

Summary
Ferroptosis is a form of regulated cell death characterized by the iron-dependent accumulation of lipid hydroperoxides to lethal levels. Emerging evidence suggests that ferroptosis represents an ancient vulnerability caused by the incorporation of polyunsaturated fatty acids into cellular membranes, and that cells have developed complex systems that exploit and defend against this vulnerability in different contexts. The sensitivity to ferroptosis is tightly linked to numerous biological processes, including amino acid, iron and polyunsaturated fatty acid metabolism, and the biosynthesis of glutathione, phospholipids, NADPH and coenzyme Q10. Ferroptosis has been implicated in the pathological cell death associated with degenerative diseases (i.e., Alzheimer's, Huntington's, and Parkinson's diseases), carcinogenesis, stroke, intracerebral hemorrhage, traumatic brain injury, ischemia-reperfusion injury, and kidney degeneration in mammals and is also implicated in heat stress in plants. Ferroptosis may also have a tumor suppressor function that could be harnessed for cancer therapy. This Primer reviews the mechanisms underlying ferroptosis, highlights connections to other areas of biology and medicine, and recommends tools and guidelines for studying this emerging form of regulated cell death.
Keywords: Ferroptosis, cell death, iron, metabolism, ROS, peroxidation, glutathione, neurodegeneration, cancer, PUFA
Introduction
Cell death is critical in diverse aspects of mammalian development, homeostasis, and disease, and it is tightly integrated with other biological processes. The Nomenclature Committee on Cell Death (NCCD) recognizes a distinction between accidental cell death, which is caused by severe physical, chemical and mechanical insults and cannot be reversed by molecular perturbations, and regulated cell death, which can be modulated pharmacologically and genetically, and is therefore under the control of specific intrinsic cellular mechanisms. Moreover, the NCCD defines that programmed cell death is a subset of regulated cell death that is predestined to occur in normal physiological contexts such as development (Galluzzi et al., 2015).
Caspase-dependent apoptosis was the first regulated and programmed form of cell death to be characterized at the molecular level. In recent years, there has been a growing appreciation for the importance of regulated cell death mechanisms beyond apoptosis in explaining the molecular processes that control the demise of cells (Conrad et al., 2016), although the extent to which these other forms of regulated cell death are also programmed to occur under normal physiological conditions remains largely unknown. We focus in this Primer on ferroptosis, an iron-dependent form of regulated cell death that involves lethal, iron-catalyzed lipid damage. As described below, ferroptotic death is a form of regulated cell death, as it is dramatically modulated by pharmacological perturbation of lipid repair systems involving glutathione and GPX4, and is dependent on a set of positive-acting enzymatic reactions, including biosynthesis of PUFA-containing phospholipids, which are the substrates of pro-ferroptotic lipid peroxidation products, by ACSL4 and LPCAT3, and selective oxygenation of PUFA-phosphatidylethanolamines by lipoxygenases.
The term ferroptosis was coined in 2012 (Dixon et al., 2012) to describe the form of cell death induced by the small molecule erastin, which inhibits the import of cystine, leading to glutathione depletion and inactivation of the phospholipid peroxidase glutathione peroxidase 4 (GPX4) (Yang et al., 2014). GPX4 converts potentially toxic lipid hydroperoxides (L-OOH) to non-toxic lipid alcohols (L-OH) (Ursini et al., 1982) (Figure 1). Inactivation of GPX4 through depletion of GSH with erastin, or with the direct GPX4 inhibitor (1S,3R)-RSL3 (hereafter referred to as RSL3), ultimately results in overwhelming lipid peroxidation that causes cell death (Table 1). Thus, suppression of lipid peroxidation is required in mammalian cells to prevent the onset of ferroptosis, which may occur by default under conditions where GPX4 is inactivated. Since the initial description of this process, additional compounds and regulatory mechanisms have been identified (Xie et al., 2016a; Yang and Stockwell, 2016) (Tables 1 and 2), and this process has been implicated in a variety of pathological contexts and therapeutic strategies, described below.
Figure 1. GPX4 uses glutathione to eliminate lipid peroxides formed in phospholipids containing polyunsaturated fatty acids.
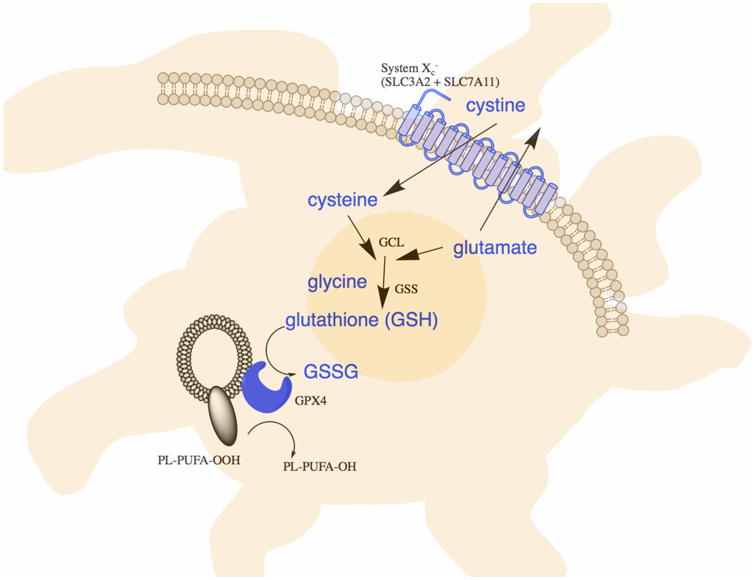
System xc- imports cystine, which is reduced to cysteine and used to synthesize glutathione, a necessary cofactor of GPX4 for eliminating lipid peroxides.
Table 1. Reagents that modulate ferroptosis.
Numerous reagents have been found to modulate sensitivity to ferroptosis.
| Reagent | Target/function | Impact on ferroptosis | |
|---|---|---|---|
| Inducers/ Sensitizers | Erastin, PE, IKE, other erastin analogs | System xc- | Prevents cystine import, causes GSH depletion |
| Sulfasalazine | System xc- | Low potency inhibitor that prevents cystine import, causes GSH depletion | |
| Glutamate | System xc- | High extracellular glutamate concentrations prevent cystine import, causes GSH depletion | |
| Sorafenib | System xc- | Indirectly blocks system xc- activity resulting in GSH depletion | |
| (1S,3R)-RSL3 | GPX4 | Covalent inhibitor of GPX4 that causes accumulation of lipid hydroperoxides | |
| ML162, DPI compounds 7,10, 12, 13, 17, 18, 19 | GPX4 | Covalent inhibitor of GPX4 that causes accumulation of lipid hydroperoxides | |
| Cystine/cysteine deprivation, BSO, DPI2, cisplatin | GSH | Deplete GSH | |
| FIN56 | SQS and GPX4 | Depletes CoQ10 via SQS-mevalonate pathway, and causes decrease in GPX4 protein abundance | |
| FINO2 | Lipid peroxidation | Induces lipid peroxidation and loss of GPX4 activity | |
| Statins (e.g., cerivastatin, simvastatin) | HMGCR | Block CoQ10 biosynthesis via mevalonate pathway | |
| Cysteinase | Cysteine | Depletes cysteine, resulting in GSH depletion | |
| Silica-based nanoparticles | GSH, iron | Deliver iron into cells and reduce GSH abundance | |
| CCl4 | Unknown | Unknown | |
| Ferric ammonium citrate | Iron | Increased iron abundance | |
| Trigonelline, brusatol | NRF2 | Blocks NRF2 | |
| Inhibitors | Vitamin E, alpha-tocopherol, Trolox, tocotrienols | Lipid peroxidation | Blocks propagation of lipid peroxidation, may inhibit lipoxygenases |
| Deuterated polyunsaturated fatty acids (D-PUFAs) | Lipid peroxidation | Blocks initation and propagation of lipid peroxidation | |
| Butylated hydroxytoluene, butylated hydroxyanisole | Lipid peroxidation | Blocks lipid peroxidation | |
| Ferrostatins, liproxstatins | Lipid peroxidation | Blocks lipid peroxidation | |
| CoQ10, Idebenone | Lipid peroxidation | Blocks lipid peroxidation | |
| XJB-5-131 | Lipid peroxidation | Nitroxide antioxidant that suppresses lipid peroxidation | |
| Deferoxamine, cyclipirox, deferiprone | Iron | Depletes iron and prevents iron-dependent lipid peroxidation | |
| Glutamine deprivation, Glutaminolysis inhibitor | Glutaminolysis | Unknown | |
| CDC, Baicalein, PD-146176, AA-861, Zileuton | Lipoxygenases | Block lipoxygenase-induced lipid peroxidation | |
| Cycloheximide | Protein synthesis | Suppresses ferroptosis induced by system xc- inhibitors | |
| beta-mercaptoethanol | Reducing agent | Suppresses ferroptosis induced by system xc- inhibitors by reducing extracellular cystine to cysteine, bypassing system xc- | |
| Dopamine | Neurotransmitter | Blocks GPX4 degradation | |
| Selenium | Selenoproteins | Increases abundance of selenoproteins | |
| Vildagliptin, alogliptin and linagliptin | DPP4 | Blocks DPP4-mediated lipid peroxidation |
Table 2. Genes involved in ferroptosis.
Numerous genes have been found to regulate ferroptosis or to serve as markers of ferroptosis.
| Gene | Name | Function |
|---|---|---|
| ACSL4 | Acyl-CoA Synthetase Long-Chain Family Member 4 | Converts free fatty acids into fatty CoA ester, required for ferroptosis |
| AKR1C1-3 | Aldo-Keto Reductase Family 1 Member C1 | Involved in eliminating end products of lipid peroxidation |
| ALOXs | Lipoxygenases | Involved in peroxidation of polyunsaturated fatty acids in ferroptosis contexts |
| ATP5G3 | ATP synthase, H+transporting, mitochondrial Fo complex subunitC3(subunit9) | Knockdown suppresses erastin-induced ferroptosis |
| CARS | Cysteinyl tRNA synthetase | Knockdown causes increased transsulfuration pathway activity, and resistance to ferroptosis induced by system xc- inhibitors |
| CBS | Cystathionine beta synthase | Marker of transsulfuration pathway activity |
| CD44v | CD44 molecule | Stabilizes system xc- expression |
| CHAC1 | ChaC Glutathione Specific Gamma-Glutamylcyclotransferase 1 | Induced by system xc-inhibitors |
| CISD1 | CDGSH iron sulfur domain 1 | Regulates mitochondrial iron abundance |
| CS | Citrate synthase | Knockdown suppresses erastin-induced ferroptosis |
| DPP4 | Dipeptidyl-dippeptidase-4 | Promotes lipid peroxidation through NOXs |
| FANCD2 | Fanconi anemia complementation group D2 | Suppresses ferroptosis |
| GCLC/GCLM | Glutamate-cysteine ligase | Biosynthesis of glutathione |
| GLS2 | Glutaminase 2 | Required for glutaminolysis and ferroptosis |
| GPX4 | Glutathione peroxidase 4 | Reduces membrane phospholipid hydroperoxides to suppress ferroptosis |
| GSS | Glutathione synthetase | Biosynthesis of glutathione |
| HMGCR | 3-Hydroxy-3-Methylglutaryl-CoA Reductase | Catalyzes rate-limiting step in mevalonate-derived terpene biosynthesis, including CoQ10 |
| HSPB1/5 | Heat shock protein beta 1/5 | Regulates iron uptake an GPX4 abundance |
| KOD | Kiss of death | Induced during ferroptosis in response to heat stress in Arabidopsis |
| LPCAT3 | Lysophosphatidylcholine Acyltransferase 3 | Involved in biosynthesis of phospholipids, required for ferroptosis |
| MT1G | Metallothionein-1G | Metal-binding protein |
| NCOA4 | Nuclear Receptor Coactivator 4 | Involved in ferritinophagy and control of free iron abundance; promotes ferroptosis |
| NFE2L2 | Nuclear factor, erythroid 2 like 2 | Encodes NRF2, a master regulator of anti-oxidant responses that drives resistance to ferroptosis |
| PTGS2 | Prostaglandin-Endoperoxide Synthase 2 | Induced by GPX4 inhibitors |
| RPL8 | Ribosomal protein L8 | Knockdown suppresses erastin-induced ferroptosis |
| SAT1 | Spermidine/Spermine N1-Acetyltransferase 1 | Promotes lipid peroxidation, possibly through lipoxygeneses |
| SLC7A11 | Solute carrier family 7 member 11 | Encodes a component of system xc-, requires for cystine import; also induced in response to system xc- inhibitors |
| SQS | Squalene synthase | Involved in biosynthesis of mevalonate-derived terpenes; impacts coQ10 synthesis |
| TFRC | Transferrin receptor | Imports iron into cells; promotes ferroptosis |
| TP53 | Tumor protein 53 | Represses SLC7A11 to promote ferroptosis as a tumor suppression mechanism |
| TTC35/EMC2 | ER membrane protein complex subunit 2 | Knockdown suppresses erastin-induced ferroptosis |
Definition of ferroptosis and potential physiological functions
Ferroptosis describes a form of regulated cell death that occurs as a consequence of lethal lipid peroxidation. Cell death occurring exclusively by ferroptosis can be suppressed by iron chelators, lipophilic antioxidants, inhibitors of lipid peroxidation, and depletion of polyunsaturated fatty acids (PUFAs), and correlates with the accumulation of markers of lipid peroxidation.
A normal physiological function for ferroptosis as an adaptive and programmed form of cell death (e.g., during development) has not been established. However, ferroptosis has several connections to pathological cell death. A complex set of processes, described below, can drive or suppress lethal lipid peroxidation, suggesting that Nature regulates this vulnerability in numerous contexts. For example, some degenerative pathologies appear to be caused by overwhelming the capacity to repair peroxidized lipids, resulting in cell death; emerging evidence also suggests that ferroptosis might serve a tumor suppressor function in removing cells that lack access to critical nutrients in their environment, or that have been compromised by infection or environmental stress, although these studies are still preliminary. The bulk of studies to date suggest that ferroptosis is triggered by degenerative processes or may be induced therapeutically in some cancers, but few studies have explored its natural functions. It is conceivable, but not yet demonstrated, that ferroptosis could be triggered during development or normal homeostatic tissue turnover by the accumulation of glutamate, iron, or PUFA-phospholipids, or by depletion of endogenous inhibitors of ferroptosis, such as GSH, NADPH, GPX4 or vitamin E. Examining such possibilities will be an important area of investigation in the future to determine whether ferroptosis is genetically programmed to occur, or is primarily a vulnerability caused by pathologies and exploited by potential therapeutics.
Early history: studies linking thiol metabolism to oxidative cell death
Reduced glutathione (γ-L-glutamyl-L-cysteinylglycine, GSH) is an essential intracellular antioxidant synthesized from glutamate, cysteine and glycine in two steps by the ATP-dependent cytosolic enzymes glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS) (Figure 1); the rate of glutathione synthesis is limited by cysteine availability. Early studies identified extracellular cysteine and cystine as essential for growth of HeLa and other cells in culture (Eagle, 1955). Cysteine is required for cell growth, and to prevent cell death: human fibroblasts cultured in cystine-free medium die due to glutathione depletion, and this death is prevented by the lipophilic antioxidant α-tocopherol (Bannai et al., 1977). In addition, iron chelators, such as deferoxamine, prevent this cell death (Murphy et al., 1989). These results established the concept that extracellular cystine and intracellular cysteine are required to maintain the biosynthesis of glutathione and to suppress a type of cell death in mammalian cells that is also preventable by treatment with iron chelators or lipophilic antioxidants. Glutathione levels were observed to be key regulators of viability in some contexts in plants as well: abortion of premature pollen due to abnormal microspore formation in the sterile rice plant Oryza sativa L was found to involve depletion of GSH and NADPH during meiosis, and consequent accumulation of ROS and cell death, which is intriguingly reminiscent of ferroptosis (Wan et al., 2007). However, when these historical studies were performed, a framework for ferroptosis was lacking, and these observations were not yet interpreted as evidence for a unique form of regulated cell death.
The biochemical control of ferroptosis
Ferroptosis initiation and execution lies at the intersection of amino acid, lipid and iron metabolism (Figure 2), but ferroptosis sensitivity is also modulated by several other pathways and processes.
Figure 2. Pathways controlling ferroptosis.

The indicated pathways regulate ferroptosis sensitivity.
Amino acid and glutathione metabolism
Amino acid metabolism is tightly linked to the regulation of ferroptosis (Angeli et al., 2017). Because cysteine availability limits the biosynthesis of glutathione, some cells make use of the transsulfuration pathway to biosynthesize cysteine from methionine and therefore bypass the requirement for cystine import via the cystine/glutamate antiporter system xc-; consequently, these cells are resistant to ferroptosis induced by system xc- inhibitors (Figure 2). A genome-wide short interfering RNA (siRNA) screen for suppressors of erastin-induced ferroptosis revealed that knockdown of cysteinyl-tRNA synthetase (CARS) results in upregulation of the transsulfuration pathway and resistance to erastin-induced ferroptosis (Hayano et al., 2016). Knockdown of some, but not all, tRNA synthetases results in upregulation of the transsulfuration pathway, which can be detected by measuring the abundance of cystathionine beta synthase (CBS) mRNA (Hayano et al., 2016).
Glutamate and glutamine are also important regulators of ferroptosis (Gao et al., 2015). Glutamate is exchanged for cystine in a 1:1 ratio by system xc-, so glutamate levels impact system xc- function. Indeed, high extracellular concentrations of glutamate inhibit system xc- and induce ferroptosis, perhaps explaining toxic effects of glutamate when it accumulates to high concentrations in the nervous system (Dixon et al., 2012). Of note, system xc- knockout mice are protected from neurotoxic insults due to decreased extracellular brain glutamate levels (Massie et al., 2011). Thus, accumulation of extracellular glutamate could serve as a natural trigger for inducing ferroptosis in physiological contexts.
Glutamine is naturally present at high concentrations in human tissues and plasma, and its degradation (via glutaminolysis) provides fuel for the tricarboxylic acid (TCA) cycle and building blocks for essential biosynthetic processes, such as lipid biosynthesis. In the absence of glutamine, or when glutaminolysis is inhibited, cystine starvation and blockage of cystine import fail to induce the accumulation of reactive oxygen species (ROS), lipid peroxidation, and ferroptosis. This observation may be explained by the fact that α-ketoglutarate (αKG), a product of glutaminolysis, is required for ferroptosis (Gao et al., 2015).
Not all routes of glutaminolysis fuel ferroptosis, however. The first step of glutaminolysis involves conversion of glutamine into glutamate, a reaction catalyzed by the glutaminases GLS1 and GLS2. Although these enzymes are structurally and enzymatically similar, only GLS2 is required for ferroptosis (Gao et al., 2015). The GLS2 gene, but not GLS1, is a transcriptional target of the tumor suppressor p53, and upregulation of GLS2 contributes to p53-dependent ferroptosis (Jennis et al., 2016). In some cases, p53 can limit ferroptosis by blocking DPP4 activity in a transcription-independent manner (Xie et al., 2017). Because ferroptosis has been suggested to be a relevant cell death mechanism in tissue injury (Conrad et al., 2016; Linkermann et al., 2014), glutaminolysis-targeted therapy may be effective in treating organ damage mediated by ferroptosis. Indeed, inhibition of glutaminolysis has been shown to attenuate ischemia/reperfusion-induced heart and kidney damage and brain hemorrhage in experimental models (Gao et al., 2015; Li et al., 2017; Linkermann et al., 2014).
Lipid metabolism
Lipid metabolism is also intimately involved in determining cellular sensitivity to ferroptosis. PUFAs, which contain bis-allylic hydrogen atoms that can be readily abstracted, are susceptible to lipid peroxidation and are necessary for the execution of ferroptosis (Yang et al., 2016). Thus, the abundance and localization of PUFAs determine the degree of lipid peroxidation that occurs in cells, and hence the extent to which ferroptosis is operative. Free PUFAs are substrates for synthesis of lipid signaling mediators, yet they must be esterified into membrane phospholipids and undergo oxidation to become ferroptotic signals (Kagan et al., 2017). Lipidomic studies suggest that phosphatidylethanolamines (PEs) containing arachidonic acid (C20:4) or its elongation product, adrenic acid (C22:4), are key phospholipids that undergo oxidation and drive cells towards ferroptotic death (Doll et al., 2017; Kagan et al., 2017). Therefore, formation of coenzyme-A-derivatives of these PUFAs and their insertion into phospholipids are necessary for the production of ferroptotic death signals. This is another potential point of regulation of ferroptosis, and future investigations may suggest physiological contexts in which ferroptosis is triggered or blocked by modulating enzymes involved in the biosynthesis of PUFA-containing membrane phospholipids.
For example, two enzymes, ACSL4 and LPCAT3 (Table 2), are involved in the biosynthesis and remodeling of PUFA-PEs in cellular membranes. Loss of these gene products depletes the substrates for lipid peroxidation and increases resistance to ferroptosis (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017; Yuan et al., 2016b). Conversely, cells that are supplemented with arachidonic acid or other PUFAs are sensitized to ferroptosis (Yang et al., 2016). Hydroperoxy derivatives of PUFA-PEs also cause ferroptotic death when added to cells with inactivated GPX4 (Kagan et al., 2017). Intriguingly, proper development of the fetal immune system in humans depends on adequate PUFA dietary intake, suggesting a possible role for ferroptosis in this process, although other explanations are also possible (Enke et al., 2008).
Enzymatic effectors, such as non-heme, iron-containing proteins, including lipoxygenases (LOXs), can mediate ferroptotic peroxidation (Kagan et al., 2017; Seiler et al., 2008; Yang et al., 2016). Free PUFAs, rather than PUFA-containing phospholipids, are the preferred substrates of LOXs (Kuhn et al., 2015); PE phospholipids can, however, form a non-bilayer arrangement (van den Brink-van der Laan et al., 2004) that may facilitate pro-ferroptotic oxidation of PUFA-containing PE phospholipids by LOXs, rather than free PUFAs. Indeed, genetic depletion of LOXs protects against erastin-induced ferroptosis (Yang et al., 2016), suggesting LOXs contribute to ferroptosis. Several ferroptosis inhibitors, including members of the vitamin E family (tocopherols and tocotrienols) and flavonoids, can inhibit LOX activity in some contexts (Khanna et al., 2003; Xie et al., 2016b). Some lipoxygenases are required for normal embryonic development in vertebrates; for example, 12S-lipoxgenase is essential for the development of numerous tissues in zebrafish, suggesting that ferroptosis could be involved in these developmental processes, although further study is required to test this possibility (Haas et al., 2011).
The execution phase of ferroptosis may be a direct result of lipid peroxidation. Lipid peroxides decompose into reactive derivatives, including aldehydes and Michael acceptors, which can react with proteins and nucleic acids (Gaschler and Stockwell, 2017). The hypothesis that these reactive intermediates are responsible for cell death is supported by the observation that a cell line selected for erastin resistance showed several-hundred-fold upregulation of AKR1C family genes (Dixon et al., 2014), which encode aldoketoreductases that, among other functions, reduce reactive end-products of lipid peroxidation to unreactive compounds (MacLeod et al., 2009).
Iron metabolism
Iron is required for the accumulation of lipid peroxides and the execution of ferroptosis. Thus, iron import, export, storage, and turnover impact ferroptosis sensitivity. Transferrin and transferrin receptor, which import iron from the extracellular environment, are required for ferroptosis (Gao et al., 2015; Yang and Stockwell, 2008), while silencing of the iron metabolism master regulator IREB2 decreases sensitivity to ferroptosis (Dixon, 2012). Within cells, autophagy can modulate sensitivity to ferroptosis through its impact on iron metabolism (Gao et al., 2015; Hou et al., 2016). Selective autophagy of ferritin, referred to as ferritinophagy, enhances ferroptosis sensitivity, by controlling iron availability (Gao et al., 2016; Hou et al., 2016; Mancias et al., 2014; Wang et al., 2016b). Ferritin is recognized by the specific cargo receptor NCOA4, which recruits ferritin to autophagosomes for lysosomal degradation and the release of free iron (Mancias et al., 2014). Other proteins impacting iron metabolism in the cell (e.g., HSPB1 and CISD1) impact ferroptosis sensitivity (Sun et al., 2015; Yuan et al., 2016a). Thus, regulation of iron metabolism and ferritinophagy are additional potential points of control of ferroptosis.
Other metabolic pathways controlling ferroptosis sensitivity
Several other metabolic pathways modulate cellular sensitivity to ferroptosis. The mevalonate pathway leads to the production of coenzyme Q10 (CoQ10), which is required to shuttle electrons as part of the mitochondrial electron transport chain; however, this function is not relevant to ferroptosis (Dixon et al., 2012). Rather, CoQ10 moonlights as an endogenous inhibitor of ferroptosis by serving an antioxidant function in membranes (Shimada et al., 2016b). The ferroptosis-inducing compound FIN56 depletes CoQ10 by modulating squalene synthase activity (SQS), which in part drives accumulation of lethal lipid peroxidation (Shimada et al., 2016b). Statin drugs, which inhibit HMG CoA reductase, the rate-limiting enzyme of the mevalonate pathway, sensitize cells to ferroptosis, presumably by depleting CoQ10, and possibly by also inhibiting downstream tRNA isopentenylation via TRIT1, which is required for the biosynthesis of GPX4 (Fradejas et al., 2013; Shimada et al., 2016b; Viswanathan et al., 2017).
NADPH and selenium abundance also impact ferroptosis sensitivity. NADPH is an essential intracellular reductant needed to eliminate lipid hydroperoxides. Indeed, NADPH levels are a biomarker of ferroptosis sensitivity across many cancer cell lines (Shimada et al., 2016a). Selenium is required for the biosynthesis of GPX4, which has an active-site selenocysteine (Cardoso et al., 2017). Thus, selenium supplementation promotes ferroptosis resistance, while selenium depletion promotes ferroptosis sensitivity, presumably by modulating GPX4 abundance and activity (Cardoso et al., 2017). A number of other genes have been implicated as modulators of sensitivity to ferroptosis or markers of ferroptosis in diverse contexts: SAT1, which lies downstream of p53 (Ou et al., 2016) is involved in polyamine metabolism; kiss of death (KOD) in Arabidopsis (Distefano et al., 2017) and FANCD2 in bone marrow stromal cells are induced during ferroptosis (Song et al., 2016); TTC35, CS, ATP5G3, and RPL8 are involved in diverse processes in human cancer cells and suppress erastin-induced ferroptosis upon knockdown (Dixon et al., 2012).
The NRF2 transcription factor promotes resistance to ferroptosis (Sun et al., 2016b): NRF2 controls the expression of AKR1C, the metal-binding protein MT-1G (Sun et al., 2016a), and other antioxidant genes, the expression of key proteins in iron signaling, including ferritin and ferroportin, and the expression of enzymes in the pentose phosphate pathway, which generate the bulk of cellular NADPH. Additionally, genes encoding proteins responsible for glutathione synthesis, including SLC7A11, GCLC/GLCM, and GSS are NRF2 target genes (Kerins and Ooi, 2017).
Links between ferroptosis and pathology
Recent discoveries have revealed connections between ferroptosis and degenerative and neoplastic diseases (Conrad et al., 2016; Toyokuni et al., 2017). Historically, regulated cell death was assumed to be apoptotic in models of ischemic injury to the brain, heart, liver, kidney and intestine. When necroptosis was discovered as a RIPK3-and-MLKL-dependent form of regulated necrosis, it was suggested that this pathway is a primary contributor to ischemic injury in the heart and kidney (Linkermann et al., 2013; Newton et al., 2016). Only later was it discovered that ferroptosis is a primary driver of ischemic injury in some models (Tonnus and Linkermann, 2017).
Inhibitors of ferroptosis, such as ferrostatins and liproxstatins, protect from ischemic injury in mouse models in the liver, kidney, brain, and heart (Friedmann Angeli et al., 2014; Gao et al., 2015; Linkermann et al., 2014; Skouta et al., 2014; Tuo et al., 2017; Dixon et al, 2012). These inhibitors are also protective in models of degenerative brain disorders, including Parkinson's, Huntington's, and Alzheimer's Diseases, as well as in other forms of neurodegeneration and traumatic and hemorrhagic brain injury (Chen et al., 2015; Do Van et al., 2016; Gascon et al., 2016; Guiney et al., 2017; Hambright et al., 2017; Li et al., 2017; Skouta et al., 2014; Zille et al., 2017). Ferroptosis in some other tissues and diseases has been examined, including liver hemochromatosis (Wang et al., 2017). A number of clinicopathological features of dementia are consistent with ferroptosis (Table 3). Similar features are manifest in other neurodegenerative diseases (Belaidi and Bush, 2016).
Table 3. Features of dementia consistent with ferroptosis.
The listed features of dementia are consistent with a role for ferroptosis in the disease pathogenesis.
| Feature | Comment | References. |
|---|---|---|
| Decreased cortical GSH | Found in post-mortem and imaging assays; an association with amyloid pathology has been reported | (Chiang et al., 2017; Mandal et al., 2015) |
| Lipid peroxidation products | Hydroxynonenal, malondialdehyde, and acrolein are reported elevated; may be related to redox-catalysis by metal-Aβ | (Di Domenico et al., 2017; Williams et al., 2006) |
| Depletion of PUFAs | Decreased DHA associated with reduced levels of PE in AD hippocampus; loss of PUFAs in AD brains | (Prasad et al., 1998; Sydenham et al., 2012) |
| Excess extracellular glutamate | Glutamate trafficking/receptor systems altered in AD; NMDAR blocker, memantine, is an approved AD drug | (Greenamyre et al., 1985; Revett et al., 2013) |
| Pathology spreads | Neurodegeneration spreads along the pathways of neural connectivity; release of toxic factors (lipid radicals, PUFA oxidation products) possible | (Braak and Del Tredici, 2015; Brettschneider et al., 2015) |
| Decreased cortical GPX4 | Human brain tissue data indirectly implicate GPX4 impairment | (Cardoso et al., 2017) |
| Increased cortical p53 | Increased p53 and lipid peroxidation products in post-mortem human brain tissue | (Cenini et al., 2008) |
| Increased cortical iron | MRI studies show elevated cortical iron in AD; hippocampal iron accelerates cognitive deterioration in patients positive for amyloid pathology | (Ayton et al., 2017; Raven et al., 2013) |
| Increased 12/15 lipoxygenase activity | LOX activity explored as pharmacological target for AD | (Di Meco et al., 2017) |
| Clinical benefit of Vitamin E | A large randomized control trial of patients with mild-moderate AD showed that vitamin E slowed cognitive decline by 19% / yr; vitamin E is a low potency anti-ferroptotic agent | (Dysken et al., 2014) |
| Possible clinical benefit of iron chelation | In preclinical AD models, iron chelators rescue cognitive deficits; a phase 2 clinical trial reported that desferrioxamine delayed deterioration in AD | (Crapper McLachlan et al., 1991) |
| ACSL4 inhibitors decrease dementia risk in humans | Long-term use of pioglitazone, a recently discovered ACSL4 inhibitor, abnegates the increased risk of dementia in non-insulin dependent diabetes patients | (Heneka et al., 2015) |
Brain iron levels inevitably rise during aging and in degenerative diseases, which can be detected both in post-mortem analysis as well as in living individuals; such increases could contribute to an age-dependent risk of ferroptosis (Belaidi and Bush, 2016; Buijs et al., 2017). Genetic evidence also links brain degeneration to ferroptosis: inducible deletion of gpx4 in adult mice causes hippocampal neuronal loss with astrogliosis, as seen in AD (Yoo et al., 2012). The study of ferroptosis in nervous tissue is complicated by the presence of neuronal support cells, including astrocytes, microglia and oligodendrocytes. Damage incurred by a support cell undergoing ferroptosis could, in principle, be transmitted to neurons in a wave-like propagation of ferroptosis (Linkermann et al., 2014). In addition, treatment with the iron chelator deferiprone was recently reported to be beneficial in a randomized controlled trial for PD (Devos et al., 2014); dopamine is also reported to enhance stability of GPX4 (Wang et al., 2016a). Given the plethora of disease implications, the optimization of existing inhibitors or the identification of new inhibitors that block ferroptosis is a potential approach to treating a number of degenerative diseases.
Ferroptosis applications in neoplastic diseases
Cancer cells are susceptible to perturbations of thiol metabolism, whereas oxidative stress via excess iron is associated with carcinogenesis (Toyokuni et al., 2017). Agents that inhibit cystine uptake via the cystine/glutamate antiporter (system xc-), such as sulfasalazine, arrest tumor growth and can induce ferroptosis in some circumstances (Dixon et al., 2014; Toyokuni et al., 2017). Likewise, direct depletion of cystine from plasma using an engineered cystine-degrading enzyme conjugate (i.e., cyst(e)inase) arrests tumor growth and triggers cell death (Cramer et al., 2017). Agents that conjugate to glutathione, such as APR-246, as well as chemical or genetic inhibition of glutathione biosynthesis, disrupt tumor cell growth and induce a ferroptosis-like form of cell death (Liu et al., 2017). Typically, elevated levels of ROS are detected in response to perturbation of cysteine or glutathione metabolism and, where tested, cell death is prevented by antioxidant treatment.
Erastin and RSL3 were originally identified in phenotypic screens for compounds that are selectively lethal to engineered tumor cells. Improved analogs of erastin with increased solubility, selectivity, and potency have been created, and some have shown efficacy in xenograft tumor studies (Yang et al., 2014). Nanoparticles that induce ferroptosis have also demonstrated efficacy in xenograft studies (Kim et al., 2016). The tumor suppressor p53 has been reported to repress SLC7A11, a component of system xc-, thus inducing ferroptosis in some contexts (Jiang et al., 2015). In addition, CD44v, a cancer stem cell marker, associates with system xc-, and stabilizes this complex; this observation suggests that CD44v may be a biomarker for tumors that are sensitive to system xc- inhibitors that induce ferroptosis (Toyokuni et al., 2017).
Model systems in which ferroptosis has been observed
A variety of experimental settings—from cell culture to mice and plants—can be used to explore mechanisms of ferroptosis. Careful consideration must be given to the selection of appropriate cell lines, however, because not all cell lines and experimental systems are susceptible to this process. In addition, ex vivo cultures have been shown to be sensitive to ferroptosis, including hippocampal postnatal rat brain slices treated with glutamate (Dixon et al., 2012), striatal rat brain slices with ectopic mutant huntingtin expression (Skouta et al., 2014), and freshly isolated renal tubules (Linkermann et al., 2014).
A large-scale characterization of ferroptosis sensitivity of cancer cell lines found that cancer cell lines have highly varied sensitivity to ferroptosis (Yang et al., 2014). Cell lines such as HT-1080 fibrosarcoma cells and Panc-1 pancreatic cancer cells have robust ferroptotic responses and are frequently used as model systems to study ferroptosis mechanisms. In addition, mouse embryonic fibroblasts (MEFs) are generally sensitive to ferroptosis, even when lacking the apoptotic regulators BAX and BAK (Friedmann Angeli et al., 2014; Wolpaw et al., 2011). HT-22 hippocampal neuronal cells have been used as a model for neuronal sensitivity to ferroptosis (Xie et al., 2016a), and U937 monocytes have been used as models for studying ferroptosis in immune cells (Conrad et al., 2016). Some primary cell systems have also been used for studies of ferroptosis, including HRPTEpiCs (primary human renal proximal tubule epithelial cells), HK2 cells, mouse lung epithelial cells, human bronchial epithelial cells, and spinal motor neurons. In addition, Arabidopsis seedlings exposed to 55 °C heat stress have been used as a model for ferroptotic-like cell death in plants (Distefano et al., 2017).
Commonly used reagents for studying ferroptosis
There are four classes of ferroptosis inducers currently in use (Yang and Stockwell, 2016): (i) system xc- inhibitors (erastin and its analogs, sulfasalazine, glutamate, and sorafenib); (ii) GPX4 inhibitors, such as RSL3 and ML162; (iii) FIN56, which depletes GPX4 protein, and the lipophilic antioxidant CoQ10 (Shimada et al., 2016b); and (iv) FINO2, which indirectly inhibits GPX4 activity and stimulates lipid peroxidation (Abrams et al., 2016) (Table 1). In addition to these canonical ferroptosis inducers, several other reagents can induce ferroptosis in some contexts: buthionine sulfoximine (BSO) depletes glutathione, and in some cases is capable of inducing ferroptosis (Yang et al., 2014); CCl4 may induce ferroptosis in the liver (Guo et al., 2017), artesunate can induce ferroptosis in pancreatic cancer cells (Eling et al., 2015), cisplatin induces both ferroptosis and apoptosis in several tissues (Jennis et al., 2016), and a derivative of artemisinin (Greenshields et al., 2017) induce both ferroptosis and apoptosis.
A variety of pharmacological and genetic inhibitors of ferroptosis have also been reported (Angeli et al., 2017; Conrad et al., 2016; Doll et al., 2017). Inhibitors of lipid metabolism that suppress PUFA incorporation into phospholipid membranes have been reported, such as knockdown or knockout of ACSL4 or LPCAT3, and thiazolidinediones, which inhibit ACSL4 (Dixon et al., 2015; Doll et al., 2017). Inhibitors of lipid peroxidation, such as LOX inhibitors, also suppress ferroptosis (Yang et al., 2016). Inhibitors of iron metabolism and iron chelators (e.g., deferoxamine (DFO) and ciclopirox (CPX)) suppress ferroptosis by reducing availability of iron (Yang and Stockwell, 2008). Inhibition of glutaminolysis, as noted above, suppresses ferroptosis, although the mechanism of action is not known (Gao et al., 2015). The nitroxide XJB-5-131 is a potent inhibitor of ferroptosis, probably by blocking lipid peroxidation in relevant membranes (Krainz et al., 2016). Similarly, ferrostatins and liproxstatins inhibit lipid peroxidation, possibly by acting as radical-trapping antioxidants (Zilka et al., 2017), similarly to the lipophilic antioxidants BHT, BHA and vitamin E. In the latter case, tocotrienols are more effective than tocopherols (Kagan et al., 2017). Selectively bis-allylic deuterated PUFAs suppress propagation of lipid peroxidation and have been found to suppress ferroptosis induced by RSL3 and erastin, although they are more effective against the former than the latter (Yang et al., 2016). Finally, the protein synthesis inhibitor cycloheximide suppresses ferroptosis induced by system xc- inhibition (Yagoda et al., 2007).
Given different mechanisms of ferroptotic inhibition, the characterization of new inhibitors should be accompanied by an evaluation of antioxidant or iron-chelating activity; otherwise, the mechanism of action of inhibitors that appear to act through distinct mechanisms may be misinterpreted. For example, many LOX inhibitors and the MEK inhibitor U0126 exhibit antioxidant activity, which is probably the basis of their ability to suppress ferroptosis. In addition, the necroptosis inhibitor necrostatin-1 has off-target activity, which can suppress ferroptosis at high concentrations (Friedmann Angeli et al., 2014), but this is unrelated to necroptosis.
Measuring lipid peroxidation is essential for evaluating whether ferroptosis occurs in specific contexts. C11-BODIPY and Liperfluo are lipophilic ROS sensors that provide a rapid, indirect means to detect lipid ROS (Dixon et al., 2012). Liquid chromatography (LC) / tandem mass spectrometry (MS) analysis has lower throughput but can be used to detect specific oxidized lipids directly (Friedmann Angeli et al., 2014; Kagan et al., 2017). In addition, isoprostanes have been used to measure lipid peroxidation (Milne et al., 2007), although not yet in the context of ferroptosis.
Other useful assays for studying ferroptosis include measuring iron abundance and GPX4 activity. The former can be detected using inductively coupled plasma-MS or calcein AM quenching, as well as other specific iron probes (Hirayama and Nagasawa, 2017; Spangler et al., 2016), while the latter can be detected using phosphatidylcholine hydroperoxide reduction in cell lysates using LC-MS (Yang et al., 2014).
Since ferroptosis susceptibility is so closely tied to the metabolic state of the cell, it is important in in vivo and in vitro studies to control the composition of chow and serum composition, respectively, which can vary between lots, laboratories, and experiments, by altering the levels of key ferroptotic regulators, such as selenium, iron, vitamin E, cysteine, glutathione and PUFAs in experimental samples. In addition, redox conditions vary between cell-based/organoid cultures and in vivo conditions, which is illustrated by the frequent requirement for system xc- in cell culture, whereas system xc- knockout mice are viable (McCullagh and Featherstone, 2014). This can be addressed to some extent by measuring the impact of system xc- both in vivo and in culture, and by modulating oxygen concentrations in culture (Dixon et al., 2014).
Given the sensitivity of ferroptosis to small variations in the levels of so many different molecules and metabolites (iron, PUFAs, cysteine) it is possible that dozens or hundreds of genes and proteins could impact ferroptosis sensitivity by having small effects on the levels of one or more metabolites. Such connections do not imply a direct role in the execution of ferroptosis, and while important to document, should be distinguished from core regulators of iron-dependent lipid peroxidation, such as ACSL4, system xc-, and GPX4.
Connections between ferroptosis and other cell death pathways
Both ferroptosis and necroptosis contribute to ischemic injury, such that prevention of ferroptosis and necroptosis at the same time provides maximum benefit (Linkermann et al., 2014). Moreover, some modulators affect both apoptosis and ferroptosis, such as p53: whereas the role of p53 in apoptosis has been investigated for more than two decades, its role in ferroptosis has only been reported recently (Jiang et al., 2015). For the most part, however, ferroptosis appears to be independent of other known cell death pathways. Ferroptosis proceeds even in the absence of key effectors of apoptosis, such as BAX, BAK and caspases. Likewise, ferroptosis proceeds in the absence of the key components of necroptosis, such as MLKL, RIPK1 and RIPK3.
Assessing ferroptosis in diverse biological contexts
Ferroptosis is a process that has likely been discovered and re-discovered a number of times over the years before a detailed molecular understanding of this phenotype was solidified (Dixon and Stockwell, 2013). Since 2012, rapid progress has been made in defining the molecular regulation of ferroptosis. We anticipate that additional regulators and biological contexts will emerge for ferroptosis in the future. We suggest here basic criteria for concluding that ferroptosis is occurring in a particular experimental system, while acknowledging that these criteria may vary depending on the mechanism by which ferroptosis is triggered. In general, ferroptosis should be suppressed by both an iron chelator (e.g., DFO or CPX) and a lipophilic antioxidant (e.g., ferrostatin, liproxstatin, vitamin E, BHT), and should involve accumulation of lipid hydroperoxides. Ferroptosis induced by system xc- inhibition should be suppressed by β-mercaptoethanol, which reduces extracellular cystine to cysteine, bypassing the requirement for system xc- for import, as cysteine is imported through other mechanisms (Dixon, 2012). System xc- inhibition should result in glutathione depletion. Gene expression markers associated with cells undergoing ferroptosis include increases in CHAC1 and PTGS2 mRNA and ACSL4 protein (Table 2), but these changes may not be observed in all experiments.
Inhibitors of apoptosis (e.g., caspase inhibitors or deletion of BAX/BAK) or necroptosis (e.g., necrostatins or necrosulfonamide) should be examined to rule out these mechanisms as being required for cell death that is postulated to be ferroptosis. Cells dying by ferroptosis primarily exhibit shrunken and damaged mitochondria by electron microscopy, with few other morphological changes evident prior to the point of cell death (Abrams et al., 2016; Dixon et al., 2012; Yagoda et al., 2007); in particular, nuclei remain intact during ferroptosis (Dolma et al., 2003; Friedmann-Angeli et al, 2014). In contrast, apoptosis typically involved fragmentation and margination of chromatin, as well as generation of apoptotic bodies and plasma membrane blebbing. Necroptosis has been demonstrated to involve swelling of cells (Berghe et al., 2010) and was shown to be associated with the generation of broken pieces of plasma membrane that are released from the plasma membrane as vesicles; these “bubbles” stain positive for annexin V as a result of ESCRT-III activation and are released before plasma membrane rupture (Gong et al., 2017). Such features have not been observed during ferroptosis and therefore differentiate these two forms of regulated necrosis. Another form of regulated necrosis is pyroptosis and critically relies on the protein gasdermin D; time-lapse videos of pyroptotic cells demonstrated rapid necrotic cell death that is preceded by intensive blebbing (Ding et al., 2016). This typical morphology has not been seen during ferroptosis. Thus, ferroptosis has unique morphological characteristics.
One major outstanding question is why ferroptosis exists, and whether it is adaptive or indicative of a biochemical failure in cells. One possibility is that the incorporation of polyunsaturated fatty acids (PUFAs) into cell membranes during evolution was highly advantageous, allowing more complex physiological functions, including the development of complex neuronal circuits, modulating membrane fluidity, and allowing cells to adapt to environments with different temperatures, among other beneficial functions (Barelli and Antonny, 2016). At the same time, use of PUFAs in membranes creates a new Achilles' heel in the form of susceptibility to lethal lipid peroxidation. These fatty acids possess methylene (CH2) groups flanked on both sides by carbon–carbon double bonds, and one of these hydrogen atoms can be removed to make a highly stabilized pentadienyl radical. The ability to form stabilized radicals makes PUFAs susceptible to peroxidation in the presence of other radical species, as well as metals, oxygen, and specific enzymes. Moreover, oxygenated PUFAs biosynthesized by specialized enzymes – cyclooxygenases, lipoxygenases and cytochromes P450 – are broadly used for signaling purposes. Paradoxically, the signaling benefits of these enzymatic oxygenation reactions are also associated with a risk of generating reactive electrophiles targeting nucleophilic sites in vital proteins. It is possible, that one of the physiological roles of the ferroptotic program is to maximize the benefits and minimize the risk by eliminating cells with excessive production of electrophilic intermediates that cannot be removed.
In summary, ferroptosis represents a unique form of regulated cell death, with many of its physiological roles yet to be defined. Using the criteria and reagents described herein may allow the scientific community to determine the physiological and pathophysiological functions of ferroptosis in the years to come.
Acknowledgments
We thank The Banbury Center at Cold Spring Harbor Labs for hosting a meeting of the authors that served as the genesis of this manuscript. We thank the following funding organizations: NIH (R35CA209896 to BRS, 4R00CA166517 and 1R01GM122923 to SJD, R01CA171101 to FT, R01CA188025 to ST, CA102184 to MEM, HL114453, U19AI068021, NS076511, NS061817 to VEK and HB, R01GM115366, R01CA160417 to DT), JSPS KAKENHI (JP16K15257 and JP17H04064 to ST), ACS (RSG-16-014-01-CDD to DT), NNSFC (2016A030308011 and 31671435 to DT) and HFSP (RGP 0013/14) to MC and VEK. CSR is an employee of and has a profit interest in Collaborative Medicinal Development, LLC. KN is a paid consultant of and has a profit interest in Collaborative Medicinal Development, LLC. AIB is a shareholder in Prana Biotechnology Ltd, Mesoblast Ltd, Grunbiotics Pty Ltd, Cogstate Ltd, and is a paid consultant for and has a profit interest in Collaborative Medicinal Development, LLC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams RP, Carroll WL, Woerpel KA. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem Biol. 2016;11:1305–1312. doi: 10.1021/acschembio.5b00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol Sci. 2017;38:489–498. doi: 10.1016/j.tips.2017.02.005. [DOI] [PubMed] [Google Scholar]
- Ayton S, Fazlollahi A, Bourgeat P, Raniga P, Ng A, Lim YY, Diouf I, Farquharson S, Fripp J, Ames D, et al. Cerebral quantitative susceptibility mapping predicts β-amyloid-related cognitive decline. Brain. 2017 doi: 10.1093/brain/awx137. in press. [DOI] [PubMed] [Google Scholar]
- Bannai S, Tsukeda H, Okumura H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem Biophys Res Commun. 1977;74:1582–1588. doi: 10.1016/0006-291x(77)90623-4. [DOI] [PubMed] [Google Scholar]
- Barelli H, Antonny B. Lipid unsaturation and organelle dynamics. Curr Opin Cell Biol. 2016;41:25–32. doi: 10.1016/j.ceb.2016.03.012. [DOI] [PubMed] [Google Scholar]
- Belaidi AA, Bush AI. Iron neurochemistry in Alzheimer's disease and Parkinson's disease: targets for therapeutics. J Neurochem. 2016;139 Suppl 1:179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- Berghe TV, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain. 2015;138:2814–2833. doi: 10.1093/brain/awv236. [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16:109–120. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijs M, Doan NT, van Rooden S, Versluis MJ, van Lew B, Milles J, van der Grond J, van Buchem MA. In vivo assessment of iron content of the cerebral cortex in healthy aging using 7-Tesla T2*-weighted phase imaging. Neurobiol Aging. 2017;53:20–26. doi: 10.1016/j.neurobiolaging.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Cardoso BR, Hare DJ, Bush AI, Roberts BR. Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry. 2017;22:328–335. doi: 10.1038/mp.2016.196. [DOI] [PubMed] [Google Scholar]
- Cenini G, Sultana R, Memo M, Butterfield DA. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer's disease. Journal of cellular and molecular medicine. 2008;12:987–994. doi: 10.1111/j.1582-4934.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hambright WS, Na R, Ran Q. Ablation of ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015 Sep 23; doi: 10.1074/jbc.M115.680090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang GC, Mao X, Kang G, Chang E, Pandya S, Vallabhajosula S, Isaacson R, Ravdin LD, Alzheimer's Disease Neuroimaging, I. Shungu DC. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with 1H-MRS and Pittsburgh Compound-B PET. AJNR American journal of neuroradiology. 2017;38:1130–1137. doi: 10.3174/ajnr.A5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23:120–127. doi: 10.1038/nm.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapper McLachlan DR, Dalton AJ, Kruck TPA, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, Jonneaux A, Ryckewaert G, Garcon G, Rouaix N, et al. Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid Redox Signal. 2014;21:195–210. doi: 10.1089/ars.2013.5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, Tramutola A, Butterfield DA. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. 2017;111:253–261. doi: 10.1016/j.freeradbiomed.2016.10.490. [DOI] [PubMed] [Google Scholar]
- Di Meco A, Li JG, Blass BE, Abou-Gharbia M, Lauretti E, Pratico D. 12/15-Lipoxygenase Inhibition Reverses Cognitive Impairment, Brain Amyloidosis, and Tau Pathology by Stimulating Autophagy in Aged Triple Transgenic Mice. Biol Psychiatry. 2017;81:92–100. doi: 10.1016/j.biopsych.2016.05.023. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Erratum: Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;540:150. doi: 10.1038/nature20106. [DOI] [PubMed] [Google Scholar]
- Distefano AM, Martin MV, Cordoba JP, Bellido AM, D'Ippolito S, Colman SL, Soto D, Roldan JA, Bartoli CG, Zabaleta EJ, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 2017;216:463–476. doi: 10.1083/jcb.201605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch ME, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti N, Slusher BS, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014 doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2013;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol. 2015;10:1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, et al. Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178. doi: 10.1016/j.nbd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD, McCarten JR, Malphurs J, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311:33–44. doi: 10.1001/jama.2013.282834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H. Nutrition needs of mammalian cells in tissue culture. Science. 1955;122:501–514. doi: 10.1126/science.122.3168.501. [DOI] [PubMed] [Google Scholar]
- Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517–532. doi: 10.18632/oncoscience.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enke U, Seyfarth L, Schleussner E, Markert UR. Impact of PUFA on early immune and fetal development. Br J Nutr. 2008;100:1158–1168. doi: 10.1017/S000711450801413X. [DOI] [PubMed] [Google Scholar]
- Fradejas N, Carlson BA, Rijntjes E, Becker NP, Tobe R, Schweizer U. Mammalian Trit1 is a tRNA([Ser]Sec)-isopentenyl transferase required for full selenoprotein expression. Biochem J. 2013;450:427–432. doi: 10.1042/BJ20121713. [DOI] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73. doi: 10.1038/cdd.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascon S, Murenu E, Masserdotti G, Ortega F, Russo GL, Petrik D, Deshpande A, Heinrich C, Karow M, Robertson SP, et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell stem cell. 2016;18:396–409. doi: 10.1016/j.stem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169:286–300 e216. doi: 10.1016/j.cell.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenamyre JT, Penney JB, Young AB, D'Amato CJ, Hicks SP, Shoulson I. Alterations in L-glutamate binding in Alzheimer's and Huntington's diseases. Science. 1985;227:1496–1499. doi: 10.1126/science.2858129. [DOI] [PubMed] [Google Scholar]
- Greenshields AL, Shepherd TG, Hoskin DW. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol Carcinog. 2017;56:75–93. doi: 10.1002/mc.22474. [DOI] [PubMed] [Google Scholar]
- Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S. Ferroptosis and cell death mechanisms in Parkinson's disease. Neurochem Int. 2017;104:34–48. doi: 10.1016/j.neuint.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, Dai X, Li Z, Wu G. Ferroptosis: A Novel Anti-Tumor Action for Cisplatin. Cancer Res Treat. 2017 doi: 10.4143/crt.2016.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas U, Raschperger E, Hamberg M, Samuelsson B, Tryggvason K, Haeggstrom JZ. Targeted knock-down of a structurally atypical zebrafish 12S-lipoxygenase leads to severe impairment of embryonic development. Proc Natl Acad Sci U S A. 2011;108:20479–20484. doi: 10.1073/pnas.1117094108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17. doi: 10.1016/j.redox.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–278. doi: 10.1038/cdd.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Fink A, Doblhammer G. Effect of pioglitazone medication on the incidence of dementia. Ann Neurol. 2015;78:284–294. doi: 10.1002/ana.24439. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Nagasawa H. Chemical tools for detecting Fe ions. J Clin Biochem Nutr. 2017;60:39–48. doi: 10.3164/jcbn.16-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, 3rd, Kang R, Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30:918–930. doi: 10.1101/gad.275891.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2017;Aug 10 doi: 10.1089/ars.2017.7176. 10.1089/ars.2017.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. The Journal of biological chemistry. 2003;278:43508–43515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977–985. doi: 10.1038/nnano.2016.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainz T, Gaschler MM, Lim C, Sacher JR, Stockwell BR, Wipf P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent Sci. 2016;2:653–659. doi: 10.1021/acscentsci.6b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochimica et biophysica acta. 2015;1851:308–330. doi: 10.1016/j.bbalip.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2:e90777. doi: 10.1172/jci.insight.90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111:16836–16841. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, Pearson HB, Fisher OM, Read M, Guerra GR, et al. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. 2017;8:14844. doi: 10.1038/ncomms14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod AK, McMahon M, Plummer SM, Higgins LG, Penning TM, Igarashi K, Hayes JD. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis. 2009;30:1571–1580. doi: 10.1093/carcin/bgp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Saharan S, Tripathi M, Murari G. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer's disease. Biol Psychiatry. 2015;78:702–710. doi: 10.1016/j.biopsych.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Massie A, Schallier A, Kim SW, Fernando R, Kobayashi S, Beck H, De Bundel D, Vermoesen K, Bannai S, Smolders I, et al. Dopaminergic neurons of system x(c)(-)-deficient mice are highly protected against 6-hydroxydopamine-induced toxicity. Faseb J. 2011;25:1359–1369. doi: 10.1096/fj.10-177212. [DOI] [PubMed] [Google Scholar]
- McCullagh EA, Featherstone DE. Behavioral characterization of system xc-mutant mice. Behavioural brain research. 2014;265:1–11. doi: 10.1016/j.bbr.2014.02.010. [DOI] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–1576. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113:E6806–E6812. doi: 10.1073/pnas.1607152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer's disease. Neurochem Res. 1998;23:81–88. doi: 10.1023/a:1022457605436. [DOI] [PubMed] [Google Scholar]
- Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer's disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis. 2013;37:127–136. doi: 10.3233/JAD-130209. [DOI] [PubMed] [Google Scholar]
- Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid Abeta peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci. 2013;38:6–23. doi: 10.1503/jpn.110190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell metabolism. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol. 2016a;23:225–235. doi: 10.1016/j.chembiol.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat Chem Biol. 2016b;12:497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg P, Lo D, Weinberg J, Linkermann A, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Xie Y, Kang R, Hou W, Sun X, Epperly MW, Greenberger JS, Tang D. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun. 2016;480:443–449. doi: 10.1016/j.bbrc.2016.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler B, Morgan CW, Fontaine SD, Vander Wal MN, Chang CJ, Wells JA, Renslo AR. A reactivity-based probe of the intracellular labile ferrous iron pool. Nat Chem Biol. 2016;12:680–685. doi: 10.1038/nchembio.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016a;64:488–500. doi: 10.1002/hep.28574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016b;63:173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, Wang H, Cao L, Tang D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Onco. 2015 Mar 2; doi: 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydenham E, Dangour AD, Lim WS. Omega 3 fatty acid for the prevention of cognitive decline and dementia. Cochrane Database Syst Rev. 2012:CD005379. doi: 10.1002/14651858.CD005379.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnus W, Linkermann A. The in vivo evidence for regulated necrosis. Immunol Rev. 2017;277:128–149. doi: 10.1111/imr.12551. [DOI] [PubMed] [Google Scholar]
- Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic Biol Med. 2017;108:610–626. doi: 10.1016/j.freeradbiomed.2017.04.024. [DOI] [PubMed] [Google Scholar]
- Tuo QZ, Lei P, Jackman KA, Li XI, Xiong H, Li XL, Liuyang ZY, Roisman L, Zhang ST, Ayton S, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Molecular Psychiatry. 2017 doi: 10.1038/mp.2017.171. [DOI] [PubMed] [Google Scholar]
- Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211. doi: 10.1016/0005-2760(82)90150-3. [DOI] [PubMed] [Google Scholar]
- van den Brink-van der Laan E, Killian JA, de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochimica et biophysica acta. 2004;1666:275–288. doi: 10.1016/j.bbamem.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Li S, Wen L, Kong J, Wang K, Zhu Y. Damage of oxidative stress on mitochondria during microspores development in Honglian CMS line of rice. Plant Cell Rep. 2007;26:373–382. doi: 10.1007/s00299-006-0234-2. [DOI] [PubMed] [Google Scholar]
- Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang R, Tang D. Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun. 2016a;480:602–607. doi: 10.1016/j.bbrc.2016.10.099. [DOI] [PubMed] [Google Scholar]
- Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449–465. doi: 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, Yu P, Shi ZH, Wu WS, Gao G, et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front Aging Neurosci. 2016b;8:308. doi: 10.3389/fnagi.2016.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging. 2006;27:1094–1099. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Pe'er D, Shaik F, Bulinski JC, Stockwell BR. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc Natl Acad Sci U S A. 2011;108:E771–780. doi: 10.1073/pnas.1106149108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016a;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Song X, Sun X, Huang J, Zhong M, Lotze MT, Zeh HJ, 3rd, Kang R, Tang D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun. 2016b;473:775–780. doi: 10.1016/j.bbrc.2016.03.052. [DOI] [PubMed] [Google Scholar]
- Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell reports. 2017;20:1692–1704. doi: 10.1016/j.celrep.2017.07.055. [DOI] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by Gpx4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SE, Chen L, Na R, Liu Y, Rios C, Van Remmen H, Richardson A, Ran Q. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52:1820–1827. doi: 10.1016/j.freeradbiomed.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016a;478:838–844. doi: 10.1016/j.bbrc.2016.08.034. [DOI] [PubMed] [Google Scholar]
- Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016b;478:1338–1343. doi: 10.1016/j.bbrc.2016.08.124. [DOI] [PubMed] [Google Scholar]
- Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci. 2017;3:232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, Ratan RR. Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke. 2017;48:1033–1043. doi: 10.1161/STROKEAHA.116.015609. [DOI] [PMC free article] [PubMed] [Google Scholar]