

Abstract
Critically ill patients typically present with hyperglycemia. Treatment with conventional insulin therapy (targeting 144-180 mg/dL) improves patient survival; however, intensive insulin therapy (IIT) targeting normal blood glucose levels (81-108 mg/dL) increases the incidence of moderate and severe hypoglycemia, and increases mortality. Septic patients are especially prone to IIT-induced hypoglycemia, but the mechanism remains unknown. Here, we show that co-delivery of insulin with otherwise sublethal doses of lipopolysaccharide (LPS) induced hypoglycemic shock in mice within 1-2 hours. LPS impaired clearance of insulin, which amplified insulin receptor signaling. These effects were mediated by caspase-11, TLR4, and complement, each of which trigger eicosanoid production that potentiates insulin signaling. Finally, in an animal model of sepsis, we observed that Salmonella Typhimurium infected mice exhibited simultaneous impaired insulin clearance coexisting with insulin resistance. Our results raise the possibility that septic patients have impaired insulin clearance, which could increase their susceptibility to hypoglycemia during IIT, contraindicating its use.
Introduction
Lipopolysaccharide (LPS) is a potent microbial signature molecule that is detected by the innate immune system. TLR4 is well appreciated as the sensor for extracellular and vacuolar LPS, driving transcriptional responses to promote inflammation (1-3). Additionally, complement, although not commonly discussed as an LPS sensor, becomes activated by certain components of the LPS polysaccharide (4). More recently, LPS was found to be detected in the cytosolic compartment independently of TLR4 via inflammatory caspases (caspase-4 and -5 in humans, and caspase-11 in mice) (5-7). These inflammatory caspases initiate a form of lytic, programmed cell death termed pyroptosis. Caspase-4 and -11 (and likely -5) protect against infection by cytosol-invasive bacteria (8, 9).
Excessive activation of the immune system leads to sepsis, characterized by hypotension, organ failure, and ultimately death, all of which can be modeled by high dose (54,000 μg/kg) LPS challenge in mice. Mice deficient in TLR4 resist LPS injection (10), as do mice deficient in caspase-11, albeit incompletely (11, 12). Caspase-11 is expressed at low levels basally; however, its transcription is strongly induced by TLR and interferon signaling (5, 6, 8, 9, 13), which can be accomplished in vivo by priming with the TLR3 agonist poly(I:C) (5, 6). We and others have shown that priming with poly(I:C) rendersTlr4−/− mice susceptible to lethal challenge with LPS (we term this a “prime-challenge” model) (5, 11, 12). Casp11−/− mice are partially resistant to prime-challenge, with variable penetrance between labsor at slightly different LPS doses (6, 14). However, it should be noted that Casp11−/− mice express TLR4.
Here, we report the serendipitous discovery that LPS lethally potentiates the activity of co-delivered insulin. Insulin, normally secreted by the pancreas in response to elevated blood glucose, acts via the insulin receptor on liver, adipose, and muscle cells to restore euglycemia (15). Upon insulin receptor binding, insulin is internalized, dissociated from the receptor by endosomal acidification, and most is eventually degraded in a multistep process initiated by the insulin degrading enzyme (IDE); a portion of insulin can co-recycle to the plasma membrane with the insulin receptor (16). Perturbation of insulin degradation, for example by impairing IDE activity (17) or endosomal acidification (18, 19), enhances insulin signaling, an effect that can be dangerous when applied to super physiologic amounts of exogenous insulin. Here, we demonstrate that LPS enhances insulin activity in part by impairing its clearance, precipitating lethal hypoglycemic shock in mice. Further, mice infected with the Gram-negative pathogen Salmonella Typhimurium similarly exhibit impaired insulin clearance that coexists with insulin resistance.
Materials and Methods
Mice and in vivo challenges
Wild-type C57BL/6 (Jackson Laboratory), Casp11−/− (12), Tlr4tm1Aki/tm1Aki (3) referred to as Tlr4−/−, Casp11−/−Tlr4−/− (crossed for this study), and mpges1tm1Audoly/tm1Audoly (20) mice were used in this study. Unless indicated in Table S1, mice were female and between 20-30 g (generally >12 weeks of age). Mice were housed in a specific pathogen–free facility and fed Harlan 2920× Irradiated Regular chow. Of note: in our previous study (5), mice were fed Purina 3000 Irradiated Regular chow; mice on this diet were sensitive to significantly smaller quantities of LPS (10-100 ng/kg) when delivered in OptiMEM than mice on the Harlan 2920× diet. During all experiments, mice were allowed to feed ad libitum. All protocols were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill and met guidelines of the US National Institutes of Health for the humane care of animals.
In all non-infectious animal studies, mice were primed by intraperitoneal (i.p.) injection with poly (I:C) (10mg/kg, Invivogen, cat #tlrl-picw) in DPBS (250μL total volume) 6h before subsequent challenges. For study of lethal endotoxic shock absent insulin, primed mice were challenged via i.p. injection with LPS (54,000 μg/kg, Sigma, cat # L3024) in DPBS (500μL total volume). A 500μL injection volume was also used when the indicated quantities of LPS were delivered in DMEM (Gibco) or OptiMEM (Gibco), and when LPS was co-delivered with human insulin (100μg/kg; Thermo Fisher, cat#12585-014) in DBPS. To rescue mice challenged with LPS and insulin, animals were given an i.p. injection of 500μL 20% glucose in DPBS 30min post-injection of LPS and insulin. In some experiments, mice were injected with C-peptide (50μg/kg, Sigma, cat# C4999) together with insulin and LPS or DBPS to determine whether LPS affects insulin clearance specifically rather than protein clearance or transport from the peritoneal cavity to the serum generally (21). In other experiments, mice were injected with 500μL Evan's Blue dye solution (0.5%, Sigma, cat# 2129) with or without LPS. In pan-COX inhibition experiments, mice were given indomethacin (20mg/kg, SigmaI7378-5G) i.p. in 250μL DBPS 5.5 h after priming and 30min before challenge with LPS and insulin. To deplete complement C3 protein, mice were injected with 15 μg cobra venom factor (Complement Tech, cat # A150) in 250 μL DPBS 4 h after priming and 2h before subsequent LPS and insulin challenge. Rectal temperatures were recorded at 1h post challenge using a Micro Therma 2T thermometer (Braintree Scientific) with a lubricated RET-3 probe. Glucose concentrations were determined in tail blood using a Reli On Prime Blood Glucose Monitoring System. Mice were considered moribund when unable to right themselves or when seizures were first observed.
For study of insulin action and clearance in septic mice, animals were infected via i.p. injection with 103 colony forming units of S. Typhimurium strain 14028S. 5 days later, mice were challenged with 100 μg/kg insulin via i.p. injection; plasma and tissue samples were collected at 1 h.
Western blots
Antibodies against the following proteins were used in this study: phospho-AKT (Ser473) (Cell Signaling, clone D9E, product #4060), AKT (pan) (Cell Signaling, clone C67E7, product # 4691), phospho-Insulin Receptor β (Tyr1150/1151) (Cell Signaling, clone 19H7, product # 3024), Insulin Receptor β (Cell Signaling, clone 4B8, product # 3025).
For analysis of insulin receptor and AKT phosphorylation in ex vivo samples, tissue samples were collected from mice 1h post challenge, placed immediately on dry ice, and stored at -80°C. Frozen samples (approximately 100mg tissue) were homogenized in 1mL buffer containing 100mM KPO4 and 1% triton X-100 at pH10 using a steel ball and Retsch MM400 ball mill (30 rps for 1min or until homogenous). Samples were then centrifuged for 10min at 20,000 × g at 4°C. Total protein content of the supernatant was determined by BCA assay, and 30μg of supernatant total protein was used for Western blot analysis. Primary antibodies were used at 1:1000-1:2000 dilutions. In all figure panels, each lane represents a sample from a different mouse. Band intensities were determined using Image J software. Where quantification data are presented, phospho-AKT and phospho-insulin receptor intensities were normalized to total AKT and insulin receptor intensities, respectively; these values were then normalized to the average normalized value of control samples in an experiment so that data could be compiled from multiple experiments.
Plasma protein analysis
Cardiac blood was collected from mice at 1h post challenge (approximately 500μL), mixed with 6μL 10kU/mL heparin (Sigma, cat #H3393-10KU), and centrifuged at 20,000×g for 1 minute. From this, the serum supernatant was obtained. ELISA was used to determine insulin (Genway Biotech, product # GWB-D9BD0E) and C-peptide (Sigma, product # SE120040) concentrations according to manufacturer instructions. Notably, the insulin ELISA is specific for human insulin and therefore allows specific determination of exogenous insulin concentrations. ELISA was also used to determine IL-6, and TNF concentrations in plasma (R&D Systems, cat # DY406, and DY410, respectively).
Determination of Evan's blue dye serum concentrations
For determination of Evan's Blue concentrations, 5μL of serum collected from tail blood at the indicated time points was incubated with 45μL formamide at 55°C overnight. After incubation, samples were centrifuged at 20,000×g for 1min, supernatants collected and then measured for absorbance at 610nm; values were compared to those of a standard curve to determine the concentration of dye in serum.
Determination of plasma eicosanoids
Eicosanoid metabolites were extracted from mouse plasma by liquid:liquid extraction into ethyl acetate and quantified by HPLC-MS/MS. 200 μL plasma was acidified with 200 μL of 0.1% acetic acid in 5% methanol and spiked with internal standard 3 ng PGE2-d4 (Cayman Chemical, Detroit MI). The aqueous mixture was extracted twice with 2 ml ethyl acetate (Sigma). The organic phase from both extractions was combined into glass tubes with 6 μL 30% glycerol in methanol. Ethyl acetate was evaporated in a speed-vac at 37°C.
Liquid chromatography of extracted samples was performed with an Agilent 1200 Series capillary HPLC (Agilent Technologies, Santa Clara, CA) as previously described (22). Samples were reconstituted in 50 μl of 30% ethanol and injected in triplicate, 10 μL injections. Separations were achieved using a Halo C18 column (2.7 μm, 100×2.1 mm; MAC-MOD Analytical, Chadds Ford, PA), which was held at 50°C and a flow rate of 400 μL/min. Mobile phase A was 0.1% acetic acid in 85:15 water: acetonitrile. Mobile phase B was 0.1% acetic acid in acetonitrile. Gradient elution was used and the mobile phase was varied as follows: 20% B at 0 min, ramp from 0 to 5 min to 40% B, ramp from 5 to 7 min to 55% B, ramp from 7 to 13 min to 64% B. From 13 to 19 min the column was flushed with 100% B at a flow rate of 550 μL/min before being returned to starting conditions and equilibrated for 6 minutes.
Electrospray ionization tandem mass spectrometry was used for detection. Analyses were performed on an MDS Sciex API 3000 equipped with a TurboIon Spray source (Applied Biosystems, Foster City, CA). Turbo desolvation gas was heated to 425°C at a flow rate of 6 L/min. All analytes were monitored simultaneously in a scheduled multiple reaction monitoring experiment as negative ions at parent ion-product ion mass/charge ratio pairs and retention times. The relative response ratios of each analyte and surrogate internal standard were used to calculate concentrations compared to response ratios of a range of prostaglandin standards of known concentrations (0.1-120 pg/μL injection). Peak analysis and quantification were performed using Analyst software (SCIEX, Framingham, MA).
Hematocrit determination
Tail blood from mice was collected into heparinized capillary tubes (Fisher, cat # 22-362-566). Tubes were then sealed at one end by flame and centrifuged at 796×g for 10 minutes. Hematocrit ratio was determined as the height of the pelleted cell fraction from the sealed tube end compared to the total length of separated blood components.
Statistical analysis
All data were analyzed using Graph Pad Prism 5 software. Statistical tests were used as indicated in the figure legends. P values less than 0.05 were used to identify statistically significant differences. Where animal survival data are presented, data are pooled from at least 2 experiments as detailed in Table S1.
Results
Caspase-11 and TLR4 account for the lethality of LPS challenge in poly(I:C) primed mice
To determine whether TLR4 accounts for the residual lethality seen in prime-challenge of Casp11−/− mice, we examined survival of mice deficient in TLR4, caspase-11, or both. The various challenges and medium vehicles used throughout our studies are listed in Table S2. As expected, primed Tlr4−/− mice uniformly succumbed to a high dose 54,000 μg/kg LPS challenge delivered in DPBS (Fig. 1). Nearly all Casp11−/− mice also succumbed to this challenge, further highlighting the variable penetrance of the response to similar dosing in different labs (6, 14). In contrast, Casp11−/−Tlr4−/− mice uniformly survived (Fig. 1), although apparent clinical symptoms such as piloerection and lethargy were still observed approximately 6-12 h after LPS injection in the Casp11−/−Tlr4−/− mice. Together, these data indicate that caspase-11 and TLR4 are each sufficient to drive lethality and together account for all of the lethality of the prime-54,000 μg/kg challenge regimen, but suggest that LPS from E. coli O111:B4 can weakly activate additional immune pathways.
Fig. 1. TLR4 and caspase-11 each contribute to lethality in primed mice challenged with LPS.
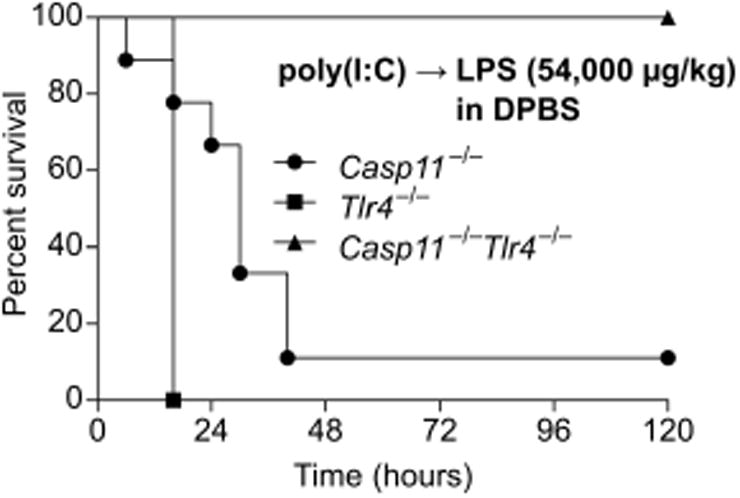
Survival of poly(I:C) primed mice challenged with LPS (54,000 μg/kg) in DPBS (without insulin). Data are pooled from 2 experiments.
Low-dose LPS induces shock when delivered in OptiMEM
In vitro, extracellular LPS does not efficiently access the cytosolic sensor caspase-11 unless it is packaged with a transfection reagent (5, 6). In an attempt to develop a novel model of sepsis where LPS is specifically delivered to the cytosol in vivo, we injected mice with LPS packaged with a transfection reagent (note that we used the tissue culture medium OptiMEM as a solution, which optimizes the packaging reaction). To our surprise, poly(I:C) primed mice challenged with even a medium dose of 400 μg/kg LPS rapidly succumbed to shock, becoming moribund within 1 to 2.5 hours of challenge (Fig. 2A). To our surprise, inclusion of the transfection reagent had no appreciable effect on this phenotype (not shown), suggesting that aberrant translocation of LPS to the cytosol is more efficient in vivo than in vitro. This result starkly contrasted the mortality kinetics of poly(I:C) primed mice challenged with high dose 54,000 μg/kg LPS in DPBS, where mice succumb in 6-48 hours (Fig. 1), suggesting OptiMEM contributed to the rapid lethality and extreme sensitization to low dose LPS. As a control, OptiMEM alone was not sufficient to precipitate shock, nor was medium dose 400 μg/kg LPS delivered in DPBS or another tissue culture medium, DMEM (Fig. 2A, B). Mice challenged with LPS in OptiMEM became severely hypothermic (Fig. 2C), and also exhibited seizures that are not typical of high dose LPS challenge in DPBS. Seizures can be caused by many factors, one of which is extreme hypoglycemia. We therefore examined the blood glucose levels of mice challenged with LPS in OptiMEM, and found it to be below the lower limit of detection (Fig. 2D).
Fig. 2. LPS delivered in OptiMEM rapidly induces shock.
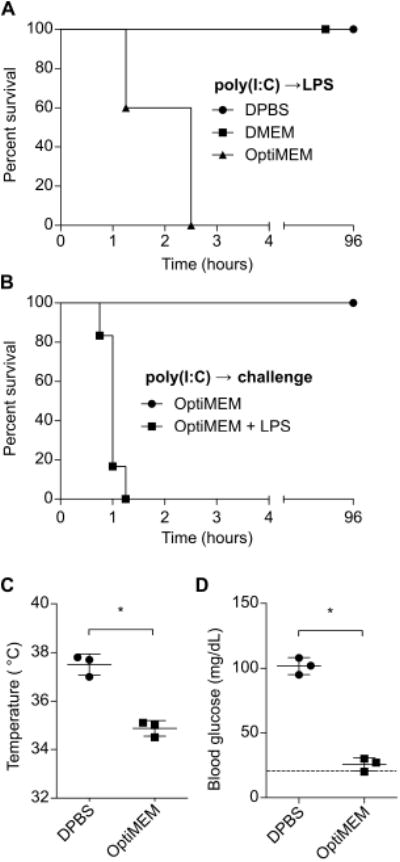
(A)Survival of mice challenged with LPS (400 μg/kg) in DPBS, DMEM, or OptiMEM. (B)Survival of mice challenged with OptiMEM with or without LPS (400 μg/kg). (C and D) Core temperatures (C) and blood glucose concentrations (D) at 1h of mice after challenge with LPS in DBPS or OptiMEM as described in (A). Survival data were pooled from 2 experiments. Temperature and glucose data are representative of 2 independent experiments. Error bars, mean ± SD. Statistically significant differences were determined by a 2-tailed unpaired t-test; * P ≤ 0.05.
LPS potentiates insulin induced hypoglycemic shock
OptiMEM is a proprietary formulation designed to culture cells in low serum conditions and contains, among other ingredients, insulin. Given the pronounced hypoglycemia we observed during challenge with LPS in OptiMEM, we hypothesized that insulin was the key factor that synergized with LPS to drive shock. Indeed, addition of insulin to LPS in DPBS recapitulated the phenotype of mice challenged with LPS in OptiMEM (Fig. 3A-C). This effect was not unique to LPS, as the TLR2 agonist Pam3CSK4 could similarly precipitate shock when co-delivered with insulin (Fig. 3D). Formal demonstration that Pam3CSK4 is signaling via TLR2 will require the use of Tlr2−/− mice in future work. Strikingly, insulin did not induce extreme hypoglycemia (below 20 mg/dL) until we administered doses 4-fold higher than employed with LPS, highlighting the potency of the interaction between LPS and insulin (Fig. S1A). Mice challenged with LPS and insulin could be rescued by administration of glucose, implicating extreme hypoglycemia as the cause of death (Fig. 3E). Together, these data demonstrate that insulin can synergize with LPS-induced inflammatory mediators to drive hypoglycemic shock.
Fig. 3. Co-delivery of LPS and insulin drives hypoglycemic shock.
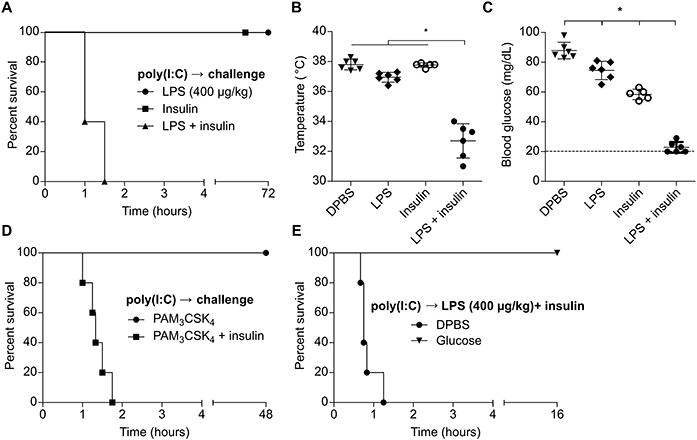
(A) Survival of mice challenged with LPS (400 μg/kg), insulin, or both. (B and C) Core temperatures (B) and blood glucose concentrations (C) of mice 1 h after challenge as in (A). (D) Survival of mice challenged with Pam3CSK4 (1 mg/kg) with or without insulin. (E) Survival of mice after challenge with LPS (400 μg/kg) and insulin and subsequent control or glucose injection. (A - E) Pooled data from 2 independent experiments. Error bars, mean ± SD. Statistically significant differences were determined by ANOVA and Tukey's multiple comparisons test; * P ≤ 0.05.
LPS impairs insulin clearance and enhances insulin receptor signaling
Various mechanisms could explain how LPS and insulin potentiated hypoglycemic shock. Insulin had no effect on LPS induced IL-6 or TNF secretion (Fig. 4A, B), suggesting that LPS acts upstream of or in parallel to insulin rather than insulin potentiating LPS signaling. LPS alone had a minimal effect on blood glucose, suggesting that an additive effect on glucose consumption by LPS and insulin signaling pathways cannot fully account for the extreme hypoglycemia (Fig. 4C). AKT is a node downstream of insulin signaling as well as multiple immune signaling pathways; however, LPS alone did not induce appreciable AKT phosphorylation in vivo (Fig. 4C), arguing against the synergistic intersection of immunological and insulin signaling pathways at the level of AKT. However, LPS amplified both insulin driven AKT and insulin receptor phosphorylation (Fig. 4C-H), indicating that the effect of LPS occurs upstream of AKT, either at or upstream of the insulin receptor. We therefore examined plasma insulin concentrations using an ELISA specific for the exogenous human insulin that we injected (this ELISA does not detect endogenous murine insulin). Insulin concentrations peaked at approximately 20 minutes in mice treated with insulin (Fig. S1B). At one hour, we observed that mice challenged with LPS and insulin together had significantly elevated plasma insulin compared to mice treated with insulin alone (Fig. S1C, Fig. 3I).
Fig. 4. LPS impairs insulin clearance and amplifies insulin receptor signaling.
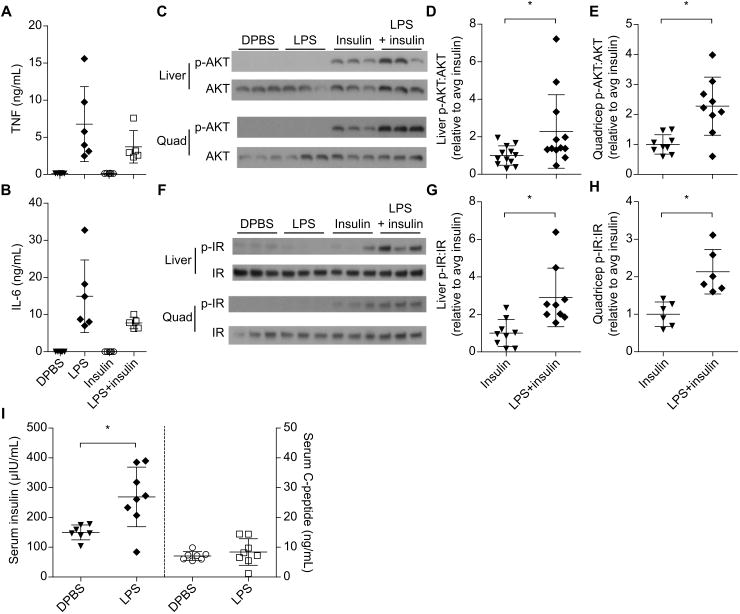
(A and B) Plasma TNF and IL-6 concentrations from mice 1 h after DBPS injection or challenge with LPS (400 μg/kg), insulin, or both. Data pooled from 2 experiments. (C - H) AKT and insulin receptor phosphorylation observed at 1 h by Western blot in mice challenged as in (A). Western blots are representative of 3 or more (liver and quadricep AKT, liver IR) or 2 (quad IR) independent experiments. Densitometry data are pooled from 3 or more (D, E, G) or 2 (H) experiments. (I) Plasma insulin and C-peptide concentrations at 1 h in mice challenged with insulin and C-peptide with or without LPS. Pooled data from 2 experiments. Error bars, mean ± SD. Statistically significant differences were determined by a 2-tailed unpaired t-test; * P ≤ 0.05.
Serum insulin concentration reflects both the rate of insulin absorption into the blood from the peritoneal cavity and the rate of clearance by tissues. Absorption of Evan's blue dye was unaffected by LPS (Fig. S1D), suggesting that LPS does not generally enhance fluid absorption from the peritoneal cavity into the blood. To further study insulin absorption and clearance, we compared serum concentrations of co-injected insulin and C-peptide (21) with or without LPS. The majority of insulin is cleared after insulin receptor binding, internalization, and subsequent intracellular degradation, much of which occurs in the liver (16). In contrast, C-peptide, a fragment of pro-insulin that is secreted with mature insulin, is cleared in an insulin receptor-independent manner primarily in the kidneys (23). Unlike its effect on plasma insulin, LPS did not affect concentrations of co-injected C-peptide (Fig. 4I). These data suggest that LPS impairs clearance of insulin semi-specifically, an effect that is linked to enhanced insulin signaling in vivo (17).
Differential contribution of caspase-11, TLR4, and complement
The inflammatory sensors and downstream mediators that are detrimental in the synergy between medium dose 400 μg/kg LPS and insulin may be the same or different from those that cause mortality after challenge with 54,000 μg/kg LPS alone. Recall that we found caspase-11 and TLR4 fully account for mortality in poly(I:C) primed mice challenged with high dose 54,000 μg/kg LPS without insulin (Fig. 1). We next studied whether both of these sensors would be similarly detrimental during co-challenge with LPS and insulin. First, as a control, we determined that Casp11−/−, Tlr4−/−, and Casp11−/−Tlr4−/− mice had normal insulin sensitivity in the absence of LPS challenge (Fig. 5A). Next, we examined the resistance of these mice to several low doses of LPS combined with insulin. Wild type mice succumb to even very low dose 2.5μg/kg LPS challenge when insulin is included (Fig. 5B). Tlr4−/− mice exhibited intermediate resistance to challenge with insulin and 2.5μg/kg LPS (Fig. 5B), but uniformly succumbed to challenge with insulin and 40 μg/kg LPS (Fig. 5C). Whereas Casp11−/− mice survived challenge with insulin and 2.5 μg/kg LPS (Fig. 5B), they were nevertheless completely susceptible to challenge with insulin and 40 μg/kg LPS (Fig. 5C). Further, liver AKT and insulin receptor phosphorylation were significantly reduced in Casp11−/− mice challenged with insulin and LPS 2.5μg/kg, but not in Tlr4−/− mice (Fig. 5D-G), again indicating that LPS acts at or upstream of the insulin receptor. Therefore, caspase-11 and TLR4 both can potentiate insulin signaling, with caspase-11 being more sensitive in this model.
Fig. 5. LPS enhances insulin signaling via caspase-11, TLR4, and complement.
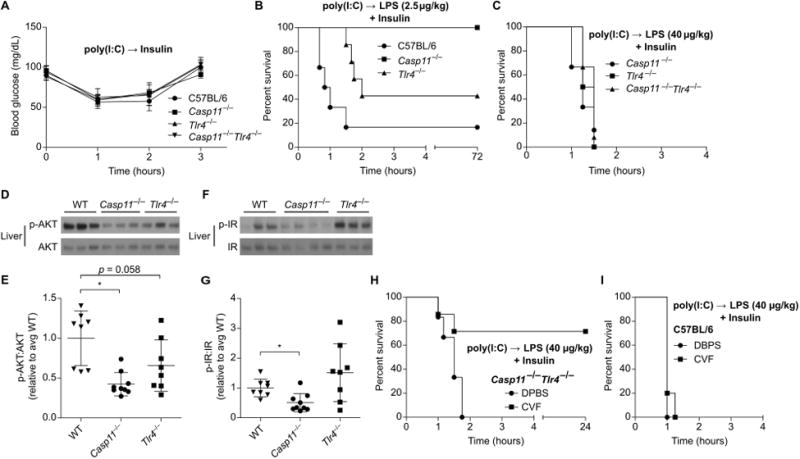
(A) Blood glucose concentrations over time in mice treated with insulin. Pooled data from 2 experiments. (B and C) Survival of mice after challenge with insulin and LPS (B, 2.5 μg/kg; C, 40 μg/kg). Pooled data from 2 experiments. (D - G) Phosphorylation of liver AKT (D, E) and insulin receptor (F, G) determined by Western blot 1 h after challenge as in (B). Densitometry data are pooled from 3 experiments. (H, I) Survival of control or CVF treated mice after challenge with LPS (40 μg/kg) and insulin. Pooled data from 2 experiments. Error bars, mean ± SD. Statistically significant differences were determined by a 2-tailed unpaired t-test; * P ≤ 0.05.
We hypothesized that Casp11−/−Tlr4−/− mice would fully resist challenge with insulin and the 40 μg/kg LPS dose that was lethal to Casp11−/− (Tlr4+/+) mice. Unexpectedly, Casp11−/−Tlr4−/− mice were also uniformly susceptible to this challenge (Fig. 5C). This result is in stark contrast to the complete resistance of Casp11−/−Tlr4−/− mice to LPS 54,000 μg/kg challenge without insulin (Fig. 1),indicating that lower dose LPS triggers additional immune pathways capable of enhancing insulin signaling that are not lethal on their own.
LPS is known to activate the complement cascade with a potency determined by the O-antigen structure (4). LPS from E. coli serotype O111:B4 is suggested to have weak but detectable complement activating properties (4). We therefore sought to block this third pathway in Casp11−/−Tlr4−/− mice using cobra venom factor, which depletes complement in vivo. Indeed, while cobra venom factor did not protect WT mice, it rescued Casp11−/−Tlr4−/− mice from challenge with insulin and 400 μg/kg LPS (Fig. 5H, I). Altogether, these data suggest that caspase-11, TLR4, and the complement cascade act in parallel as three independent LPS response systems capable of enhancing insulin signaling.
Cyclooxygenase and PGE2 enhance insulin signaling
Various cyclooxygenase (COX) dependent prostaglandins have been shown to modulate signaling by hormones involved in glucose homeostasis, such as insulin (24-26) and glucagon (27). We previously observed that inhibition of COX signaling rescues mice challenged with low dose LPS in OptiMEM (5), a phenotype we now know to be dependent on insulin in the OptiMEM. Consistently, treatment of mice with in domethacin, a COX inhibitor, prevented mortality after LPS plus insulin challenge (Fig. 6A), abrogating extreme hypothermia and hypoglycemia (Fig. 6B, C), inhibiting hyper-phosphorylation of AKT and insulin receptor (Fig. 6D-G), and improving insulin clearance (Fig. 6H). Challenge with insulin and a higher dose of 400 μg/kg LPS overwhelmed the protection afforded by COX inhibition, although mortality was significantly delayed (Fig. S2A), suggesting either incomplete inhibition or implicating additional, lower potency mediators acting in parallel.
Fig. 6. Eicosanoid signaling acts downstream of LPS to amplify insulin activity.
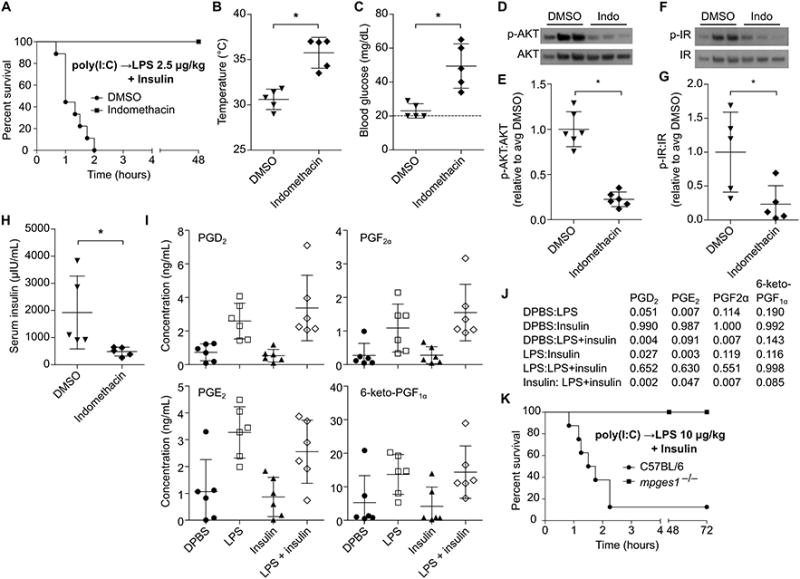
(A) Survival of DMSO or indomethacin treated mice after challenge with LPS (2.5 μg/kg) and insulin. (B and C) Core temperatures (B) and blood glucose concentrations (C) of mice 1 h after challenge as described in (A). (D - G) Phosphorylation of liver AKT (D, F) and insulin receptor (E, G) determined by Western blot 1 h after challenge as described in (A). (H) Plasma insulin concentrations of mice 1 h after treatment as in (B). (I) Plasma prostaglandin concentrations in mice 1h after challenge with DBPS, LPS (400 μg/kg), insulin, or LPS and insulin. (J) P values for the indicated comparisons of data from (J) as determined by ANOVA and Tukey's multiple comparisons test. (K) Survival of mice challenged with insulin and LPS (10 μg/kg). Data are pooled from at 3 (A, K) or 2 experiments (B – I). Error bars, mean ± SD. Statistically significant differences were determined by a 2-tailed unpaired t-test; * P ≤ 0.05.
TLR4 and complement are each known to activate COX signaling within an hour of LPS exposure (28, 29), and caspase-11 is likely to as well by analogy to caspase-1 (30). Indeed, profiling of plasma lipid signaling molecules revealed that LPS induced production of the COX metabolites prostaglandin E2 (PGE2), PGD2, PGF2α, and 6-keto-PGF1α (Fig. 6I, J) at 1h. Insulin had no effect on prostaglandin production (Fig. 6I, J), further supporting that LPS acts upstream of insulin rather than vice versa. The magnitude of LPS induced eicosanoid production was not sufficient to affect hematocrit (Fig. S2B), which is in contrast to the outcome of rapid systemic caspase-1 activation, which causes hypovolemic shock (30). Amongst prostaglandins, PGE2 has been shown to enhance insulin signaling in hepatocytes (24), skeletal muscle (25), and adipocytes (26), all important regulators of insulin induced glucose clearance. Mice deficient in Mpges1, a microsomal prostaglandin E synthase largely responsible for PGE2 production in response to LPS (20), survived challenge with insulin 10 μg/kg LPS (Fig. 6K). These Mpges1−/−and corresponding WT control mice were maintained in a different mouse facility where WT mice resisted 2.5 μg/kg challenge, but were sensitive to a slight dose escalation up to 10 μg/kg. This suggests that the microbiota or other housing factors can have a subtle effect that changes the effective dosing in the range of a 4-fold effect, consistent with variable penetrance of LPS lethality between labs (Fig. 1 and (6, 14)). Together, these data demonstrate that COX and, especially, its downstream metabolite PGE2 mediate LPS potentiation of insulin shock.
Septic mice exhibit impaired insulin clearance
In order to translate our findings to an animal model of sepsis, we studied insulin signaling and clearance in mice infected with the Gram-negative bacterial pathogen Salmonella Typhimurium. Five days post infection, at which point mice exhibit moderate clinical signs of infection such as lethargy, piloerection, and ocular discharge (but are not moribund), we injected mice with insulin and examined changes in blood glucose, insulin receptor and AKT phosphorylation, and insulin clearance. Compared to control mice, infected mice were moderately hypoglycemic prior to insulin injection; however, infected mice exhibited a blunted reduction in blood glucose in response to insulin (Fig. 7A), suggesting that these mice were insulin resistant similar to septic patients in the ICU (31). Indeed, infected mice exhibited similar levels of AKT phosphorylation to uninfected control mice (Fig 7B, D); however, infected mice had markedly enhanced insulin receptor phosphorylation (Fig. 7C, E), consistent with insulin resistance mechanisms operating downstream of the insulin receptor (16, 32). This high insulin receptor phosphorylation correlated with high serum insulin (Fig. 7F), indicating that infected mice have impaired insulin clearance. Altogether, these data demonstrate that during infection, insulin clearance defects that prolong and enhance insulin receptor activity can coexist with insulin resistance, which limits the rate and magnitude of insulin's effects. In LPS challenge models (Fig. 2-6), insulin resistance did not have time to develop, whereas in an infectious model insulin resistance did occur through a mechanism distinct from impaired insulin clearance (Fig. 7).
Fig. 7. Septic mice exhibit simultaneous insulin resistance and impaired insulin clearance.
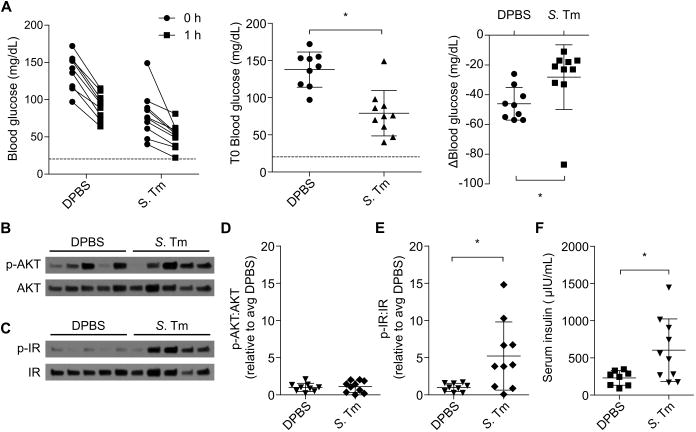
Mice were injected with PBS or S. Typhimurium. After 5 days they were challenged with insulin. (A) Change in blood glucose of control and infected mice before and 1h after insulin challenge. (B – E) Liver AKT and insulin receptor phosphorylation observed 1 h after insulin challenge in mice by Western blot. Western blots are representative of 2 independent experiments. Densitometry data are pooled from 2 experiments. (F) Plasma insulin concentrations 1 h after insulin challenge. Pooled data from 2 experiments. Error bars, mean ± SD. Statistically significant differences were determined by a 2-tailed unpaired t-test; * P ≤ 0.05.
Discussion
Herein, we described our serendipitous discovery that LPS enhances the effects of co-delivered insulin, precipitating hypoglycemic shock within 1-2 h of challenge. This may have clinical relevance to the use of insulin to treat septic patients.
Critically ill patients typically present with hyperglycemia as a result of stress hormone and inflammatory cytokine signaling, which induces peripheral insulin resistance over time (31). Mechanistically, this occurs as LPS and other pathogen signatures induce inflammatory mediators such as TNF and IL-6, which drive inhibitory serine phosphorylation of the insulin receptor downstream adaptor protein IRS-1, inhibit IRS-1 tyrosine phosphorylation, and perturb blood flow to insulin responsive organs (33). This insulin resistance is seemingly at odds with the insulin hypersensitivity observed in mice treated with LPS and insulin. We propose that the insulin clearance defect that we observe exists in critically ill patients, but was previously unappreciated because it is obscured by the simultaneous and dominant effect of peripheral insulin resistance. Indeed, mice infected with S. Typhimurium simultaneously exhibited bothinsulin resistance and impaired insulin clearance. The mechanisms by which inflammatory signals drive insulin resistance may be distinct from those that impair insulin clearance. Insulin resistance mechanisms act primarily at the post-receptor level, leaving insulin receptor surface expression, auto-phosphorylation, and internalization largely unaffected (16, 32). Consistent with this, infected mice exhibited elevated serum insulin and insulin receptor phosphorylation after insulin treatment, yet nevertheless had lower than expected AKT phosphorylation and a blunted change in blood glucose.
Treating hyperglycemia with insulin improves the survival of patients in intensive care units (ICU); however, extreme controversy has surrounded how stringently blood glucose should be controlled (34-40). In 2002, Van den Berghe and colleagues demonstrated reduced mortality in surgical ICU patients receiving insulin to maintain blood glucose between 80 and 110 mg/dL (intensive insulin therapy; IIT) compared with those receiving conventional insulin therapy (CIT) targeting 180-200 mg/dL (34). This resulted in a significant shift in clinical practice: whereas marked hyperglycemia was commonly tolerated in ICU patients before the Van den Berghe study, after it there was a major push to control blood glucose as a therapy for critically ill patients (41). However, in a later study these investigators observed equal survival rates in the medical ICU between patients receiving IIT and CIT, although they argued that IIT reduced morbidity (35).
The NICE-SUGAR clinical trial, which enrolled 6104 patients, investigated whether the benefit of IIT over CIT (targeting 144-180 mg/dL) would be reproduced in a large multicenter trial (38). Instead, IIT increased mortality. Moreover, whereas only 0.5% of patients on CIT experienced extreme hypoglycemia (<40 mg/dL), this condition occurred in 6.8% of patients receiving IIT (38); 17.6% of CIT patients experienced moderate hypoglycemia (41-70 mg/dL) compared to 74.2% of IIT patients (39). Subsequent analysis identified both moderate and extreme hypoglycemia as risk factors for death (39), which is in line with multiple concurrent studies and meta analyses (36-40). Amongst subgroups of ICU patients, those with sepsis are particularly prone to hypoglycemia during IIT (42). Why sepsis predisposes to hypoglycemia remains unknown.
In our study, LPS impaired insulin clearance and augmented insulin signaling in both liver and skeletal muscle – tissues critical to the effect of insulin to reduce blood sugar – via caspase-11, TLR4, and complement. We propose that signaling from these sensors converges upon common mediators, especially eicosanoids, which enhance insulin action. Pam3CSK4 also lethally potentiated insulin activity, presumably acting via TLR2; whether Pam3CSK4 acts via eicosanoids and perturbs insulin clearance remains to be determined. S. Typhimurium infection similarly impaired insulin clearance while simultaneously inducing insulin resistance. In the setting of a hyperinsulinemic-euglycemic clamp, it has been suggested that LPS can impair insulin clearance (43, 44); however, ours is the first study to investigate the underlying mechanisms, and more importantly its potentially dire consequences. Further, we propose that an insulin clearance defect can paradoxically coexist with insulin resistance.
COX dependent eicosanoid production is known to occur rapidly downstream of TLR4 and complement receptor activation (28, 29), with kinetics that are consistent with the 1-2 hour time frame of hypoglycemic shock observed here in mice treated with insulin and LPS. Furthermore, since activation of caspase-1 induces calcium flux that drives eicosanoid production (30), it is likely that this mechanism is conserved for caspase-11. Thus, TLR4, complement, and caspase-11 should each contribute to the net prostaglandin production we observed to occur during LPS challenge. Although prostaglandins are critical mediators of the effect upon insulin, it is important to note that the protective effect of COX blockade can be overcome at higher doses of LPS, suggesting non-prostaglandin signaling pathways also act in this model.
Insulin clearance defects during S. Typhimurium infection likely act through multiple TLRs (including TLR4, TLR2, and TLR9) and complement. Although caspase-1 and -11 are efficiently evaded by S. Typhimurium (8, 45), these may become aberrantly activated in the setting of sepsis in an immunopathologic manner.
There are multiple possible mechanisms by which inflammatory signaling pathways could impair insulin clearance. A first possible mechanism is that inflammatory mediators affect the activity of the insulin degrading enzyme (IDE) (16), which is sensitive to reactive oxygen and nitrogen species (46, 47). Indeed, administration of LPS to microglial cells in vitro abrogates IDE activity (46). Further, pharmacological inhibition of IDE in vivo impairs insulin clearance and simultaneously enhances insulin sensitivity (17), similar to what we observe here. ROS also enhances insulin signaling in vitro and in vivo by inactivating protein tyrosine phosphatases that dephosphorylate the insulin receptor and downstream signaling molecules (48, 49).
A second possible mechanism to consider is the rate of trafficking or acidification of the insulin-insulin receptor containing vacuole. Inhibition of endosomal acidification limits dissociation of insulin from the insulin receptor and subsequent insulin degradation (18); in vitro and in vivo, pharmacological inhibition of endosomal acidification enhances insulin signaling activity (19). Whether LPS impairs acidification of endosomes containing insulin: insulin receptor complexes remains to be determined, but this could conceivably occur via ROS dependent mechanisms or inhibition of PKA signaling (50).
A third mechanism could operate on the level of hepatic perfusion. Reduced blood flow through the liver should limit insulin trafficking through this major site of insulin clearance. Such a mechanism has broad implications beyond insulin as it would affect the activity and clearance of other liver-metabolized pharmacological agents administered to septic patients.
The elevated insulin concentrations we observe no doubt lead to increased glucose disposal and underlie the extreme hypoglycemia observed in mice challenged with LPS and insulin, which alone could explain the lethal hypoglycemia that we observe. However, it remains possible that LPS could simultaneously enhance insulin action via additional mechanisms. For example, several groups have demonstrated that PGE2 augments insulin signaling in key insulin responsive cell types: hepatocytes (24), skeletal muscle (25), and adipocytes (26). Furthermore, PGE2 inhibits glucagon signaling in hepatocytes by blocking cAMP generation (27). This would tip the balance towards glucose uptake and away from restoration of blood glucose by glucagon.
LPS is well known to affect glucose metabolism independently of its effects on insulin signaling. For example, LPS challenge causes transient hyperglycemia, which is associated with liver and muscle glycogen depletion (51), followed by hypoglycemia and impaired hepatocyte glucose production (52). Furthermore, despite eventually causing insulin resistance, LPS alone enhances glucose disposal in rats at early time points (53). Low dose LPS alone mildly affected the blood glucose of mice in our studies, suggesting such mechanisms may contribute somewhat to the extreme hypoglycemia we observed in mice challenged with LPS and insulin.
Our data in animal studies raise the possibility that subgroups of ICU patients, particularly those with sepsis, suffer from an impaired ability to clear insulin. The co-existence of insulin resistance and an insulin clearance defect would confound the clinical use of insulin therapy in critically ill patients. Insulin resistance and initially elevated blood glucose will buffer the effect of exogenous insulin, but a simultaneous defect in insulin clearance could steadily drive the blood concentration of insulin higher. This could be exaggerated by a loss of homeostatic responses that normally maintain blood glucose in a tight range. For example, poor perfusion of the pancreas could lead to a lag time in glucagon responses to hypoglycemia, while inflammatory effects on the liver lead to prolonged insulin circulation. There is considerable variability amongst insulin infusion protocols used in ICUs, resulting in widely differing recommendations for dosing and variable glucose control (54). Insulin administration protocols that signal a caregiver to increase the dose of insulin before already administered insulin has had time to act (where the protocol assumes rapid insulin clearance) will increase the risk of extreme hypoglycemia. This effect could be overlaid upon other mechanisms that may contribute to hypoglycemia in septic patients, including inadequate nutrition, glycogen depletion (55), and enhanced insulin-independent glucose utilization (56). Optimized protocols should circumvent these problems by taking into account the possibility of an underlying insulin clearance defect (54).
Understanding how insulin clearance kinetics differ between sub-populations of critically ill patients may reveal that insulin administration protocols need to be modified for different patients depending upon the underlying etiology of their illness and how it affects the kinetics of insulin clearance. This could significantly impact the management of blood glucose levels in critically ill patients.
Supplementary Material
Acknowledgments
J.A.H and E.A.M. designed the study, analyzed all data, and prepared the manuscript. J.A.H performed or contributed to all experiments. M.L.E. and F.B.L. performed experiments and provided critical technical expertise. L.T., B.H.K., B.A.C., and D.C.Z. provided critical reagents and intellectual and technical contributions. The authors thank V. Dixit and S. Akira for sharing key mouse strains (Casp11−/− mice were provided under a materials transfer agreement with Genentech). We thank D. Mao, T. Atherton, and D. Trinh for managing mouse colonies. The authors have no competing financial interests to disclose. The data presented here are tabulated in the main paper and the supplementary materials.
This work was supported by NIH grants AI007273 (J.A.H.), AI097518 (E.A.M.), AI057141 (E.A.M.), AR061491 (B.H.K.), and Z01 ES025034 (D.C.Z.).
References and Notes
- 1.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 2.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 3.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 4.Zhao L, Ohtaki Y, Yamaguchi K, Matsushita M, Fujita T, Yokochi T, Takada H, Endo Y. LPS-induced platelet response and rapid shock in mice: contribution of O-antigen region of LPS and involvement of the lectin pathway of the complement system. Blood. 2002;100:3233–3239. doi: 10.1182/blood-2002-01-0252. [DOI] [PubMed] [Google Scholar]
- 5.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 7.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 8.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JPY, Coers J, Aderem A, Buxbaum JD, Miao EA. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host Microbe. 2015;18:320–332. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 12.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 13.Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem. 2002;277:41624–41630. doi: 10.1074/jbc.M207852200. [DOI] [PubMed] [Google Scholar]
- 14.Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity. 2015;43:923–932. doi: 10.1016/j.immuni.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 16.Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19:608–624. doi: 10.1210/edrv.19.5.0349. [DOI] [PubMed] [Google Scholar]
- 17.Maianti JP, McFedries A, Foda ZH, Kleiner RE, Du XQ, Leissring MA, Tang WJ, Charron MJ, Seeliger MA, Saghatelian A, Liu DR. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature. 2014;511:94–98. doi: 10.1038/nature13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doherty JJ, Kay DG, Lai WH, Posner BI, Bergeron JJ. Selective degradation of insulin within rat liver endosomes. J Cell Biol. 1990;110:35–42. doi: 10.1083/jcb.110.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bevan AP, Krook A, Tikerpae J, Seabright PJ, Siddle K, Smith GD. Chloroquine extends the lifetime of the activated insulin receptor complex in endosomes. J Biol Chem. 1997;272:26833–26840. doi: 10.1074/jbc.272.43.26833. [DOI] [PubMed] [Google Scholar]
- 20.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 22.Zha W, Edin ML, Vendrov KC, Schuck RN, Lih FB, Jat JL, Bradbury JA, DeGraff LM, Hua K, Tomer KB, Falck JR, Zeldin DC, Lee CR. Functional characterization of cytochrome P450-derived epoxyeicosatrienoic acids in adipogenesis and obesity. J Lipid Res. 2014;55:2124–2136. doi: 10.1194/jlr.M053199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zavaroni I, Deferrari G, Lugari R, Bonora E, Garibotto G, Dall'Aglio E, Robaudo C, Gnudi A. Renal metabolism of C-peptide in man. J Clin Endocrinol Metab. 1987;65:494–498. doi: 10.1210/jcem-65-3-494. [DOI] [PubMed] [Google Scholar]
- 24.Takasuga S, Katada T, Ui M, Hazeki O. Enhancement by adenosine of insulin-induced activation of phosphoinositide 3-kinase and protein kinase B in rat adipocytes. J Biol Chem. 1999;274:19545–19550. doi: 10.1074/jbc.274.28.19545. [DOI] [PubMed] [Google Scholar]
- 25.Nesher R, Karl IE, Kipnis DM. Dissociation of effects of insulin and contraction on glucose transport in rat epitrochlearis muscle. Am J Physiol. 1985;249:C226–32. doi: 10.1152/ajpcell.1985.249.3.C226. [DOI] [PubMed] [Google Scholar]
- 26.Olefsky JM. Interaction between insulin receptors and glucose transport: effect of prostaglandin E2. Biochem Biophys Res Commun. 1977;75:271–276. doi: 10.1016/0006-291x(77)91039-7. [DOI] [PubMed] [Google Scholar]
- 27.Brass EP, Alford CE, Garrity MJ. Inhibition of glucagon-stimulated cAMP accumulation and fatty acid oxidation by E-series prostaglandins in isolated rat hepatocytes. Biochim Biophys Acta. 1987;930:122–126. doi: 10.1016/0167-4889(87)90164-9. [DOI] [PubMed] [Google Scholar]
- 28.Perlik V, Li Z, Goorha S, Ballou LR, Blatteis CM. LPS-activated complement, not LPS per se, triggers the early release of PGE2 by Kupffer cells. Am J Physiol Regul Integr Comp Physiol. 2005;289:R332–R339. doi: 10.1152/ajpregu.00567.2004. [DOI] [PubMed] [Google Scholar]
- 29.Casteleijn E, Kuiper J, Van Rooij HC, Kamps JA, Koster JF, Van Berkel TJ. Endotoxin stimulates glycogenolysis in the liver by means of intercellular communication. J Biol Chem. 1988;263:6953–6955. [PubMed] [Google Scholar]
- 30.Moltke Van J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, Van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson BT. Glucose control in sepsis. Clin Chest Med. 2008;29:713–20x. doi: 10.1016/j.ccm.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 32.Draznin B. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: the two sides of a coin. Diabetes. 2006;55:2392–2397. doi: 10.2337/db06-0391. [DOI] [PubMed] [Google Scholar]
- 33.Marette A. Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr Opin Clin Nutr Metab Care. 2002;5:377–383. doi: 10.1097/00075197-200207000-00005. [DOI] [PubMed] [Google Scholar]
- 34.van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- 35.Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Van Wijngaerden E, Bobbaers H, Bouillon R. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449–461. doi: 10.1056/NEJMoa052521. [DOI] [PubMed] [Google Scholar]
- 36.Wiener RS, Wiener DC, Larson RJ. Benefits and risks of tight glucose control in critically ill adults: a meta-analysis. JAMA. 2008;300:933–944. doi: 10.1001/jama.300.8.933. [DOI] [PubMed] [Google Scholar]
- 37.Brunkhorst FM, Engel C, Bloos F, Meier-Hellmann A, Ragaller M, Weiler N, Moerer O, Gruendling M, Oppert M, Grond S, Olthoff D, Jaschinski U, John S, Rossaint R, Welte T, Schaefer M, Kern P, Kuhnt E, Kiehntopf M, Hartog C, Natanson C, Loeffler M, Reinhart K German Competence Network Sepsis (SepNet) Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358:125–139. doi: 10.1056/NEJMoa070716. [DOI] [PubMed] [Google Scholar]
- 38.NICE-SUGAR Study Investigators. Finfer S, Chittock DR, Su SYS, Blair D, Foster D, Dhingra V, Bellomo R, Cook D, Dodek P, Henderson WR, Hébert PC, Heritier S, Heyland DK, McArthur C, McDonald E, Mitchell I, Myburgh JA, Norton R, Potter J, Robinson BG, Ronco JJ. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283–1297. doi: 10.1056/NEJMoa0810625. [DOI] [PubMed] [Google Scholar]
- 39.NICE-SUGAR Study Investigators. Liu B, Chittock DR, Norton R, Myburgh JA, McArthur C, Mitchell I, Foster D, Dhingra V, Henderson WR, Ronco JJ, Bellomo R, Cook D, McDonald E, Dodek P, Hébert PC, Heyland DK, Robinson BG. Hypoglycemia and risk of death in critically ill patients. N Engl J Med. 2012;367:1108–1118. doi: 10.1056/NEJMoa1204942. [DOI] [PubMed] [Google Scholar]
- 40.Park S, Kim DG, Suh GY, Kang JG, Ju YS, Lee YJ, Park JY, Lee SW, Jung KS. Mild hypoglycemia is independently associated with increased risk of mortality in patients with sepsis: a 3-year retrospective observational study. Crit Care. 2012;16:R189. doi: 10.1186/cc11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malhotra A. Intensive insulin in intensive care. N Engl J Med. 2006;354:516–518. doi: 10.1056/NEJMe058304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krinsley JS, Grover A. Severe hypoglycemia in critically ill patients: risk factors and outcomes. Crit Care Med. 2007;35:2262–2267. doi: 10.1097/01.CCM.0000282073.98414.4B. [DOI] [PubMed] [Google Scholar]
- 43.Mulligan KX, Morris RT, Otero YF, Wasserman DH, McGuinness OP. Disassociation of muscle insulin signaling and insulin-stimulated glucose uptake during endotoxemia. PLoS ONE. 2012;7:e30160. doi: 10.1371/journal.pone.0030160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otero YF, Mulligan KX, Barnes TM, Ford EA, Malabanan CM, Zong H, Pessin JE, Wasserman DH, McGuinness OP. Enhanced Glucose Transport, but not Phosphorylation Capacity, Ameliorates Lipopolysaccharide-Induced Impairments in Insulin-Stimulated Muscle Glucose Uptake. Shock. 2016;45:677–685. doi: 10.1097/SHK.0000000000000550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ralat LA, Ren M, Schilling AB, Tang WJ. Protective role of Cys-178 against the inactivation and oligomerization of human insulin-degrading enzyme by oxidation and nitrosylation. J Biol Chem. 2009;284:34005–34018. doi: 10.1074/jbc.M109.030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malito E, Ralat LA, Manolopoulou M, Tsay JL, Wadlington NL, Tang WJ. Molecular bases for the recognition of short peptide substrates and cysteine-directed modifications of human insulin-degrading enzyme. Biochemistry. 2008;47:12822–12834. doi: 10.1021/bi801192h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, Stepto N, Wu B, Mitchell CA, Tonks NK, Watt MJ, Febbraio MA, Crack PJ, Andrikopoulos S, Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7:1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breton S, Brown D. Regulation of luminal acidification by the V-ATPase. Physiology (Bethesda) 2013;28:318–329. doi: 10.1152/physiol.00007.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Virkamäki A, Yki-Järvinen H. Mechanisms of insulin resistance during acute endotoxemia. Endocrinology. 1994;134:2072–2078. doi: 10.1210/endo.134.5.8156907. [DOI] [PubMed] [Google Scholar]
- 52.Raetzsch CF, Brooks NL, Alderman JM, Moore KS, Hosick PA, Klebanov S, Akira S, Bear JE, Baldwin AS, Mackman N, Combs TP. Lipopolysaccharide inhibition of glucose production through the Toll-like receptor-4, myeloid differentiation factor 88, and nuclear factor kappa b pathway. Hepatology. 2009;50:592–600. doi: 10.1002/hep.22999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lang CH, Spolarics Z, Ottlakan A, Spitzer JJ. Effect of high-dose endotoxin on glucose production and utilization. Metab Clin Exp. 1993;42:1351–1358. doi: 10.1016/0026-0495(93)90137-d. [DOI] [PubMed] [Google Scholar]
- 54.Wilson M, Weinreb J, Hoo GWS. Intensive insulin therapy in critical care: a review of 12 protocols. Diabetes Care. 2007;30:1005–1011. doi: 10.2337/dc06-1964. [DOI] [PubMed] [Google Scholar]
- 55.Liu MS, Kang GF. Liver glycogen metabolism in endotoxin shock. I. Endotoxin administration decreases glycogen synthase activities in dog livers. Biochem Med Metab Biol. 1987;37:61–72. doi: 10.1016/0885-4505(87)90010-7. [DOI] [PubMed] [Google Scholar]
- 56.Lang CH, Dobrescu C. Sepsis-induced increases in glucose uptake by macrophage-rich tissues persist during hypoglycemia. Metab Clin Exp. 1991;40:585–593. doi: 10.1016/0026-0495(91)90048-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.