev Cancer. 2004;4:45. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 15.a) Nath S, Mukherjee P. Trends Mol Med. 2014;20:332. doi: 10.1016/j.molmed.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miedler J, Abdul-Karim F, Wang N, Baar J. Cancer Research. 2009;69:3009. [Google Scholar]
- 16.Pillai K, Pourgholami MH, Chua TC, Morris DL. American Journal of Clinical Oncology. 2015;38:108. doi: 10.1097/COC.0b013e31828f5a07. [DOI] [PubMed] [Google Scholar]
- 17.Heublein S, Mayr D, Egger M, Karsten U, Goletz S, Angele M, Gallwas J, Jeschke U, Ditsch N. Journal of Experimental & Clinical Cancer Research. 2015;34:1. doi: 10.1186/s13046-015-0152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira CSM, Matthews CS, Missailidis S. Tumor Biology. 2006;27:289. doi: 10.1159/000096085. [DOI] [PubMed] [Google Scholar]
- 19.a) Cutler JI, Auyeung E, Mirkin CA. Journal of the American Chemical Society. 2012;134:1376. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]; b) Seferos DS, Prigodich AE, Giljohann DA, Patel PC, Mirkin CA. Nano Letters. 2009;9:308. doi: 10.1021/nl802958f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Massich MD, Giljohann DA, Seferos DS, Ludlow LE, Horvath CM, Mirkin CA. Molecular Pharmaceutics. 2009;6:1934. doi: 10.1021/mp900172m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harmsen S, Bedics MA, Wall MA, Huang R, Detty MR, Kircher MF. Nat Commun. 2015:6. doi: 10.1038/ncomms7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrault SD, Chan WCW. Journal of the American Chemical Society. 2009;131:17042. doi: 10.1021/ja907069u. [DOI] [PubMed] [Google Scholar]
- 22.Patel PC, Hao L, Au Yeung WS, Mirkin CA. Molecular Pharmaceutics. 2011;8:1285. doi: 10.1021/mp200084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiessling F, Mertens ME, Grimm J, Lammers T. Radiology. 2014;273:10. doi: 10.1148/radiol.14131520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sefah K, Shangguan D, Xiong X, O’Donoghue MB, Tan W. Nat Protocols. 2010;5:1169. doi: 10.1038/nprot.2010.66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.